

Abstract
Cytokines present during low-grade inflammation contribute to β-cell dysfunction and diabetes. Cytokine signaling disrupts β-cell glucose-stimulated Ca2+ influx (GSCI) and endoplasmic reticulum (ER) Ca2+ ([Ca2+]ER) handling, leading to diminished glucose-stimulated insulin secretion (GSIS). However, cytokine-mediated changes in ion channel activity that alter β-cell Ca2+ handling remain unknown. Here we investigated the role of K+ currents in cytokine-mediated β-cell dysfunction. Kslow currents, which control the termination of intracellular Ca2+ ([Ca2+]i) oscillations, were reduced following cytokine exposure. As a consequence, [Ca2+]i and electrical oscillations were accelerated. Cytokine exposure also increased basal islet [Ca2+]i and decreased GSCI. The effect of cytokines on TALK-1 K+ currents were also examined as TALK-1 mediates Kslow by facilitating [Ca2+]ER release. Cytokine exposure decreased KCNK16 transcript abundance and associated TALK-1 protein expression, increasing [Ca2+]ER storage while maintaining 2nd phase GSCI and GSIS. This adaptive Ca2+ response was absent in TALK-1 KO islets, which exhibited decreased 2nd phase GSCI and diminished GSIS. These findings suggest that Kslow and TALK-1 currents play important roles in altered β-cell Ca2+ handling and electrical activity during low-grade inflammation. These results also reveal that a cytokine-mediated reduction in TALK-1 serves an acute protective role in β-cells by facilitating increased Ca2+ content to maintain GSIS.
Introduction
Failure of β-cells to secrete sufficient insulin precedes the onset of type 2 diabetes mellitus (T2DM)1. As the incidence of T2DM is rapidly increasing, it is important to identify better therapeutic options for reducing β-cell failure during the pathogenesis of the disease. Low-grade inflammation is a key contributor to β-cell dysfunction in T2DM1–8. Conditions of over-nutrition and inactivity result in low-grade systemic inflammation during which pro-inflammatory cytokine concentrations (e.g. tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interferon-γ (IFN-γ)) increase several fold over basal levels1–4,8–10. For example, in a rat model of T2DM pancreatic cytokine levels were all elevated above nontreated controls (e.g. TNF-α increased from 24.3 ± 3.6 pg/mg protein to 47.9 ± 3.5 pg/mg protein (P < 0.05), IL-1β increased from 25.5 ± 2.7 pg/mg protein to 29.2 ± 1.7 pg/mg protein (P < 0.05), and IFN-γ increased from 49.4 ± 4.2 pg/mg protein to 65.1 ± 6.7 pg/mg protein (P < 0.05))11. The presence of these cytokines contributes to insulin resistance and diminished β-cell function5. Under stressful conditions (e.g. glucolipotoxicity) β-cells are also capable of secreting pro-inflammatory cytokines, which damage islet function4,12. Cytokine-mediated islet dysfunction correlates with increased basal intracellular Ca2+ ([Ca2+]i), reduced glucose-stimulated Ca2+ influx (GSCI), increased [Ca2+]i oscillation frequency, altered endoplasmic reticulum (ER) Ca2+ ([Ca2+]ER) storage, and increased apoptotic signaling5–7,13. While chronic low-grade inflammation leads to β-cell dysfunction in T2DM, the mechanisms responsible remain unresolved. Understanding how cytokines disrupt islet Ca2+ handling may illuminate therapeutic targets for preventing β-cell failure during T2DM.
Calcium enters β-cells through voltage-dependent Ca2+ channels (VDCCs) that are controlled by ion channel-mediated changes in plasma membrane potential (Vm)14,15. Thus, cytokine-mediated perturbations in Ca2+ handling would be predicted to result from changes in ion channel activity or expression. For example, accelerated islet [Ca2+]i oscillations may be influenced by changes in K+ currents that control [Ca2+]i oscillations such as Ca2+-activated K+ (KCa), ATP-sensitive K+ (KATP) channels, and/or the two-pore domain K+ (K2P) channel TALK-114,16,17. Kslow is a KCa current controlled in part by [Ca2+]ER release that senses and controls [Ca2+]i by hyperpolarizing β-cell Vm16,18–22. In β-cells it is believed to be composed of intermediate conductance (IK) KCa, apamin-insensitive small conductance (SK) KCa, and/or KATP channels18,21,22. TALK-1, while not sensitive to Ca2+, tunes islet [Ca2+]i oscillation frequency, partly by facilitating [Ca2+]ER release and activating Kslow14,23–27. Interestingly, the transcript abundance of some of these K+ channels is decreased following cytokine exposure, including KCNK16 (gene encoding TALK-1) and ABCC8 (gene encoding sulfonylurea receptor 1 (SUR1))28. Likewise, mitochondrial function is reduced following cytokine exposure, which decreases ATP production and would be predicted to activate KATP channels29. This suggests that cytokine-induced changes in K+ channel function modulate [Ca2+]i oscillation frequency during the pathogenesis of T2DM.
To further reveal how cytokines dysregulate β-cell [Ca2+]i we investigated the electrophysiological mechanisms responsible for defective β-cell Ca2+ handling during low-grade inflammation. A cytokine-mediated increase in β-cell electrical oscillations was identified, which resulted from Vm depolarization and shorter interburst interval duration. Furthermore, these results demonstrated that Kslow and TALK-1 currents modify β-cell Ca2+ handling to preserve GSIS during acute low-grade inflammation representative of T2DM.
Results
Cytokine exposure decreases KCNK16 transcript abundance and associated TALK-1 protein expression
To investigate the effect of low-grade inflammation on islet TALK-1 transcript and protein expression, islets were treated for 24 hrs with a low concentration of cytokines. Quantitative RT-PCR revealed a loss of KCNK16 (encodes TALK-1) and ATP2A2 (encodes sarco/endoplasmic reticulum Ca2+-ATPase 2b (SERCA2b)) transcript in mouse islets following cytokine exposure (KCNK16: 95.63 ± 5.89% reduction and ATP2A2: 62.81 ± 11.95% reduction, Fig. 1a, P < 0.01). Western blotting of human islets also showed a cytokine-mediated reduction in TALK-1 protein (63.79 ± 14.81% reduction, Fig. 1b, c, P < 0.05). Similarly, cytokine exposure reduced TALK-1 immunofluorescence in mouse and human islet slices (mouse: 42.05 ± 17.33% reduction and human: 57.26 ± 17.25% reduction, Fig. 1d–g, P < 0.05).
Figure 1.

Cytokine exposure reduces islet KCNK16 transcript abundance and associated TALK-1 protein. (a) qRT-PCR analysis of KCNK16 (encodes TALK-1) and ATP2A2 (encodes SERCA2b) transcript relative to GAPDH in nontreated (gray) and cytokine treated (black) WT mouse islets (N = 4 animals), (b) western blot analysis of human islet TALK-1 protein content with (black) and without (gray) cytokines (N = 3 donors), (c) images of human islet TALK-1 western blots for all donors, (d) representative immunofluorescent images of nontreated (upper panels) and cytokine treated (lower panels) mouse islet slices (TALK-1 - green, insulin - red, and nucleus - blue; scale bars are 20 µm), (e) average TALK-1 fluorescence intensity in nontreated (gray) and cytokine treated (black) mouse islet slices (N ≥ 3 islet slices), (f) representative immunofluorescent images of nontreated (upper panels) and cytokine treated (lower panels) human islet slices, and (g) average TALK-1 fluorescence intensity in nontreated (gray) and cytokine treated (black) human islet slices (N ≥ 5 islet slices). Statistical analysis was conducted using unpaired two-tailed t-tests and uncertainty is expressed as SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
Cytokine exposure alters islet Ca2+ handling
As TALK-1 in part controls β-cell [Ca2+]i oscillation frequency and GSCI, we examined the role of the channel in cytokine-mediated changes to islet Ca2+ handling14,26,27. Oscillations in [Ca2+]i were slower in WT than in TALK-1 KO islets (WT: 1.18 ± 0.07 oscillations/min and TALK-1 KO: 1.51 ± 0.16 oscillations/min, Fig. 2a–d, P < 0.01) and cytokine exposure accelerated [Ca2+]i oscillations in both equivalently (WT: 2.01 ± 0.15x, P < 0.001 and TALK-1 KO: 1.77 ± 0.20x, P < 0.001). Cytokine exposure increased basal [Ca2+]i in WT islets (11.36 ± 2.53% increase, Fig. 2e, P < 0.01) while basal [Ca2+]i in TALK-1 KO islets was trending higher (6.15 ± 4.08% increase, Fig. 2e, P = 0.085). Cytokine exposure reduced 1st phase GSCI in WT and TALK-1 KO islets (WT: 64.55 ± 5.99% decrease and TALK-1 KO: 68.12 ± 4.79% decrease, Fig. 2f, P < 0.001). In the absence of cytokines 2nd phase GSCI was higher in TALK-1 KO islets than in WT islets (48.31 ± 19.55% increase, Fig. 2g, P < 0.05). Following cytokine exposure 2nd phase GSCI in TALK-1 KO islets was reduced to WT levels (60.03 ± 4.89% decrease, Fig. 2g, P < 0.001) while 2nd phase GSCI in WT islets was unaffected.
