

Abstract
Background and Purpose
SQSTM1/p62 is a multifunctional, stress‐induced, scaffold protein involved in multiple cellular processes including autophagic clearance, regulation of inflammatory responses and redox homeostasis. Its altered function has been associated with different human pathologies, such as neurodegenerative, metabolic and bone diseases (down‐regulation), and cancerogenesis (up‐regulation). However, its role in the off‐target effects of clinically used drugs is still not understood.
Experimental Approach
We evaluated the expression of p62 in cultured Hep3B cells and their derived ρ° cells (lacking mitochondria), along with markers of autophagy and mitochondrial dysfunction. The effects of efavirenz were compared with those of known pharmacological stressors, rotenone, thapsigargin and CCCP, and we also used transient silencing with siRNA and p62 overexpression. Western blotting, quantRT‐PCR and fluorescence microscopy were used to assay these effects and their underlying mechanisms.
Key Results
In Hep3B cells, efavirenz augmented p62 protein content, an effect not observed in the corresponding ρ° cells. p62 up‐regulation followed enhanced SQSTM1 expression mediated through the transcription factor CHOP/DDIT3, while other well‐known regulators (NF‐kB and Nrf2) were not involved. Inhibition of autophagy with 3MA or with transient silencing of Atg5 did not affect SQSTM1 expression in efavirenz‐treated cells while p62 overexpression ameliorated the deleterious effect of efavirenz on cell viability.
Conclusion and Implications
In our model, p62 exerted a specific, autophagy‐independent role and protected against efavirenz‐induced mitochondrial ROS generation and activation of the NLRP3 inflammasome. These findings add to the multifunctional nature of p62 and may help to understand the off‐target effects of clinically useful drugs.
Abbreviations
- ΔΨm
mitochondrial membrane potential
- CCCP
carbonyl cyanide 3‐chlorophenylhydrazone
- CHOP
C/EBP homologous protein
- EFV
efavirenz
- HIV
human immunodeficiency virus
- Nrf2
nuclear factor erythroid 2 [NF‐E2]‐related factor 2
- Tg
thapsigargin
Introduction
Sequestosome 1 (SQSTM1)/p62/A170 or ZIP3 (referred to in this article as p62) is a stress‐induced 62‐kDa protein expressed in most tissues and encoded by an immediate‐early response gene activated by a variety of extracellular signals involved in cell proliferation, differentiation and, particularly, oxidative stress (Ishii et al., 1997). It displays many biological activities and can be considered a complex metabolic hub (Katsuragi et al., 2015). In recent years, evidence has been growing of its involvement in human pathologies including Alzheimer's disease (Ramesh Babu et al., 2008) and Parkinson's disease (Nakaso et al., 2004), bone diseases (Laurin et al., 2002) and neuronal disorders (Rea et al., 2014). Enhanced p62 activity has been linked to tumourigenesis both due to its over‐regulation (Duran et al., 2008; Puissant et al., 2012) and by its abnormal accumulation via impaired autophagy (Mathew et al., 2009). This protein is also considered an important metabolic regulator, as mice lacking this protein, sqstm1 −/− mice, develop mature‐onset obesity, leptin resistance and insulin intolerance (Rodriguez et al., 2006). Several clinically useful drugs have been shown to alter p62 activity. Verapamil induces p62‐dependent autophagic Keap1 degradation which then activates Nrf2 (Lee et al., 2017) while anaesthetic drugs induce p62 transcription and promote autophagy in vitro (Zhuang et al., 2015). At present, however, the role of p62 in the off‐target effects of clinically used drugs, particularly in regard to autophagy‐independent roles, is still not well understood.
p62 is a notoriously multifunctional protein. In its role as a scaffold or adapter protein, it contains a series of motifs which enable its interaction with a plethora of selective signal transduction pathways involved in cell proliferation and survival/death through regulation of transcription factor activities. For example, NF‐κB activation (Sanz et al., 1999) and the activation of the Keap‐Nrf2 pathway through a positive feedback loop in which SQSTM1 itself is transcriptionally up‐regulated by Nrf2 (Jain et al., 2010; Puissant et al., 2012).
A second role of p62 is to coordinate ubiquitin‐mediated regulatory processes (Shin, 1998; Puissant et al., 2012). This function derives from its ability to non‐covalently interact with ubiquitin through its C‐terminal ubiquitin‐binding domain, thus facilitating proteasome‐dependent degradation of ubiquitinated target proteins (Harper and Schulman, 2006). The participation of p62 in autophagy is a multilevel process. It enables mTORC1 activation on lysosomes, possesses LC3‐interacting region and can also act as a cargo receptor for the autophagy of ubiquitinated proteins aggregated into so‐called p62 bodies (Bjørkøy et al., 2005). Moreover, p62 is itself a substrate for selective autophagy and is often used as a marker of autophagic flux, as its accumulation can indicate inefficient (or blocked) autophagy (Puissant et al., 2012; Katsuragi et al., 2015). However, the participation of p62 in autophagy and particularly in regard to its correlation with the autophagic flux is suggested to be largely cell‐ and condition‐specific (Liu et al., 2016). Mounting evidence associates p62 with the signalling pathways that control inflammation; numerous recent articles have addressed its connection to inflammasomes, multi‐protein oligomer platforms of which Nod‐like receptor protein 3 (NLRP3) is the most extensively studied. In accordance with its multiple intracellular roles, the implication of p62 in inflammasome regulation is multifaceted; it has been shown to be essential for the activation of NLRP3 in different circumstances (Harijith et al., 2014), while it is also considered a crucial factor in the inhibition of inflammasome activity (through its participation in autophagy enabling its degradation).
We have previously described a complex effect of the antiretroviral drug efavirenz, a member of the non‐nucleoside analogue reverse transcriptase inhibitor family, on human hepatic cells in culture, manifested in both mitochondrial dysfunction – diminished O2 consumption, decreased mitochondrial membrane potential (ΔΨm) and increased mitochondrial ROS generation (Apostolova et al., 2010) and the presence of ER stress with activation of the three arms of the unfolded protein response (UPR) (Apostolova et al., 2013). In line with evidence obtained in other cellular models (Dong et al., 2013; Weiß et al., 2016), we have shown that under these conditions, efavirenz also up‐regulates autophagy, and autophagic flux is normal at moderate efavirenz concentrations (10 and 25 μM) (Apostolova et al., 2011). Higher concentrations of efavirenz (50 μM) were associated with inhibited autophagic flux and induction of apoptosis (Apostolova et al., 2010; Apostolova et al., 2011). In this model, autophagy is protective as its pharmacological inhibition worsens the effect of efavirenz.
In the present study, we have sought to explore the role of p62 in this model and the specificities of its activation, for which we compared the effects of efavirenz on p62 with those exerted by sublethal concentrations of other specific stressors, chosen due to the similaritites of their actions with those of efavirenz. In order to better understand the role of mitochondria in the effect of efavirenz , we assessed the levels of p62 in a model of ρ° cells generated in Hep3B background, employed as a robust approach to mimic a scenario of non‐fully functional mitochondria. We have found that efavirenz induces p62 transcriptionally via the transcription factor C/EBP homologous protein (CHOP) and that this increase is relevant for the efavirenz‐induced mitochondrial ROS generation and inflammasome activation. The fact that p62 is up‐regulated by an antiretroviral drug is a finding that not only adds to the existing knowledge of p62 regulation but also has potential clinical implication.
Methods
Cell culture and transfection experiments
Hep3B cells (human hepatoma, ATCC HB‐8064, Manassas, VA, USA) were cultured as described elsewhere (Apostolova et al., 2010). ρ° cells were obtained by treating wild‐type cells (8 weeks) with the mutagen, ethidium bromide (100 ng·mL−1), to deplete mitochondrial DNA (mtDNA) in medium supplemented with 50 μg·mL−1 uridine. The presence of Hep3B ρ° phenotype was verified by assessment of O2 consumption in an intact cell suspension using Clark type O2 electrode (ρ° cells displayed approximately 15% basal respiration compared to WT), as well as by monitoring cell proliferation in culture medium containing glucose or galactose and the protein expression of mtDNA‐encoded proteins (Apostolova et al., 2013). Primary human hepatocytes were purchased from Life Technologies (Carlsbad, CA, USA) and were seeded in type I collagen‐coated plates following the manufacturer's instructions using William's E medium without phenol red, supplemented with 5% heat‐inactivated (iFBS), 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin and primary hepatocyte maintenance supplement (cocktail solution of 0.8 mg·mL−1 dexamethasone, 6.25 μg·mL−1 human recombinant insulin, 6.25 μg·mL−1 human transferrin, 6.25 ng·mL−1 selenium complex, and 5.35 μg·mL−1 linoleic acid, GlutaMAX™ and HEPES).
All treatments were performed in complete cell culture medium over 24 h, unless indicated otherwise. The effect of efavirenz was compared to that of two classic mitochondrial stressors, rotenone and carbonyl cyanide 3‐chlorophenylhydrazone (CCCP). Rotenone is a widely used inhibitor of mitochondrial respiration and, like efavirenz, inhibits complex I of the electron transport chain, leading to a drop in ∆Ψm and enhancement of the mitochondrial superoxide production (Ishiguro et al., 2001; Li et al., 2003). CCCP is a protonophore used as a potent chemical uncoupler of oxidative phosphorylation. Unlike efavirenz and rotenone, it stimulates mitochondrial respiration while dissipating ∆Ψm.
