

Abstract
Background and Purpose
Opioids may inhibit the 5‐HT transporter (SERT) and the noradrenaline transporter (NET). NET inhibition may contribute to analgesia, and SERT inhibition or interactions with 5‐HT receptors may cause serotonergic toxicity. However, the effects of different opioids on the human SERT, NET and 5‐HT receptors have not been sufficiently studied.
Experimental Approach
We determined the potencies of different opioids to inhibit the SERT and NET in vitro using human transporter‐transfected HEK293 cells. We also tested binding affinities at 5‐HT1A, 5‐HT2A and 5‐HT2C receptors. Additionally, we assessed clinical cases of the serotonin syndrome associated with each opioid reported by PubMed and a World Health Organization database.
Key Results
Dextromethorphan, l(R)‐methadone, racemic methadone, pethidine, tramadol and tapentadol inhibited the SERT at or close to observed drug plasma or estimated brain concentrations in patients. Tapentadol was the most potent NET inhibitor. Pethidine, tramadol, l(R)‐methadone, racemic methadone, dextromethorphan and O‐desmethyltramadol also inhibited the NET. 6‐Monoacetylmorphine, buprenorphine, codeine, dihydrocodeine, heroin, hydrocodone, hydromorphone, morphine, oxycodone and oxymorphone did not inhibit the SERT or NET. Fentanyl interacted with 5‐HT1A receptors and methadone, pethidine and fentanyl with 5‐HT2A receptors, in the low micromolar range. Opioids most frequently associated with the serotonin syndrome are tramadol, fentanyl, tapentadol, oxycodone, methadone and dextromethorphan.
Conclusions and Implications
Some synthetic opioids interact with the SERT and NET at potentially clinically relevant concentrations. SERT inhibition by tramadol, tapentadol, methadone, dextromethorphan and pethidine may contribute to the serotonin syndrome. Direct effects on 5‐HT1A and/or 5‐HT2A receptors could be involved with methadone and pethidine.
Abbreviations
- DAT
dopamine transporter
- ICSR
Individual Case Safety Report
- MDMA
3,4‐methylenedioxymethamphetamine
- NET
noradrenaline transporter
- SERT
5‐HT transporter
Introduction
Opioids primarily activate opioid receptors, but some atypical synthetic opioids have also been shown to interact with the noradrenaline transporter (NET) and/or the 5‐HT (serotonin) transporter (SERT) (Codd et al., 1995; Barann et al., 2015). The effects of opioids on noradrenaline and 5‐HT transport have previously been studied using rat brain synaptosomes (Larsen and Hyttel, 1985; Driessen et al., 1993; Codd et al., 1995; Frink et al., 1996; Giusti et al., 1997; Tzschentke et al., 2007). However, only one study of which we are aware assessed a larger group of opioids using the same rat transporter assay (Codd et al., 1995). Additionally, no data have been reported on the effects of different clinically used opioids on the human SERT, NET and dopamine transporter (DAT) using the same method, thus hindering direct comparisons of their transporter inhibition potencies. Only one recent study assessed the effects of a few opioids on the human SERT and showed SERT inhibition by tramadol and pethidine but not morphine, hydromorphone, fentanyl and alfentanil (Barann et al., 2015). More information on the non‐opioid effects of various opioids is needed because these analgesic substances are very widely used. Dopamine and the DAT are involved in addiction. Inhibition of NET may contribute to the analgesic effects of synthetic opioids, such as the newly marketed dual‐mechanism analgesic tapentadol (Tzschentke et al., 2007; Bee et al., 2011; Schroder et al., 2011). SERT inhibition may have analgesic effects but may also increase the risk of 5‐HT toxicity (Boyer and Shannon, 2005). The adverse effects of 5‐HT can be mild and include nausea, vomiting and insomnia. However, a more severe manifestation of 5‐HT toxicity is the potentially fatal serotonin syndrome, which includes a triad of effects: mental state changes (delirium, agitation, confusion and coma), autonomic stimulation (hyperthermia, tachycardia and diaphoresis) and neuromuscular excitation (tremor, hyperreflexia and rigidity; Gillman, 2005). The serotonin syndrome has been associated with several opioids or combinations of opioids with other serotonergic drugs in numerous case reports (Schwartz et al., 2008; Guo et al., 2009; Monte et al., 2010; Rastogi et al., 2011; Walter et al., 2012; Shakoor et al., 2014; Abadie et al., 2015). In a small analysis of pharmacovigilance data from a single country, tramadol was the opioid that was most frequently associated with the serotonin syndrome (Chassot et al., 2012). However, a larger and more representative analysis of such spontaneous adverse‐effect reporting data is needed.
The primary aim of the present study was to investigate and compare the potencies of a larger group of representative and widely used opioids to inhibit the human SERT, NET and DAT in vitro. We also tested whether opioids that interact with one of these monoamine transporters induce transporter‐mediated monoamine release. Furthermore, activity at 5‐HT1A, 5‐HT2A and 5‐HT2C receptors may be involved in animal models of the serotonin syndrome (Martin et al., 1991; Van Oekelen et al., 2002; Tao et al., 2003; Fox et al., 2009). Therefore, we determined the affinities of opioids to bind directly to these 5‐HT receptors. Finally, we collected data on the frequency of reports of 5‐HT toxicity associated with opioids and sought to establish links between the in vitro data and clinical data.