Figure 2.

Cytokine exposure elevates basal islet [Ca2+]i and disrupts GSCI. (a) Representative Fura-2 AM recordings (F340/F380) of changes in β-cell [Ca2+]i for nontreated (gray) and cytokine treated (black) WT mouse islets (the lines above the figure indicate glucose concentrations), (b) average [Ca2+]i oscillation frequency for nontreated (gray) and cytokine treated (black) WT mouse islets with 11 mM glucose (N ≥ 12 islets), (c) representative Fura-2 AM recordings of changes in β-cell [Ca2+]i for nontreated (gray) and cytokine treated (black) TALK-1 KO mouse islets, (d) average [Ca2+]i oscillation frequency for nontreated (gray) and cytokine treated (black) TALK-1 KO mouse islets with 11 mM glucose (N ≥ 30 islets), (e) average basal [Ca2+]i AUC (0–200 sec) for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO islets (N = 3 animals), (f) average 1st phase GSCI AUC (200–600 sec) for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO islets (N = 3 animals), and (g) average 2nd phase GSCI AUC (600–1000 sec) for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO islets (N = 3 animals). Statistical analysis was conducted using 1-way ANOVA and uncertainty is expressed as SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
Cytokine-mediated downregulation of TALK-1 increases β-cell [Ca2+]ER storage
Cytokines alter β-cell [Ca2+]ER in addition to [Ca2+]i5. Because TALK-1 modulates [Ca2+]ER, we examined whether a cytokine-mediated effect on the channel tunes β-cell [Ca2+]ER27. First, [Ca2+]ER was examined by blocking SERCA with CPA to induce [Ca2+]ER release, then [Ca2+]i was quantified. Under these conditions peak [Ca2+]ER release was higher in TALK-1 KO than in WT β-cells (3.37 ± 1.37% increase, Fig. 3a–c, P < 0.05). Cytokine exposure increased peak [Ca2+]ER release in WT (6.42 ± 2.12% increase, Fig. 3c, P < 0.05) but not TALK-1 KO β-cells. This was verified in whole islets using an adenoviral-based genetically-encoded, β-cell specific (under an insulin promoter) ER Ca2+ indicator (D4ER, Fig. 3d). This approach also indicated that [Ca2+]ER in TALK-1 KO β-cells was higher in than in WT β-cells (9.12 ± 2.36% increase, Fig. 3e, P < 0.05). Furthermore, cytokine exposure again increased [Ca2+]ER in WT (10.44 ± 3.57% increase, Fig. 3e, P < 0.05) but not TALK-1 KO β-cells.
Figure 3.

Cytokine exposure increases β-cell [Ca2+]ER. (a) Representative Fura-2 AM CPA responses of nontreated (gray) and cytokine treated (black) WT mouse β-cells (left; black bars correspond to the addition of CPA) and relative cytokine-induced change in [Ca2+]i AUC (right; N = 3 animals), (b) representative Fura-2 AM CPA responses of nontreated (gray) and cytokine treated (black) TALK-1 KO mouse β-cells (left) and relative cytokine-induced change in [Ca2+]i AUC (right; N = 3 animals), (c) average peak CPA responses ([F340/380]max/F[340/380]min) for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO mouse β-cells (N = 3 animals), (d) representative fluorescent images of the D4ER genetically encoded, β-cell specific [Ca2+]ER indicator in WT (top) and TALK-1 KO (bottom) islets (green: F535, blue: F475, cyan: merge; scale bar is 50 µm), and (e) average D4ER intensity (F535/F475) of WT and TALK-1 KO mouse β-cells from intact islets (N = 4 animals). Statistical analysis was conducted using 1-way ANOVA and uncertainty is expressed as SEM (*P < 0.05, **P < 0.01).
Cytokine exposure depolarizes β-cell Vm and increases electrical activity
To further probe cytokine-mediated TALK-1 channel effects on β-cell function, β-cell K2P currents and Vm were determined. For K2P current recording a whole-cell voltage-clamp technique was employed. β-cell Vm was ramped from −120 mV to 60 mV and the resulting whole-cell β-cell currents were measured. K2P currents were isolated by blocking KATP currents with tolbutamide, KV currents with TEA, and KCa currents by removing Ca2+ and including EGTA in the bath solution. Cytokine exposure reduced K2P currents in WT (Δ−30 mV: −0.90 ± 0.30 pA/pF, Δ0 mV: −4.26 ± 1.97 pA/pF, Δ30 mV: −8.33 ± 3.50 pA/pF, Δ60 mV: −12.94 ± 5.00 pA/pF, Fig. 4a, b, P < 0.05) but not TALK-1 KO β-cells (Fig. 4c, d). The whole-cell K2P conductance of WT and TALK-1 KO β-cells was also analyzed between 0 and 60 mV. Cytokine exposure decreased WT β-cell K2P conductance (nontreated: 0.46 ± 0.02 pS/pF and cytokine treated: 0.31 ± 0.02 pS/pF, P < 0.001) but had no effect on TALK-1 KO β-cell K2P conductance (nontreated: 0.32 ± 0.01 pS/pF and cytokine treated: 0.31 ± 0.04 pS/pF). Together, these data indicate that cytokine exposure affects β-cell K2P currents in a TALK-1 specific manner.
Figure 4.

Cytokine exposure decreases TALK-1 K2P currents. (a) Representative whole-cell voltage-clamp recordings of K2P currents in nontreated (gray) and cytokine treated (black) WT mouse β-cells, (b) average K2P currents in nontreated (gray) and cytokine treated (black) WT mouse β-cells at selected voltages (N = 4 animals), (c) representative whole cell voltage ramp recordings of K2P currents in nontreated (gray) and cytokine treated (black) TALK-1 KO mouse β-cells, and (d) average K2P currents in nontreated (gray) and cytokine treated (black) TALK-1 KO mouse β-cells at selected voltages (N = 3 animals). Statistical analysis was conducted using paired two-tailed t-tests and uncertainty expressed as SEM (*P < 0.05, **P < 0.01).
Changes in β-cell Vm were monitored in current-clamp mode using a perforated-patch technique to maintain the integrity of intracellular metabolism and signaling30. Cytokine exposure accelerated electrical oscillation frequency in WT β-cells more than in TALK-1 KO β-cells (WT: 12.29 ± 0.79-fold increase and TALK-1 KO: 6.27 ± 0.84-fold increase, Fig. 5a–e, P < 0.001). As the oscillation frequency of WT and TALK-1 KO islets were indistinguishable following cytokine exposure this difference was likely due to the fact that nontreated TALK-1 KO islets oscillate more rapidly than WT islets14. The impact of cytokine exposure on β-cell Vm was studied under basal (2 mM glucose, resting Vm) and stimulatory conditions (11 mM glucose, interburst Vm (between plateau potentials) and plateau Vm (during plateau potentials)) in whole islets. Cytokines depolarized WT β-cell resting Vm (8.44 ± 2.12 mV, Fig. 5f, P < 0.01) and interburst Vm (7.60 ± 2.80 mV, Fig. 5g, P < 0.05) but not plateau Vm. Cytokine exposure did not alter TALK-1 KO β-cell resting and interburst Vm, but hyperpolarized plateau Vm (7.35 ± 3.03 mV, Fig. 5h, P < 0.05). TALK-1 KO β-cell resting Vm, was likely not affected by cytokine exposure because they are already more depolarized that WT β-cells (5.82 ± 2.16 mV, Fig. 5f, P < 0.05).