Thapsigargin, a highly selective inhibitor of the sarco/endoplasmic reticulum Ca2+‐ATPase, provokes a depletion of Ca2+ in the ER lumen and is employed as a known inducer of ER stress (Thastrup et al., 1990). Thapsigargin also displays direct and indirect effects on mitochondria. While moderately increased cytosolic Ca2+ concentrations lead to enhanced mitochondrial Ca2+ uptake, O2 consumption and enhanced ∆Ψm, more pronounced effects of thapsigargin comprise severe mitochondrial alterations, such as Ca2+‐mediated mitochondrial fission and apoptosis (Hom et al., 2007). Given that thapsigargin is both an inducer of ER stress and a mitochondrial stressor, it was of interest to compare and contrast its action with that of efavirenz.
Transient silencing of SQSTM1/p62, DDIT3/CHOP/GADD153 and ATG5 was carried out by RNA interference. siRNAs (sip62 and siCHOP) were from Santa Cruz Biotechnology (Heidelberg, Germany) while siATG5 was from Dharmacon (Lafayette, CO, USA). SignalSilence® unconjugated control siRNA (Cell Signaling Technology, Danvers, MA, USA) was used for control experiments. Transient transfection was performed in t‐75 flasks using Lipofectamine™ 2000 transfection reagent (ThermoFisher Scientific, Walthman, MA, USA), according to the manufacturer's instructions. Transfections were performed in serum‐free OptiMEM (ThermoFisher Scientific) containing siRNA (750 pmol of siRNA) and transfection reagent (37.5 μL of Lipofectamine™ 2000). After 24 h, the specific treatments were performed as indicated.
To overexpress p62, cells were transfected with mCherry‐p62 (a kind gift from Prof. Terje Johansen, Department of Medical Biology, The Arctic University of Norway) while pDEST plasmid (Invitrogen) was employed for control experiments. Transient transfection was performed in t‐25 flasks using Lipofectamine™ 2000 transfection reagent, according to the manufacturer's instructions. Transfection was performed in serum‐free OptiMEM (ThermoFisher Scientific) containing 2.63 μg plasmid DNA and transfection reagent (7.9 μL of Lipofectamine™ 2000). After 7 h, medium was refreshed and treatments (24 h) were initiated 30 h after transfection.
Protein extraction and Western blot analysis
Whole‐cell, mitochondrial‐enriched and cytosolic protein extracts were obtained, quantified and immunoblotted as described elsewhere (Apostolova et al., 2010). After blocking for 1 h (5% skimmed milk or bovine serum albumin), the membranes were incubated overnight at 4°C with primary antibodies: p62/SQSTM1 (sc‐28 359), Nrf2 (sc‐722) (both from Santa Cruz Biotechnology), CHOP/DDIT3 (ab11419), GRP78 (ab21685), Porin/VDAC1 (ab15895) (all three from Abcam, Cambridge, UK), NF‐κB (p65) (#339900, Life Technologies, Rockford, IL, USA); LC3 (L8918), β‐Actin (A5060), GAPDH (G9545) (all three from Sigma‐Aldrich), Atg5 (D5F5U) (#12994), SAPK/JNK (#9252) and Phospho‐SAPK/JNK (Thr183/Tyr185) (#9251) (all three from Cell Signaling Technology). Secondary antibodies employed are as follows: peroxidase‐labelled anti‐mouse (#32230, ThermoFisher Scientific) and anti‐rabbit IgG (PI‐1000, Vector laboratories, Burlingame, CA, USA). The reactive signals were visualized using ECL™ Western Blotting Detection Reagents (GE Healthcare, Buckinghamshire, UK), Luminata™ Crescendo Western HRP Substrate (Millipore, Billerica, MA, USA) or SuperSignal WestFemto (Pierce Chemicals, Boulder, CO, USA) and visualized with a digital luminescent image analyser (FUJIFILM LAS3000, Fujifilm, Japan). Densitometric analyses were performed using MultiGauge software V3.0.
Quantitative RT‐PCR
Quantitative RT‐PCR (in duplicate) was performed using mRNA obtained from a t‐25 flask or 10cm2 cell cultures treated with vehicle, efavirenz (10, 25 μM) or the control stressor (Tg, Rot or CCCP). Total RNA was extracted (RNeasy Mini Kit (Qiagen, Hilden, Germany), eluted in 30 μL of water and quantified (NanoDrop® ND‐1000 spectrophotometer, NanoDrop Technologies, Wilmington, DE, USA). For first strand cDNA synthesis (SuperScript™ III Reverse Transcriptase, Invitrogen), 1 μg of total RNA was reverse‐transcribed in a final volume of 20 μL and the reverse transcriptase was inactivated (70°C, 15 min). PCR reactions (LightCycler® 96 Real Time PCR System, Roche Applied Biosystems) were performed mixing 1 μL of cDNA with SYBR® Premix Ex Taq™ (Takara Biotechnology, Kusatsu, Japan). Primers 1 μM (ACTB employed as a housekeeping gene, SQSTM1, NLRP3, IL‐1β, IL6 and TNFa from Integrated DNA Technologies, Leuven, Belgium) were added in final reaction volume of 10 μL. The reactions were as follows: 30 s‐95°C; 5 s‐95°C, 20 s‐60°C (50 cycles); 1 s‐95°C, 15 s‐65°C, 1 s‐95°C; and 30 s‐40°C. The specificity of the amplified products was verified by melting curve analysis. Normalized results (interpolated values for each sample divided by the corresponding value for ACTB) were expressed as percentage of expression of the specific gen/ACTB ratio versus control (100%).
| Gene Symbol | Sequence direction | Sequence | Band size (bp) | Annealing Tª (°C) |
|---|---|---|---|---|
| NLRP3 | Sense | 5′‐CTTCTCTGATGAGGCCCAAG‐3′ | 200 | 50 |
| Antisense | 5′‐GCAGCAAACTGGAAAGGAAG‐3′ | |||
| IL‐1β | Sense | 5′‐TTCGACACATGGGATAACGAGG‐3′ | 84 | 52 |
| Antisense | 5′‐TTTTTGCTGTGAGTCCCGGAG‐3′ | |||
| IL‐6 | Sense | 5′‐CACTGGTCTTTTGGAGTTTGAGG‐3′ | 169 | 51 |
| Antisense | 5′‐ ATTTGTGGTTGGGTCAGGGG‐3′ | |||
| TNFα | Sense | 5′‐AGCCGAATCGCCGTCTCCTA‐3′ | 124 | 58 |
| Antisense | 5′‐CAGCGCTGAGTCGGTCACCC‐3′ | |||
| SQSTM1 | Sense | 5′‐GGTTGCCTTTTCCAGTGACG‐3′ | 243 | 52 |
| Antisense | 5′‐TCGCAGACGCTACACAAGTC‐3′ | |||
| ACTB | Sense | 5′‐AGCACGGCATCGTCACCAACT‐3′ | 183 | 58 |
| Antisense | 5′‐ACATGGCTGGGGTGTTGAAGG −3′ |
Fluorescence microscopy and static cytometry
Fluorescence was detected with an IX81 Olympus microscope (Hamburg, Germany) and quantified by static cytometry software ‘ScanR’ version 2.03.2 (Olympus). Cells were seeded (30 000 cells per well), allowed to attach and treated for 24 h (in duplicate, 48‐well plates). The corresponding fluorochromes (2.5 μM Hoechst 33 342 to stain nuclei from Sigma‐Aldrich and 2.5 μM MitoSOX or 5 μM tetramethylrhodamine methyl ester to assess mitochondrial ROS or ΔΨm respectively, both from ThermoFisher Scientific) were added for the last 30 min of the treatment. Cells were then washed in HBSS and live cell images (25 images per well) were recorded immediately. To assess cell proliferation or survival, cells were counted according to Hoechst fluorescence.
Confocal fluorescence microscopy
Treatment was performed in multi‐well coverslips (ThermoFisher Scientific), and cells were then fixed with 4% formaldehyde (15 min, room temperature), washed (three times in PBS), blocked (60 min, room temperature) and incubated (overnight, 4°C) with primary antibodies: anti‐p62 (mouse antibody, SantaCruz Biotechnology) at 1:150 and anti‐TOM20 (rabbit antibody, BD Biosciences, NJ, USA) at 1:350 to mark mitochondria. Samples were washed and incubated (1 h, room temperature) with secondary antibodies (goat anti‐rabbit Alexa Fluor 488 at 1:500 or goat anti‐mouse Alexa Fluor 594 at 1:600, both from ThermoFisher Scientific) and 5 μM of the fluorochrome Hoechst 33 342 (to mark nuclei) was added for the last 30 min.
After washing with PBS, images were acquired with a Leica TCS‐SP2 confocal laser scanning unit with argon and helium‐neon laser beams and attached to a Leica DMIRBE inverted microscope. Images were captured at 63× magnification with a HCX PL APO 40.0 × 1.32 oil UV objective. Technical replicates – duplicates – were employed to ensure the reliability of single values.
