

Hereditary spastic paraplegias (HSPs) and ataxias are genetically heterogeneous disorders, with more than 70 genes implicated in each group. A smaller fraction of disorders from both groups manifest both with spastic paresis and ataxia, and recognizing this phenotype helps narrowing down the differential diagnosis.1 Recently, homozygous and compound heterozygous mutations in CAPN1, which encode for the neuronal cysteine protease calpain, have been described as a cause of HSP (SPG76, MIM#616907).2 Here, we report 3 patients from 2 families with homozygous CAPN1 mutations who are characterized with slowly progressive lower limb spasticity with mild ataxia. Review of all patients with CAPN1 mutations so far supports the strong association of cerebellar involvement with this disorder and delineates several additional disease characteristics.
Family 1
A 37-year-old woman was referred to our laboratory with spastic ataxia. Her symptoms started with gait difficulty at 21 years, followed by dysarthria 8 years later, and eventually, she was unable to walk without aid at age 32 years (table and figure e-1, http://links.lww.com/NXG/A23). Neurologic examination revealed dysarthria, spasticity in lower >> upper extremities, mild weakness in proximal leg muscles, Achilles tendon contracture, mild dysmetria, dysdiadochokinesia, and a spastic-ataxic gait. Cranial MRI and nerve conduction studies were unremarkable.
Table.
Clinical features and mutations of 3 Turkish patients with CAPN1 mutations
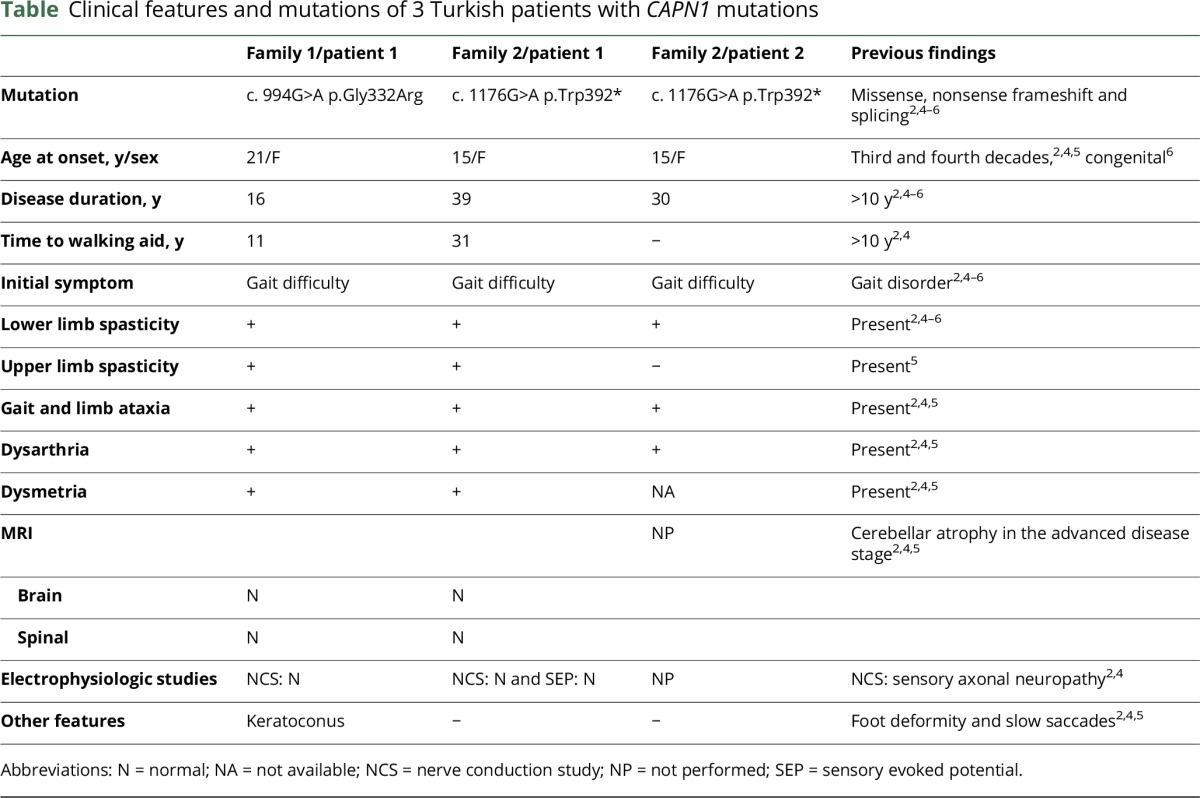
Whole-exome sequencing revealed a homozygous p.Gly332Arg mutation in the CAPN1 gene (e-Methods, http://links.lww.com/NXG/A26). The variation is located at a conserved site, with a GERP++ score of 4.63 (figure e-2, http://links.lww.com/NXG/A24), next to the start of the β-strand of the protease domain of calpain-1 protein, conformationally in proximity to the active site at position 296, the critical Ca2+ binding site (figure e-2).3 The variation was not present in the Exome Aggregation Consortium (ExAC) browser and was seen twice in a heterozygous state in the Genome Aggregation Database (gnomAD) (minor allele frequency [MAF] = 8.204e-6).