Figure 5.

Cytokine exposure increases β-cell electrical excitability. (a) Representative perforated-patch current-clamp Vm recording of a nontreated WT β-cell (lines above figure indicate glucose concentrations), (b) representative Vm recording of a cytokine treated WT β-cell, (c) representative Vm recording of a nontreated TALK-1 KO β-cell, (d) representative Vm recording of a cytokine treated TALK-1 KO β-cell, (e) average electrical oscillation frequency for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO β-cells (N ≥ 6 cells), (f) average resting Vm for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO β-cells (N ≥ 8 cells), (g) average interburst Vm for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO β-cells (N ≥ 7 cells), (h) average plateau Vm for nontreated (gray) and cytokine treated (black) WT and TALK-1 KO β-cells (N ≥ 8 cells). Statistical analysis was conducted using 1-way ANOVA and uncertainty is expressed as SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
Cytokine exposure reduces β-cell Kslow currents
As Kslow also contributes to islet [Ca2+]i oscillations and is controlled by [Ca2+]i influx as well as [Ca2+]ER release, this current was next investigated as a possible contributor to cytokine-mediated elevated β-cell electrical excitability16,19–22. As previously described, Ca2+ influx was activated using a voltage command to sequentially generate 26 APs, then the subsequent outward K+ currents were recorded (Fig. 6a–c)19. Kslow currents were fit to a model of two-phase exponential decay using GraphPad Prism (Table 1) and the related kinetic parameters were determined (Y0: peak Kslow amplitude, % fast: % of Kslow occurring in the fast phase, Kf: fast phase rate constant, Ks: slow phase rate constant, t1/2, f: fast phase half-life, t1/2, s: slow phase half-life, τf: fast phase time constant, and τs: slow phase time constant). The resulting Kslow currents were biphasic, composed of a rapidly decaying fast phase followed by a gradually declining slow phase (Fig. 6d, e). In the absence of cytokines peak Kslow amplitude was lower in TALK-1 KO β-cells and the current decayed more rapidly than in WT β-cells. In WT β-cells cytokine exposure reduced peak Kslow amplitude and accelerated decay of the current. This reduction in Kslow would be predicted to increase β-cell electrical excitability22.
Figure 6.

Cytokine exposure reduces β-cell Kslow currents. (a) Overview of Kslow perforated-patch voltage-clamp recording protocol, (b) representative nontreated WT β-cell Kslow trace (average of 6 cells from one animal), (c) representative cytokine treated WT Kslow trace (average of 6 cells from one animal), (d) average Kslow currents for nontreated WT (dark green) and TALK-1 KO (red) β-cells (N ≥ 15 cells), and (e) average Kslow currents for nontreated (dark green) and cytokine treated (light green) WT β-cells (N ≥ 31 cells).
Table 1.
Kinetic parameters for the two-phase exponential decay of Kslow.
| Parameter | (A) WT Nontreated (N = 43) | (B) WT Cytokines (N = 31) | (C) TALK-1 KO Nontreated (N = 15) | P-value A vs B | P-value A vs C | P-value B vs C |
|---|---|---|---|---|---|---|
| Y0 | 4.427 ± 0.028 | 4.100 ± 0.026 | 3.418 ± 0.030 | *** | *** | *** |
| % fast | 58.80 ± 0.39 | 68.58 ± 0.48 | 72.71 ± 0.43 | *** | *** | *** |
| Kf | 14.26 ± 0.35 | 14.20 ± 0.32 | 13.94 ± 0.39 | ns | ns | ns |
| Ks | 0.778 ± 0.012 | 1.301 ± 0.026 | 0.669 ± 0.024 | *** | ** | *** |
| t1/2, f | 0.049 ± 0.001 | 0.049 ± 0.001 | 0.050 ± 0.001 | ns | ns | ns |
| t1/2, s | 0.892 ± 0.014 | 0.533 ± 0.011 | 1.037 ± 0.038 | *** | *** | *** |
| τf | 0.070 ± 0.002 | 0.070 ± 0.002 | 0.072 ± 0.002 | ns | ns | ns |
| τs | 1.286 ± 0.020 | 0.769 ± 0.015 | 1.496 ± 0.055 | *** | *** | *** |
Statistical analysis was conducted using 1-way ANOVA and uncertainty is expressed as SEM (**P < 0.01, ***P < 0.001).
Cytokine-induced loss of TALK-1 diminishes insulin expression and eventually results in reduced GSIS
Cytokine-mediated effects on GSIS were investigated. Data are presented as a fold increase compared to insulin secretion from nontreated islets at 2 mM glucose (un-normalized data shown for nontreated islets at 2 mM glucose). Insulin secretion at 2 mM glucose was indistinguishable in WT and TALK-1 KO islets (WT 7.26 ± 1.19 pg/islet/hr and TALK-1 KO: 7.67 ± 0.84 pg/islet/hr) and cytokine exposure increased basal secretion equivalently in WT and TALK-1 KO islets (WT: 2.24 ± 0.52x, P < 0.05 and TALK-1 KO: 2.23 ± 0.35x, Fig. 7a, P < 0.05). Under stimulatory glucose conditions (7 and 11 mM glucose) TALK-1 KO islets secreted more insulin than WT islets (7 mM glucose- WT: 5.47 ± 0.52x and TALK-1 KO: 14.67 ± 2.11x, P < 0.05; 11 mM glucose- WT: 18.78 ± 2.49x and TALK-1 KO: 43.83 ± 5.40x, Fig. 7a, P < 0.05). Cytokine exposure had no effect on GSIS from WT or TALK-1 KO islets at 7 mM glucose but reduced GSIS from TALK-1 KO islets at 11 mM glucose (22.02 ± 6.00x, Fig. 7a, P < 0.01). Next, insulin protein and transcript levels were investigated to determine whether the cytokine-induced reduction in GSIS from TALK-1 KO islets was due to reduced insulin expression or defective secretion. TALK-1 KO islets contained less insulin protein than WT islets (WT: 49.03 ± 3.68 ng insulin/islet and TALK-1 KO: 34.08 ± 2.31 ng insulin/islet, Fig. 7b, P < 0.05). Ins2 transcript was also lower in TALK-1 KO islets (61.06 ± 6.19% decrease, Fig. 7c, P < 0.01) while Ins1 and proprotein convertase 1 (PCSK1) transcript levels were indistinguishable in WT and TALK-1 KO islets. The insulin content of WT islets decreased to a level similar to TALK-1 KO islets following cytokine exposure (28.09 ± 1.80 ng insulin/islet, Fig. 7b, P < 0.01) but did not change in TALK-1 KO islets. After cytokine exposure Ins1, Ins2, and PCSK1 transcripts decreased to similar levels in both WT and TALK-1 KO islets (Fig. 7c, P < 0.05). These results suggest that decreased GSIS from TALK-1 KO islets is due to a dysregulation of insulin secretion rather than insulin expression.
Figure 7.
