

Abstract
Objective
Immune myopathies with perimysial pathology (IMPP) have a combination of damage to perimysial connective tissue and muscle fiber necrosis, more prominent near the perimysium. We studied the clinical and laboratory correlates of patients with pathologically defined IMPP.
Methods
This is a retrospective chart and pathology review of 57 consecutive patients with IMPP myopathology and, for comparison, 20 patients with dermatomyositis with vascular pathology (DM-VP).
Results
Compared with DM-VP, IMPP patients more commonly had interstitial lung disease (ILD) (p < 0.01), Raynaud phenomenon (p < 0.05), mechanic's hands (p < 0.05), arthralgias (p < 0.001), and a sustained response to immunomodulatory therapy (p < 0.05), and less frequently had a concurrent malignancy (p < 0.01). IMPP patients had higher serum creatine kinase values (p < 0.05), more frequent serum Jo-1 (p < 0.03) or SSA/SSA52 autoantibodies (p < 0.05), and less frequent antinuclear antibodies (p < 0.01). IMPP patients with serum Jo-1/antisynthetase antibodies were more likely to have ILD (p < 0.05) and inflammatory arthritis (p < 0.05) than IMPP patients without these antibodies.
Conclusions
IMPP myopathology is associated with an increased risk of ILD, Raynaud phenomenon, mechanic's hands, and inflammatory arthritis when compared with another immune myopathy (DM-VP). IMPP patients require regular screening for ILD, particularly those with antisynthetase antibodies. The absence of myositis-specific autoantibodies in a large percentage of IMPP patients emphasizes the important role for myopathology in identifying patients at higher risk of severe comorbid conditions such as ILD.
Acquired immune and inflammatory myopathies (IIMs) are a heterogeneous group of disorders. Classification schemes have been based on clinical, autoantibody, or myopathologic features.1–5 Serum antibodies to aminoacyl-tRNA synthetases are associated with a multisystem syndrome that includes IIM. Clinical manifestations include myopathy, interstitial lung disease (ILD), arthritis, Raynaud phenomenon, and skin rash. The commonest antisynthetase antibody, anti-Jo-1, is directed against histidyl tRNA synthetase. Myopathology in patients with anti-Jo-1 antibodies includes damage to perimysial connective tissue and muscle fibers.4,6,7 Perimysial connective tissue pathology includes damaged structures with fragmentation and scattered histiocytic cells. Muscle fiber pathology includes necrosis and regeneration, more prominent in regions neighboring the perimysium. We have termed myopathies with this combined pattern of damage, immune myopathies with perimysial pathology (IMPP).4 IMPP also occurs with serum antibodies to hydroxy-methyl-glutaryl-coenzyme A reductase (HMGCR),8 a nonaminoacyl-tRNA synthetase antigen. Other patients with IMPP have no associated myositis-specific antibodies (MSA).
This study examined clinical and laboratory features of a cohort of consecutive patients with IMPP evaluated at our institution. We compared IMPP patients with a cohort of patients with a different immune myopathy syndrome, dermatomyositis with vascular pathology (DM-VP). DM-VP differs pathologically from IMPP, as it is a myovascular disorder. Muscle fiber changes are generally atrophy, rather than the necrosis seen in IMPP.4,9 Our results show that IMPP is associated with myopathy syndromes having multisystem features involving muscle, lungs, skin, and joints.
Methods
Patients
We retrospectively reviewed charts, laboratory data, and muscle biopsies from 57 consecutive patients with muscle biopsies interpreted as having an IMPP (table 1 and figure) who had been clinically evaluated at Washington University School of Medicine in Saint Louis between 1990 and 2013. IMPP biopsies had acid phosphatase positive cells in perimysial connective tissue and evidence of myopathy defined as one of the following: abnormal variation in the muscle fiber size, necrosis or regeneration, or major histocompatibility complex (MHC) Class I upregulation by muscle fibers. Some patients in this group were previously reported.6,10 For comparison, we reviewed a consecutive series of 20 patients with biopsies interpreted as DM-VP (Table 1 and figure). DM-VP biopsies had vascular pathology, with abnormal endomysial capillaries or perivascular lymphocytic foci and perifascicular myopathy with muscle fiber atrophy, reduced cytochrome oxidase staining, or increased MHC Class I expression.