Methods
Inhibition of 5‐HT, dopamine and noradrenaline uptake
Inhibition of the human NET, DAT and SERT was assessed in HEK 293 cells (Invitrogen, Zug, Switzerland) stably transfected with the respective human transporter as previously described (Tatsumi et al., 1997; Hysek et al., 2012; Luethi et al., 2017b). The cells were cultured in DMEM (Gibco, Life Technologies, Zug, Switzerland) with 10% fetal bovine serum (Gibco) and 250 μg·mL−1 Geneticin (Gibco) to 70–90% confluence, detached and then resuspended (3 × 106 cells·mL−1) in Krebs‐Ringer bicarbonate buffer (Sigma‐Aldrich, Buchs, Switzerland). For [3H]‐dopamine uptake experiments, the uptake buffer was supplemented with 0.2 mg·mL−1 ascorbic acid. The cell suspension (100 μL) was incubated with 25 μL buffer containing the test drugs, vehicle control or monoamine‐specific inhibitors (10 μM nisoxetine for NET, 10 μM mazindol for DAT and 10 μM fluoxetine for SERT) for 10 min in a round bottom 96‐well plate at room temperature by shaking at 450 rotations min−1 on a rotary shaker. Monoamine uptake transport was then initiated by adding 50 μL of [3H]‐noradrenaline (13.1 Ci·mmol−1; PerkinElmer), [3H]‐dopamine (30.0 Ci·mmol−1, PerkinElmer) or [3H]‐5‐HT (80.0 Ci·mmol−1; Anawa, Zurich, Switzerland) dissolved in buffer at a final concentration of 5 nM for an additional 10 min. Thereafter, 100 μL of the cell suspension was transferred to 500 μL microcentrifuge tubes that contained 50 μL of 3 M KOH and 200 μL silicon oil (1:1 mixture of silicon oil types AR 20 and AR 200; Sigma‐Aldrich). To separate the cells from the uptake buffer, they were centrifuged through silicone oil for 3 min at 16 550 × g, and the tubes were frozen in liquid nitrogen immediately afterward. The cell pellet was then cut into 6 mL scintillation vials (Perkin‐Elmer) that contained 0.5 mL lysis buffer (0.05 M TRIS–HCl, 50 mM NaCl, 5 mM EDTA and 1% NP‐40 in water). The samples were shaken for 1 h before 5 mL scintillation fluid (Ultimagold, Perkin Elmer, Schwerzenbach, Switzerland) was added. Monoamine uptake was then quantified by liquid scintillation counting on a Packard 1900 TR Tri‐Carb Liquid Scintillation Counter (Packard Instrument Company). Non‐specific uptake that was determined in the presence of selective inhibitors was subtracted from the total counts, and monoamine uptake was compared with the vehicle control.
Transporter‐mediated monoamine release
Substances that inhibit the monoamine uptake may also be monoamine transporter substrates and release monoamines via the transporter. The potential of the drugs which inhibited the uptake to also initiate transporter‐mediated noradrenaline or 5‐HT efflux was assessed in HEK 293 cells that overexpressed the respective human transporter as previously described (Simmler et al., 2013, 2014; Luethi et al., 2017b). Briefly, 100 000 cells per well were cultured overnight in a poly‐D‐lysine coated XF24 cell culture microplate (Seahorse Biosciences, North Billerica, MA, USA). Thereafter, the cells were preloaded with 10 nM [3H]‐noradrenaline, [3H]‐dopamine or [3H]‐5‐HT diluted in 85 μL Krebs‐HEPES buffer (130 mM NaCl, 1.3 mM KCl, 2.2 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 10 mM HEPES, 10 mM D‐glucose, pH 7.5) containing 10 μM pargyline and 0.2 mg·mL−1 ascorbic acid for 20 min at 37°C, washed twice and treated with 1000 μL Krebs‐HEPES buffer containing 100 μM of the test drugs for 15 min (DAT and SERT) or 45 min (NET) at 37°C by shaking at 300 rotations min−1 on a rotary shaker. The cells were then washed again with cold buffer and lysed in 50 μL lysis buffer during 1 h. Thereafter, 40 μL of the cell lysate was transferred into 4 mL scintillation vials with 3.5 mL scintillation fluid, and the radioactivity inside the cells was quantified by liquid scintillation counting as described for the monoamine uptake inhibition assay. Monoamine transporter blockers (10 μM nisoxetine for NET, 10 μM mazindol for DAT and 10 μM citalopram for SERT) were included in the experiment to determine ‘pseudo‐efflux’ caused by non‐specific monoamine release and subsequent reuptake inhibition (Scholze et al., 2000). Thus, these uptake inhibitors served as negative control conditions. 3,4‐Methylenedioxymethamphetamine (MDMA) was used as comparator compound that is known to induce monoamine release in this assay (positive control in each experiment; Hysek et al., 2012; Simmler et al., 2014). All of the conditions were normalized to radioactive counts of the assay buffer control condition. The use of a single high concentration and the release durations were based on kinetic evaluation of the release‐over‐time curves for substrate‐releasers in previous studies (Hysek et al., 2012; Simmler et al., 2014).
5‐HT1A and 5‐HT2A receptor radioligand binding assays
For membrane preparations, HEK293 cells, transiently transfected with the 5‐HT1A or 5‐HT2A receptor, were released from culture flasks using trypsin/EDTA, harvested, washed twice with ice‐cold PBS (without Ca2+ and Mg2+), pelleted at 210 × g for 5 min at 4°C, frozen and stored at −80°C (Luethi et al., 2017a). Frozen pellets were suspended in 20 mL HEPES‐NaOH (20 mM, pH 7.4) containing 10 mM EDTA and homogenized with a Polytron (PT 6000, Kinematica, Lucerne, Switzerland) at 14 000 rpm for 20 s. The homogenates were centrifuged at 48 000 × g for 30 min at 4°C. Subsequently, the supernatants were removed and discarded, and the pellets resuspended in 20 mL HEPES‐NaOH (20 mM, pH 7.4) containing 0.1 mM EDTA using the Polytron (20 s at 14 000 rpm). This procedure was repeated and the final pellets resuspended in HEPES‐NaOH containing 0.1 mM EDTA and homogenized using the Polytron. Typically, aliquots of 2 mL membrane portions were stored at −80°C. With each new membrane batch, the K D was determined by a saturation curve.
For the competitive binding assays, 1.39 nM [3H]8‐hydroxy‐2‐(di‐n‐propylamine)tetralin ([3H]‐8‐OH‐DPAT) and 0.45 nM [3H]‐ketanserin were used as 5‐HT1A and 5‐HT2A receptor radioligands, respectively, at concentrations equal or close to the K D values (1 and 0.45 nM, respectively). Specific binding of the radioligands to the target receptors was defined as the difference between total binding (binding buffer alone) and non‐specific binding determined in the presence of 10 μM pindolol (for the 5‐HT1A receptor radioligand) or 10 μM spiperone (for the 5‐HT2A receptor radioligand). The compounds were tested at a broad range of concentrations (30 pM to 30 μM) in duplicates. The test compounds were diluted in binding assay buffer at pH 7.4 (50 mM Tris/HCl, 10 mM MgCl2 and 1 mM EGTA), and dilution curves were made in assay microplates (Greiner, 96 well, U‐bottom, PS). A total of 50 μL of radioligand and 100 μL of membrane suspension were added to the assay plates (final volume in each well, 200 μL) that were incubated and shaken for 30 min at room temperature. Incubations were terminated by rapid filtration through Unifilter‐96 plates (Packard Instrument Company, PerkinElmer, Schwerzenbach, Switzerland) and glass filters GF/C (PerkinElmer) presoaked for a minimum of 1 h in polyethylenimine (0.3%) and washed three times with ice‐cold washing buffer (50 mM Tris/HCl, pH 7.4). After the addition of Microscint 40 (45 μL per well, PerkinElmer), the Unifilter‐96 plates were sealed. After 1 h, radioactivity was counted using a TopCount Microplate Scintillation Counter (Packard Instrument Company).