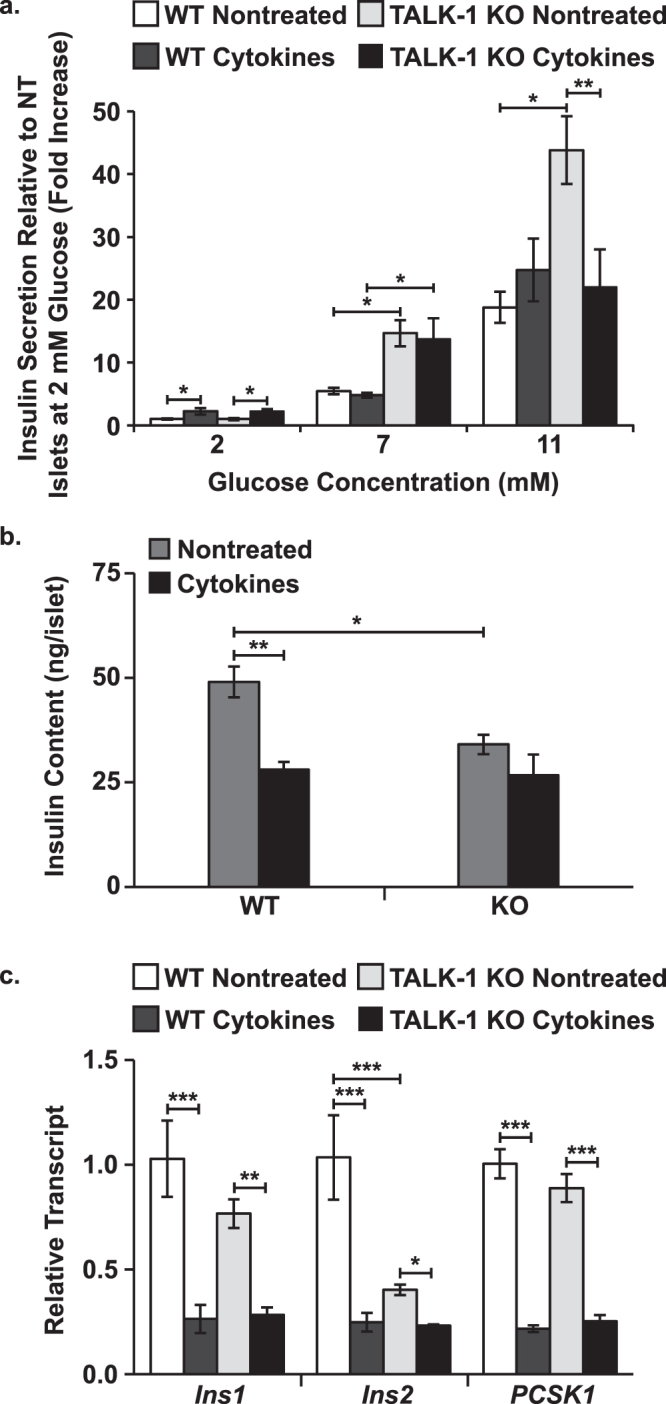
Cytokine exposure decreases insulin protein and related transcripts in WT and TALK-1 KO islets but only disrupts GSIS from TALK-1 KO islets. (a) Average basal (2 mM glucose) and glucose-stimulated (7 and 11 mM glucose) insulin secretion from nontreated and cytokine treated WT and TALK-1 KO islets (N ≥ 3 animals), (b) average insulin content of nontreated (gray) and cytokine treated (black) WT and TALK-1 KO islets (N = 3 animals), and (c) preproinsulin (Ins1 and Ins2) and proprotein convertase 1 (PCSK1) transcript relative to GAPDH in nontreated and cytokine treated WT and TALK-1 KO islets (N = 3 animals). Statistical analysis was conducted using 1-way ANOVA and uncertainty is expressed as SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
Statistical Analysis
All experimental data presented here are shown as mean values ± SEM. Unpaired two-tailed t-tests, paired two-tailed t-tests, and 1-way analysis of variance (ANOVA) tests were used to determine statistical significance between test groups as appropriate. P < 0.05 was considered statistically significant and P < 0.10 was considered to be trending toward significance.
Discussion
Here we demonstrated that both Kslow and TALK-1 currents are important contributors to cytokine-mediated changes in β-cell electrical activity and Ca2+ handling. Cytokine exposure reduced Kslow, which was accompanied by increased electrical excitability and [Ca2+]i oscillation frequency. These cytokine-induced changes were caused in part by reduced [Ca2+]ER release, which resulted from diminished TALK-1 expression. Moreover, during low-grade inflammation, downregulation of TALK-1 preserved GSIS presumably by maintaining 2nd phase GSCI. However, β-cells without functional TALK-1 channels failed to undergo cytokine-induced compensatory Ca2+ changes leading to defective GSCI and GSIS. Together these data suggest that TALK-1 and Kslow play important roles in β-cell adaptation to acute low-grade inflammation.
Although it has been established that low-grade inflammation perturbs β-cell Ca2+ handling, the electrical signaling mechanisms responsible for these changes have not been characterized5–7,13. Many ion channels contribute to physiological control of β-cell electrical excitability and some of these channels become perturbed during conditions of low-grade inflammation5,6,28,31. For example, TALK-1 K+ flux plays a key role in modulating β-cell electrical excitability and expression of the gene that encodes TALK-1 (KCNK16) is reduced during inflammation14,28. As predicted based on knockout of the channel, cytokine-mediated loss of TALK-1 currents depolarized β-cell Vm, which was accompanied by elevated electrical and [Ca2+]i oscillation frequency. TALK-1 KO β-cells displayed a similar cytokine-induced increase in excitability, but Vm was not changed and 2nd phase GSCI was decreased. The frequency of β-cell [Ca2+]i oscillations controls insulin secretion32. Therefore, by lowering TALK-1 expression β-cells were able to increase [Ca2+]i oscillation frequency and acutely maintain physiological [Ca2+]i levels during 2nd phase GSCI in response to low-grade inflammation33,34. However, when [Ca2+]i oscillations become too rapid (e.g. in TALK-1 KO islets following cytokine exposure) the amplitude of [Ca2+]i oscillations begins to decrease35. Thus, in TALK-1 KO islets [Ca2+]i levels declined during 2nd phase GSCI along with [Ca2+]i oscillation amplitude. Disruption of islet [Ca2+]i oscillations is a hallmark of metabolic stress in obese rodent models like those on a high fat diet (HFD) or without leptin receptors36,37. The deterioration of orderly [Ca2+]i oscillations in islets is also believed to contribute to the loss of pulsatile insulin secretion observed in T2DM, which leads to insulin resistance and hyperglycemia38,39. Thus, β-cells accelerated [Ca2+]i oscillations to maintain GSCI during acute low-grade inflammation; however, with chronic inflammation the Ca2+ response continued to change, eventually resulting in reduced islet [Ca2+]i and GSIS. Although TALK-1 plays a role in the adaptation of β-cells to low-grade inflammation, [Ca2+]i oscillations were still accelerated in TALK-1 KO islets following cytokine exposure, which implicates other ion channels in these cytokine-induced β-cell effects.
Activation of a Kslow current is the primary contributor to β-cell Vm repolarization following the slow waves of depolarization that mediate [Ca2+]i oscillations16,19–22. Following cytokine exposure β-cell Vm became depolarized during the electrically silent periods between plateau potentials and the frequency of electrical oscillations increased. This suggests that cytokines affect one or more KCa channels that make up the Kslow current. Indeed, pharmacological inhibition of Kslow accelerates islet [Ca2+]i oscillations similar to cytokine exposure22. As anticipated, our results showed that low level cytokine exposure was sufficient to significantly reduce the magnitude and duration of Kslow in WT β-cells. The Kslow current was biphasic in nature, likely because the current is comprised of multiple K+ channels with varying degrees of Ca2+ sensitivity as well as differing activation and inactivation kinetics. Furthermore, it has been established that [Ca2+]ER release contributes significantly to Kslow activation21. Therefore, Kslow kinetics may be influenced by the temporal profile of ER Ca2+-induced Ca2+ release (CICR) or the proximity of KCa channels to [Ca2+]ER release sites40. TALK-1 has been shown to act as a counter-current for Ca2+ release from the ER of β-cells and indeed genetic ablation of TALK-1 increases β-cell [Ca2+]ER storage and reduces the magnitude of Kslow27. Thus, cytokine-mediated reduction in ER TALK-1 likely contributes to reduced Kslow currents by preventing efficient release of Ca2+ from ER stores. Likewise, under conditions of β-cell stress (e.g. low-grade inflammation) SERCA expression is reduced, which would be expected to perturb [Ca2+]ER and further diminish Kslow currents19–21,41. Cytokine-induced reduction of Kslow caused more frequent and shorter interburst silent phases in β-cell electrical activity during glucose stimulation. Due to these effects on electrical activity, islet [Ca2+]i oscillations were accelerated, amplitude was reduced, and [Ca2+]i was elevated at the nadir between oscillations. In the future, it will be important to identify the KCa channel(s) responsible for the reduction in Kslow currents following cytokine exposure and to determine if these channels can be manipulated to improve β-cell function during low-grade inflammation present during the pathogenesis of T2DM.