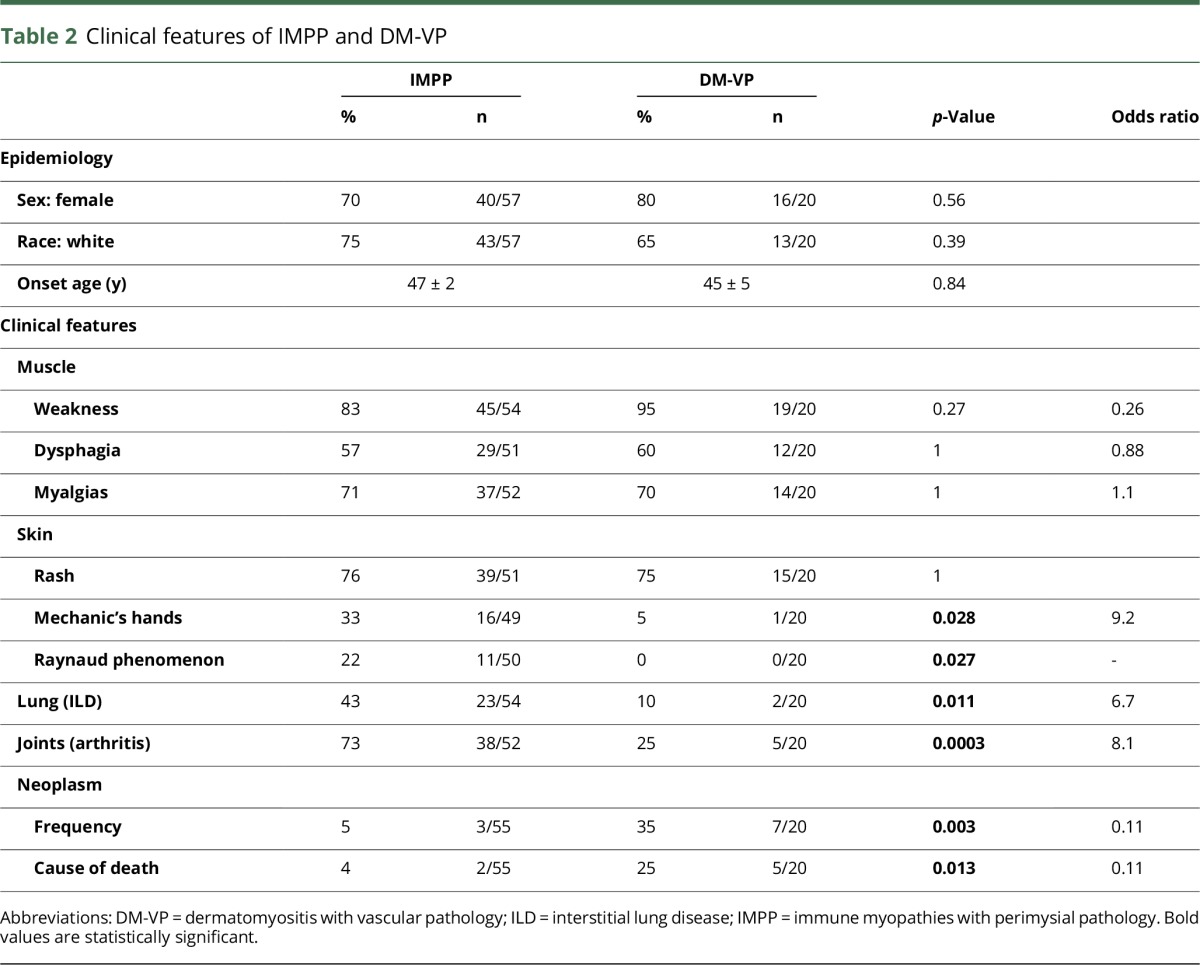
Table 1.