5‐HT2C receptor radioligand binding assay
Substances that showed binding at the 5‐HT1A and 5‐HT2A receptors were also tested at the 5‐HT2C receptor (many compounds that bind to the 5‐HT2A receptor also bind to the 5‐HT2C receptor). For membrane preparations, HEK293 cells, transiently transfected with the 5‐HT2C receptor, were released from culture flasks using trypsin/EDTA, harvested, washed twice with ice‐cold PBS (without Ca2+ and Mg2+), pelleted at 210 × g for 5 min at 4°C, frozen and stored at −80°C (Luethi et al., 2017a). Frozen pellets were suspended in 20 mL HEPES/NaOH (20 mM, pH 7.4) containing 10 mM EDTA and homogenized with a Polytron (PT 6000, Kinematica, Lucerne, Switzerland) at 14 000 rpm for 20 s. The homogenates were centrifuged at 48 000 × g for 30 min at 4°C. Subsequently, the supernatants were removed and discarded, and the pellets resuspended in 20 mL HEPES‐NaOH (20 mM, pH 7.4) containing 0.1 mM EDTA using the Polytron (20 s at 210 × g). This procedure was repeated and the final pellets resuspended in HEPES/NaOH containing 0.1 mM EDTA and homogenized using the Polytron. Typically, aliquots of 2 mL membrane portions were stored at −80°C. With each new membrane batch, the K D was determined by a saturation curve.
For the competitive binding assay, [3H]‐mesulergine was used as 5‐HT2C receptor radioligand at 1.6 nM, a concentration equal to the K D value. Specific binding of the radioligand to the target receptor was defined as the difference between total binding (binding buffer alone) and non‐specific binding determined in the presence of 10 μM mianserin. The compounds were tested at a broad range of concentrations (30 pM to 30 μM) in duplicates. The test compounds were diluted in binding assay buffer at pH 7.4 (50 mM Tris/HCl, 10 mM MgCl2, 1 mM EGTA and 10 μM pargyline), and dilution curves were made in 96‐well white polystyrene assay plates (Sigma‐Aldrich, Buchs, Switzerland). Membrane stocks were thawed and resuspended to a concentration of approximately 0.04 mg protein mL−1 binding assay buffer using a Polytron tissue homogenizer. The membrane homogenate (40 μg·mL−1) was then lightly mixed for 5–30 min with YSi‐poly‐l‐lysine (PerkinElmer, Schwerzenbach, Switzerland) at 0.5 mg beads per well. A total of 50 μL of the membranes/beads mixture was added to each well of the assay plate that contained the radioligand (50 μL) and the test compounds (final volume in each well, 200 μL) to start the assay. The assay plates were sealed, incubated for 2 h at room temperature with agitation and then counted in the PVT SPA counting mode of a TopCount Microplate Scintillation Counter (Packard Instrument Company, PerkinElmer, Schwerzenbach, Switzerland).
Cytotoxicity
To confirm cell integrity during the pharmacological assays, cytotoxicity was assessed using the ToxiLight bioassay (Lonza, Basel, Switzerland) according to the manufacturer's instructions and as described previously (Rickli et al., 2015). The assay quantitatively measures the release of adenylate kinase from damaged cells, providing a highly sensitive method of measuring cytolysis (Crouch et al., 1993).
Database searches for opioids associated with serotonin syndrome
The Medline PubMed database and VigiBase™ World Health Organization (WHO) Global Database of Individual Case Safety Reports (ICSRs) were searched using VigiLyze™ as the search tool for cases of serotonin syndrome associated with opioids. Published cases and case series of serotonin syndrome were searched in Medline using the terms ‘serotonin syndrome’ AND each of the opioids investigated in the present study in vitro. All publications up to 31 August 2016 were included. The reports were manually searched for relevance, and the drugs were classified as ‘suspected among other drugs’ or ‘the only suspected drug’ (according to the assessment of the authors of the case reports). Review articles that did not report actual patient data were not considered.
The WHO database search was performed on 18 April 2016. For each opioid, we identified all spontaneous reports and filtered the results using the Medical Dictionary for Regulatory Activities (MedDRA) adverse reaction term ‘serotonin syndrome’. Only ICSRs in which the opioid was reported as ‘suspected’ or ‘interacting’ were included. ICSRs that reported that the opioid were ‘concomitantly’ used but without a time relationship with the adverse reaction were excluded.
Data and statistical analysis
The data and statistical analysis complied with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Calculations were performed using Prism 7.0a software (GraphPad, San Diego, CA, USA). Monoamine transporter inhibition data were fit by nonlinear regression to variable‐slope sigmoidal dose–response curves, and IC50 values were determined. The SERT/NET ratio is expressed as (1/SERT IC50):(1/NET IC50). Compound‐induced release from five independent experiments was compared with negative controls using ANOVA followed by Dunnett's test. Values of P < 0.05 were considered statistically significant. The substances were considered a monoamine releaser if they caused significantly higher efflux than the negative controls. IC50 values of radioligand binding were determined by calculating nonlinear regression curves for a one‐site model using three independent 10‐point concentration–response curves, run in duplicate, for each compound. Ki (affinity) values, which correspond to the K Ds, were determined using the Cheng‐Prusoff equation: K i = IC 50 / (1 + [S]/K M ). K i values are presented as means ± SD (in μM).
Materials
Buprenorphine, citalopram, codeine, dextromethorphan, dihydrocodeine, fentanyl, heroin (diacetylmorphine, diamorphine), 6‐acetylmorphine (6‐mono‐acetylmorphine), hydrocodone, hydromorphone, mazindol, MDMA, methadone, morphine, oxycodone, oxymorphone, pethidine (meperidine), tramadol, O‐desmethyl‐cis‐tramadol, tapentadol, venlafaxine and fluoxetine were purchased from Lipomed (Arlesheim, Switzerland). Mianserin, nisoxetine, pargyline, pindolol and spiperone were supplied by Sigma‐Aldrich (Buchs, Switzerland). D(S)‐methadone and l(R)‐methadone were obtained from Alsachim (Illkirch Graffenstaden, France). The HPLC purity of all of the substances was >98%. [3H]‐8‐OH‐DPAT, [3H]‐ketanserin and [3H]‐mesulergine were supplied by Perkin‐Elmer.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Inhibition of 5‐HT, dopamine and noradrenaline uptake
IC50 values for SERT, DAT and NET inhibition are shown in Table 1, and the full inhibition curves are shown in Supporting Information Figure S1. Dextromethorphan, l(R)‐methadone and racemic methadone potently inhibited the SERT, with concentrations that are likely to be reached in the human brain when these drugs are used in patients (Table 3). Dextromethorphan was as potent as fluoxetine. Pethidine, tramadol, tapentadol and d(S)‐methadone also inhibited the SERT at low micromolar concentrations, with IC50 values of 1–10 μM (Table 1) and at concentrations similar to or close to those reached in human brain at therapeutic doses (Table 3). The SERT inhibition potency and SERT/NET ratio values of tramadol and tapentadol were in the same order of magnitude (Table 1).
