

Abstract
Accumulating evidences have indicated that lncRNAs play an important role in various human complex diseases. However, known disease-related lncRNAs are still comparatively small in number, and experimental identification is time-consuming and labor-intensive. Therefore, developing a useful computational method for inferring potential associations between lncRNAs and diseases has become a hot topic, which can significantly help people to explore complex human diseases at the molecular level and effectively advance the quality of disease diagnostics, therapy, prognosis and prevention. In this paper, we propose a novel prediction of lncRNA-disease associations via lncRNA-disease-gene tripartite graph (TPGLDA), which integrates gene-disease associations with lncRNA-disease associations. Compared to previous studies, TPGLDA can be used to better delineate the heterogeneity of coding-non-coding genes-disease association and can effectively identify potential lncRNA-disease associations. After implementing the leave-one-out cross validation, TPGLDA achieves an AUC value of 93.9% which demonstrates its good predictive performance. Moreover, the top 5 predicted rankings of lung cancer, hepatocellular carcinoma and ovarian cancer are manually confirmed by different relevant databases and literatures, affording convincing evidence of the good performance as well as potential value of TPGLDA in identifying potential lncRNA-disease associations. Matlab and R codes of TPGLDA can be found at following: https://github.com/USTC-HIlab/TPGLDA.
Introduction
Long non-coding RNAs (lncRNAs) are a new class of transcripts, with the length longer than 200nt1–3, which have been implicated in a number of normal physiological processes at every stage of life, from embryonic development and cellular cell fate determination to physiological homoeostasis of entire organisms4. Accumulating studies have indicated that a large quantity of lncRNAs are critical in many important biological processes such as chromatin modification, transcriptional and post-transcriptional regulation, genomic splicing, differentiation, immune responses, cell cycle control and so on3,5,6. Especially, it has been demonstrated that a large number of lncRNAs are involved in numerous complex human diseases3,4, such as neurological disorders7, coronary artery diseases8, cardiovascular diseases9, and various cancers10. Accordingly, inferring potential associations between lncRNAs and diseases can help us understand the pathogenesis of complex diseases at the molecular level and benefit biomarker identification for disease diagnosis, therapy, prognosis and monitoring5.
Up to now, a large amount of lncRNA-related biological data has been distributed in different public databases11–13 and only few associations between lncRNAs and diseases have been reported. Collecting and integrating these data from a great number of literatures and databases is costly. Moreover, many biological experiments are time-consuming and expensive. Accordingly, researchers have started to focus on developing computational prediction approaches based on the existing datasets, which can quantify the associations’ probability between lncRNAs and diseases, and the most promising lncRNA-disease associations are used for further biological experimental validation. In this case, the time and cost of biological experiments can be effectively reduced14.
In recent years, some computational models have been proposed to identify potential associations between lncRNAs and diseases. For example, based on important assumption that similar diseases are often associated with functionally similar lncRNAs5, Chen and Yan propose a computational approach of LRLSLDA to identify potential disease–related lncRNAs. LRLSLDA is a novel semi-supervised learning method in Laplacian regularized least squares framework. Moreover, this method does not require negative samples and can produce reliable results based on lncRNA expression profile and known lncRNA-disease associations. Subsequently, based on the finding that functionally related genes are often associated with phenotypically similar diseases15,16, Sun et al.17. Construct an lncRNA-lncRNA functional similarity network. Then, they develop a global network-based computational approach, RWRlncD, to identify potential lncRNA–disease associations by integrating disease similarity network, lncRNA functional similarity network and experimentally verified lncRNA-disease associations. Based on the same assumption, Ganegoda et al.18. Further propose a kernel based random walk with restart in heterogeneous network model (KRWRH), which incorporates with lincRNA tissue specific information, disease phenotype information and experimentally validated disease-lincRNA associations. KRWRH uses Gaussian interaction profile kernel to calculate the similarities of diseases and lincRNAs, and random walk with restart method is utilized for final prediction. The good experimental results highlight the importance and effectiveness of inferring potential disease-lincRNA associations using different biological information18.
Despite the success achieved by aforementioned methods, another important factor contributing to infer potential lncRNA-disease associations lies in the fact that coding and non-coding genes are often cooperated in human diseases, which has been demonstrated in many previous studies19–22 For example, Sahu et al.23 demonstrate that coding gene-TAF1D and lncRNA-SNHG1 are highly co-expressed in neuroblastoma. At the same time, rich information about gene-disease associations is available in database such as DisGeNET24 and PsyGeNET25. Therefore, if effectively used, such information may be of great help to infer potential associations between lncRNAs and diseases. Recently, Yang et al.22 conduct a pioneer study in which the authors integrate coding gene-disease associations and propose a propagation algorithm to infer potential lncRNA-disease associations based on a bipartite graph of coding-non-coding genes-disease. The authors show integrating coding gene-disease associations with lncRNA-disease associations can significantly enhance its prediction performance. Despite its low complexity and effectiveness, the bipartite graph model used in this study treats coding and non-coding genes without distinction and therefore cannot fully account for the heterogeneity of coding-non-coding genes-disease association. In addition, it cannot work for the lncRNAs and diseases without any known associations (hereafter isolated nodes), due to the fact that this method depends on the topological structure of bipartite graph and in consequence isolated nodes cannot get any information22. Another pioneer study called ncPred uses a tripartite network to infer potential ncRNA-disease association by integrating ncRNA-target associations and disease-target associations26. In this excellent work, the target information gives rise to a bridge connecting the ncRNA and diseases which significantly improve its predictive performance.