Myopathology: IMPP and DM-VP groups
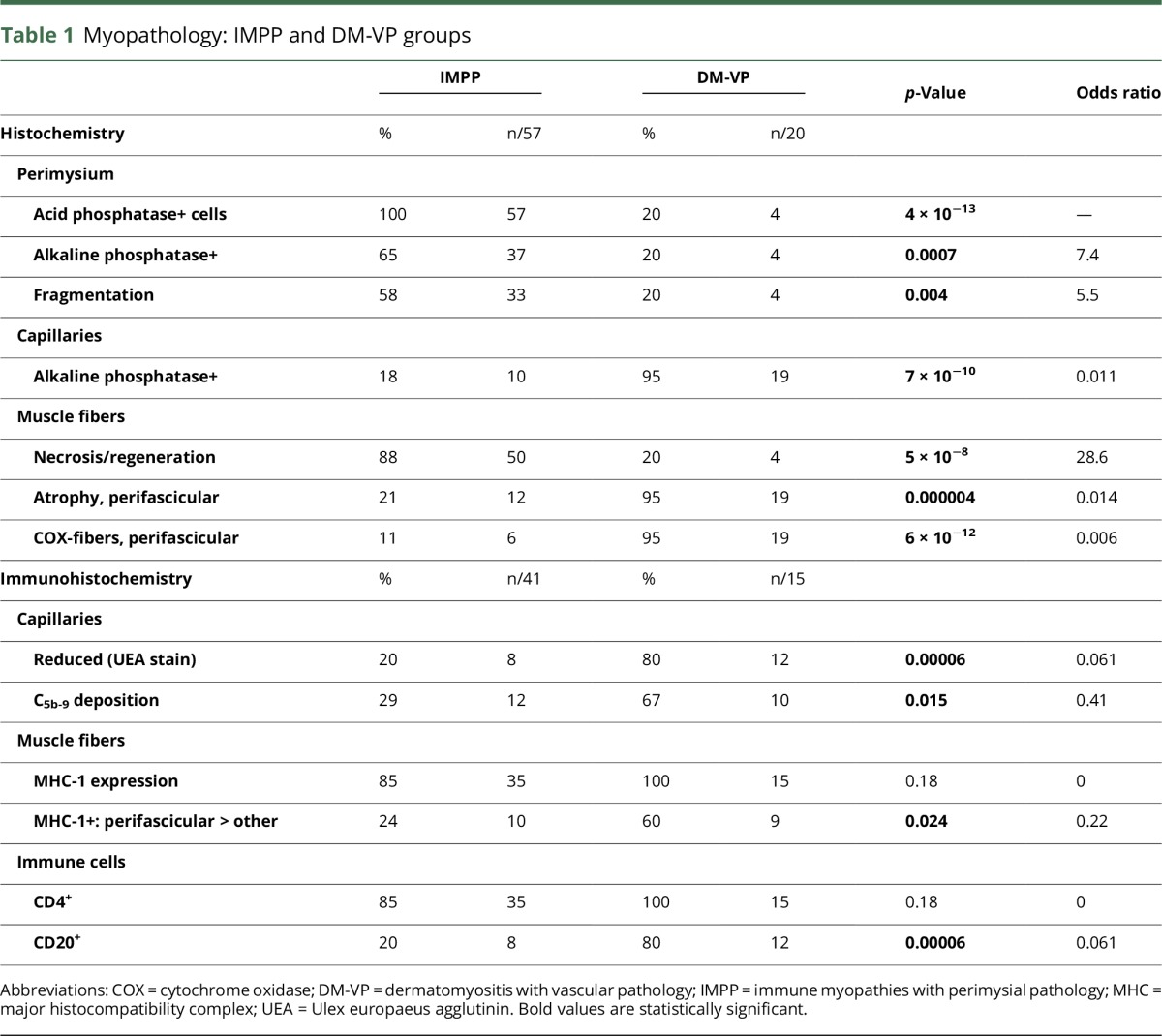
Figure. Comparative myopathology of immune myopathies with perimysial pathology (IMPP) and dermatomyositis with vascular pathology (DM-VP).