Chromatin immunoprecipitation (ChIP) assay
Hep3B cells treated with efavirenz for 24 h were cross‐linked with 1% formaldehyde at room temperature for 10 min followed by 0.125% glycine for 2 min. Subsequently, cells were washed three times with ice‐cold PBS and collected into PBS and centrifuged for 5 min at 500 g. Cells were then resuspended in 0.3 mL of SDS sonication buffer (1% SDS, 5 mM EDTA, 50 mM Tris–HCl pH 8.0 and protease inhibitor cocktail; Roche Diagnostics GmbH, Mannheim, Germany) and sonicated three times, for 20 s each time at maximum speed (Branson, Digital Sonifier, Emerson Electric Co., MO, USA) followed by centrifugation at 16 000 rcf (10 min, 4°C). Supernatants were collected and the immunoprecipitation was performed overnight at 4°C with anti‐Nrf2 (sc‐722, Santa Cruz Biotechnology), anti‐NF‐κB (p65) (#339900, ThermoFisher Scientific) anti‐DDIT3 antibody (ab11419, Abcam) or with control IgG antibody (ThermoFisher Scientific). After immunoprecipitation, 60 μL protein A‐Sepharose beads (GE Healthcare Life Science) were added to the supernatants and incubated overnight at 4°C. Precipitates were washed sequentially, once with low‐salt wash buffer (1% Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl pH 8, 150 mM NaCl), twice with high‐salt wash buffer (1% Triton X‐100, 2 mM EDTA, 20 mM Tris–HCl pH 8, 500 mM NaCl) and once with LiCl wash buffer (0.25 mM LiCl, 1% NP‐40, 0.1% Tween 20, 1 mM EDTA, 10 mM Tris–HCl pH 8.0), 5 min each. Precipitates were then washed twice with TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA) and extracted twice with elution buffer (1% SDS, 0.1 M NaHCO3). Eluates were pooled and heated at 65°C for 16 h to reverse the formaldehyde cross‐linking in the presence of 0.25 M NaCl. DNA fragments were purified with a PureLink™ Quick PCR purification kit (ThermoFisher Scientific). PCR was performed using TaKaRa Taq™ (Takara) with specific primers to amplify the binding site of Nrf2 (Jain et al., 2010), NF‐κB or CHOP/DDIT3 on the SQSTM1 promoter (35 cycles). PCR products were separated by electrophoresis in 2% agarose gel.
| Transcription factor | Sequence direction | Sequence | Band size (bp) |
|---|---|---|---|
| NF‐κB | Sense | 5′‐ATTACGACAGCGGTCATGGG‐3′ | 116 |
| Antisense | 5′‐CTCCCGGAGGGTAAACAAGG‐3′ | ||
| Nrf2 | Sense | 5′‐CTCTCAGGCGCCTGGGCTGCTGAG‐3′ | 152 |
| Antisense | 5′‐CGGCGGTGGAGAGTGGAAAATGCC‐3′ | ||
| CHOP | Sense | 5′‐CACCCACACATCACCTTCGC‐3′ | 238 |
| Antisense | 5′‐ACCAGAGTAACCTGTCCGCC‐3′ |
Data and statistical analysis
The data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All experiments were repeated independently at least five times. Technical replicates in a run (at least in duplicate) were averaged and yielded a value for a biological replicate. The study was performed in vitro using cell lines, and all the samples were analysed and quantified objectively, without randomization of samples or blinding of the operator due to technical limitations and a large number of assays. Data are shown as % of control, with untreated cells set to 100%. Data (mean ± SEM) were analysed using GraphPad Prism v.6. software with a one‐way ANOVA multiple comparison test followed by a Newman–Keuls test or Student's t‐test. Mitochondrial and cytosolic fractions were analysed by two‐way ANOVA multiple comparison test followed by a Sidak's test. For all analyses, P<0.05 was taken to show statistical significance
Materials
Unless stated otherwise, chemicals were from Sigma‐Aldrich (Steinheim, Germany). Efavirenz (Sequoia Research Products, Pangbourne, UK) was dissolved in methanol (stock solution of 3 mg·mL−1) and employed at clinically relevant plasma concentrations (10–50 μM). Its solvent, methanol, was used as a control and had no effect on any of the parameters evaluated. Control treatments:‐ rotenone (25 μM), (CCCP; 10 μM), thapsigargin (2 μM), the autophagy inhibitors 3‐methyladenine (3MA, 2.5 mM) and bafilomycin A1 (20 nM), the selective calcium chelator BAPTA‐AM (10 μM, Abcam, Cambridge, UK) and the inhibitor of gene transcription actinomycin D (2 μg·mL−1) were dissolved in DMSO.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
p62 expression is increased at mRNA and protein level upon exposure to mitochondrial and ER stress stimuli
Firstly, we analysed the protein expression of p62 by Western blotting in total cell extracts obtained after 24 h treatment with efavirenz and compared it with several mitochondrial and ER stressors. As shown in Figure 1A, efavirenz (10, 25 and 50 μM) led to an increase in the protein expression of p62, a finding that was corroborated by confocal fluorescence microscopy (Figure 2E). Both thapsigargin and rotenone, but not CCCP, enhanced p62 levels. We also assessed the expression of LC3, as its conversion of the soluble from LC3‐I (18 kDa) to the autophagosome‐linked LC3‐II (16 kDa) is commonly used as a general indicator of autophagic induction. As shown earlier (Apostolova et al., 2011), 24 h of efavirenz treatment increased LC3‐II levels. In order to explore the timing of the efavirenz effect on p62 expression, we studied it 4, 8, 24 and 48 h of treatment. We observed that p62 content had already increased at 4 h (statistically significant with 25μM efavirenz), peaked at 24 h exposure and continued at 48 h (Figure 1B). The observation that the levels detected at 48 h were lower than those observed at 24 h is likely due to a feedback loop in the regulation of the synthesis of p62 and/or the fact that efavirenz is metabolized by hepatic cells and therefore its concentration is diminished after 48 h. We also assessed the levels of p62 in ρ° cells generated in Hep3B background, which are less vulnerable to efavirenz than WT cells (Polo et al., 2015), a finding compatible with the crucial participation of mitochondria in the cellular effect of efavirenz. Interestingly, as shown in Figure 1C, the increase in p62 protein content was greatly reduced (efavirenz) or completely abolished (rotenone) in respiration‐deficient cells (ρ°), while there was no difference after treatment with thapsigargin. Of note, ρ° cells under basal conditions showed a significantly decreased p62 expression compared to WT cells. These findings demonstrate that mitochondrial function plays a role in the activation of p62. Lastly, qRT‐PCR analysis revealed that efavirenz or thapsigargin produced a significant increase in p62 mRNA. Of note, there were no changes when cells were exposed to CCCP, which is in accordance with the lack of effect on the protein level of p62 (Figure 1D). A similar efavirenz‐induced increase of p62 mRNA was also detected in primary human hepatocytes (Figure 1E), which rules out the possibility that the effect was related to the cancerous nature of Hep3B and confirms our previous findings that Hep3B cells are a reliable cell line for this type of pharmacological study (Apostolova et al., 2011, 2013). Moreover, cells co‐treated with efavirenz and actinomycin D, an inhibitor of gene transcription (Perry and Kelley, 1970), did not display augmented p62 protein expression, suggesting that the up‐regulation of p62 occurred at transcriptional level (Figure 1F). This result points to the accumulation of p62 as a result of increased SQSTM1 expression and not as a consequence of altered degradation in the cytosol or other processes related to protein stability and turn‐over.
Figure 1.