Inspired by the above methods, in this paper we present a novel computational approach of a Tripartite Graph for potential LncRNA-Disease Association identification (TPGLDA) by integrating gene-disease associations and lncRNA-disease associations. The method begins with an lncRNA-disease-gene tripartite graph to delineate the heterogeneity of coding-non-coding genes-disease association. Subsequently, an effective resource allocation algorithm is proposed to accurately identify potential lncRNA-disease associations. Furthermore, lncRNA expression similarity and disease semantic similarity are introduced into TPGLDA to make inference for isolated nodes14,27. To perform a proper evaluation of our proposed method, we utilize leave-one-out cross validation experiment to demonstrate its superior performance compared with existing approaches. Besides, the analyses of several important cancers (i.e. lung cancer, hepatocellular carcinoma, ovarian cancer, etc.) effectively support the practical application of our method. We then use TPGLDA to infer potential lncRNA–disease associations and some high-ranking results are successfully verified by related literatures and databases, such as Lnc2Cancer28, LncRNA2Target29 and MNDR30. These results afford convincing evidence of the good performance of TPGLDA as well as potential value in supporting further biological experiments and promoting research productivity.
Results
Overview of proposed method
The overview of TPGLDA in identifying potential lncRNA-disease associations can be simple summarized as following four steps (Fig. 1). First, we construct lncRNA-disease and gene-disease adjacency matrix by using known lncRNA-disease associations and known gene-disease associations. For an isolated node, we need to calculate the interaction profile (Eq. 12) and then integrate this vector into adjacency matrix for further resource allocation. Second, we construct lncRNA-disease-gene tripartite graph. Third, the process of resource-allocation on tripartite graph builds the potential lncRNA-disease associations. Finally, the resource score (Rscore) of each potential disease-related candidate lncRNA is calculated in turn. We rank all candidates’ Rscore for each disease in descending order, and a higher score will have greater possibility for further verification.
Figure 1.

The flowchart of TPGLDA. (a) Construct lncRNA-disease and gene-disease adjacency matrix. Calculate interaction profile for isolated nodes and integrate into adjacency matrix for further resource allocation. (b) Construct lncRNA-disease-gene tripartite graph. (c) Resource allocation on tripartite graph and build the potential lncRNA-disease associations. (d)Calculate the resource score (Rscore) of candidate lncRNAs and rank all candidates’ Rscore for each disease in descending order.
Evaluation of prediction performance
Leave-one-out cross validation (LOOCV) is implemented on our gold standard dataset to evaluate the performance of TPGLDA in inferring potential associations between lncRNAs and diseases5,18,22. At each step of the LOOCV experiment, each known lncRNA-disease association is removed from the training samples in turn as test sample, whereas other known associations are taken as training samples for model learning5,14,22. Then, the receiver operating characteristics (ROC) curve is utilized to demonstrate the predictive performance of our proposed method and other methods mentioned in this paper by plotting the true positive rate (Sn, sensitivity) and the false positive rate (1-Sp, 1-specificity) at different cutoff points. Here, sensitivity refers to the ratio of positive cases which can be correctly identified, and specificity represents the percentage of negative cases which can be correctly predicted. The value of AUC is calculated from the corresponding area under ROC curve, and the perfect performance appears in AUC = 1 while the random performance emerges in AUC = 0.514,31,32. Besides, we also adopt other evaluation measures such as accuracy (Acc), precision (Pre), and Matthews’s correlation coefficient (MCC)31,33. The definitions can be obtained as follows:
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
where TP means true positives, FP refers to false positives, TN is true negatives, and FN represents false negatives.
Compared with other methods
In order to comprehensively assess the predictive ability of TPGLDA to predict lncRNA–disease associations, we compare our method with two state-of-the-art methods: LRLSLDA5 and KRWRH18, and the corresponding ROC curves of different methods are shown in Fig. 2. Note that Yang’s method22 is not assessed here as it requires the node degree of each candidate ≥2. As a result, both LRLSLDA and KRWRH achieve reliable performance with AUC values of 82.2% and 83.8%, respectively, and TPGLDA has improved with an AUC value of 93.9%. Besides, other common performance evaluation measures, including Sn, Sp, Pre, Acc, and MCC, are also used to measure the predictive performance of these methods, and the results are shown in Table 1. Here, we adopt two stringency levels to measure the predictive performance31,34. At medium stringency level of specificity (Sp = 95.0%), KRWRH achieves the values of Sn, Pre, Acc, and MCC are 42.6%, 18.7%, 93.6% and 25.4%, respectively, which performs slightly better than LRLSLDA. By contrast, the corresponding values of TPGLDA are 76.8%, 29.4%, 94.5%, 45.4%, respectively. When the stringency level of specificity enlarges to 99.0%, the performance of our proposed method and the other two methods are consistently improved across all measures. Overall, these assessments generally confirm the good performance of TPGLDA in recovering experimentally verified lncRNA-disease associations.
Figure 2.

Performance comparison between TPGLDA, LRLSLDA and KRWRH in terms of AUC and ROC curve based on LOOCV. As a result, TPGLDA achieves the highest AUCs of 0.939. The base line indicates random performance.
Table 1.