Table 1.
Monoamine transporter inhibition and 5‐HT receptor binding by different opioids and known SERT/NET inhibitors (antidepressants)
| – | NET | DAT | SERT | SERT/NET ratio | 5‐HT1A | 5‐HT2A | 5‐HT2C |
|---|---|---|---|---|---|---|---|
| IC50 [μM] (95% CI) | IC50 [μM] (95% CI) | IC50 [μM] (95% CI) | (95% CI) | Receptor binding Ki ± SD [μM] | Receptor binding Ki ± SD [μM] | Receptor binding Ki ± SD [μM] | |
| Opioids | |||||||
| Dextromethorphan | 5.8 (3.9–8.6) | >100 | 0.068 (0.047–0.100) | 85 (39–186) | >17 | >13 | NA |
| Methadone | 4.1 (2.7–6.3) | >100 | 0.23 (0.16–0.32) | 18 (8–39) | >17 | 0.61 ± 0.03 | 2.2 ± 0.3 |
| l(R)‐methadone | 2.5 (1.7–3.7) | >100 | 0.28 (0.21–0.37) | 9 (5–18) | >17 | 0.72 ± 0.61 | 2.6 ± 0.3 |
| Pethidine | 1.6 (1.0–2.4) | >100 | 1.6 (0.95–2.5) | 1.0 (0.4–2.6) | >17 | 3.6 ± 0.35 | 15 ± 0.1 |
| Tramadol | 2.1 (1.4–3.1) | 100 (67–148) | 3.3 (2.7–4.1) | 0.62 (0.34–1.1) | >17 | >13 | NA |
| Tapentadol | 1.3 (1.0–1.6) | 78 (60–102) | 3.3 (2.3–4.8) | 0.39 (0.21–0.70) | >17 | 6.3 ± 0.2 | 12 ± 3.2 |
| d(S)‐methadone | 69 (42–113) | >100 | 5.6 (3.9–8.0) | 12 (5–29) | >17 | 0.52 ± 0.11 | 1.9 ± 0.2 |
| O‐desmethyltramadol | 6.1 (4.6–8.1) | >100 | 24 (16–36) | 0.26 (0.13–0.52) | >17 | >13 | NA |
| Fentanyl | 52 (40–69) | >100 | >100 | NA | 2.1 ± 0.20 | 1.3 ± 0.12 | >15 |
| Buprenorphine | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Codeine | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Dihydrocodeine | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Heroin | >100 | >100 | >100 | NA | >17 | >13 | NA |
| 6‐Acetylmorphine | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Hydrocodone | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Hydromorphone | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Morphine | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Oxycodone | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Oxymorphone | >100 | >100 | >100 | NA | >17 | >13 | NA |
| Example of SERT/NET inhibitors used for the treatment of depression | |||||||
| Fluoxetine | NA | NA | 0.092 (0.076–0.121) | NA | >17 | 0.13 ± 0.01 | 0.17 ± 0.03 |
| Citalopram | >20 | >20 | 0.038 (0.031–0.046) | >20 | NA | NA | NA |
| Duloxetine | 0.12 (0.10–0.15) | NA | 0.044 (0.037–0.053) | NA | NA | NA | NA |
| Venlafaxine | 0.41 (0.30–0.56) | NA | NA | NA | >17 | >13 | >15 |
Uptake values are means of three independent experiments and 95% confidence intervals (CI); NA, not assessed; SERT/NET ratio, 1/SERT IC50/1/NET IC50.
Table 3.
Estimates of human plasma and brain concentrations of opioids when used clinically
| SERT inhibition in vitro | Ctotal,plasma [μM] | Human drug concentrations in vivo | Single and (daily) doses | References | |||
|---|---|---|---|---|---|---|---|
| IC50 [μM] | fu,plasma | Cu,plasma [μM] | Cu,brain f [μM] | mg | |||
| Dextromethorphan | 0.07 | 0.01–1.4c | 75–100 p.o | (Chen et al., 1990; Pope et al., 2004; Zawertailo et al., 2010) | |||
| 0–5.6d | 0–0.44d | −400 p.o. | (Steinberg et al., 1996) | ||||
| Methadone | 0.23 | 0.3–7.3b | 0.14 | 0.04–1.0 | 0.03–0.8f , g | 60–120 (10–430) p.o. | (Eap et al., 1990; Eap et al., 2007; Kalvass et al., 2007) |
| l(R)‐methadone | 0.28 | 0.2–3.7b | 0.13 | 0.03–0.5 | 0.03–0.5f , g | 30–60 p.o. | (Eap et al., 1990; Foster et al., 2000; Eap et al., 2007; Kalvass et al., 2007; Meini et al., 2015) |
| Pethidine | 1.6 | 0.7–6e | 0.38h | 0.3–2.3 | 0.7–5.4h | 50–150 (100–500) i.m. | (Erstad et al., 1997; Kalvass et al., 2007) |
| Tramadol | 3.3 | 0.1–2.5a | 0.80 | 0.1–2.0 | 0.1–4.6j | (100–400 p.o.) | (Saarikoski et al., 2013; Kitamura et al., 2014; Saarikoski et al., 2015; de Moraes et al., 2016; Tanaka et al., 2016) |
| Tapentadol | 4.3 | 0.1–1.9 | 0.80 | 0.1–1.5 | 0.2–3.5l | (100–600 p.o.) | (Xu et al., 2010; Zannikos et al., 2013; Kitamura et al., 2014) |
| d(S)‐methadone | 5.6 | 0.2–3.6b | 0.10 | 0.02–0.4 | 0.02–0.3f , g | metabolite | (Eap et al., 1990; Foster et al., 2000; Eap et al., 2007; Kalvass et al., 2007) |
| O‐desmethyltramadol | 24 | 0.05–0.6 | NA | NA | 0.1–1.1k | metabolite | (Saarikoski et al., 2013; Saarikoski et al., 2015; Tanaka et al., 2016) |
| Fentanyl | 154 | 0.002–0.03 | 0.17 | 0.0003–0.005 | 0.0003–0.005i | 12–200 μg·h−1 s.c. | (Kalvass et al., 2007; Heiskanen et al., 2015) |
| Morphine | >100 | 0–0.1 | 0.76 | 0–0.08 | 0.02–0.04j | 5–40 (20–150) p.o. | (Kalvass et al., 2007; De Gregori et al., 2014; Schou et al., 2015) |
| 0–0.5 | 0–0.4 | 0.1j | (30–380 p.o.) | (Wolff et al., 1995; Friden et al., 2009) | |||
| Oxycodone | >100 | 0.3 | 0.6 | 0.2 | 0.2 | (10–160 p.o.) | (Friden et al., 2009) |
IC50 values are reproduced from Table 1 and are means of three experiments; human plasma concentrations are ranges observed in patients at steady state or in healthy subjects after single dose administration.