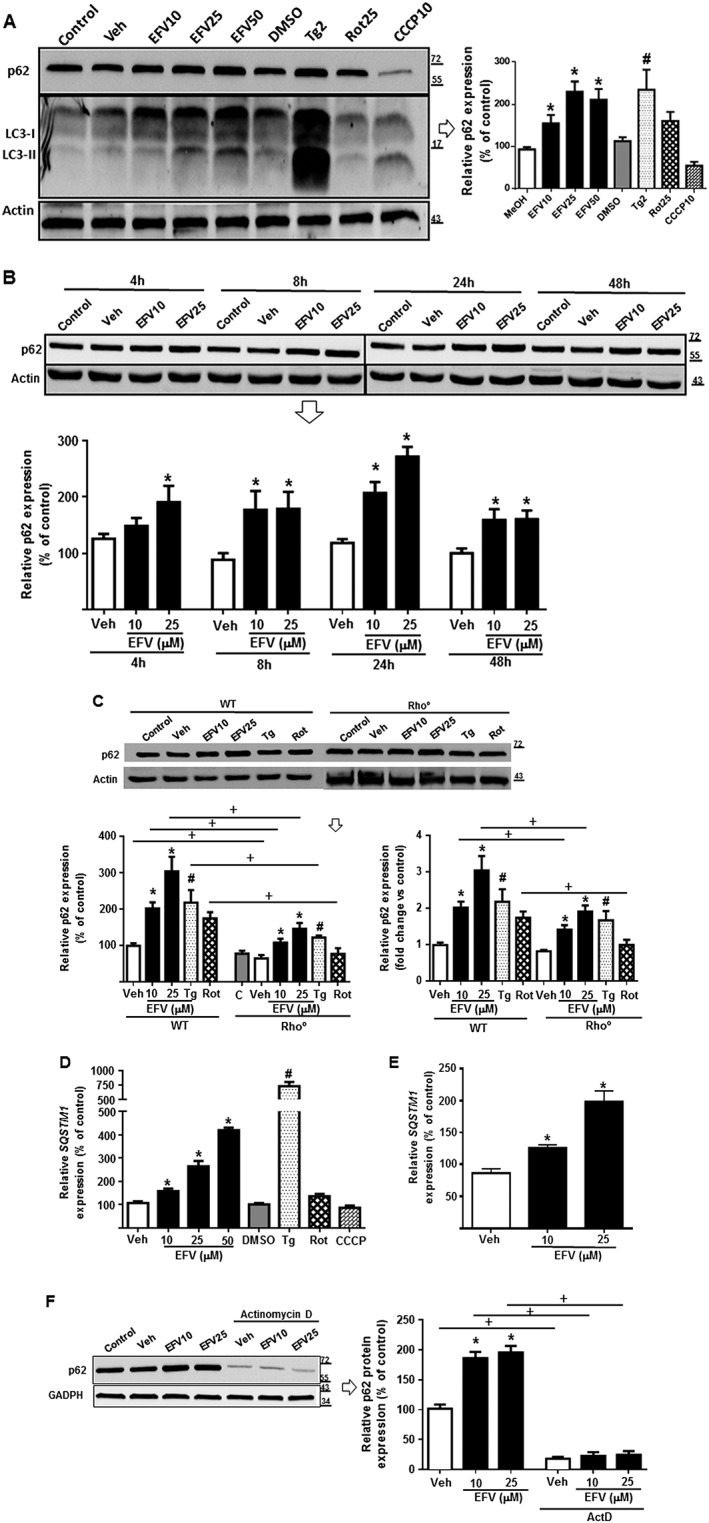
p62 expression is increased at protein and mRNA level upon exposure to mitochondrial and ER stress stimuli. Cells were treated for 24 h (A, C, D, E and F) or 4, 8, 24 and 48 h (B) with increasing concentrations of efavirenz (EFV), vehicle (MeOH or DMSO), thapsigargin (Tg) 2 μM, rotenone (Rot) 25 μM or CCCP 10 μM. Data (mean ± SEM) are expressed as relative protein or mRNA content in relation to that of untreated cells (control, considered 100%) after normalization with expression of the housekeeping protein (β‐actin or GAPDH) or gene (ACTB). (A) Immunoblot analysis of total cell extracts showing a representative Western blot image and summary of densitometry data of p62 expression (n = 9 except for EFV50 and rotenone n = 8, CCCP, n = 7, thapsigargin, n = 6). (B) Time‐course immunoblot analysis of total cell extracts showing representative Western blot image and densitometry data expressing quantification of p62 protein levels at the indicated treatment times (n = 7). p62 expression in untreated cells (at all time points) was considered 100%. (C) Immunoblot analysis of total cell extracts showing representative Western blot image of p62 and summary of densitometry data in ρ+ and ρ° cells (n = 6). p62 expression in untreated wild‐type (ρ+) cells was considered 100% (left panel) while p62 expression in untreated (ρ+ and ρ°; shown as Rho) cells was considered 1 (right panel). (D) Relative mRNA expression levels of SQSTM1 in Hep3B cells (n = 8, except for DMSO, thapsigargin, rotenone and CCCP n = 5) and (E) human hepatocytes (n = 2) were analysed by quantitative RT‐PCR. Data were normalized versus the housekeeping gene β‐Actin (ACTB). (F) Immunoblot analysis of total cell extracts showing representative Western blot image of p62 and summary of densitometry data in cells co‐treated with efavirenz and actinomycin D (2 μg·mL−1) (an inhibitor of gene transcription) (n = 7 except for EFV10 and EFV25, n = 5). Data (mean ± SEM) were calculated as percentage of control (untreated cells). *P < 0.05 for efavirenz; #P < 0.05 for thapsigargin, rotenone, CCCP; significantly different from vehicle; one‐way ANOVA followed by a Newman–Keuls test. +P < 0.05, significantly different as indicated; Student's t‐test.
Figure 2.
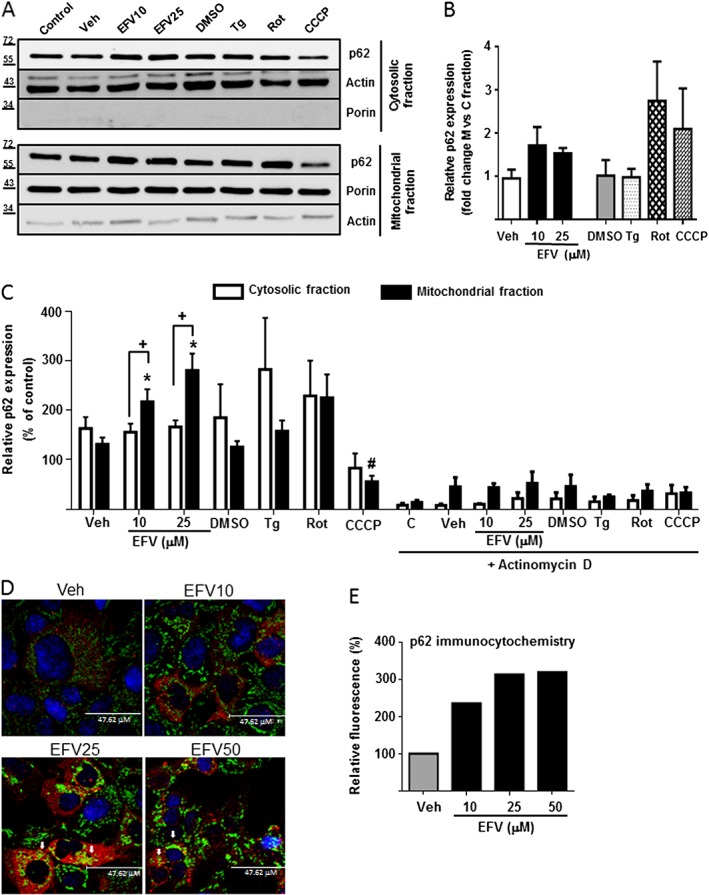
Differential sub‐cellular expression of p62. Representative Western blot image (A) of p62 and summary of densitometry data (B, C) in mitochondria‐enriched and cytosolic extracts obtained from Hep3B cells treated with increasing concentrations of efavirenz (EFV), vehicle (MeOH or DMSO), thapsigargin (Tg) 2 μM, rotenone (Rot) 25 μM or CCCP 10 μM for 24 h in the presence or absence of 2 μg·mL−1 actinomycin D (n = 5). Equal amounts of protein (30 μg) from mitochondrial and cytosolic protein extracts were loaded. Actin and porin expression was assessed to ensure the purity of the extracts. Data (mean ± SEM) were calculated as percentage of control (untreated cells were considered 100% in both extracts). *P < 0.05, for efavirenz and #P < 0.05 for thapsigargin, rotenone or CCCP, significantly different from vehicle; one‐way ANOVA followed by a Newman–Keuls test. +P < 0.05, significantly different as indicated; two‐way ANOVA followed by a Sidak's test. (D) Representative confocal fluorescence microscopy images (63× 3.0 digital zoom) of cells treated with Veh or EFV (10, 25 or 50 μM) and immunostained for p62 (red) and mitochondria (TOM20, green). Nuclei were stained with the fluorochrome Hoechst (blue). Arrows show points of colocalization. (E) Quantification of the fluorescence signal of p62 expressed as %.
p62 localization is differentially modified upon treatment with different stimuli of mitochondrial and ER stress
Having assessed the modified p62 content in whole‐cell extracts, we next explored the presence of this protein in different subcellular compartments by assessing the level of p62 protein expression in mitochondria‐enriched fraction and comparing it to that in the cytosolic fraction obtained of cells treated for 24 h (Figure 2A–C). The purity of these fractions was corroborated with the expression of two marker proteins, porin and actin. Interestingly, efavirenz produced a greater increase of p62 content in the mitochondrial extract compared to the cells exposed to an inducer of ER stress, thapsigargin. Rotenone only produced an increase in p62 in the mitochondrial fraction whereas, as suggested previously, CCCP did not lead to an increase in p62 content; instead, a significant decrease was observed both in the mitochondria‐enriched and cytosolic fraction (Figure 2A–C). Of note, both cytosolic and mitochondrial levels of p62 were largely diminished in cells co‐treated with actinomycin D (Figure 2C). EFV‐induced increase in the mitochondrial content of p62 was also observed by immunocytochemistry experiments in which cells exposed to EFV25 or EFV50 displayed enhanced colocalization between p62 and TOM20 (mitochondrial outer membrane protein) fluorescent signals (Figure 2D).
The increase in p62 expression is CHOP‐dependent
Our next aim was to analyse the mechanism through which p62 was up‐regulated. Both efavirenz and thapsigargin induce an increase in cytosolic Ca2+ concentration in Hep3B cells (Apostolova et al., 2013), and we suspected that this increase contributed to p62 up‐regulation. In order to explore this possibility, we treated cells with efavirenz, thapsigargin or rotenone in the presence of the intracellular Ca2+ chelator BAPTA‐AM for 24 h. The results shown in Figure 3A reveal that Ca2+ plays a partial role in the increased p62 gene expression, triggered by efavirenz or thapsigargin, as the increase in p62 was partly reversed in the presence of BAPTA‐AM. The complexity of the effect was again evident, as the increase of p62 induced by rotenone was not reversed by BAPTA‐AM.
Figure 3.