IMPP myopathology includes perimysium that is widened and damaged (A; H&E stain), occupied by histiocytic cells (C; acid phosphatase stain), and stained by alkaline phosphatase (E). Muscle fiber damage includes necrosis, (membrane attack complex [C5b-9] deposited in fiber cytoplasm [G]), more prominent near the perimysium. DM-VP myopathology includes muscle fiber pathology with perifascicular atrophy without necrosis (B; H&E stain) and metabolic changes, including reduced staining on cytochrome oxidase (H). DM-VP vascular pathology includes perivascular (perimysial) lymphocytic inflammation (D; Congo red stain) and endomysial capillary changes with increased alkaline phosphatase staining (F). Scale bar = 100 μM (Panels A–H).
Definition of clinical features
ILD was defined by imaging features, pulmonary function tests, biopsy data, and/or expert opinion of a pulmonologist. Inflammatory arthritis was defined by imaging features and/or expert opinion of a rheumatologist. Dermatologic features were defined by clinical description (i.e., mechanic's hands, Gottron sign), biopsy data, and/or expert opinion of a dermatologist. Myalgias and Raynaud phenomenon were defined by the clinicians' documentation as present or absent. Dysphagia was defined by objective testing (modified barium swallow or endoscopic evaluation of swallowing) and/or expert opinion of a speech therapist. Paraneoplastic syndromes were defined as a concurrent cancer diagnosis within a 5-year period preceding or subsequent to the myopathy diagnosis.
Laboratory data
The initial serum creatine kinase and aldolase values (IU/L) are reported. Panels of serum MSA were evaluated by clinical laboratories in Oklahoma or California. HMGCR antibodies were tested at Washington University in St. Louis.8 Electromyographic evidence of an “irritable” myopathy was defined as the presence of increased insertional or spontaneous activity (in the form of fibrillations, positive sharp waves, or myotonic/repetitive discharges), motor unit potentials of reduced amplitude and duration, and early recruitment.
Histochemical and immunohistochemical evaluation
Cryostat sections of rapidly frozen muscle were processed as previously described.6,11 Immunohistochemical stains were performed on muscle cryosections with paired controls on the same glass slide. Primary antibodies used in this study were CD4, CD20, C5b-9 complement (membrane attack complex) (Sigma-Aldrich, St. Louis, MO), and class I human MHC (US Biological, Swampscott, MA). Ulex europaeus agglutinin I lectin (UEA, Sigma-Aldrich) was used to visualize the vascular endothelium. In most cases, the pathologists interpreting biopsies (A.P. and R.C.B.) were blinded to results of MSA testing.
Statistical analysis
The Fisher exact test was used to compare categorical variables. Independent 2-tailed t-tests were used to compare and report quantitative variables. A p value <0.05 was considered statistically significant between groups. Because this was a retrospective exploratory study, we did not correct for multiple comparisons. When clinical or laboratory data were unavailable, the subject was not included in the analysis for that feature.
Standard protocol approvals, registrations, and patient consents
The Human Studies Committee of Washington University in Saint Louis approved all procedures.
Results
Pathology of IMPP and DM-VP
IMPP patients were chosen based on the presence of perimysial pathology, highlighted by acid phosphatase positive perimysial cells and alkaline phosphatase staining of the perimysium, and these features were less common in the DM-VP group (table 1; figure). DM-VP patients were chosen based on the presence of vascular pathology, as defined by any of the following features, lymphocytic foci surrounding larger vessels in vascular perimysium, alkaline phosphatase staining or C5b-9 deposition on endomysial capillaries, or loss of endomysial capillaries on UEA, and these features were less common in the IMPP group. Muscle fiber necrosis and regeneration, features not part of the selection process, were more common in the IMPP group. The most common pathologic change in muscle fibers in the DM-VP group was atrophy. Lymphocyte foci containing CD20 B-cells were more common in DM-VP. CD8 positive T-lymphocytes were uncommon in both DM-VP and IMPP.