Trough concentrations at steady state at median oral daily doses of 112.5 mg divided into three doses (Tanaka et al., 2016).
Peak concentrations at steady state after oral doses of 10–430 mg methadone daily (Eap et al., 2007).
NA, not available
Peak concentrations at steady state after oral daily doses of 60 mg dextromethorphan with or without inhibition of its metabolism (Pope et al., 2004)
Maximum concentration after administration of dextromethorphan at high doses up to 400 mg (Steinberg et al., 1996). The maximum total concentration in cerebrospinal fluid was 0.44 μM (Steinberg et al., 1996).
Maximal concentration after a single dose of pethidine of 50 mg intramuscularly was 2 μM (Erstad et al., 1997). The drug label indicates threefold higher maximal single doses of 150 mg i.m. resulting in threefold higher estimated concentrations up to 6 μM.
Cu,brain = (fu,brain × Kp × Cu,plasma)/fu,plasma (Schou et al., 2015); Cu,brain, unbound concentration in brain, surrogate for brain interstitial fluid concentrations; fu,brain, fraction unbound in brain; fu,plasma, fraction unbound in plasma; Kp, total brain to total plasma ratio; Cu,plasma, unbound concentration in plasma.
fu,brain and Kp values (0.03 and 4, respectively) were taken from mice and from racemic methadone (Kalvass et al., 2007).
fu,brain, Kp and fu,plasma values (0.13, 6.8 and 0.38, respectively) were taken from mice (Kalvass et al., 2007).
fu,brain, Kp and fu,plasma values (0.07, 2.4 and 0.17, respectively) were taken from mice (Kalvass et al., 2007).
fu,brain, Kp and fu,plasma values (0.5, 0.36 and 0.76, respectively) were from mice and monkeys (Schou et al., 2015).
Cu,brain was calculated using Cu,plasma and the Kp,uu value of 2.3 from a rat microdialysis study (Kitamura et al., 2014) which higher than that using CSF in rats (0.6) (Sheikholeslami et al., 2016) or humans (1.4) (Friden et al., 2009)
Tapentadol was the most potent NET inhibitor, which was almost as potent as venlafaxine (Table 1). Pethidine, tramadol, l(R)‐methadone, methadone, dextromethorphan and O‐desmethyltramadol also inhibited the NET at low micromolar concentrations (1–10 μM; Table 1). Typical phenanthrene opioids, including 6‐acetylmorphine, buprenorphine, codeine, dihydrocodeine, heroin, hydrocodone, hydromorphone, morphine, oxycodone and oxymorphone, did not inhibit the SERT or NET (all IC50 values >100 μM). None of the opioids tested inhibited the DAT (all IC50 values ≥60 μM).
Transporter‐mediated release of 5‐HT and noradrenaline
Opioids that inhibited monoamine uptake were also tested with regard to transporter‐mediated monoamine release. None of the opioids acted as a releaser of 5‐HT or noradrenaline at a high concentration of 100 μM (Supporting Information Figure S2). Only the positive control MDMA induced significantly greater 5‐HT and noradrenaline release than citalopram and nisoxetine respectively. Dopamine release was not assessed because none of the opioids interacted with the DAT in the uptake assay.
5‐HT1A, 5‐HT2A and 5‐HT2C receptor binding affinity
None of the opioids exhibited relevant affinity for the 5‐HT1A receptor (Ki > 17 μM) with the exception of fentanyl (Table 1). In contrast, methadone, pethidine and fentanyl showed affinity for the 5‐HT2A receptor at low micromolar concentrations (Table 1) that were in the range of those concentrations observed in plasma or estimated to be present in the brain in humans treated therapeutically with methadone (Table 3). Methadone but none of the other opioids also showed very low affinity for the 5‐HT2C receptor (Table 1).
Cytotoxicity
None of the opioids showed cytotoxicity.
Opioids associated with the serotonin syndrome
The PubMed search yielded 99 patient cases (Supporting Information Table S1) that involved 114 administrations of opioids (Table 2). Twelve cases involved two opioids, and three cases involved three opioids. In the few cases providing detailed diagnostic information, serotonin syndrome was typically diagnosed according to the criteria of Hunter (Dunkley et al., 2003) or Sternbach (Sternbach, 1991). The opioids that were most frequently reported to be associated with serotonin syndrome (>10 cases) were fentanyl and tramadol, followed by oxycodone and dextromethorphan (Table 2). However, five of these cases involved both fentanyl and oxycodone. All of the cases, with the exception of one case that was associated with tramadol overdose (Marechal et al., 2011) and one case that was associated with therapeutic doses of dextromethorphan (Kinoshita et al., 2011), involved other drugs in addition to the opioid. In most cases, SERT inhibitors (SSRIs) were also involved, leading to serotonergic toxicity usually shortly after dose escalation or the addition of another serotonergic medication. SSRIs inhibited the SERT more potently than the opioids as shown for some examples in Table 1. These SSRIs were also frequently reported (>10 cases/drug) as potential causes of clinical serotonin syndrome cases (Table 2).
Table 2.