Because [Ca2+]ER homeostasis is essential for many biological processes including protein expression, folding, and transport, dysregulation of this equilibrium could also lead to unfolded protein response (UPR) and ER stress42. In fact, pro-inflammatory cytokine exposure has been shown to cause ER stress, hence it is plausible that cytokine-induced disruption of [Ca2+]ER handling contributes to β-cell dysfunction and destruction43. Interestingly, the islets of TALK-1 KO mice on a long-term (20 week) HFD displayed reduced ER stress markers27. Cytokine exposure increased [Ca2+]ER levels to those observed in TALK-1 deficient β-cells, which may help mitigate β-cell ER stress during acute low-grade inflammation. However, it is well-established that prolonged cytokine exposure depletes β-cell [Ca2+]ER levels6,7,44. This results in part from diminished islet SERCA expression observed under low-grade inflammatory conditions and in T2DM patients45,46. Reduced SERCA expression should reduce [Ca2+]ER refilling and thus lower [Ca2+]ER stores. These data suggest that loss of TALK-1 expression preserves [Ca2+]ER levels during acute inflammation, but may not compensate for the loss of [Ca2+]ER uptake during chronic low-grade inflammation. Further studies are required to fully determine the importance of TALK-1-mediated elevation of β-cell [Ca2+]ER levels in response to acute and chronic low-grade inflammation.
In addition to accelerating islet [Ca2+]i and electrical oscillations, low-grade inflammation resulted in increased basal [Ca2+]i at 2 mM glucose and decreased 1st phase GSCI at 11 mM glucose. While these changes in β-cell Ca2+ handling could be partly due to impaired [Ca2+]ER uptake (e.g. reduced SERCA activity), other mechanisms are anticipated to play a significant role5–7. For example, cytokine exposure diminishes mitochondrial ATP production, which could decrease 1st phase GSCI by enhancing KATP channel activity and thus limiting β-cell Vm depolarization29,47. Cytokine exposure also activates voltage-dependent Ca2+ channels (VDCCs), which would be predicted to elevate basal β-cell [Ca2+]i levels5,6,31. This suggests that the global changes in ion channel activity (e.g. KATP and VDCC activation) only modestly depolarize β-cell Vm. However, cytokine-induced depolarization of β-cells at rest (2 mM glucose) diminishes glucose-stimulated Vm depolarization. This decreased glucose-stimulated ΔVm leads to reduced GSCI following cytokine exposure. Interestingly, islets of T2DM patients display elevated basal insulin secretion and a defective 1st phase GSIS response, which would be projected to result from increased basal islet [Ca2+]i and decreased 1st phase GSCI48,49. In the future, it will be important to determine how these cytokine-mediated changes to islet Ca2+ handling contribute to perturbations in GSIS during the pathogenesis of T2DM.
Not only do cytokines affect β-cell electrical excitability and Ca2+ handling they also modulate insulin expression and secretion50,51. Genetic ablation of islet TALK-1 augments GSIS by depolarizing β-cell Vm, thus it was theorized that a cytokine-mediated drop in TALK-1 expression would also increase GSIS14. Acute cytokine exposure increased basal insulin secretion at 2 mM glucose, which is likely due to elevated basal [Ca2+]i resulting in part from VDCC activation5,6,31. GSIS from WT islets was unaffected by acute cytokine exposure, but GSIS from TALK-1 KO islets was decreased by approximately 50% at 11 mM glucose. This suggests that acute augmentation of β-cell [Ca2+]i by reducing TALK-1 expression maintains physiological GSIS following cytokine exposure. However, this adaptive GSIS response becomes less robust when TALK-1 is absent for an extended period of time (i.e. TALK-1 KO), which impairs 2nd phase GSCI and GSIS. TALK-1 KO β-cells may compensate for channel loss over time, and as a result they become more sensitive to low-grade inflammation. TALK-1 KO β-cells are also more sensitive to glucose, displaying higher GSIS at lower glucose concentrations14. Thus, it is possible that the readily releasable insulin granule pool was depleted under these conditions, resulting in decreased GSIS. Interestingly, without cytokines TALK-1 KO islets contained less insulin protein and Ins2 transcript than WT islets even though their GSIS was greater. The enhanced secretory ability of TALK-1 KO islets likely feeds back and limits insulin expression. As insulin content becomes equivalent in WT and TALK-1 KO islets following cytokine exposure, decreased GSIS from TALK-1 KO islets was not likely due to defective insulin expression. While WT islets responded to cytokines with increased secretory capacity, TALK-1 KO islets may have been maximally active and thus not capable of further amplifying GSIS. It is probable that changes in islet GSIS under acute low-grade inflammatory conditions are related to the Ca2+-modulatory function of islet TALK-1 and Kslow currents. By downregulating TALK-1 channels and inhibiting KCa channel activity, normal GSIS is maintained in the short-term.
In conclusion, Kslow currents are reduced during low-grade inflammation resulting in increased β-cell electrical and [Ca2+]i oscillation frequency. Cytokine-induced downregulation of TALK-1 contributes to decreased Kslow activity by reducing [Ca2+]ER release. Importantly, these changes acutely preserve normal β-cell [Ca2+]i levels during 2nd phase GSCI and thus help maintain GSIS during low-grade inflammation. These results also predict that during chronic inflammation loss of TALK-1 impairs β-cell Ca2+ handling resulting in defective GSIS.
Methods
Materials and reagents
Unless otherwise stated all research materials were obtained from Thermo Fisher Scientific and Sigma-Aldrich.
Mouse islet and β-cell isolation and culture
Animals were handled in compliance with the Guide for the care and use of laboratory animals, Eighth edition (2011) (http://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf) and in line with guidelines approved by the Vanderbilt University Animal Care and Use Committee (protocol # M1600063-00). Wild type (WT) and TALK-1 knockout (KO) islets were isolated from the pancreata of age-matched 6- to 12-week old C57B16/J mice as previously described52. Islets were dispersed using 0.0075% trypsin-EDTA and cultured overnight on poly-d-lysine coated 35 mm glass-bottom dishes in RPMI 1640 supplemented with 15% FBS, 100 IU∙ml−1 penicillin, and 100 mg∙ml−1 streptomycin (islet media) at 37 °C, 5% CO2. Alternatively, whole islets were cultured overnight on poly-d-lysine coated glass-bottom dishes in islet media. Unless otherwise specified, after overnight incubation all samples were supplemented with either fresh islet media or islet media with cytokines (0.1 ng/mL TNF-α, 0.05 ng/mL IL-1β, and 10 ng/mL IFN-γ) and used for testing within 48 hrs.