Clinical features of IMPP and DM-VP
Both IMPP (70%) and DM-VP (80%) groups were predominantly female. Onset ages varied widely in both IMPP (16–83 years) and DM-VP (7–78 years) groups (table 2). There were no differences in age, sex, race, or frequencies of weakness, dysphagia, and skin rash comparing the IMPP and DM-VP groups. Systemic features common in anti-Jo-1 antibody syndromes were all more common in IMPP patients than DM-VP patients. Features more common in IMPP included mechanic's hands, Raynaud phenomenon, ILD, joint disorders, and serum antisynthetase antibodies. IMPP patients were less likely to have an associated malignancy or to die of a malignancy, compared with DM-VP. Causes of death in the IMPP group were ILD (2), neoplasm (2), and congestive heart failure (1). All deaths (5) in the DM-VP group were associated with neoplasm. ILD and arthritis were more common in IMPP patients with antisynthetase antibodies compared to those without antisynthetase antibodies (ILD = 63% vs 26%, respectively, p = 0.03; arthritis = 94% and 61%, respectively, p = 0.03). Arthritis was also more common in antisynthetase antibody-negative IMPP compared with DM-VP (61% vs 25%, p = 0.03). ILD showed a trend toward greater frequency in antisynthetase antibody-negative IMPP compared with DM-VP (26% and 10%, respectively), but the difference was not statistically significant. Other clinical, laboratory, electrodiagnostic, and myopathologic features were similar in antisynthetase antibody-negative and antibody-positive IMPP subgroups (data not shown).
Table 2.
Clinical features of IMPP and DM-VP
Laboratory features of IMPP and DM-VP
Serum creatine kinase and aldolase were 8-fold and 4-fold higher (p = 0.04) in the IMPP group (table 3), consistent with the muscle fiber necrosis and regeneration that is common in IMPP but rare in DM-VP (Table 1 and Figure A-H). Jo-1 (p < 0.03) and SSA (p < 0.05) antibodies were more common in IMPP patients. Forty-six patients in the IMPP cohort and 17 patients in the DM-VP underwent MSA testing. In addition to Jo-1 and HMGCR, the only other MSA present in the IMPP cohort were EJ (2) and PL-12 (1), whereas Mi-2 (1) was the only additional MSA identified in the DM-VP cohort. ANA was more commonly positive in DM-VP than IMPP patients. C-reactive protein and erythrocyte sedimentation rate values did not differ between the 2 groups. Electromyograms showing “irritable” myopathy were similarly frequent in the 2 groups.
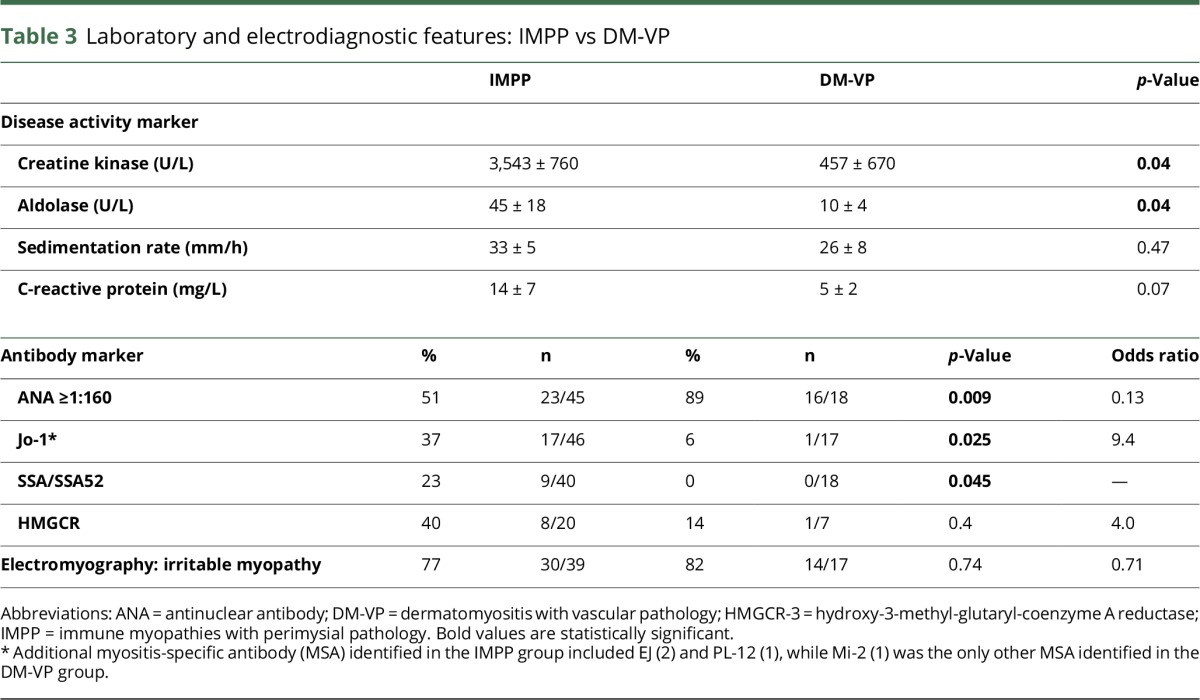
Table 3.