Cases of serotonin syndrome reported, classified by opioid associated with report
| Drug | WHO database | Medline database | |
|---|---|---|---|
| Only suspected cause or among others | Only suspected cause | Number of published cases | |
| Opioid | |||
| Tramadol | 647 | 62 | 26 |
| Tapentadol | 115 | 42 | 1 |
| Fentanyl | 363 | 19 | 45 |
| Dextromethorphan | 86 | 7 | 12 |
| Pethidine | 66 | 6 | 5 |
| Hydromorphone | 41 | 3 | 2 |
| Buprenorphine | 20 | 3 | 1 |
| Oxycodone | 101 | 2 | 13 |
| Methadone | 93 | 2 | 3 |
| Morphine | 64 | 1 | 4 |
| Codeine | 32 | 0 | 0 |
| Hydrocodone | 6 | 0 | 2 |
| Dihydrocodeine | 4 | 0 | 0 |
| Oxymorphone | 3 | 0 | 0 |
| Serotonergic drugs for the treatment of depression | |||
| Fluoxetine | 641 | 181 | 54 |
| Citalopram | 777 | 178 | 57 |
| Duloxetine | 993 | 550 | 20 |
| Venlafaxine | 859 | 240 | 75 |
The WHO database search yielded a total of 1641 ICSRs with at least one of the opioids noted as the suspected drug or an interacting drug and 147 ICSRs with the opioid as the only suspected cause (Table 2). The opioids that were most frequently reported in association with serotonin syndrome either alone or in combination with other drugs (e.g. SSRIs) were tramadol, fentanyl, tapentadol, oxycodone, methadone and dextromethorphan (Figure 1, Table 2). The single suspected opioids that were most frequently linked to serotonin syndrome were (in decreasing order) tramadol, tapentadol, fentanyl, dextromethorphan and pethidine (Table 2). In the majority of cases, serotonin syndrome occurred within the labelled dose range with overdose reported in less than 10% of the cases. Serotonergic drugs for the treatment of depression were reported to be suspected or interacting drugs in the majority of these ICSRs involving opioids. As expected, a separate WHO database search showed that the serotonergic drugs for the treatment of depression were also frequently reported as suspected or interacting drugs associated with serotonin syndrome (positive control, Table 2).
Figure 1.
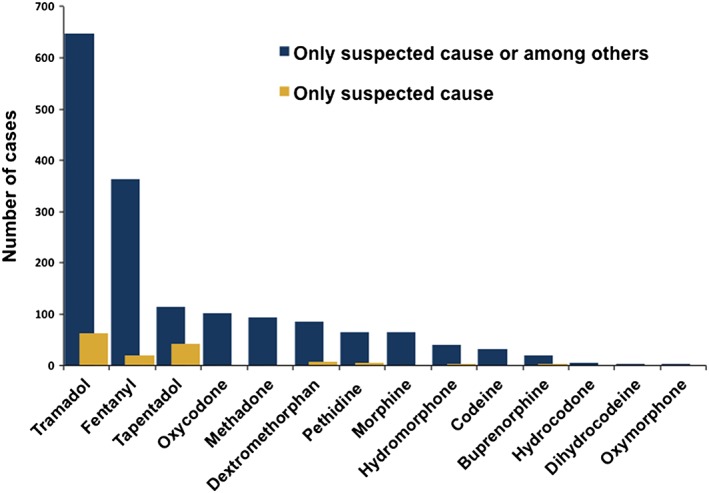
Number of spontaneous ICSR of serotonin syndrome in the VigiBase™ WHO Global Database per opioid reported as the suspected cause among other drugs or the only suspected cause.
Discussion
The present in vitro study showed that the synthetic atypical opioids dextromethorphan, methadone, pethidine, tramadol and tapentadol acted as SERT and NET inhibitors at or close to clinically observed free drug plasma and estimated free human brain concentrations (Table 3). Dextromethorphan preferentially inhibited the SERT versus NET. Tapentadol and tramadol were 2.6‐ and 1.6‐fold more potent inhibitors of the NET versus SERT respectively. Consistent with the present findings, tramadol and pethidine inhibited the human SERT in vitro, whereas morphine, hydromorphone and fentanyl were inactive (Barann et al., 2015). Also consistent with the present findings, dextromethorphan, methadone, pethidine, tramadol and tapentadol have previously been shown to block the rat SERT and NET in rat brain synaptosome in vitro assays (Larsen and Hyttel, 1985; Driessen et al., 1993; Codd et al., 1995; Frink et al., 1996; Giusti et al., 1997; Tzschentke et al., 2007). Morphine and codeine did not block the rat SERT or NET (Codd et al., 1995; Frink et al., 1996; Tzschentke et al., 2007) as shown here for the human transporter. In rats, both tramadol and tapentadol increased extracellular 5‐HT and noradrenaline levels in the brain, measured by in vivo microdialysis (Tzschentke et al., 2007; Bloms‐Funke et al., 2011).
The assumption that formed the basis of the present study was that opioids may increase the risk of serotonergic toxicity by inhibiting the SERT similarly to antidepressants and possibly at higher concentrations. In fact, SERT inhibition in vitro was found at opioid concentrations that were similar to those observed in vivo in human plasma and estimated to be present in the brain when the respective opioids were used clinically (Table 3).
We also found that opioids that were SERT inhibitors in vitro were also among those that were most frequently reported to be associated with serotonin syndrome in patients, including tramadol, tapentadol, methadone and dextromethorphan. However, fentanyl and oxycodone were also linked to serotonin syndrome but did not interact with the SERT in vitro, suggesting SERT‐independent effects on the 5‐HT system in vivo. Therefore, some opioids may also directly interact with 5‐HT receptors, such as 5‐HT1A and 5‐HT2A, that have been implicated in animal models of serotonin syndrome (Martin et al., 1991; Van Oekelen et al., 2002; Tao et al., 2003; Fox et al., 2009), or indirectly activate 5‐HT release via opioid receptor stimulation (Tao and Auerbach, 1995; Tao and Auerbach, 2002; Benade et al., 2017). In fact, the present study showed that fentanyl directly bound to 5‐HT1A, as previously shown (Martin et al., 1991) and, also to 5‐HT2A receptors, although both at concentrations clearly higher than those observed in human plasma. Methadone and pethidine also showed relevant affinity for the 5‐HT2A receptor at or near human plasma concentrations during therapeutic use of these opioids (Table 3). Tramadol was also reported to induce 5‐HT efflux in the rat raphe nucleus, possibly independently from its action as a SERT inhibitor (Bamigbade et al., 1997). None of the opioids that were tested in the present study was a DAT inhibitor, consistent with previous studies of the rat DAT (Frink et al., 1996). Thus, unlike amphetamines and cocaine that inhibited the DAT in the assay used in the present study (Simmler et al., 2013, 2014), opioids indirectly stimulate the dopaminergic system in vivo (Benade et al., 2017), which is the basis for their reinforcing properties.