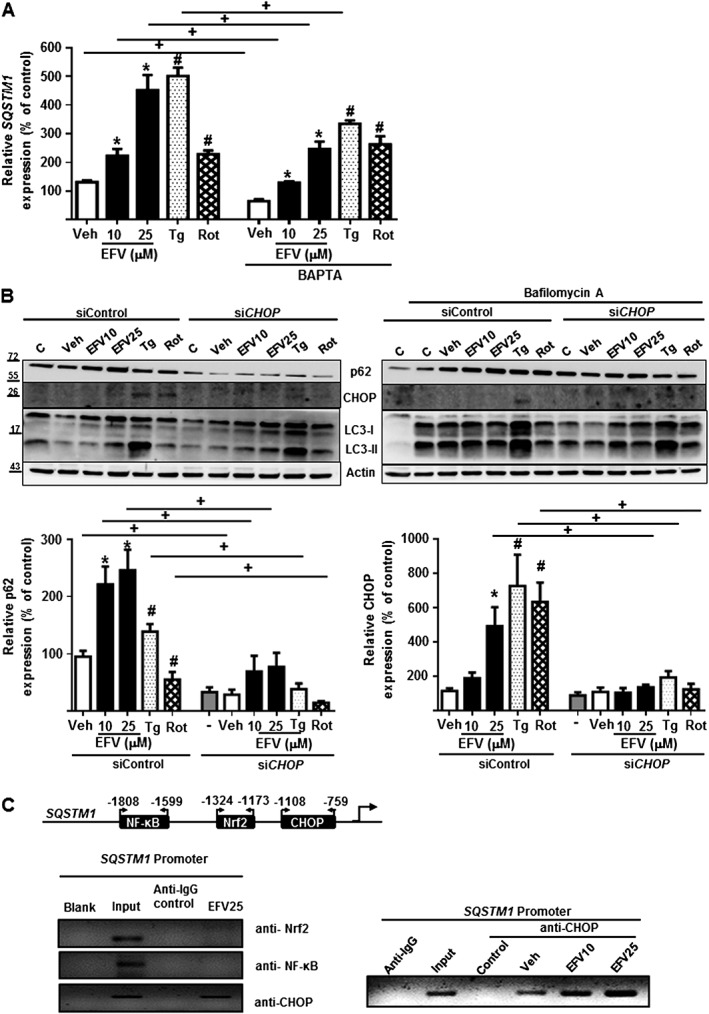
p62 expression is calcium‐ and CHOP‐dependent. Hep3B cells were treated for 24 h with increasing concentrations of efavirenz (EFV), vehicle (MeOH), thapsigargin (Tg) 2 μM or rotenone (Rot) 25 μM in the presence or absence of the inhibitor of autophagy, bafilomycin A1 20 nM. (A) Relative mRNA expression levels of SQSTM1, in presence or absence of the Ca2+ chelator BAPTA‐AM (10 μM), was analysed by quantitative RT‐PCR. Data were normalized versus the housekeeping gene β‐Actin (ACTB) and SQSTM1 expression in untreated cell (without BAPTA‐AM) was considered 100% (mean ± SEM, n = 5). (B) Representative WB image of p62, LC3 and CHOP, and summary of densitometry data in cells transfected with siControl or siCHOP. Data (mean ± SEM, n = 5) were calculated as percentage of untreated siControl cells (considered 100%). *P < 0.05, for efavirenz and #P < 0.05 for thapsigargin, rotenone or CCCP, significantly different from vehicle; one‐way ANOVA followed by a Newman–Keuls test. +P < 0.05, significantly different as indicated; Student's t‐test. (C) CHOP is associated with SQSTM1 promoter in presence of efavirenz, but not with Nrf2 or NF‐κB. A representative image of PCR bands after chromatin immunoprecipitation (ChIP) assay performed in Hep3B cells treated with efavirenz (25 μM), where chromatin was immunoprecititated with anti‐Nrf2, anti‐NF‐κB or anti‐CHOP antibody and PCR assay was carried out to amplify the immunoprecipitated chromatin using primers flanking the binding site of Nrf2, NF‐κB or CHOP on the SQSTM1 promoter (left panel); representative image of ChIP assay showing SQSTM1 promoter occupancy by CHOP in Hep3B cells treated with vehicle and efavirenz (10 and 25 μM) (right panel). A non‐related antibody anti‐IgG and input control were employed as negative and positive controls respectively.
Several transcription factors have been reported to trans‐regulate the gene expression of p62 (Puissant et al., 2012). Taking into account the specific dual effect of ER stress and mitochondrial dysfunction in our model, we investigated whether the up‐regulation of p62 was due to the transcription factor CHOP, known to be activated by both ER stress and oxidants. To this aim, we transiently silenced CHOP by means of RNA interference and assessed the effect of efavirenz, thapsigargin and rotenone in these cells. Interestingly, in cells with constitutively knocked‐down expression of CHOP, the up‐regulation of p62 was greatly reduced, an effect that occurred with all the stimuli, thus hinting that the increase in p62 was largely CHOP‐mediated (Figure 3B). The implication of CHOP in the regulation of p62 transcription by efavirenz was assessed by means of a ChiP assay (Figure 3C) ruled out the participation of NF‐κB and Nrf2, other transcription factors of p62. p62 activation has also been described to be mediated by JNK, a member of the MAPK family whose activation by a variety of cellular stresses stimulated the process of autophagy (Puissant et al., 2012). In order to assess whether such a regulation may exist in our model, we analysed the phosphorylation of JNK by Western blotting after 24 h treatment with EFV or the other stress stimuli (Supporting Information Figure S1). Interestingly, there was an increase in the level of p‐JNK in cells exposed to efavirenz which was not detected with the other stressors. However, the content of total JNK upon efavirenz exposure was also slightly increased which led to a lack of increase in the pJNK/JNK ratio.
p62 up‐regulation occurs independently of autophagy but is related to mitochondrial function and inflammasome activation
As previously mentioned, efavirenz (10 and 25 μM) induced autophagy in the model of cultured hepatic cells we used. Taking into account that p62 is degraded by autophagy and bearing in mind that it is commonly employed as a marker of autophagy, we aimed to analyse the participation of p62 in the autophagic process induced by efavirenz. Firstly, we observed that while CHOP silencing led to a significant decrease in p62 expression as mentioned previously, it did not affect LC3 levels (Figure 3B). Moreover, we co‐treated cells with bafilomycin A1, an inhibitor of vacuolar H+ ATPase (V‐ATPase) employed to prevent fusion between autophagosomes and lysosomes, thus disabling the last stage of autophagy. Co‐treatment with bafilomycin A1 did not impede p62 up‐regulation triggered by efavirenz or the other two stumuli, as seen by Western blotting (Figure 3B, right panel). Secondly, we transiently silenced p62 (using siRNA) to almost abolish p62 and then assessed the levels of LC3 using thapsigargin for comparison, as it was the stimulus that showed most similarities with efavirenz. As shown in Figure 4A, p62 silencing had no effect on LC3 expression in efavirenz‐ or thapsigargin‐treated cells and did not affect basal levels of LC3. These findings reflect the lack of influence of p62 on autophagosome formation and on the normal autophagic flux (there is no accumulation of LC3‐II). The absence of a correlation between autophagy and p62 was also demonstrated in the opposite direction, in cells were the early stages of autophagy were inhibited. This was achieved with the application of 3MA, a pharmacological inhibitor of class III PI3K, or with transient silencing of Atg5, a protein which, in combination with Atg12, functions as an E1‐like activating enzyme in a ubiquitin‐like conjugating system involved in autophagosome formation. In both cases, the inhibition of autophagy did not affect p62 gene expression in efavirenz‐treated cells (Figure 4B, C).
Figure 4.
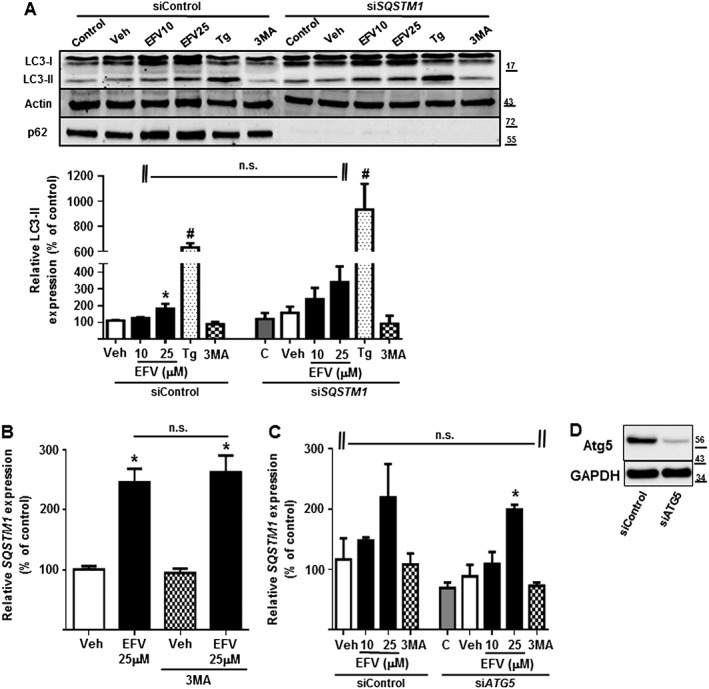
p62 up‐regulation occurs independently of autophagy. Hep3B cells were treated with vehicle (MeOH), EFV (10 or 25 μM), Tg 2 μM or the inhibitor of autophagy 3‐methyladenine (3MA) 2.5 mM for 24 h. (A) Representative Western blot image of LC3 and summary of densitometry data after normalization with the expression of β‐Actin in cells transiently transfected with siControl or siSQSTM1 where the expression of LC3‐II in untreated siControl cells was considered 100%. The absence of p62 confirmed the efficacy of SQSTM1 silencing. (B) Relative mRNA levels of SQSTM1, in absence or presence of 2.5 mM 3MA, were analysed by quantitative RT‐PCR and normalized versus the housekeeping gene β‐Actin (ACTB). (C) Relative mRNA levels of SQSTM1, in siControl and siATG5 cells, were analysed by quantitative RT‐PCR and normalized versus the housekeeping gene β‐Actin (ACTB). Data (mean ± SEM, n = 5) were calculated as percentage of control (untreated cells). *P < 0.05, for efavirenz and #P < 0.05 for thapsigargin, significantly different from vehicle; one‐way ANOVA followed by a Newman–Keuls test. n.s., no significant effects of siRNA or 3MA; Student's t‐test. (D). The expression of ATG5 in order to verify its silencing was studied by Western blotting (representative image) and the expression of GAPDH was employed as a reference.