Laboratory and electrodiagnostic features: IMPP vs DM-VP
Response to treatment.
The myopathy in all but 1 IMPP patient (98%) had a sustained beneficial response to immunomodulatory therapies, as measured by strength testing, improvement/resolution of myalgias, and/or reduction in muscle enzymes. Immunomodulatory therapies used in the IMPP group included oral corticosteroids (n = 47); oral methotrexate (n = 25); pulse intravenous methylprednisolone (n = 13); azathioprine (n = 12); mycophenolate mofetil (n = 7); rituximab (n = 7); and intravenous immunoglobulin (n = 4). The myopathy in DM-VP patients was less responsive to immunomodulatory therapy with only 14 (70%) subjects demonstrating a sustained beneficial response (p = 0.002). The immunomodulatory therapies used in the DM-VP cohort were comparable to those used in the IMPP cohort. Five of the 6 nonresponders in the DM-VP cohort had paraneoplastic myopathies, suggesting that this factor likely accounted for the difference in treatment responsiveness between groups.
Discussion
We have proposed that IIMs are usefully classified according to patterns of involvement of different tissues in muscle and types of cellular and humoral immune features.4 In this study, we compared patients with IMPP, whose distinctive myopathologic features include damage to perimysial connective tissue, muscle fiber necrosis, and histiocytic inflammatory cellularity, to DM-VP, a disorder with large and small vessel damage, muscle fiber atrophy, but not necrosis, and lymphocytic perivascular inflammation (Table 1; figure). The distinctive pathologic features of IMPP and DM-VP in this study are consistent with previously reported patterns.4,6,7,9,12
Several systemic and laboratory features of patients with IMPP differed from DM-VP. IMPP patients more often had joint disorders and ILD. Neoplasm occurrence and cancer-related deaths were less frequent in IMPP. The type of skin pathology differed between the groups. Mechanic's hands and Raynaud phenomenon were nearly exclusive to IMPP. Serum Jo-1 antibodies were also more frequent in IMPP (37% vs 6% in DM-VP).
Clinical syndromes in patients with IMPP myopathology showed some features that were also common in DM-VP and immune myopathies in general. These features included female predominance, wide variability in age at onset, weakness that is proximal greater than distal and symmetric, dysphagia, and myalgias. The disease course was subacute or chronic, with durations over months to years.
The frequency of skin disorders was similar in IMPP and DM-VP groups. Clinically, approximately 75% of both groups would have been characterized as dermatomyositis by commonly used criteria. Differences in muscle pathology suggest that immune dermatomyopathies may actually comprise several distinct clinical and pathologic syndromes. IMPP dermatomyopathy syndromes seem likely to be due to connective tissue damage, while DM-VP is probably a vascular disorder. Another dermatomyopathy syndrome, regional ischemic immune myopathy (RIIM), has prominent vascular pathology with muscle fiber necrosis occurring in border-zone regions of muscle, a different pattern from IMPP and DM-VP.13 Both of these vascular dermatomyopathies (RIIM and DM-VP) are associated with an increased frequency of neoplasms and likely account for a substantial proportion of the established association between dermatomyositis (as defined by other criteria) and malignancy.14,15 Overall, it is likely that the dermatomyopathies, or “dermatomyositis,” should be considered a group of disorders with varying comorbid conditions beyond skin and muscle, rather than a single homogeneous disease.