Drugs that inhibit the NET have analgesic properties when administered alone but also potentiate opioid‐induced analgesia (Luccarini et al., 2004; Hall et al., 2011). While opioids primarily produce their analgesic effects via μ‐opioid receptor stimulation, noradrenergic systems may also be critically involved in the analgesic properties of some compounds (Sawynok and Reid, 1987; Schroder et al., 2010, 2011; Benade et al., 2017). NET knockout did not significantly alter morphine‐induced analgesia in mice indicating no major role of the NET in the analgesic response to morphine (Hall et al., 2011), which showed no NET inhibition in the present study. However, noradrenaline clearly contributes to the analgesic effects of tapentadol, in addition to its opioidergic properties (Tzschentke et al., 2007; Bee et al., 2011; Schroder et al., 2011). In the present study, tapentadol was the most potent human NET inhibitor among all of the opioids tested. Tapentadol also inhibited noradrenaline uptake into rat synaptosomes and increased extracellular brain concentrations of noradrenaline (Tzschentke et al., 2007; Benade et al., 2017). In the present study, tapentadol inhibited the human NET approximately threefold more potently than the human SERT, confirming data from a study of rat transporters (Tzschentke et al., 2007). Apart from tapentadol, tramadol was the only other opioid that more potently inhibited the NET versus SERT, although it was almost equipotent at these two transporters. Additionally, the SERT/NET ratio did not differ relevantly between tapentadol and tramadol, indicating that there may not be a robust or large difference between the two compounds in terms of NET or SERT inhibition. Finally, the present study showed that pethidine, tramadol and l(R) methadone also inhibited the NET at concentrations within or close to the range that is present in human plasma and brain (Table 3). Thus, noradrenaline may contribute to the analgesic effects not only of tapentadol but also of pethidine, tramadol and l(R)‐methadone.
The present clinical data analysis showed that serotonergic drugs for the treatment of depression were also involved in the majority of serotonin syndrome cases associated with opioids. In another analysis, most cases of serotonin syndrome resulted from the combined use of more than one serotonergic drug (Chassot et al., 2012) indicating a higher risk of serotonin syndrome when opioids are used with other serotonergic substances. The combined use of opioid analgesics with serotonergic antidepressants is very common in the treatment of chronic pain. For example, in a recent analysis among 433 multimorbid hospital patients with chronic pain, 71% of the patients received opioids and 35% received antidepressants and potential interactions between opioids and serotonergic antidepressants (SSRIs, SNRIs) were identified in 57 (13%) of all patients (Siebenhuener et al., 2017).
The present study has important limitations. The WHO data may mainly reflect the frequency of reporting rather than the true incidence of serotonin syndrome per opioid. Underreporting is common, and the true incidence of serotonin syndrome cannot be estimated from the present data. Additionally, we did not account for differences in the prescribing frequency or time on market. Adverse effects may be more frequently reported in the case of a novel medication. For example, tramadol and tapentadol were first reported in 1997 and 2010 respectively. Additionally, most cases were reported during the last 10 years, possibly reflecting changes in reporting and/or potential bias that resulted from post‐marketing studies of more recently marketed opioids. Furthermore, the diagnosis of serotonin syndrome may not be correct in some cases or is at least often not well documented in many spontaneous reports. There was no qualitative analysis of the reported cases, and some were only poorly documented, and some symptoms could have been other adverse effects of the opioids not meeting all the criteria of serotonin syndrome. Additionally, the reported associations are possible or likely but not definitive. In the majority of the reports of serotonin syndrome, opioids were co‐used with other substances and they were infrequently noted as the only suspected cause. The present spontaneous report data can only generate signals of possible adverse reactions that need further observation but cannot be used as prevalence markers or confirmation of a causal relationship. Nevertheless, the opioids that were more frequently reported to be associated with serotonin syndrome in the WHO database were generally the same opioids that were more frequently mentioned in case reports in PubMed.
In conclusion, we have characterized the effects of a series of opioids on the human SERT, NET, DAT, 5‐HT1A and 5‐HT2A receptors using the same method, thus allowing direct comparisons between substances. Several synthetic opioids inhibited the NET and SERT, which may contribute to their analgesic properties but may also increase the risk of 5‐HT toxicity. Serotonin syndrome may result from SERT inhibition by tramadol, tapentadol, methadone, dextromethorphan and pethidine, especially when combined with other serotonergic medications, but there may also be SERT‐independent effects with other opioids, such as fentanyl and oxycodone. These mechanisms and the risk of serotonin syndrome need to be further investigated.
Author contributions
A.R. and M.E.L. designed the research. A.R., E.L. and M.C.H. performed the research. A.R., E.L., M.C.H. and M.E.L. analysed data. A.R., E.L. and M.E.L. wrote the manuscript with input from all of the other authors.
Conflict of interest
M.C.H. is an employee of F. Hoffmann‐La Roche. The other authors do not have any conflicts of interest to declare for this work.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Monoamine uptake inhibition in stably transfected HEK 293 cells that expressed the human NET, DAT, or SERT. The data are presented as the mean ± SEM of three independent experiments. Curves were fitted by non‐linear regression, and corresponding IC50 values are shown in Table 1. DAT inhibition curves were not performed for substances that did not inhibit the DAT at 100 μM.
Figure S2 None of the opioids released serotonin (5‐HT) or norepinephrine (NE). Monoamine release was induced by 100 μM of the compounds after preloading HEK 293 cells that expressed the human NET or SERT with radiolabeled monoamine. The dashed line marks nonspecific ‘pseudo‐efflux’ that arises from monoamine diffusion and subsequent reuptake inhibition. Substances that caused significantly more monoamine efflux (***P < 0.001) than non‐releasing uptake inhibitors (open bars) were determined to be monoamine releasers. 3,4‐Methylenedioxymethamphetamine (MDMA) served as positive control known to release 5‐HT and NE. There was a significant main effect of 5‐HT and NE release (F9,64 = 89.13, P < 0.001 and F10,78 = 21.46, P < 0.001, respectively) but only the positive control MDMA induced significantly greater 5‐HT and NE release compared with citalopram and nisoxetine (both P < 0.001) respectively. The data are presented as the mean and SEM of five independent experiments.
Table S1 Cases of opioid‐associated serotonin syndrome reported by PubMed.
Acknowledgements
This work was supported by the University Hospital Basel. The authors thank Sylvie Chaboz for technical assistance and Michael Arends for text editing.
Rickli, A. , Liakoni, E. , Hoener, M. C. , and Liechti, M. E. (2018) Opioid‐induced inhibition of the human 5‐HT and noradrenaline transporters in vitro: link to clinical reports of serotonin syndrome. British Journal of Pharmacology, 175: 532–543. doi: 10.1111/bph.14105.