Comparison with other computational approaches at two stringency levels (Sp = 99.0% and Sp = 95.0%).
| TPGLDA | KRWRH | LRLSLDA | |
|---|---|---|---|
| Sp=99.0% | |||
| Sn | 53.5% | 11.7% | 10.7% |
| Acc | 97.8% | 96.7% | 96.7% |
| Pre | 59.2% | 24.1% | 22.6% |
| MCC | 55.2% | 15.2% | 14.0% |
| Sp=95.0% | |||
| Sn | 76.9% | 42.6% | 35.2% |
| Acc | 94.5% | 93.6% | 93.4% |
| Pre | 29.4% | 18.7% | 16.0% |
| MCC | 45.4% | 25.4% | 20.7% |
Furthermore, considering the importance of top portion of the prediction results32, the corresponding AUC values within the top k candidates of ranking lists are measured and the detailed results are shown in Fig. 3. In the top 20 ranking lists, the corresponding AUC values achieved by LRLSLDA and KRWRH are 63.3% and 58.9%, respectively, whereas TPGLDA achieves an AUC value of 84.4%. For the results in the top 100, LRLSLDA achieves good performance with an AUC of 85.6%. By contrast, TPGLDA obtains a better AUC value of 92.2%. Besides, we also report the corresponding recall rate (Fig. 4), which measures the number of known lncRNA-disease association pairs that can be correctly identified within the top k candidates of ranking lists31,32. In the top 20 candidates, our method can successfully rank about 76% of known lncRNA-disease association pairs. When the rank threshold reaches to 100, the recall values of LRLSLDA and KRWRH are improved to 72.6% and 78.7%, respectively, and the corresponding value obtained by TPGLDA is 93.6%. Taken together, TPGLDA achieves decent recall in above different top k ranking lists, suggesting that our method can infer the largest number of positive samples based on different cutoffs.
Figure 3.

The average AUCs across all the diseases at different top k cutoffs.
Figure 4.
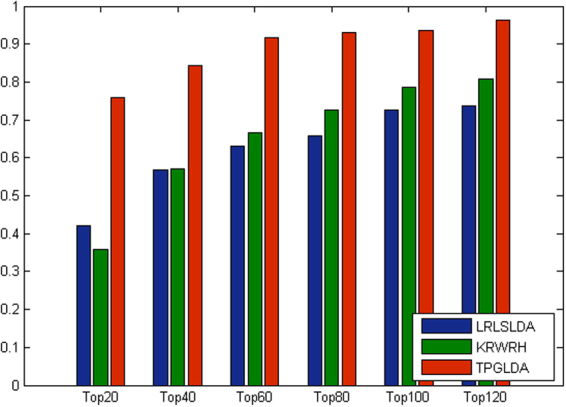
The average recall across all the diseases at different top k cutoffs.
Similar to previous studies31,32, we apply the LOOCV experiment on 15 diseases for demonstrating the practical predictive ability of different methods, and the corresponding AUC values are shown in Table 2. As a result, TPGLDA compares favorably with KRWRH and LRLSLDA in terms of AUC values. For example, for bladder cancer, KRWRH and LRLSLDA achieve the corresponding AUC value of 77.4% and 76.5%, respectively, and in comparison TPGLDA obtains an AUC value of 88.3%. Also, the AUC value of TPGLDA for breast cancer reaches 85.2%, which is more than 15% better than the other methods investigated in this paper. Furthermore, the average AUC values of TPGLDA, KRWRH and LRLSLDA for all 15 diseases are calculated, and the results are 87.4%, 76.7% and 74.1%, respectively. Besides, we further report the Friedman rank sum test on our dataset to show the statistical significance in performance improvement of TPGLDA (Supplementary Table S6). These examinations demonstrate that TPGLDA has practical ability to predict various potential lncRNA-disease associations.
Table 2.
Prediction results for TPGLDA, KRWRH and LRLSLDA utilizing leave-one-out cross validation experiment on 15 diseases.
| Disease name | No. of Associated lncRNAs | AUC | ||
|---|---|---|---|---|
| TPGLDA | KRWRH | LRLSLDA | ||
| Gastric Cancer | 24 | 0.893 | 0.832 | 0.756 |
| Colorectal Cancer | 21 | 0.884 | 0.782 | 0.687 |
| Breast Cancer | 20 | 0.852 | 0.675 | 0.655 |
| Hepatocellular Carcinoma | 20 | 0.911 | 0.891 | 0.751 |
| Non-Small Cell | ||||
| Lung Cancer | 15 | 0.799 | 0.759 | 0.765 |
| Prostate Cancer | 13 | 0.886 | 0.807 | 0.758 |
| Esophageal Squamous | ||||
| Cell Carcinoma | 13 | 0.822 | 0.835 | 0.739 |
| Ovarian Cancer | 12 | 0.892 | 0.731 | 0.768 |
| Bladder Cancer | 11 | 0.883 | 0.774 | 0.765 |
| Lung Cancer | 9 | 0.828 | 0.737 | 0.750 |
| Melanoma | 9 | 0.939 | 0.627 | 0.815 |
| Glioma | 9 | 0.820 | 0.710 | 0.808 |
| Tumor | 8 | 0.950 | 0.786 | 0.625 |
| Schizophrenia | 8 | 0.860 | 0.854 | 0.630 |
| Papillary Thyroid Carcinoma | 7 | 0.892 | 0.700 | 0.835 |
In addition to our gold dataset used in performance evaluation, Chen et al. (2013)35 dataset used in ncPred26 is applied to make comparison with ncPred (Supplementary Table S7). By applying a 10-fold cross-validation procedure repeated 30 times, we calculate the corresponding averaged AUC value and the result obtained by TPGLDA is 0.7586 ± 0.0306, which is comparable with the result (0.7566 ± 0.0218) reported by ncPred26. At the same time, the Friedman rank sum test’s result is also reported in Supplementary Table S8. The above two results indicate that there is no statistically significant difference between ncPred and TPGLDA predictions in Chen et al. dataset.
At the same time, the time complexity of resource allocation in lncRNA-disease association and gene-disease are O(n2m) and O(nrm), respectively. Considering the fact that the number of disease-related genes is an order of larger than the number of lncRNAs and diseases, the overall time complexity of resource allocation in tripartite graph is O(nrm), which is more efficient to that of ncPred (O(r2m))26. However, the parallelization and optimization techniques can greatly reduce the differences in computational speed. Also, we compare the running time of different methods and the results show the running time of TPGLDA is 0.6 second in average, which is comparable with other methods (Supplementary Table S9).