We wished to explore further the effect of p62 silencing in efavirenz‐treated cells and so analysed representative parameters of both ER stress and mitochondrial function. In terms of ER stress, transient p62 silencing did not affect CHOP or GRP78 up‐regulation, which were both increased upon efavirenz treatment, as with the classic ER stressor thapsigargin (Figure 5A). However, an assessment of mitochondrial function revealed that, while the dissipation of ΔΨm recorded in efavirenz‐treated cells did not depend on p62, the increase in mitochondrial ROS was more prominent in cells in which p62 had been silenced (Figure 5B, C). This finding is another indication that supports the idea of efavirenz‐triggered mitochondrial dysfunction as the primary inducer of p62 expression. p62 silencing, however, did not enhance the deleterious action of the higher concentration of efavirenz (25 μM) on cell number (Figure 5C). Of note, although it did not reach statistical significance, the combination of p62 silencing with the treatment of 3MA enhanced both the effect of efavirenz on cell number and that on mitochondrial superoxide production (Mitosox fluorescence) (Figure 5C).
Figure 5.
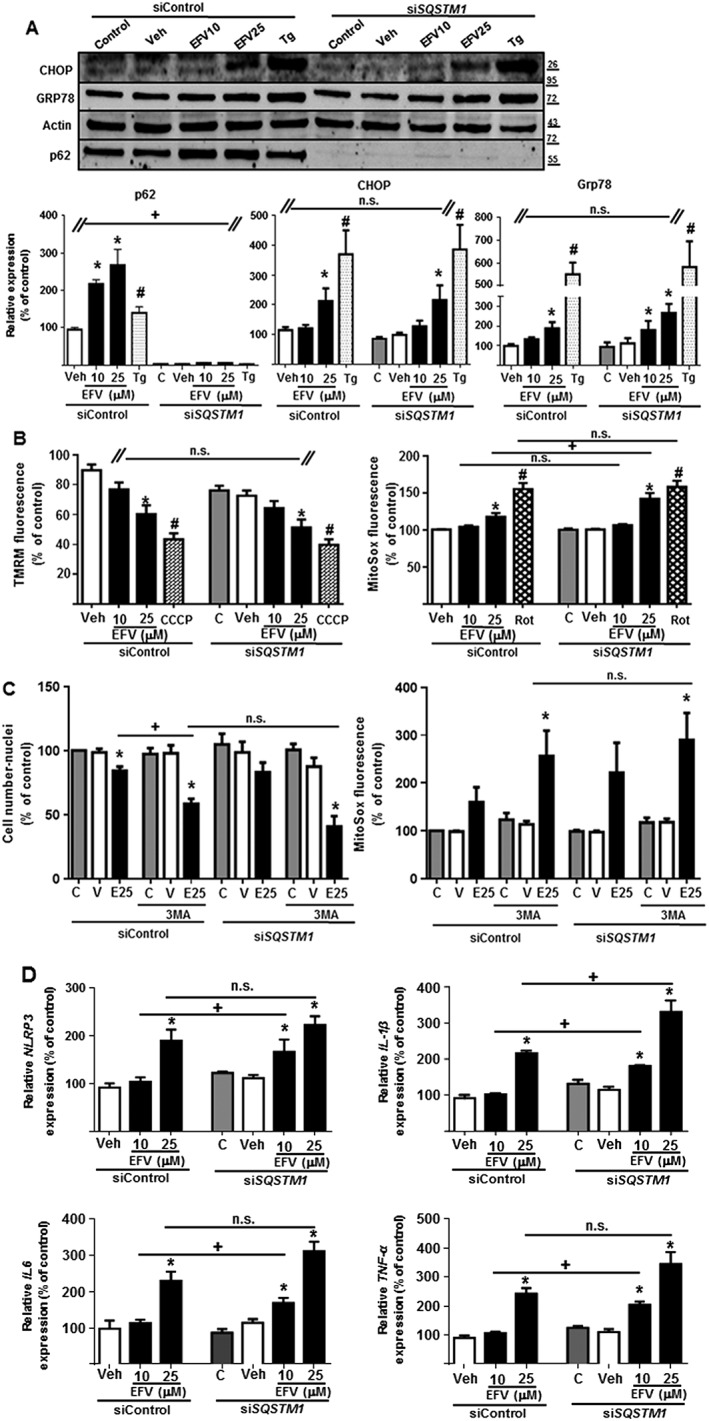
SQSTM1 silencing enhanced the mitochondrial dysfunction and inflammatory response induced by efavirenz but not the UPR. Hep3B cells transiently transfected with siControl or SQSTM1 were treated for 24 h with increasing concentrations of efavirenz (EFV), vehicle, thapsigargin (Tg) 2 μM, rotenone (Rot) 25 μM or CCCP 10 μM, in the absence or presence of the autophagic inhibitor 3MA. (A) Representative Western blot image of p62, CHOP and GRP78 and summary of densitometry data (n = 5). The absence of p62 confirmed the efficacy of SQSTM1 silencing. (B) Quantitative analysis of ΔΨm (tetramethylrhodamine methyl ester fluorescence) and mitochondrial superoxide production (MitoSOX fluorescence) by live cell fluorescence microscopy coupled with static cytometry (n = 7). (C) Quantitative analysis of the cell number (Hoechst fluorescence) and the mitochondrial superoxide production (MitoSOX fluorescence) by live cell fluorescence microscopy coupled with static cytometry. (D) Relative mRNA expression levels of inflammation‐related genes were analysed by quantitative RT‐PCR (n = 5). Data (mean ± SEM) were calculated as percentage of control (untreated SiControl cells). *P < 0.05, for efavirenz and #P < 0.05 for thapsigargin, rotenone or CCCP, significantly different from vehicle; one‐way ANOVA followed by a Newman–Keuls test. +P < 0.05, significantly different as indicated; Student's t‐test.
Inflammasomes can be activated by a great variety of stressful stimuli, with the inflammasome NLRP3 being specifically triggered in the presence of ER stress, mitochondrial dysfunction and/or oxidative stress (Sutterwala et al., 2014). With this in mind, we next assessed the expression of NLRP3 in our model and found that 24 h treatment with efavirenz led to a significant and concentration‐dependent up‐regulation of NLRP3 gene expression. Importantly, p62 silencing further enhanced this effect (Figure 5D). This result was supported by the study of three other markers of inflammation: IL‐1β, IL‐6 and TNF‐α (Figure 5D). Efavirenz (25 μM) significantly increased expression of all three cytokines and the effect was enhanced (statistical significance was now reached with 10 μM) when p62 was silenced. Altogether, these results rule out a prominent role of p62 in autophagy or ER homeostasis, while suggesting a protective function of p62 regarding mitochondrial oxidative stress and inflammasome activation in this complex drug‐induced model of dual mitochondrial dysfunction and ER stress. In order to confirm the protective role of p62 up‐regulation in the present model, we transiently overexpressed p62 using mCherry‐p62 reporter protein and assessed the effect of efavirenz or thapsigargin on the cell number. Importantly, as shown in Figure 6, the deleterious effect of efavirenz on cell viability was decreased when p62 was overexpressed.
Figure 6.
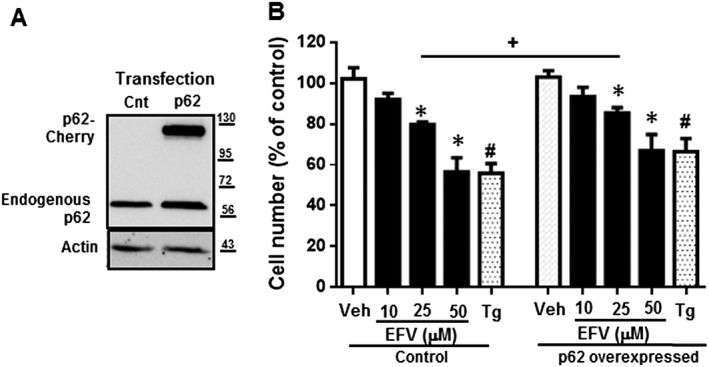
p62 overexpression is protective regarding cellular viability. Hep3B cells transiently transfected with Control plasmid or SQSTM1‐cherry were treated for 24 h with increasing concentrations of EFV, vehicle or thapsigargin (Tg) 2 μM. The efficacy of SQSTM1 overexpression was confirmed by WB (A). Quantitative analysis of the cell number (Hoechst fluorescence) by live cell fluorescence microscopy coupled with static cytometry (B). Data (mean ± SEM) were calculated as percentage of control (untreated Control cells). *P < 0.05, for efavirenz and #P < 0.05 for thapsigargin, significantly different from vehicle; one‐way ANOVA followed by a Newman–Keuls test. +P < 0.05, significantly different as indicated; Student's t‐test.