Mouse islet qRT-PCR
WT or TALK-1 KO mouse islets were cultured for 24 hrs at 37 °C, 5% CO2 in 6-cm petri dishes in islet media or in islet media with cytokines. RNA was isolated with TRIzol Reagent as per manufacturer’s instructions. Islet cDNA was prepared from RNA with SuperScript IV Reverse Transcriptase using an S1000 Thermal Cycler (Bio-Rad). Transcript abundance relative to GAPDH was determined by qRT-PCR using primers specific to TALK-1 (KCNK16; forward, 5′-CAG CTC TGG CTG CTC AGT AGG-3′; reverse, 5′-CTC ATG CAG AGA TGG GGA TCT T-3′), SERCA2b (ATP2A2; forward, 5′-AGG GAC TGC AGT GGC TAA GA-3′; reverse, 5′-GCC ACA ATG GTG GAG AAG TT-3′), preproinsulin 1 (Ins1; forward, 5′-AGC AAG CAG GTC ATT GTT CC-3′; reverse, 5′-GAC GGG ACT TGG GTG TGT AG-3′), preproinsulin 2 (Ins2; forward, 5′-TCT TCT ACA CAC CCA TGT CCC-3′; reverse, 5′-GGT GCA GCA CTG ATC CAC-3′), proprotein convertase 1 (PCSK1; forward, 5′-CCT CCT ACA GCA GTG GTG ATT ACA-3′; reverse, 5′-GGG TCT CTG TGC AGT CAT TGT-3′), and GAPDH (forward, 5′-GAG AAT GGG AAG CTT GTC ATC AAC-3′; reverse, 5′-ACT CCA CGA CAT ACT CAG CAC CAG-3′ using iTaq Universal SYBR Green Supermix (Bio-Rad) with a CFX Real time PCR Instrument (Bio-Rad).
Human islet western blotting
Human islets were provided through an approved protocol by the Integrated Islet Distribution Program (IIDP) (https://iidp.coh.org/). The IIDP obtained informed consent for deceased donors in accordance with NIH guidelines prior to reception of human islets for our studies. Work detailed here was approved by the Vanderbilt University Health Sciences Committee Institutional Review Board (IRB# 110164). Islet donor information is provided in Table 2. The islets were cultured overnight in islet media or islet media with cytokines. The islets were washed with ice cold phosphate buffered saline (PBS) and resuspended in ice cold radioimmunoprecipitation buffer (RIPA) with (in mmol/L) 50.0 Tris pH 7.4, 150.0 NaCl, and 1.0 EDTA (Corning) with 1.0 (w/v%) NP-40 and 1x protease inhibitor cocktail (Roche, Penzberg, Germany). Islets were sonicated on ice with a sonic dismembrator model 100, incubated at 4 °C for 30 min, and centrifuged for 10 min at 12,000 RPM, 4 °C. Protein concentration was determined by BCA assay as per manufacturer’s instructions. Samples (40 µg/lane) were resolved under denaturing conditions on a NuPAGE 4–12% Bis-Tris Gel then transferred to a nitrocellulose membrane. The membrane was incubated with rabbit anti-TALK-1 (1:500) overnight at 4 °C then TALK-1 bands were visualized with HRP-conjugated donkey anti-rabbit secondary (1:1500; Jackson ImmunoResearch Laboratories) and SuperSignal™ West Pico Chemiluminescent Substrate with a Bio-Rad ChemiDoc Imaging System.
Table 2.
Human islet donor information. Summary of relevant information for human donors of islets used for immunofluorescence microscopy and immunoblotting.
| Donor | 1 | 2 | 3 |
|---|---|---|---|
| Age | 24 | 41 | 27 |
| BMI | 34.8 | 28.5 | 31.2 |
| Sex | Male | Male | Female |
| Ethnicity | Caucasian | Caucasian | Caucasian |
| HbA1c | 5.4% | 5.3% | 5.6% |
Mouse and human islet immunofluorescent analysis
WT mouse islets or human islets were cultured overnight in islet media or islet media with cytokines. The islets were washed with cold PBS and resuspended in 150 µL of 2.25 mg/mL rat tail collagen I supplemented with 1x DMEM, 200 mM HEPES, and 75 mg/mL sodium bicarbonate on ice. The samples were incubated for 40 min in a 96 well-plate at 37 °C, 5% CO2 to cross-link the collagen mixture. The collagen-embedded islets were fixed for 10 min on ice with 4% paraformaldehyde (PFA, Electron Microscopy Sciences), then the PFA was removed and the gels were transferred to 3 mL of cold PFA in a 12 well plate for 15 min (Corning). After fixation the gels were embedded in paraffin and processed into 5 µm slices. Rehydrated slices were stained with rabbit anti-TALK-1 (1:175) and guinea pig anti-insulin (1:500, Dako) followed by secondary antibodies (1:500 anti-rabbit Alexa Fluor 488 and 1:500 anti-guinea pig Alexa Fluor 647 (Jackson ImmunoResearch Laboratories)). Immunofluorescence was imaged using a Nikon Eclipse TE2000-U microscope equipped with an epifluorescence illuminator (Sutter Instrument Company), a CCD camera (HQ2; Photometrics, Inc), and Nikon Elements software (Nikon, Inc). TALK-1 immunofluorescence was quantified using the ImageJ Fiji image processing pack.
Fura-2 AM imaging of islet glucose-stimulated Ca2+ influx
WT and TALK-1 KO mouse islets were loaded with 2 µM Fura-2-acetoxymethyl ester (AM) for 25 min at 37 °C, 5% CO2. The islets were washed twice with Krebs-Ringer–HEPES buffer (KRHB) with (in mmol/L) 119.0 NaCl, 2.0 CaCl2, 4.7 KCl, 25.0 HEPES, 1.2 MgSO4, 1.2 KH2PO4 (adjusted to pH 7.4 with NaOH) then incubated 20 min in KRHB with 2.0 mM glucose (KRHB-2mM) at 37 °C, 5% CO2. Islet [Ca2+]i was measured every 5 sec as a ratio of Fura-2 AM fluorescence at 340 and 380 nm (F340/F380) with a Nikon Eclipse TE2000-U microscope and the data was analyzed using Nikon Elements software. The islets were perifused at 37 °C with a flow of 2 mL/min KRHB-2mM for 2 min then perifused under identical conditions with KRHB with 11.0 mM glucose (KRHB-11mM) for 30 min.
Fura-2 AM imaging of β-cell [Ca2+]ER
WT and TALK-1 KO mouse islet cells were loaded with Fura-2 AM, washed twice with Ca2+-free KRHB-11mM with 125 µM diazoxide (Enzo), then incubated 10 min in Ca2+-free KRHB-11mM with 125 µM diazoxide. Islet [Ca2+]i was measured every 5 sec as a ratio of Fura-2 AM fluorescence at 340 and 380 nm (F340/F380) with a Nikon Eclipse TE2000-U microscope and the data was analyzed using Nikon Elements software. The cells were perifused at 37 °C with a flow of 2 mL/min KRHB-11mM with 100 µM diazoxide for 2 min then perifused with KRHB-11mM with 100 µM diazoxide and 50 µM cyclopiazonic acid (CPA) for 3 min to release [Ca2+]ER into the cytoplasm. Flow was then stopped and the Fura-2 AM imaging allowed to continue for 15 min.
D4ER imaging of β-cell [Ca2+]ER
WT and TALK-1 KO mouse islets were cultured for 2 hrs with a 1000x multiplicity of inflection (MOI) of the ER-targeted Ca2+ probe D4ER then transferred to islet media in poly-D-lysine coated glass-bottom dishes for 48 hrs at 37 °C, 5% CO2. After 48 hrs, the islet media was replaced with fresh islet media or islet media with cytokines and the islets were cultured an additional 24 hrs at 37 °C, 5% CO2. The islets were washed twice with KRHB-11mM with 100 µM diazoxide then incubated 10 min in KRHB-11mM with 100 µM diazoxide. [Ca2+]ER concentrations were measured every minute as a ratio of D4ER fluorescence at 535 and 475 nm (F535/F475) using a Zeiss Confocal Laser Scanning Microscope equipped with Zeiss ZEN software.