Case studies
In addition to LOOCV experiment, we also employ TPGLDA to rank all candidate lncRNAs investigated in this paper, and these predictions are used for further analysis in this study. In consistence with previous studies5,14,22, all experimentally validated lncRNA-disease associations are utilized as training sample. Subsequently, the resource score for each potential lncRNA-disease association is calculated in turn, and the predicted results are listed in Supplementary Table S3 in descending order. Higher Rscore indicates greater potential association between lncRNA and disease. In order to further verify the ability of TPGLDA in discovering potential lncRNA–disease associations, the case studies of three diseases: hepatocellular carcinoma, lung cancer and ovarian cancer are reviewed in detail. Here, the top 5 predicted disease-related lncRNAs computed by TPGLDA and their evidences are listed in Table 3. Predictive results are supported by relevant literatures and databases, and the detailed cases can be interpreted by the examples as follow.
Table 3.
The top 5 predictions computed by TPGLDA for Lung Cancer, Hepatocellular Carcinoma and Ovarian Cancer and the confirmation for their associations by related databases.
| LncRNA | TPGLDA’s rank | Evidences (PMID) | Description |
|---|---|---|---|
| Lung Cancer | |||
| GAS5 | 1 | 25925741,24357161 | Lnc2Cancer,LncRNA2Target |
| CDKN2B-AS1 | 2 | 21489289,26408699 | MNDR,Lnc2Cancer |
| UCA1 | 3 | 26380024 | Lnc2Cancer |
| PVT1 | 4 | 26493997;26493997 | Lnc2Cancer,literature |
| HNF1A-AS1 | 5 | 25863539 | literature |
| Hepatocelluar Carcinoma | |||
| GAS5 | 1 | 26404135, 26163879 | Lnc2Cancer, literature |
| SOX2-OT | 2 | 26097588 | Lnc2Cancer |
| PVT1 | 3 | 25624916 | Lnc2Cancer |
| LINC00152 | 4 | 27351280, 26356260 | Lnc2Cancer, literature |
| UCA1 | 5 | 27215316, 27167190 | Lnc2Cancer, literature |
| Ovarian Cancer | |||
| MEG3 | 1 | 24859196 | Lnc2Cancer,LncRNA2Target |
| GAS5 | 2 | 26503132 | Lnc2Cancer |
| CCAT2 | 3 | 27283598 | Lnc2Cancer |
| BANCR | 4 | unconfirmed | unconfirmed |
| CDKN2B-AS1 | 5 | 27095571 | Lnc2Cancer |
Lung cancer is one of the most common cancers worldwide which has extremely high mortality rate36. In the United States, lung cancer ranks second only to the highest cancer deaths according to the Estimated New Cancer Cases of the ten leading cancer types37. Among the top 5 lung cancer-related candidates ranked by TPGLDA, all 5 potential lncRNAs are verified to be associated with lung cancer by related literatures and databases. For example, the lncRNA-CDKN2B-AS1 promotes NSCLC cell proliferation and inhibits apoptosis by suppressing KLF2 and P21 expression38. Furthermore, recent research demonstrates that upregulated lncRNA-UCA1 contributes to progression of lung carcinoma, and lncRNA-UCA1 holds great promise as a potential predictive biomarker in clinical diagnosis for lung cancer39.
Hepatocellular carcinoma is predominant component of the primary liver cancer, which is the fifth most common cancer around the world as well as the third most common cause of cancer mortality40. The top three hepatocellular carcinoma-related candidates, lncRNA-GAS5, lncRNA-SOX2-OT and lncRNA-PVT1, are all confirmed by recent experimental reports. LncRNA-GAS5, acting as a proto-oncogene, is revealed to be critical to hepatocellular carcinoma and the deletion allele is significantly correlated with higher expression of lncRNA-GAS5 in hepatocellular carcinoma tissues41. Besides, the up-regulation of lncRNA-SOX2-OT is reported to facilitate hepatocellular carcinoma cell metastasis and high expression of lncRNA-SOX2-OT is demonstrated to be associated with histological grade, TNM stage and vein invasion42. Furthermore, Ding et al.43 point out that lncRNA-PVT1 is associated with tumor progression and can serve as a novel biomarker for predicting tumor recurrence in hepatocellular carcinoma-related patients.
Ovarian cancer is a major case of cancer deaths in women, especially for ovarian epithelial carcinoma44,45. Currently, this cancer is generally detected in the late diagnosis, and the etiology of ovarian cancer is poorly understood for us45. Among the top 5 predictions of ovarian cancer, most lncRNAs can be confirmed by related literatures. For example, a recent experimental result shows that lncRNA-MEG3 may play a significant role as a tumor suppressor in ovarian cancer cells46. Furthermore, LncRNA-GAS5 is verified to be related to ovarian cancer cell apoptosis by means of the mitochondria-mediated apoptosis pathway, which can be used as a new therapeutic target and has an important role in disease progression47.