IMPP muscle pathology is associated with a consistent multisystem disorder, involving muscle, skin, lungs and joints, which is similar to that associated with serum anti-Jo-1, and, more generally, antisynthetase, antibodies. However, most of our patients did not have antisynthetase antibodies, rendering muscle biopsy considerably more sensitive for defining these syndromes than serum antibody testing.
Although antisynthetase antibodies commonly occur in a subset of IMPP, the relationship of these antibodies directed against intracellular antigens to disease pathogenesis remains unclear. The pattern of muscle pathology suggests that the antigenic targets in IMPP are likely related to connective tissue, which could explain the pattern of multisystem involvement. Acid phosphatase–positive perimysial histiocytes in IMPP are a notable myopathologic difference from the perivascular lymphocytes present in DM-VP. It is possible that these acid phosphatase positive histiocytes, a signature feature of IMPP, are ultimately responsible for the muscle fiber necrosis typical of this condition, perhaps via cytokine-mediated pathways different from those associated with muscle fiber atrophy and regeneration in other forms of immune myopathy.16 It is well established in hereditary disorders that abnormalities in connective tissue proteins can produce severe myopathies.17–20 Further search for the specific tissue autoantigens (possibly in connective tissue), and how perimysial pathology leads to muscle fiber necrosis, in IMPP will be important.
Our results are the outcome of a retrospective study of muscles from patients, most of whom presented to neurologists with primary complaints of weakness. Prospective studies would be useful to test our findings that IMPP is associated with distinctive multisystem disorders in adults and children. Studies of the frequency and types of IMPP myopathy and lung and skin pathology in patients presenting with ILD or skin disorders could provide further information about systemic immune disorders affecting connective tissue. Larger series could also evaluate whether antisynthetase and other serum autoantibodies in IMPP patients such as anti-HMGCR are associated with specific subgroups of pathologic changes and clinical system involvement. A limitation of our study, given its retrospective nature, is that we could not guarantee that diagnostic testing, including MSA testing, was uniform across all subjects. While over 80% of the subjects in both cohorts did undergo some form of MSA testing, this study includes subjects evaluated over a 24-year period that coincided with a substantial increase in the number of myositis-specific autoantibodies available for clinical testing. Future prospective studies, ideally with serum banking to allow for retrospective testing of newly identified autoantibodies, are ultimately necessary to determine the actual prevalence of MSA in these disorders.
In any case, IMPP syndromes appear to be a distinctive subgroup of disorders that do not fit well into the current classifications of immune myopathies as polymyositis and dermatomyositis. The absence of myositis-specific autoantibodies in a large percentage of IMPP patients emphasizes an important role for myopathology in identifying these patients at an increased risk of ILD and distinguishing them from IIM patients with an increased risk of malignancy.
Glossary
- DM-VP
dermatomyositis with vascular pathology
- HMGCR
hydroxy-methyl-glutaryl-coenzyme A reductase
- IIM
immune and inflammatory myopathy
- ILD
interstitial lung disease
- IMPP
immune myopathies with perimysial pathology
- MHC
major histocompatibility complex
- MSA
myositis-specific antibody
- RIIM
regional ischemic immune myopathy
- UEA
Ulex europaeus agglutinin
Author Contributions
Dr. Bucelli: designed the study, performed the data collection/analysis and statistical analysis, and wrote the paper. Dr. Pestronk: designed the study, performed the data collection/analysis, and wrote the paper.
Study Funding
No targeted funding reported.
Disclosure
A. Pestronk holds patents and receives payments for TS-HDS antibody, GALOP antibody, GM1 ganglioside antibody, and sulfatide antibody; received speaker honoraria from Athena; received research support from Genzyme, Insmed, Knopp, Ultragenyx, ISIS, Sanofi, Cytokinetics, GSK, Biogen, CSL Behring, NIH, Washington University Neuromuscular Research Fund, CINRG Children's Hospital Washington DC, and Muscular Dystrophy Association; holds stock in Johnson & Johnson; receives payments for antibody testing from Athena; and served on the advisory board of and received travel funding from Myositis Foundation. R.C. Bucelli receives an annual gift from a patient's family for Parsonage-Turner research; served one time on the advisory board of MT Pharma; and has equity in Neuroquestions, LLC. Go to Neurology.org/nn for full disclosure forms.