β-cell voltage clamp recordings
WT and TALK-1 KO mouse islet cells were washed twice with KRHB-11mM. A whole-cell patch clamp technique was employed to record TALK-1-like K2P currents in single β-cells using an Axopatch 200B amplifier in voltage clamp mode with pCLAMP10 software (Molecular Devices). Cells were patched in KRBS-11mM, then in order to isolate K2P channel currents flow was switched to Ca2+-free KRHB-11mM with (in mmol/L) 0.2 tolbutamide (MP Biomedicals), 10.0 tetraethylammonium chloride hydrate (TEA), and 1.0 ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA) (adjusted to pH 7.4 with NaOH). Patch electrodes (6–12 MΩ) were filled with voltage clamp intracellular solution (IC) with (in mmol/L) 140.0 KCl, 1.0 MgCl2, 10.0 EGTA, 10.0 HEPES, and 8.0 Mg-ATP (adjusted to pH 7.2 with KOH). For all voltage-clamp recordings the command voltage was held at −80 mV for 15 sec then increased from −120 mV to +60 mV over a period of 1 sec.
β-cell Vm recordings
WT and TALK-1 KO mouse islet clusters were washed twice with KRHB-11mM then incubated in KRHB-11mM for 20 min. Patch electrodes (4–6 MΩ) were filled with Vm IC with (in mmol/L) 140.0 KCl, 1.0 MgCl2, and 5.0 HEPES (adjusted to pH 7.2 with KOH) supplemented with 20 µg/mL amphotericin B. The Vm of individual β-cells within islet clusters (10–20 cells) was recorded in current clamp mode using an Axopatch 200B amplifier with pCLAMP10 software. The electrical activity of patched β-cells was recorded for at least 10 min in KRHB-11mM then flow was changed to KRHB-2mM until action potential (AP) firing ceased for at least 3 min and baseline Vm stabilized. Cells were identified as β-cells if electrical activity ceased with 2 mM glucose.
β-cell Kslow current recordings
Mouse islet cells were washed twice with KRHB-11mM. Single cells were patched in KRBS-11mM and a perforated patch clamp technique was employed to record β-cell Kslow currents using an Axopatch 200B amplifier with pCLAMP10 software. Patch electrodes (6–12 MΩ) were filled with Kslow IC with (in mmol/L) 76.0 K2SO4, 10.0 KCl, 10.0 NaCl, 1.0 MgCl2, and 5.0 HEPES (adjusted to pH 7.35 with KOH). For all Kslow electrophysiological recordings the command voltage was held at −40 mV for 7 sec, ramped between −40 and 20 mV for 5.2 sec (wave form: triangle, train rate: 5 Hz, pulse width: 0.2 sec), and returned to −40 mV for 20 sec.
Islet insulin content and glucose-stimulated insulin secretion
WT and TALK-1 KO mouse islets were cultured overnight in islet media (supplemented with 0.5 mg/mL BSA) or islet media with cytokines at 37 °C, 5% CO2. Islets were cultured in equilibration media (DMEM (no glucose) with 10% FBS, 0.5 mg/mL BSA, 10 mM HEPES, and 0.5 mM CaCl2) supplemented with 5.6 mM glucose for 1 hr at 37 °C, 5% CO2. For insulin secretion, 20 islets were picked on ice into a 24-well plate (Corning) containing 500 µL of secretion media (DMEM (no glucose) with 0.5 mg/mL BSA, 10 mM HEPES, and 0.5 mM CaCl2) supplemented with 2 mM or 11 mM glucose, then insulin secretion was initiated for 1 hr at 37 °C, 5% CO2. After 1 hr, the plate was transferred to ice for 10 min to halt secretion and supernatants were collected in low retention 1.6 mL centrifuge tubes, which were stored at −20 °C until analyzed. Supernatants were supplemented with 1:100 mammalian protease inhibitor cocktail (MPIC). Whole islet insulin was extracted overnight at 4 °C into 400 µL of acid ethanol. Islet secretion supernatants and protein extracts were analyzed by the Vanderbilt Hormone Assay and Analytical Services Core (supported by NIH grants DK059637 and DK020593).
Data Availability
All data are available upon request from the corresponding author.
Electronic supplementary material
Acknowledgements
We would like to thank the Integrated Islet Distribution Program (IIDT) for providing healthy human islets and the Vanderbilt Translational Pathology Shared Resource (supported by NCI/NIH Cancer Center Support Grant 2P30 CA068485-14 and the Vanderbilt Mouse Metabolic Phenotyping Center Grant 5U24DK059637-13) for their assistance in preparing mouse and human islet slices. MTD was supported by the Vanderbilt Integrated Training in Engineering and Diabetes (ITED) grant T32DK101003 and NIH grant DK-097392. In addition, research in the laboratory of DAJ was supported by NIH grant DK-081666, ADA grant 1-17-IBS-024, and a Vanderbilt University Diabetes Research Training Center (DRTC) Pilot and Feasibility grant P60-DK-20593.
Author Contributions
M.T.D. contributed to the design of experiments, collected, analyzed, and interpreted data, and wrote the manuscript. A.M.B., M.K.A., S.C.M., K.L.J., and P.K.D. performed experiments and assisted in data analysis. D.A.J. conceived and designed these studies, oversaw data collection, interpreted results, and helped to write the manuscript. All authors approved the final version of the manuscript. D.A.J. is the guarantor of this work.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-19600-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cnop M, et al. Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes. Diabetes. 2005;54:S97–S107. doi: 10.2337/diabetes.54.suppl_2.S97. [DOI] [PubMed] [Google Scholar]
- 2.Al-Shukaili, A. et al. Analysis of inflammatory mediators in type 2 diabetes patients. International journal of endocrinology2013 (2013). [DOI] [PMC free article] [PubMed]
- 3.Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nature reviews Drug discovery. 2014;13:465–476. doi: 10.1038/nrd4275. [DOI] [PubMed] [Google Scholar]
- 4.Donath MY, Böni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic β-cell in type 2 diabetes. Physiology. 2009;24:325–331. doi: 10.1152/physiol.00032.2009. [DOI] [PubMed] [Google Scholar]
- 5.Dula SB, et al. Evidence that low-grade systemic inflammation can induce islet dysfunction as measured by impaired calcium handling. Cell calcium. 2010;48:133–142. doi: 10.1016/j.ceca.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qureshi FM, Dejene EA, Corbin KL, Nunemaker CS. Stress-induced dissociations between intracellular calcium signaling and insulin secretion in pancreatic islets. Cell calcium. 2015;57:366–375. doi: 10.1016/j.ceca.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramadan JW, Steiner SR, O’Neill CM, Nunemaker CS. The central role of calcium in the effects of cytokines on beta-cell function: implications for type 1 and type 2 diabetes. Cell calcium. 2011;50:481–490. doi: 10.1016/j.ceca.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spranger J, et al. Inflammatory cytokines and the risk to develop type 2 diabetes. Diabetes. 2003;52:812–817. doi: 10.2337/diabetes.52.3.812. [DOI] [PubMed] [Google Scholar]
- 9.Alexandraki K, et al. Inflammatory process in type 2 diabetes. Annals of the New York Academy of Sciences. 2006;1084:89–117. doi: 10.1196/annals.1372.039. [DOI] [PubMed] [Google Scholar]
- 10.King GL. The role of inflammatory cytokines in diabetes and its complications. Journal of periodontology. 2008;79:1527–1534. doi: 10.1902/jop.2008.080246. [DOI] [PubMed] [Google Scholar]
- 11.Hazman Ö, Ovalı S. Investigation of the anti-inflammatory effects of safranal on high-fat diet and multiple low-dose streptozotocin induced type 2 diabetes rat model. Inflammation. 2015;38:1012–1019. doi: 10.1007/s10753-014-0065-1. [DOI] [PubMed] [Google Scholar]
- 12.Maedler K, et al. Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. The Journal of clinical investigation. 2002;110:851–860. doi: 10.1172/JCI200215318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma PR, et al. An islet-targeted genome-wide association scan identifies novel genes implicated in cytokine-mediated islet stress in type 2 diabetes. Endocrinology. 2015;156:3147–3156. doi: 10.1210/en.2015-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vierra NC, et al. Type 2 diabetes-associated K+ channel TALK-1 modulates ß-cell electrical excitability, second-phase insulin secretion, and glucose homeostasis. Diabetes. 2015;64:3818–3828. doi: 10.2337/db15-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gandasi NR, et al. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. The Journal of clinical investigation. 2017;127:2353. doi: 10.1172/JCI88491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobson DA, et al. Calcium‐activated and voltage‐gated potassium channels of the pancreatic islet impart distinct and complementary roles during secretagogue induced electrical responses. The Journal of physiology. 2010;588:3525–3537. doi: 10.1113/jphysiol.2010.190207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stancill, J. S. et al. Chronic β-Cell Depolarization Impairs β-Cell Identity by Disrupting a Network of Ca2+-Regulated Genes. Diabetes, db161355 (2017). [DOI] [PMC free article] [PubMed]