Discussion
Although drug‐induced toxicity is a major cause of morbidity and mortality worldwide, the off‐target effects of many clinically used agents are still largely unknown. This is particularly relevant in complex clinical situations involving long‐term treatments, the presence/appearance of concomitant diseases that require additional therapies and ageing of the patient population. Drug‐induced toxicity can be triggered or developed by different mechanisms that often involve the malfunction of specific subcellular compartments such as the mitochondrion (mitochondrial dysfunction) or the ER (ER stress). The latter phenomenon is provoked by accumulation of unfolded proteins in the ER lumen, which triggers the so‐called UPR. This complex cascade of events, aiming to restore ER homeostasis, is orchestrated by three ER transmembrane receptors: protein kinase (RNA)‐like endoplasmic reticulum kinase (PERK), inositol requiring protein 1 (IRE1) and activating transcription factor 6 (ATF6). Both PERK and the ATF6 arm of the UPR can activate CHOP/GADD153, a 29 kDa protein of the C/EBP family of transcriptional regulators. CHOP is known to induce growth arrest and regulates ER‐stress‐mediated apoptosis (Oyadomari and Mori, 2004; Gow and Wrabetz, 2009), but overall, CHOP‐induced signalling is not well understood. A detailed molecular analysis using Mouse embryonic fibroblasts (MEFs) revealed that CHOP functions as a master regulator of protein synthesis during conditions of ER stress and that, of note, p62 is one of the genes it directly targets (Han et al., 2013). Importantly, CHOP is also induced by classical mitochondrial toxins such as 6‐OHDA, rotenone (Omura et al., 2013; Goswami et al., 2016) and MPP+ (Zhang et al., 2015). Previous research by our group has shown that efavirenz enhances CHOP expression in human hepatic cells (Apostolova et al., 2013).
The present work explores the regulation of its expression and the role of p62 in an in vitro model of hepatotoxicity induced by the antiretroviral drug efavirenz. Hepatic cells exposed to efavirenz display altered mitochondrial function and ER stress concomitantly. Importantly, in this model, autophagy is activated and the increase in p62 content is not associated with inhibited autophagic flux. Also, the fact that silencing p62 does not affect autophagy in a significant way may be due to the presence of other adaptor proteins such as NBR1 that can compensate for the absence of p62, as has been shown in other models (Johansen and Lamark, 2011).
The present results provide several new features of p62 regulation in the context of drug‐induced toxicity: (i) ER stress and/or mitochondrial dysfunction up‐regulate p62 transcription; (ii) increased p62 expression occurs in a CHOP‐dependent manner; (iii) p62 does not affect autophagy; and (iv) increased p62 levels protect against drug‐induced mitochondrial ROS‐generation and proinflammatory signalling.
Enhanced p62 expression has been demonstrated in other models of drug‐induced toxicity, though how it is mediated is unclear. In addition to xenobiotic‐triggered transcription activation through the antioxidant response element in its promoter, there is mounting evidence that points to the up‐regulation of p62 during ER stress. For instance, methotrexate treatment leads to pro‐survival signalling in JEG/MTXR cells (methotrexate‐resistant choriocarcinoma cells), which involves ROS‐mediated JNK/p62 activation and downstream PERK‐related autophagic activities (Shen et al., 2015). Interestingly, in this model, the IRE1/JNK pathway was specifically activated by methotrexate but was not affected by ROS activation, indicating that ROS activation mediates PERK signalling without regulating IRE/JNK signalling. RNAi‐mediated depletion of p62 promoted CHOP expression, whereas the overexpression of p62 reduced the level of CHOP in JEG/MTXR cells. Moreover, in HCT116 colon cancer cells and MDA‐MB‐468 breast cancer cells, ER stress induced by brefeldin A or tunicamycin results in the transcriptional up‐regulation of p62, due mainly to the PERK arm of the UPR (and perhaps also to IRE‐1, though to a lower extent) (Deegan et al., 2015).
A very interesting finding of the present work is the association of p62 with the NLRP3‐linked proinflammatory effect of efavirenz. Treatment of human hepatic cells with efavirenz increases mitochondrial superoxide generation (Apostolova et al., 2010), and the present study provides evidence of a protective role of p62 in this regard. Moreover, efavirenz treatment enhanced the mitochondrial presence of p62 which is in line with the suggested mitochondria‐specific role of p62 – in dynamics, import and genome integrity (Seibenhener et al., 2013). As the principal generator and target of ROS in the cell, mitochondria are crucial arbitrators of the pro‐inflammatory status, as they modulate innate immunity via redox‐sensitive inflammatory pathways or direct activation of the inflammasomes, of which NLRP3 is the best known (Harijith et al., 2014). This multimeric protein complex is an important cellular signalling hub that converts extracellular (such as pathogens and their products) and intracellular stress stimuli (such as oxidative stress, altered ion homeostasis and disturbed protein synthesis and folding) into proinflammatory reactions through distinct canonical and non‐canonical pathways. In this sense, it activates caspase‐1, thereby allowing for cleavage and subsequent activation of the proinflammatory cytokines IL‐1β and IL‐18 (Strowig et al., 2012). Here, we show that efavirenz induces the expression of NLRP3 and its targets, and that this effect is more pronounced in the absence of p62. The novelty in this regard is the fact that this action occurs in context of dual pharmacologically induced stress (ER/mitochondria) in which p62 up‐regulation is mediated by CHOP and not NF‐κB. Although there is evidence of a clear link between p62 and inflammasome activation, it has mainly been described in relation with autophagy or mitophagy (Zhong et al., 2016), as p62 alleviates inflammation through degradation of specific proteins, including NLRP3, and enabled the elimination of damaged mitochondria through mitophagy. In this regard, we report a novel autophagy‐independent functional connection between p62 and NLRP3. This result is in line with recent evidence of the capacity of p62 to inactivate inflammasomes by directly binding NLR proteins and inhibiting their self‐oligomerization. Our finding may be of relevance given that dysregulated inflammasome activity has been implicated in the pathogenesis of inflammatory disorders and in certain neurodegenerative diseases and shown to exacerbate symptoms of infectious diseases (Strowig et al., 2012). Moreover, there is increasing evidence of protective autophagy‐independent action of p62, related with the elimination of protein aggregates. Actually, the incorporation of GFP‐LC3 into protein aggregates has been found to depend on its interaction with p62 – a protein largely associated with protein aggregates (Shvets and Elazar, 2008). Given the fact that there is ER stress in our model, this may be the case. Additionally, the removal of damaged mitochondrial fragments and proteins in our model may be carried out through p62. The specific mitochondrial action of p62 in this model is supported by the findings that (i) efavirenz increased the mitochondrial content of p62 and (ii) p62 is protective against efavirenz‐induced mitochondrial superoxide production.
In conclusion, the present study provides evidence of specific and autophagy‐independent regulation of p62 by an antiretroviral drug, a finding that may have far‐reaching dimensions due to the fact that p62 could be directly related to human immunodeficiency virus (HIV), as it has been described as a potential receptor for HIV‐1 gp41 (Yang et al., 2001) and HIV‐2 gp36 binding (Chen et al., 2000). More recently, p62 has been reported to enhance Tripartite Motif Containing Protein (TRIM)5alpha‐mediated retroviral restriction, which protects mammalian cells from retroviral infections (O'Connor et al., 2010). Given the growing evidence of p62 involvement in human diseases, p62 needs to be assessed as a promising effector candidate in the off‐target effects induced by drugs. This is of particular relevance in the case of antiretroviral drugs, as HIV infection is a chronic inflammatory disease and p62 behaves as a negative regulator of inflammasome activity. Nevertheless, the findings presented here were exclusively obtained in vitro, and it would be of great interest to reproduce them in an in vivo experimental setting which would provide conclusions of greater physiological and clinical relevance.
Author contributions
F.A., M.P., A.B.M. and A.M.‐R. performed experiments, analysed data and prepared the figures; N.A., A.B.‐G. and J.V.E. designed experiments, critically revised data, drafted and wrote the MS.
Conflict of interest
J.V.E. has collaborated with Gilead, Abbvie, MSD and Pfizer. The rest of authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Hep3B cells were treated with vehicles (MeOH or DMSO), Efavirenz (EFV, 10 or 25 μM), thapsigargin (Tg) 2 μM, rotenone (Rot) 25 μM or CCCP 10 μM for 24 h. (A) Representative WB image of JNK and pJNK, and (B) Summary of densitometry data after normalization with the expression of GAPDH considered a housekeeping protein. Data (mean ± SEM, n = 5) were calculated as percentage of control (untreated cells) and analysed by a one‐way ANOVA multiple comparison test followed by a Newman–Keuls test (*P < 0.05 for EFV and #P < 0.05 for CCCP versus vehicle).
Acknowledgements
The authors thank Brian Normanly for his English language editing and Prof. Terje Johansen (The Arctic University of Norway) for the gift of mCherry‐p62 plasmid.
Grants PI14/00312 and CIBER CB06/04/0071 (both from Instituto de Salud Carlos III, Ministerio de Economía, Industria y Competitividad), PROMETEOII/2014/035 and GV/2014/118 (both from Generalitat Valenciana). F.A., M.P. and A.M.‐R. are recipients of Predoctoral Trainee Research Grants (FI12/00198, Instituto de Salud Carlos III, Ministerio de Economía y Competitividad; ACIF/2013/136, Generalitat Valenciana; and FPU13/00151, Ministerio de Educación, Cultura y Deporte respectively). A.B.‐G. is recipient of a Juan de la Cierva contract (ref. JCI‐2012‐15124, Ministerio de Economía y Competitividad). A.B.M. is the recipient of Postgraduate Trainee Research Grant from Fundación Juan Esplugues. We would also like to acknowledge the support of the TRANSAUTOPHAGY COST Action, CA15138.