In addition to the above mentioned diseases, TPGLDA also achieves decent prediction results for other diseases. For examples, lncRNA-TUG1 is ranked first in our prediction list of colorectal cancer-related lncRNAs, and recent study indicates that the upregulation of lncRNA-TUG1 is closely related to the survival time of colorectal cancer patients48. Besides, we find that lncRNA-GHET1 is ranked second in our prediction list, which is shown to be significantly upregulated in colorectal cancer tissues and can serve as a therapeutic target for colorectal cancer expression patterns49. As for the prediction list of bladder cancer, lncRNA-PVT1 and lncRNA-ANRIL are ranked first and seventh, respectively, which have been shown to be up-regulated in bladder cancer50,51. In addition, LncRNA-CCAT1, ranking fourth in our breast cancer-related prediction list, is found to be up-regulated in breast cancer52, and lncRNA-TUG1, ranking first in our predicted list of gastric cancer, can promote the transferring and invading capacity of gastric cancer53. The top 10 predictions of these disease-related lncRNAs are listed in Supplementary Table S4. Furthermore, we further use TPGLDA to simultaneously rank all candidate lncRNA-disease associations5, and the top 20 potential associations are also shown in Supplementary Table S5. 12 of the top 20 predictions are confirmed by different related databases or literatures. From aforementioned case studies, we find that many predictions are confirmed by recent experimental results. For example, recent study demonstrates that lncRNA-PVT1 (rank 16th) is overexpressed in osteosarcoma and can decrease the survival rate of osteosarcoma patients54. Therefore, with the progress of the biological experiments, it is anticipated that more unconfirmed associations in our predictive results will be verified, and those potential lncRNA-disease pairs which have higher ranks will be given reasonable priority for subsequent experimental research. In conclusion, these case studies further suggest that TPGLDA is useful for inferring potential associations between lncRNAs and disease in practice.
Discussions
Accumulating evidences have highlighted the important role of developing a powerful computational method to infer potential associations between lncRNAs and diseases, which can significantly help people to explore complex diseases at the molecular level and improve the quality of various disease diagnosis, therapy, prognosis and prevention. In this paper, we propose a novel computational method, TPGLDA, to identify the underlying lncRNA-disease associations by integrating experimentally verified gene-disease associations and lncRNA-disease associations. Compared with previous methods, we develop an lncRNA-disease-gene tripartite graph to better delineate the heterogeneity of coding-non-coding genes-disease associations. For the sake of better performance, we subsequently develop an effective resource allocation algorithm on the constructed lncRNA–disease-gene tripartite graph to rank candidates. In addition, TPGLDA can be applied to the isolated nodes by integrating lncRNA similarities and disease similarities. Our method firstly demonstrates its good performance by LOOCV experiment. Furthermore, the measures of AUC values and recall values within the top k ranking lists show that TPGLDA has powerful predictive ability to infer the largest number of positive samples. Finally, the analyses of case studies further demonstrate that TGPLDA is useful for identifying potential lncRNA-disease associations in practice.
The good results achieved by TPGLDA can be largely ascribed to following factors: firstly, motivated by cooperation between non-coding genes and coding genes in human diseases14,18,22,55, we effectively construct the associations among lncRNAs, diseases and genes, and develop an lncRNA-disease-gene tripartite graph to better delineate the heterogeneity of coding-non-coding genes-disease associations. The tripartite graph integrates a large number of disease-related genes as collaborative prediction of underlying association between lncRNAs and diseases, which enrich diseases information during the process of resource allocation56. Subsequently, the contributions of resource moved in both directions are taken into consideration by a consistence-based resource allocation algorithm57, which effectively reduces the unaware biases in resource allocation process57,58 and further improves TPGLDA’s predictive performance. Finally, by adopting different biological information including disease-related genes, lncRNA expression profile and disease semantic information in our method, potential candidates can acquire more information from other diseases and lncRNAs. In summary, TPGLDA shows a decent performance and complements the detection results of the existing approaches in inferring potential associations between lncRNAs and diseases. Nevertheless, the assessment measures are not sufficient to indicate a criticism of other computational approaches. Instead, different methods show the difference between whether considering the information of disease-related genes in the heterogeneous associations or not. As a novel computational method, it is anticipated that TPGLDA has potential value in biomedical research for comprehending the pathogenesis of diseases, which can further advance the quality of disease diagnostics, therapy, prognosis and prevention.
Despite the promising results achieved by TPGLDA, some limitations still should be acknowledged for further investigation. Firstly, TPGLDA depends on the tripartite graph topology and in consequence the incompleteness of the data may limit its performance. Therefore, it may be useful to further expand the method by integrating gene-lncRNA associations or additional biological information that have been successfully adopted in existing methods such as ncPred26, which can make our proposed method more accurate and reliable. Secondly, our method focuses on unweighted tripartite graph, it will be improved by a refined algorithm with accurately defined weights on lncRNA-disease as well as gene-disease56. Finally, the experimentally available lncRNA-disease associations are still comparatively small in number. With the continuous development of biotechnology, the performance of TPGLDA is expected to further increases when more experiment verified associations are available.
Materials and Methods
Human lncRNA–disease associations and gene-disease associations
The recent version of lncRNA-disease associations are downloaded from the LncRNADisease35 database which integrates 687 experimentally validated lncRNA–disease associations between 246 diseases and 369 lncRNAs. We further filter out diseases without Disease Ontology (http://disease-ontology.org/) information and lncRNAs without expression profiles in ArrayExpress59 (http://www.ebi.ac.uk/arrayexpress/), and eventually obtain 540 experimentally verified lncRNA-disease associations. This dataset is utilized as the gold standard dataset in the leave-one-out cross validation experiment and as the training dataset for inference of lncRNA–disease association5. Using the information of lncRNA-related diseases, we further collect 5212 gene–disease associations from DisGeNET database24 (http://www.disgenet.org/web/DisGeNET/) and construct a lncRNA-disease-gene tripartite graph, which includes 115 lncRNAs, 178 diseases and 1415 genes.
LncRNA expression similarity
We obtain LncRNA expression profiles from ArrayExpress59 which contains more than 60000 expression profiles across 16 human tissues generated through RNA-Seq technology. Following previous approaches5,60, we calculate the lncRNA expression similarity (Supplementary Table S1) as the absolute Spearman correlation coefficient between the expression profiles of each lncRNA pair, and use matrix to denote the lncRNA expression similarity matrix between lncRNA and lncRNA .