References
- 1.Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015;372:1734–1747. [DOI] [PubMed] [Google Scholar]
- 2.De Bleecker JL, De Paepe B, Aronica E, et al. 205th ENMC International Workshop: pathology diagnosis of idiopathic inflammatory myopathies part II 28–30 March 2014, Naarden, The Netherlands. Neuromuscul Disord 2015;25:268–272. [DOI] [PubMed] [Google Scholar]
- 3.Danielsson O, Lindvall B, Gati I, Ernerudh J. Classification and diagnostic investigation in inflammatory myopathies: a study of 99 patients. J Rheumatol 2013;40:1173–1182. [DOI] [PubMed] [Google Scholar]
- 4.Pestronk A. Acquired immune and inflammatory myopathies: pathologic classification. Curr Opin Rheumatol 2011;23:595–604. [DOI] [PubMed] [Google Scholar]
- 5.Zong M, Lundberg IE. Pathogenesis, classification and treatment of inflammatory myopathies. Nat Rev Rheumatol 2011;7:297–306. [DOI] [PubMed] [Google Scholar]
- 6.Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000;68:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mescam-Mancini L, Allenbach Y, Hervier B, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain 2015;138:2485–2492. [DOI] [PubMed] [Google Scholar]
- 8.Alshehri A, Choksi R, Bucelli R, Pestronk A. Myopathy with anti-HMGCR antibodies: perimysium and myofiber pathology. Neurol Neuroimmunol Neuroinflamm 2015;2:e124 doi: 10.1212/NXI.0000000000000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pestronk A, Schmidt RE, Choksi R. Vascular pathology in dermatomyositis and anatomic relations to myopathology. Muscle Nerve 2010;42:53–61. [DOI] [PubMed] [Google Scholar]
- 10.Nozaki K, Pestronk A. High aldolase with normal creatine kinase in serum predicts a myopathy with perimysial pathology. J Neurol Neurosurg Psychiatry 2009;80:904–908. [DOI] [PubMed] [Google Scholar]
- 11.Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002;73:420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stenzel W, Preusse C, Allenbach Y, et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology 2015;84:1346–1354. [DOI] [PubMed] [Google Scholar]
- 13.Cai C, Alshehri A, Choksi R, Pestronk A. Regional ischemic immune myopathy: a paraneoplastic dermatomyopathy. J Neuropathol Exp Neurol 2014;73:1126–1133. [DOI] [PubMed] [Google Scholar]
- 14.Andras C, Ponyi A, Constantin T, et al. Dermatomyositis and polymyositis associated with malignancy: a 21-year retrospective study. J Rheumatol 2008;35:438–444. [PubMed] [Google Scholar]
- 15.Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis 2007;66:1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arouche-Delaperche L, Allenbach Y, Amelin D, et al. Pathogenic role of anti-signal recognition protein and anti-3-hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol 2017;81:538–548. [DOI] [PubMed] [Google Scholar]
- 17.Lampe AK, Flanigan KM, Bushby KM, Hicks D. Collagen type VI-related disorders. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R). Seattle, WA: University of Washington: 1993. [Google Scholar]
- 18.Muntoni F, Torelli S, Wells DJ, Brown SC. Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies. Curr Opin Neurol 2011;24:437–442. [DOI] [PubMed] [Google Scholar]
- 19.Sparks SE, Quijano-Roy S, Harper A, et al. Congenital Muscular dystrophy Overview. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews(R). Seattle, WA: University of Washington: 1993. [Google Scholar]
- 20.Stum M, Davoine CS, Fontaine B, Nicole S. Schwartz-Jampel syndrome and perlecan deficiency. Acta Myol 2005;24:89–92. [PubMed] [Google Scholar]