Alegre, F. , Moragrega, Á. B. , Polo, M. , Marti‐Rodrigo, A. , Esplugues, J. V. , Blas‐Garcia, A. , and Apostolova, N. (2018) Role of p62/SQSTM1 beyond autophagy: a lesson learned from drug‐induced toxicity in vitro . British Journal of Pharmacology, 175: 440–455. doi: 10.1111/bph.14093.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova N, Gomez‐Sucerquia LJ, Moran A, Alvarez A, Blas‐Garcia A, Esplugues JV (2010). Enhanced oxidative stress and increased mitochondrial mass during efavirenz‐induced apoptosis in human hepatic cells. Br J Pharmacol 160: 2069–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolova N, Gomez‐Sucerquia LJ, Gortat A, Blas‐Garcia A, Esplugues JV (2011). Compromising mitochondrial function with the antiretroviral drug efavirenz induces cell survival‐promoting autophagy. Hepatology 54: 1009–1019. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Gomez‐Sucerquia LJ, Alegre F, Funes HA, Victor VM, Barrachina MD et al (2013). ER stress in human hepatic cells treated with Efavirenz: mitochondria again. J Hepatol 59: 780–789. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A et al (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. J Cell Biol 171: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Xiao Y, Wu W, Wang Q, Luo G, Dierich MP (2000). HIV‐2 transmembrane protein gp36 binds to the putative cellular receptor proteins P45 and P62. Immunobiology 201: 317–322. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deegan S, Koryga I, Glynn SA, Gupta S, Gorman AM, Samali A (2015). A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem Biophys Res Commun 456: 305–311. [DOI] [PubMed] [Google Scholar]
- Dong Q, Oh JE, Yi JK, Kim RH, Shin KH, Mitsuyasu R et al (2013). Efavirenz induces autophagy and aberrant differentiation in normal human keratinocytes. Int J Mol Med 31: 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz‐Meco MT et al (2008). The signaling adaptor p62 is an important NF‐κB mediator in tumorigenesis. Cancer Cells 13: 343–354. [DOI] [PubMed] [Google Scholar]
- Goswami P, Gupta S, Biswas J, Joshi N, Swarnkar S, Nath C et al (2016). Endoplasmic reticulum stress plays a key role in rotenone‐induced apoptotic death of neurons. Mol Neurobiol 53: 285–298. [DOI] [PubMed] [Google Scholar]
- Gow A, Wrabetz L (2009). CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr Opin Neurobiol 19: 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J et al (2013). ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harijith A, Ebenezer DL, Natarajan V (2014). Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol 5: 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Schulman BA (2006). Structural complexity in ubiquitin recognition. Cell 124: 1133–1136. [DOI] [PubMed] [Google Scholar]
- Hom JR, Gewandter JS, Michael L, Sheu SS, Yoon Y (2007). Thapsigargin induces biphasic fragmentation of mitochondria through calcium‐mediated mitochondrial fission and apoptosis. J Cell Physiol 212: 498–508. [DOI] [PubMed] [Google Scholar]
- Ishiguro H, Yasuda K, Ishii N, Ihara K, Ohkubo T, Hiyoshi M et al (2001). Enhancement of oxidative damage to cultured cells and Caenorhabditis elegans by mitochondrial electron transport inhibitors. IUBMB Life 51: 263–268. [DOI] [PubMed] [Google Scholar]
- Ishii T, Yanagawa T, Yuki K, Kawane T, Yoshida H, Bannai S (1997). Low micromolar levels of hydrogen peroxide and proteasome inhibitors induce the 60‐kDa A170 stress protein in murine peritoneal macrophages. Biochem Biophys Res Commun 232: 33–37. [DOI] [PubMed] [Google Scholar]
- Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A et al (2010). p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element‐driven gene transcription. J Biol Chem 285: 22576–22591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T, Lamark T (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7: 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuragi Y, Ichimura Y, Komatsu M (2015). p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 282: 4672–4678. [DOI] [PubMed] [Google Scholar]
- Laurin N, Brown JP, Morissette J, Raymond V (2002). Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 70: 1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Park JS, Lee YS, Sung SH, Lee YH, Bae SH (2017). The hypertension drug, verapamil, activates Nrf2 by promoting p62‐dependent autophagic Keap1 degradation and prevents acetaminophen‐induced cytotoxicity. BMB Rep 50: 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA et al (2003). Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 278: 8516–8525. [DOI] [PubMed] [Google Scholar]
- Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL et al (2016). p62 links the autophagy pathway and the ubiqutin‐proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 21: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY et al (2009). Autophagy suppresses tumorigenesis through elimination of p62. Cell 137: 1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaso K, Yoshimoto Y, Nakano T, Takeshima T, Fukuhara Y, Yasui K et al (2004). Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson's disease. Brain Res 1012: 42–51. [DOI] [PubMed] [Google Scholar]
- O'Connor C, Pertel T, Gray S, Robia SL, Bakowska JC, Luban J et al (2010). p62/sequestosome‐1 associates with and sustains the expression of retroviral restriction factor TRIM5alpha. J Virol 84: 5997–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Kaneko M, Okuma Y, Matsubara K, Nomura Y (2013). Endoplasmic reticulum stress and Parkinson's disease: the role of HRD1 in averting apoptosis in neurodegenerative disease. Oxid Med Cell Longev 2013: 239854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Mori M (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11: 381–389. [DOI] [PubMed] [Google Scholar]
- Perry RP, Kelley DE (1970). Inhibition of RNA synthesis by actinomycin D: characteristic dose‐response of different RNA species. J Cell Physiol 76: 127–139. [DOI] [PubMed] [Google Scholar]
- Polo M, Alegre F, Funes HA, Blas‐Garcia A, Victor VM, Esplugues JV et al Mitochondrial (dys)function ‐ a factor underlying the variability of efavirenz‐induced hepatotoxicity? Br J Pharmacol 2015; 172: 1713–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A, Fenouille N, Auberger P (2012). When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2: 397–413. [PMC free article] [PubMed] [Google Scholar]
- Ramesh Babu J, Lamar Seibenhener M, Peng J, Strom AL, Kemppainen R, Cox N et al (2008). Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem 106: 107–120. [DOI] [PubMed] [Google Scholar]
- Rea SL, Majcher V, Searle MS, Layfield R (2014). SQSTM1 mutations – bridging Paget disease of bone and ALS/FTLD. Exp Cell Res 325: 27–37. [DOI] [PubMed] [Google Scholar]
- Rodriguez A, Durán A, Selloum M, Champy MF, Diez‐Guerra FJ, Flores JM et al (2006). Mature‐onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab 3: 211–222. [DOI] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz‐Meco MT, Moscat J (1999). The interaction of p62 with RIP links the atypical PKCs to NF‐kB activation. EMBO J 18: 3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Du Y, Diaz‐Meco MT, Moscat J, Wooten MC, Wooten MW (2013). A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim Biophys Acta 1833: 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yang J, Zhao J, Xiao C, Xu C, Xiang Y (2015). The switch from ER stress‐induced apoptosis to autophagy via ROS‐mediated JNK/p62 signals: a survival mechanism in methotrexate‐resistant choriocarcinoma cells. Exp Cell Res 334: 207–218. [DOI] [PubMed] [Google Scholar]
- Shin J (1998). P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm Res 21: 629–633. [DOI] [PubMed] [Google Scholar]
- Shvets E, Elazar Z (2008). Autophagy‐independent incorporation of GFP‐LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy 4: 1054–1056. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strowig T, Henao‐Mejia J, Elinav E, Flavell R (2012). Inflammasomes in health and disease. Nature 481: 278–286. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Haasken S, Cassel SL (2014). Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319: 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP (1990). Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+‐ATPase. Proc Natl Acad Sci U S A 87: 2466–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiß M, Kost B, Renner‐Müller I, Wolf E, Mylonas I, Brüning A (2016). Efavirenz causes oxidative stress, endoplasmic reticulum stress, and autophagy in endothelial cells. Cardiovasc Toxicol 16: 90–99. [DOI] [PubMed] [Google Scholar]
- Yang H, Xiao Y, Lu Y, Chen YH (2001). Characterization of interaction between C‐domain on HIV‐1 gp41 and the putative receptor protein p62. Immunobiology 203: 778–785. [DOI] [PubMed] [Google Scholar]
- Zhang GF, Zhang Y, Zhao G (2015). Crocin protects PC12 cells against MPP(+)‐induced injury through inhibition of mitochondrial dysfunction and ER stress. Neurochem Int 89: 101–110. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Umemura A, Sanchez‐Lopez E, Liang S, Shalapour S, Wong J et al (2016). NF‐κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164: 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang H, Hu D, Singer D, Walker JV, Nisr RB, Tieu K et al (2015). Local anesthetics induce autophagy in young permanent tooth pulp cells. Cell Death Dis 1: 15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Hep3B cells were treated with vehicles (MeOH or DMSO), Efavirenz (EFV, 10 or 25 μM), thapsigargin (Tg) 2 μM, rotenone (Rot) 25 μM or CCCP 10 μM for 24 h. (A) Representative WB image of JNK and pJNK, and (B) Summary of densitometry data after normalization with the expression of GAPDH considered a housekeeping protein. Data (mean ± SEM, n = 5) were calculated as percentage of control (untreated cells) and analysed by a one‐way ANOVA multiple comparison test followed by a Newman–Keuls test (*P < 0.05 for EFV and #P < 0.05 for CCCP versus vehicle).