Disease semantic similarity
Recently, disease semantic similarity has been used in predicting potential ncRNA-disease associations and its effectiveness has been demonstrated in previous studies14,32,61. In this paper, the disease semantic similarity is calculated in the same way as described in previous study62, in which a disease is represented as a directed acyclic graph (DAG) including all related annotation terms which can be obtained from the U.S. National Library of Medicine (MeSH, http://www.nlm.nih.gov/mesh). Based on their DAGs, the similarities between diseases are measured and the detailed calculations are illustrated in the DOSE package63. We eventually calculate the semantic similarities (Supplementary Table S2) among all diseases and the corresponding similarity matrix is denoted as .
TPGLDA
Inspired by previous study using tripartite graph of users, items and tags for recommendation56, in this paper we first construct an lncRNA-disease-gene tripartite graph T (L, D, G, E), where L = {}, D = { and G = {} are the nodes set of n long non-coding RNAs, the nodes set of m diseases and the node set of r genes, respectively. E denotes the interactions (edges) set between nodes in L with D and D with G. The tripartite graph can also be represented by two adjacency matrices and , where if lncRNA is associated with disease and otherwise = 0 indicating that the pair of lncRNA and disease is unknown association. Analogously, we set = 1 if disease is associated with gene , otherwise 0.
We model the prediction process of lncRNA–disease associations as resource allocation56 on the lncRNA-disease-gene tripartite graph. In order to help readers better understand the procedure of our model, a simple example of resource allocation in the tripartite graph is shown in Fig. 5. For a specific lncRNA , the initial resources, located on disease , is defined as:
| 6 |
Figure 5.

Operating principle of resource allocation in an lncRNA-disease-gene tripartite graph consisted of three lncRNAs, five diseases, and four genes. The blue circles, green squares and purple triangles represent lncRNAs in L, disease in D and genes in G, respectively. (a) For target lncRNA, the initial resources (1, 1, 0, 0, 1) locate on. (b) In the first step, each disease averagely distributes its resource to both sides of neighboring nodes based on the degree of each disease. (c) In the second step, the resources flow back to D from L and G, and final resource vector locate on D are and.
The initial resource vector is then denoted as = (1, 1, 0, 0, 1) if we choose lncRNA as a target lncRNA (Fig. 5a). The resource allocation process in TGPLDA includes two steps. In the first step of the allocation, the initial resource is simultaneously allocated from nodes in D to those in L and G, respectively. In the second step, the resource is transferred back from nodes in L and G to the D nodes. We use the corresponding weight matrix = to represent the process of resource allocation between lncRNAs and diseases, as below:
| 7 |
where is the contribution of resource moved from j-th node to i-th node in L, and can be described as the similarity58 between lncRNA and lncRNA . is the number of related diseases for lncRNA which is called the degree of . Similarly, represents the degree of node in D. We further modify the resource allocation algorithm by considering the level of consistency between the contribution of resource moved in both directions57, which reflects the impact of co-selection () between the contribution of resource from to and the contribution of resource from to , as more consistency of two objects suggests higher similarity58. Accordingly, we define a consistence-based resource allocation to represent lncRNA-disease association as follows:
| 8 |
where denotes the sum of contribution from resource allocation between i-th node and j-th node in L. The corresponding weight matrix is then rewritten as = . Combining adjacent matrix and weight matrix , the final resource located on D nodes is defined as:
| 9 |
With respect to resource allocation between genes and diseases, the same initial resource located on D nodes56 are allocated from nodes in D to nodes in G and then transferred back, and the final resource vector located on D nodes can be calculated as:
| 10 |
where = represents the degree of gene in G and = is the number of related genes for disease . By weighting both and , the final resource score used to measure potential lncRNA-related diseases are defined as follows:
| 11 |
where parameter [0, 1] is tunable and used to balance the contribution between lncRNAs and genes. In this paper, TPGLDA achieves the best prediction performance when = 0.6. In fact, our method is robust and insensitive to the selection of (see Supplementary Fig. S1).
The inference of the isolated node is implemented by following procedure proposed in previous study27, which can be summarized as follows. First, we calculate the similarity between an isolated node (e.g. a new lncRNA) and its neighbors, which is calculated by lncRNA expression similarity for an isolated lncRNA or disease semantic similarity for an isolated disease. Second, we calculate the interaction profile by the following form:
| 12 |
where is an interaction profile vector and is used to reflect potential relationships between the isolated node and diseases by considering its neighbors interactions with diseases27, which is then integrated into tripartite graph for further resource allocation.
Electronic supplementary material
Acknowledgements
This work is supported by National Natural Science Foundation of China (61471331 and 61571414); University of Science and Technology of China, USTC. We appreciate the valuable suggestions from any reviewers. We also thank Binghua Wang and Bo Ma for many helpful discussions and suggestions.