- 18.Düfer M, et al. Enhanced glucose tolerance by SK4 channel inhibition in pancreatic β-cells. Diabetes. 2009;58:1835–1843. doi: 10.2337/db08-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goforth P, et al. Calcium-activated K+ Channels of Mouse β-cells are Controlled by Both Store and Cytoplasmic Ca2+ The Journal of general physiology. 2002;120:307–322. doi: 10.1085/jgp.20028581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Göpel SO, et al. Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β cells. The Journal of general physiology. 1999;114:759–770. doi: 10.1085/jgp.114.6.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanno T, Rorsman P, Göpel S. Glucose‐dependent regulation of rhythmic action potential firing in pancreatic β‐cells by kATP‐channel modulation. The Journal of physiology. 2002;545:501–507. doi: 10.1113/jphysiol.2002.031344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang M, Houamed K, Kupershmidt S, Roden D, Satin LS. Pharmacological properties and functional role of Kslow current in mouse pancreatic β-Cells. The Journal of general physiology. 2005;126:353–363. doi: 10.1085/jgp.200509312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han J, Kang D, Kim D. Functional properties of four splice variants of a human pancreatic tandem-pore K+ channel, TALK-1. American Journal of Physiology: Cell Physiology. 2003;285:C529–C538. doi: 10.1152/ajpcell.00601.2002. [DOI] [PubMed] [Google Scholar]
- 24.Kang D, Kim D. Single-channel properties and pH sensitivity of two-pore domain K+ channels of the TALK family. Biochemical and biophysical research communications. 2004;315:836–844. doi: 10.1016/j.bbrc.2004.01.137. [DOI] [PubMed] [Google Scholar]
- 25.Duprat F, Girard C, Jarretou G, Lazdunski M. Pancreatic two P domain K+ channels TALK‐1 and TALK‐2 are activated by nitric oxide and reactive oxygen species. Journal of physiology. 2005;562:235–244. doi: 10.1113/jphysiol.2004.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickerson MT, Vierra NC, Milian SC, Dadi PK, Jacobson DA. Osteopontin activates the diabetes-associated potassium channel TALK-1 in pancreatic β-cells. PloS one. 2017;12:e0175069. doi: 10.1371/journal.pone.0175069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vierra, N. C. et al. TALK-1 channels control β-cell endoplasmic reticulum Ca2+ homeostasis. Science Signaling10, 10.1126/scisignal.aan2883 (2017). [DOI] [PMC free article] [PubMed]
- 28.Eizirik DL, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012;8:e1002552. doi: 10.1371/journal.pgen.1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang, C., Guan, Y. & Yang, J. Cytokines in the progression of pancreatic β-cell dysfunction. International journal of endocrinology2010 (2010). [DOI] [PMC free article] [PubMed]
- 30.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of neuroscience methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-T. [DOI] [PubMed] [Google Scholar]
- 31.Wang L, et al. A Low Voltage-Activated Ca2+ Current Mediates Cytokine-Induced Pancreatic β-Cell Death. Endocrinology. 1999;140:1200–1204. doi: 10.1210/endo.140.3.6556. [DOI] [PubMed] [Google Scholar]
- 32.Berggren P-O, et al. Removal of Ca2+ channel β 3 subunit enhances Ca2+ oscillation frequency and insulin exocytosis. Cell. 2004;119:273–284. doi: 10.1016/j.cell.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, et al. Long lasting synchronization of calcium oscillations by cholinergic stimulation in isolated pancreatic islets. Biophysical journal. 2008;95:4676–4688. doi: 10.1529/biophysj.107.125088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertram, R., Sherman, A. & Satin, L. S. In The Islets of Langerhans 261–279 (Springer, 2010).
- 35.Arredouani A, Henquin J-C, Gilon P. Contribution of the endoplasmic reticulum to the glucose-induced [Ca2+]c response in mouse pancreatic islets. American Journal of Physiology-Endocrinology and Metabolism. 2002;282:E982–E991. doi: 10.1152/ajpendo.00347.2001. [DOI] [PubMed] [Google Scholar]
- 36.Falcão VTF, et al. Reduced insulin secretion function is associated with pancreatic islet redistribution of cell adhesion molecules (CAMs) in diabetic mice after prolonged high-fat diet. Histochemistry and cell biology. 2016;146:13–31. doi: 10.1007/s00418-016-1428-5. [DOI] [PubMed] [Google Scholar]
- 37.Corbin KL, Waters CD, Shaffer BK, Verrilli GM, Nunemaker CS. Islet hypersensitivity to glucose is associated with disrupted oscillations and increased impact of proinflammatory cytokines in islets from diabetes-prone male mice. Endocrinology. 2016;157:1826–1838. doi: 10.1210/en.2015-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gylfe E, et al. Signaling underlying pulsatile insulin secretion. Upsala journal of medical sciences. 2000;105:35–51. doi: 10.1517/03009734000000054. [DOI] [PubMed] [Google Scholar]
- 39.Satin LS, Butler PC, Ha J, Sherman AS. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Molecular aspects of medicine. 2015;42:61–77. doi: 10.1016/j.mam.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiological reviews. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- 41.Tong X, Kono T, Evans-Molina C. Nitric oxide stress and activation of AMP-activated protein kinase impair β-cell sarcoendoplasmic reticulum calcium ATPase 2b activity and protein stability. Cell death & disease. 2015;6:e1790. doi: 10.1038/cddis.2015.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bravo R, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. International review of cell and molecular biology. 2013;301:215. doi: 10.1016/B978-0-12-407704-1.00005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods in enzymology. 2011;490:71. doi: 10.1016/B978-0-12-385114-7.00004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harbor perspectives in biology. 2011;3:a004317. doi: 10.1101/cshperspect.a004317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cardozo AK, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54:452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 46.Tong X, et al. SERCA2 Deficiency Impairs Pancreatic β-Cell Function in Response to Diet-Induced Obesity. Diabetes. 2016;65:3039–3052. doi: 10.2337/db16-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collier JJ, Fueger PT, Hohmeier HE, Newgard CB. Pro-and antiapoptotic proteins regulate apoptosis but do not protect against cytokine-mediated cytotoxicity in rat islets and β-cell lines. Diabetes. 2006;55:1398–1406. doi: 10.2337/db05-1000. [DOI] [PubMed] [Google Scholar]
- 48.Nagaraj V, et al. Elevated basal insulin secretion in type 2 diabetes caused by reduced plasma membrane cholesterol. Molecular Endocrinology. 2016;30:1059–1069. doi: 10.1210/me.2016-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guillausseau P-J, et al. Abnormalities in insulin secretion in type 2 diabetes mellitus. Diabetes & metabolism. 2008;34:S43–S48. doi: 10.1016/S1262-3636(08)73394-9. [DOI] [PubMed] [Google Scholar]
- 50.Hasnain SZ, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nature medicine. 2014;20:1417–1426. doi: 10.1038/nm.3705. [DOI] [PubMed] [Google Scholar]
- 51.Hostens K, et al. Exposure of human islets to cytokines can result in disproportionately elevated proinsulin release. Journal of Clinical Investigation. 1999;104:67. doi: 10.1172/JCI6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Philipson LH, et al. Delayed rectifier K+ channel overexpression in transgenic islets and beta-cells associated with impaired glucose responsiveness. Journal of Biological Chemistry. 1994;269:27787–27790. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available upon request from the corresponding author.