Family 2
A 54-year-old female proband presented with slowly progressive walking difficulty and extreme stiffness in the legs (table). She could not tolerate exercise since teen age, but her symptoms started to affect her daily life after her thirties. She had a scissoring and wide-based gait and needed unilateral support to walk. Her speech was explosive. Neurologic examination revealed severe spasticity in the lower and mild spasticity in the upper extremities, hyperactive deep tendon reflexes, bilateral extensor plantar responses, and patellar clonus. The Hoffmann sign was positive. She had bilateral mild dysmetria and dysdiadochokinesia. Nerve conduction studies, EMG, sensory evoked potential tests, and MRI did not reveal any pathology (figure e-3, http://links.lww.com/NXG/A25). Her parents were second-degree cousins, and one of her younger sisters had similar complaints and also a slow disease progression (figure e-1, http://links.lww.com/NXG/A23).
Among 18 homozygous variants shared by the patient and her affected sister, a single nucleotide substitution in exon 10 of the CAPN1 gene leading to a premature stop codon at position 392 was detected (e-Methods, http://links.lww.com/NXG/A26, figure e-2, http://links.lww.com/NXG/A24). The p.Trp392* variant was seen heterozygously once in the ExAC database (MAF = 8.513e-06) and was not present in the gnomAD.
Discussion
We report 3 adult-onset patients from 2 families who have disease onset with spastic paraparesis, emergence of cerebellar symptoms during disease course, slow disease progression, and homozygous mutations in the CAPN1 gene. These characteristics are in accordance with the previous reports and point out to a characteristic phenotype.2,4,5 The first study reported 8 patients from 3 families with pyramidal signs starting during young adulthood (range 19–39 years). All except for 1 patient had cerebellar findings during follow-up, and 1 patient required walking aid at the age of 40.2 In the second study, 6 patients from 4 families had slowly progressive spastic ataxia starting at young adulthood. One patient was using a wheelchair at the age of 30, and 2 patients needed walking aid in their forties.4 In a third report, 2 patients had a similar spastic-ataxia phenotype with onset around thirties.5 It is important that the range of age at onset was expanded by a recent report of a 16-year-old patient with congenital onset and pure spastic paraplegia.6
Our study, together with the rapidly expanding number of reported cases, validates the observation that homozygous or compound heterozygous mutations in calpain-1 cause a spasticity-dominant phenotype and emphasize its association with cerebellar symptoms. Accordingly, calpain-1 deficiency in mice disrupts cerebellar development causing cerebellar ataxia, and missense mutations in CAPN1 are associated with spinocerebellar ataxia in Parson Russell Terrier dogs.4,7 The cerebellar findings in CAPN1 patients reported so far include ataxic gait, dysarthria, and mild dysmetria/dysdiadochokinesia. Slow saccades were remarkable in 2 patients.2,4–6 In light of these observations, we suggest that CAPN1-based disease should be considered among the emerging group of ataxia-spasticity spectrum disorders, especially in adult patients.1
Author contributions
Cemile Kocoglu performed the analyses of whole-exome sequencing data and compiled the manuscript. Asli Gundogdu screened for repeat expansions in ataxia and performed the validation and segregation experiments. Gulsen Kocaman, Pinar Kahraman-Koytak, Kayihan Uluc, and Gunes Kiziltan evaluated the patients. Ahmet Okay Caglayan, and Kaya Bilguvar performed the analyses of whole-exome sequencing data. Atay Vural contributed to the manuscript. A. Nazli Basak designed the study and worked on the manuscript.
Study funding
Supported by the Suna and Inan Kirac Foundation and Bogazici University Research Fund.
Disclosure
C. Kocoglu has received research support from the Suna and Inan Kirac Foundation. A. Gündogdu, G. Kocaman, and P. Kahraman-Koytak report no disclosures. K. Uluc is an employee of the Marmara University Faculty of Medicine, Department of Neurology. G. Kiziltan reports no disclosures. A.O. Caglayan has served on the editorial boards of Annals of Molecular and Genetic Medicine, Annals of Neurology and Neurosciences, Austin Journal of Autism and Related Disorders, Austin Journal of Clinical Neurology, and Frontiers in Child and Neurodevelopmental Psychiatry. K. Bilguvar reports no disclosures. A. Vural has received research support from the Scientific and Technological Research Council of Turkey (TUBITAK), the European Academy of Neurology, the Alexander von Humboldt Foundation, and the Georg-Forster Experienced Researcher Fellowship. A.N. Basak reports no disclosures. Funding information and disclosures are provided at the end of the article. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Synofzik M, Schüle R. Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord 2017;0:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gan-Or Z, Bouslam N, Birouk N, et al. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am J Hum Genet 2016;98:1038–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khorchid A, Ikura M. How calpain is activated by calcium. Nat Struct Biol 2002;9:239–241. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Hersheson J, Lopez D, et al. Defects in the CAPN1 gene result in alterations in cerebellar development and cerebellar ataxia in mice and humans. Cell Rep 2016;16:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tadic V, Klein C, Hinrichs F, et al. CAPN1 mutations are associated with a syndrome of combined spasticity and ataxia. J Neurol 2017;264:1008–1010. [DOI] [PubMed] [Google Scholar]
- 6.Travaglini L, Bellacchio E, Aiello C, et al. Expanding the clinical phenotype of CAPN1-associated mutations: a new case with congenital-onset pure spastic paraplegia. J Neurol Sci 2017;378:210–212. [DOI] [PubMed] [Google Scholar]
- 7.Forman OP, De Risio L, Mellersh CS. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS One 2013;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]