Author Contributions
L.D. and M.W. wrote the main manuscript text and prepared all Tables and Figures. L.A. and D.S. provided valuable suggestions and guidance. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Liang Ding and Minghui Wang contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-19357-3.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Esteller M. Non-coding RNAs in human disease. Nature Reviews Genetics. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 2.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends in cell biology. 2011;21:354–361. doi: 10.1016/j.tcb.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Harries LW. Long non-coding RNAs and human disease. Biochemical Society Transactions. 2012;40:902–906. doi: 10.1042/BST20120020. [DOI] [PubMed] [Google Scholar]
- 5.Chen, X. & Yan, G.-Y. Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics, btt426 (2013). [DOI] [PubMed]
- 6.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature Reviews Genetics. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 7.Johnson R. Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiology of disease. 2012;46:245–254. doi: 10.1016/j.nbd.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Ishii N, et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. Journal of human genetics. 2006;51:1087–1099. doi: 10.1007/s10038-006-0070-9. [DOI] [PubMed] [Google Scholar]
- 9.Congrains A, et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis. 2012;220:449–455. doi: 10.1016/j.atherosclerosis.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 10.Li J, Xuan Z, Liu C. Long non-coding RNAs and complex human diseases. International journal of molecular sciences. 2013;14:18790–18808. doi: 10.3390/ijms140918790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amaral PP, Clark MB, Gascoigne DK, Dinger ME, Mattick J. S. lncRNAdb: a reference database for long noncoding RNAs. Nucleic acids research. 2011;39:D146–D151. doi: 10.1093/nar/gkq1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bu, D. et al. NONCODE. v3. 0: integrative annotation of long noncoding RNAs. Nucleic acids research, gkr1175 (2011). [DOI] [PMC free article] [PubMed]
- 13.Dinger ME, et al. NRED: a database of long noncoding RNA expression. Nucleic acids research. 2009;37:D122–D126. doi: 10.1093/nar/gkn617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Scientific reports. 2015;5:16840. doi: 10.1038/srep16840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ideker T, Sharan R. Protein networks in disease. Genome research. 2008;18:644–652. doi: 10.1101/gr.071852.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu M, et al. An analysis of human microRNA and disease associations. PloS one. 2008;3:e3420. doi: 10.1371/journal.pone.0003420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun J, et al. Inferring novel lncRNA–disease associations based on a random walk model of a lncRNA functional similarity network. Molecular BioSystems. 2014;10:2074–2081. doi: 10.1039/C3MB70608G. [DOI] [PubMed] [Google Scholar]
- 18.Ganegoda GU, Li M, Wang W, Feng Q. Heterogeneous network model to infer human disease-long intergenic non-coding RNA associations. IEEE transactions on nanobioscience. 2015;14:175–183. doi: 10.1109/TNB.2015.2391133. [DOI] [PubMed] [Google Scholar]
- 19.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nature Reviews Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 20.Liu M-X, Chen X, Chen G, Cui Q-H, Yan G-Y. A computational framework to infer human disease-associated long noncoding RNAs. PloS one. 2014;9:e84408. doi: 10.1371/journal.pone.0084408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang P, et al. Identification of biomarkers for the detection of early stage lung adenocarcinoma by microarray profiling of long noncoding RNAs. Lung Cancer. 2015;88:147–153. doi: 10.1016/j.lungcan.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Yang X, et al. A network based method for analysis of lncRNA-disease associations and prediction of lncRNAs implicated in diseases. PloS one. 2014;9:e87797. doi: 10.1371/journal.pone.0087797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sahu D, et al. Co-expression analysis identifies long noncoding RNA SNHG1 as a novel predictor for event-free survival in neuroblastoma. Oncotarget. 2016;7:58022–58037. doi: 10.18632/oncotarget.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piñero J, et al. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database. 2015;2015:bav028. doi: 10.1093/database/bav028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gutiérrez-Sacristán, A. et al. PsyGeNET: a knowledge platform on psychiatric disorders and their genes. Bioinformatics, btv301 (2015). [DOI] [PMC free article] [PubMed]
- 26.Alaimo, S., Giugno, R. & Pulvirenti, A. ncPred: ncRNA-disease association prediction through tripartite network-based inference. Frontiers in bioengineering and biotechnology2 (2014). [DOI] [PMC free article] [PubMed]
- 27.Mei J-P, Kwoh C-K, Yang P, Li X-L, Zheng J. Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics. 2013;29:238–245. doi: 10.1093/bioinformatics/bts670. [DOI] [PubMed] [Google Scholar]
- 28.Ning S, et al. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic acids research. 2016;44:D980–D985. doi: 10.1093/nar/gkv1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Q, et al. LncRNA2Target: a database for differentially expressed genes after lncRNA knockdown or overexpression. Nucleic acids research. 2015;43:D193–D196. doi: 10.1093/nar/gku1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, et al. Mammalian ncRNA-disease repository: a global view of ncRNA-mediated disease network. Cell Death and Disease. 2013;4:e765. doi: 10.1038/cddis.2013.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun D, Li A, Feng H, Wang M. NTSMDA: prediction of miRNA–disease associations by integrating network topological similarity. Molecular BioSystems. 2016;12:2224–2232. doi: 10.1039/C6MB00049E. [DOI] [PubMed] [Google Scholar]
- 32.Xuan, P. et al. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics, btv039 (2015). [DOI] [PubMed]
- 33.Liu Z-P, Wu L-Y, Wang Y, Zhang X-S, Chen L. Prediction of protein–RNA binding sites by a random forest method with combined features. Bioinformatics. 2010;26:1616–1622. doi: 10.1093/bioinformatics/btq253. [DOI] [PubMed] [Google Scholar]
- 34.Li, A., Ge, M., Zhang, Y., Peng, C. & Wang, M. Predicting long noncoding RNA and protein interactions using heterogeneous network model. BioMed research international2015 (2015). [DOI] [PMC free article] [PubMed]
- 35.Chen G, et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic acids research. 2013;41:D983–D986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenlee RT, Murray T, Bolden S, Wingo PA. Cancer statistics, 2000. CA: a cancer journal for clinicians. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 37.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 38.Nie F-q, et al. Long noncoding RNA ANRIL promotes non–small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Molecular cancer therapeutics. 2015;14:268–277. doi: 10.1158/1535-7163.MCT-14-0492. [DOI] [PubMed] [Google Scholar]
- 39.Wang H-M, Lu J-H, Chen W-Y, Gu A-Q. Upregulated lncRNA-UCA1 contributes to progression of lung cancer and is closely related to clinical diagnosis as a predictive biomarker in plasma. International journal of clinical and experimental medicine. 2015;8:11824. [PMC free article] [PubMed] [Google Scholar]
- 40.El–Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 41.Tao R, et al. Association between indel polymorphism in the promoter region of lncRNA GAS5 and the risk of hepatocellular carcinoma. Carcinogenesis. 2015;36:1136–1143. doi: 10.1093/carcin/bgv099. [DOI] [PubMed] [Google Scholar]
- 42.Shi X-M, Teng F. Up-regulation of long non-coding RNA Sox2ot promotes hepatocellular carcinoma cell metastasis and correlates with poor prognosis. International journal of clinical and experimental pathology. 2015;8:4008. [PMC free article] [PubMed] [Google Scholar]
- 43.Ding C, et al. Long non-coding RNA PVT1 is associated with tumor progression and predicts recurrence in hepatocellular carcinoma patients. Oncology letters. 2015;9:955–963. doi: 10.3892/ol.2014.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holschneider, C. H. & Berek, J. S. inSeminars in surgical oncology. 3-10 (Wiley Online Library). [DOI] [PubMed]
- 45.Miller NL, et al. An Rgnef (p190RhoGEF/Arhgef28) signaling axis regulates ovarian cancer progression. Cancer Research. 2014;74:3157–3157. doi: 10.1158/1538-7445.AM2014-3157. [DOI] [Google Scholar]
- 46.Sheng X, et al. Promoter hypermethylation influences the suppressive role of maternally expressed 3, a long non-coding RNA, in the development of epithelial ovarian cancer. Oncology reports. 2014;32:277–285. doi: 10.3892/or.2014.3208. [DOI] [PubMed] [Google Scholar]
- 47.Gao J, et al. Long non-coding RNA growth arrest-specific transcript 5 is involved in ovarian cancer cell apoptosis through the mitochondria-mediated apoptosis pathway. Oncology reports. 2015;34:3212–3221. doi: 10.3892/or.2015.4318. [DOI] [PubMed] [Google Scholar]
- 48.Sun J, et al. The long non-coding RNA TUG1 indicates a poor prognosis for colorectal cancer and promotes metastasis by affecting epithelial-mesenchymal transition. Journal of translational medicine. 2016;14:42. doi: 10.1186/s12967-016-0786-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou J, et al. Knockdown of long noncoding rna ghet1 inhibits cell proliferation and invasion of colorectal cancer. Oncology Research Featuring Preclinical and Clinical Cancer Therapeutics. 2016;23:303–309. doi: 10.3727/096504016X14567549091305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu H, et al. Long non-coding RNA ANRIL is up-regulated in bladder cancer and regulates bladder cancer cell proliferation and apoptosis through the intrinsic pathway. Biochemical and biophysical research communications. 2015;467:223–228. doi: 10.1016/j.bbrc.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 51.Zhuang C, et al. Tetracycline-inducible shRNA targeting long non-coding RNA PVT1 inhibits cell growth and induces apoptosis in bladder cancer cells. Oncotarget. 2015;6:41194. doi: 10.18632/oncotarget.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang X-F, Liu T, Li Y, Li S. Overexpression of long non-coding RNA CCAT1 is a novel biomarker of poor prognosis in patients with breast cancer. International journal of clinical and experimental pathology. 2015;8:9440. [PMC free article] [PubMed] [Google Scholar]
- 53.Ji T-T, Huang X, Jin J, Pan S-H, Zhuge X-J. Inhibition of long non-coding RNA TUG1 on gastric cancer cell transference and invasion through regulating and controlling the expression of miR-144/c-Met axis. Asian Pacific journal of tropical medicine. 2016;9:508–512. doi: 10.1016/j.apjtm.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 54.Zhou Q, et al. Long non-coding RNA PVT1 promotes osteosarcoma development by acting as a molecular sponge to regulate miR-195. Oncotarget. 2016;7:82620–82633. doi: 10.18632/oncotarget.13012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou M, et al. Prioritizing candidate disease-related long non-coding RNAs by walking on the heterogeneous lncRNA and disease network. Molecular BioSystems. 2015;11:760–769. doi: 10.1039/C4MB00511B. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Z-K, Zhou T, Zhang Y-C. Personalized recommendation via integrated diffusion on user–item–tag tripartite graphs. Physica A: Statistical Mechanics and its Applications. 2010;389:179–186. doi: 10.1016/j.physa.2009.08.036. [DOI] [Google Scholar]
- 57.Zhu X, Tian H, Zhang P, Hu Z, Zhou T. Personalized recommendation based on unbiased consistence. EPL (Europhysics Letters) 2015;111:48007. doi: 10.1209/0295-5075/111/48007. [DOI] [Google Scholar]
- 58.Zhu X, Tian H, Cai S. Personalized recommendation with corrected similarity. Journal of Statistical Mechanics: Theory and Experiment. 2014;2014:P07004. doi: 10.1088/1742-5468/2014/07/P07004. [DOI] [Google Scholar]
- 59.Parkinson H, et al. ArrayExpress—a public database of microarray experiments and gene expression profiles. Nucleic acids research. 2007;35:D747–D750. doi: 10.1093/nar/gkl995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang Y-A, Chen X, You Z-H, Huang D-S, Chan K. ILNCSIM: improved lncRNA functional similarity calculation model. Oncotarget. 2016;7:25902–25914. doi: 10.18632/oncotarget.8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X, et al. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Scientific reports. 2015;5:11338. doi: 10.1038/srep11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010;26:1644–1650. doi: 10.1093/bioinformatics/btq241. [DOI] [PubMed] [Google Scholar]
- 63.Yu, G. & Wang, L.-G. Disease ontology semantic and enrichment analysis. (2012). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.