References
- Abadie D, Rousseau V, Logerot S, Cottin J, Montastruc JL, Montastruc F (2015). Serotonin Syndrome: analysis of cases registered in the French pharmacovigilance database. J Clin Psychopharmacol 35: 382–388. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The concise guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174 (Suppl 1): S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017b). The concise guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174 (Suppl 1): S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamigbade TA, Davidson C, Langford RM, Stamford JA (1997). Actions of tramadol, its enantiomers and principal metabolite, O‐desmethyltramadol, on serotonin (5‐HT) efflux and uptake in the rat dorsal raphe nucleus. Br J Anaesth 79: 352–356. [DOI] [PubMed] [Google Scholar]
- Barann M, Stamer UM, Lyutenska M, Stuber F, Bonisch H, Urban B (2015). Effects of opioids on human serotonin transporters. Naunyn Schmiedebergs Arch Pharmacol 388: 43–49. [DOI] [PubMed] [Google Scholar]
- Bee LA, Bannister K, Rahman W, Dickenson AH (2011). Mu‐opioid and noradrenergic alpha(2)‐adrenoceptor contributions to the effects of tapentadol on spinal electrophysiological measures of nociception in nerve‐injured rats. Pain 152: 131–139. [DOI] [PubMed] [Google Scholar]
- Benade V, Nirogi R, Bhyrapuneni G, Daripelli S, Ayyanki G, Irappanavar S et al (2017). Mechanistic evaluation of Tapentadol in reducing the pain perception using in‐vivo brain and spinal cord microdialysis in rats. Eur J Pharmacol 809: 224–230. [DOI] [PubMed] [Google Scholar]
- Bloms‐Funke P, Dremencov E, Cremers TI, Tzschentke TM (2011). Tramadol increases extracellular levels of serotonin and noradrenaline as measured by in vivo microdialysis in the ventral hippocampus of freely‐moving rats. Neurosci Lett 490: 191–195. [DOI] [PubMed] [Google Scholar]
- Boyer EW, Shannon M (2005). The serotonin syndrome. N Engl J Med 352: 1112–1120. [DOI] [PubMed] [Google Scholar]
- Chassot M, Munz T, Livio F, Buclin T (2012). [Serotonin syndrome: review and case series from the Swiss pharmacovigilance system]). Rev Med Suisse 8: 2086–2090. [PubMed] [Google Scholar]
- Chen ZR, Somogyi AA, Bochner F (1990). Simultaneous determination of dextromethorphan and three metabolites in plasma and urine using high‐performance liquid chromatography with application to their disposition in man. Ther Drug Monit 12: 97–104. [DOI] [PubMed] [Google Scholar]
- Codd EE, Shank RP, Schupsky JJ, Raffa RB (1995). Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther 274: 1263–1270. [PubMed] [Google Scholar]
- Crouch SP, Kozlowski R, Slater KJ, Fletcher J (1993). The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods 160: 81–88. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gregori S, Minella CE, De Gregori M, Tinelli C, Ranzani GN, Govoni S et al (2014). Clinical pharmacokinetics of morphine and its metabolites during morphine dose titration for chronic cancer pain. Ther Drug Monit 36: 335–344. [DOI] [PubMed] [Google Scholar]
- Driessen B, Reimann W, Giertz H (1993). Effects of the central analgesic tramadol on the uptake and release of noradrenaline and dopamine in vitro . Br J Pharmacol 108: 806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM (2003). The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 96: 635–642. [DOI] [PubMed] [Google Scholar]
- Eap CB, Cuendet C, Baumann P (1990). Binding of d‐methadone, l‐methadone, and dl‐methadone to proteins in plasma of healthy volunteers: role of the variants of alpha 1‐acid glycoprotein. Clin Pharmacol Ther 47: 338–346. [DOI] [PubMed] [Google Scholar]
- Eap CB, Crettol S, Rougier JS, Schlapfer J, Sintra Grilo L, Deglon JJ et al (2007). Stereoselective block of hERG channel by (S)‐methadone and QT interval prolongation in CYP2B6 slow metabolizers. Clin Pharmacol Ther 81: 719–728. [DOI] [PubMed] [Google Scholar]
- Erstad BL, Meeks ML, Chow HH, Rappaport WD, Levinson ML (1997). Site‐specific pharmacokinetics and pharmacodynamics of intramuscular meperidine in elderly postoperative patients. Ann Pharmacother 31: 23–28. [DOI] [PubMed] [Google Scholar]
- Foster DJ, Somogyi AA, Dyer KR, White JM, Bochner F (2000). Steady‐state pharmacokinetics of (R)‐ and (S)‐methadone in methadone maintenance patients. Br J Clin Pharmacol 50: 427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MA, Jensen CL, Murphy DL (2009). Tramadol and another atypical opioid meperidine have exaggerated serotonin syndrome behavioural effects, but decreased analgesic effects, in genetically deficient serotonin transporter (SERT) mice. Int J Neuropsychopharmacol 12: 1055–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friden M, Winiwarter S, Jerndal G, Bengtsson O, Wan H, Bredberg U et al (2009). Structure‐brain exposure relationships in rat and human using a novel data set of unbound drug concentrations in brain interstitial and cerebrospinal fluids. J Med Chem 52: 6233–6243. [DOI] [PubMed] [Google Scholar]
- Frink MC, Hennies HH, Englberger W, Haurand M, Wilffert B (1996). Influence of tramadol on neurotransmitter systems of the rat brain. Arzneimittelforschung 46: 1029–1036. [PubMed] [Google Scholar]
- Gillman PK (2005). Monoamine oxidase inhibitors, opioid analgesics and serotonin toxicity. Br J Anaesth 95: 434–441. [DOI] [PubMed] [Google Scholar]
- Giusti P, Buriani A, Cima L, Lipartiti M (1997). Effect of acute and chronic tramadol on [3H]‐5‐HT uptake in rat cortical synaptosomes. Br J Pharmacol 122: 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo SL, Wu TJ, Liu CC, Ng CC, Chien CC, Sun HL (2009). Meperidine‐induced serotonin syndrome in a susceptible patient. Br J Anaesth 103: 369–370. [DOI] [PubMed] [Google Scholar]
- Hall FS, Schwarzbaum JM, Perona MT, Templin JS, Caron MG, Lesch KP et al (2011). A greater role for the norepinephrine transporter than the serotonin transporter in murine nociception. Neuroscience 175: 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiskanen T, Langel K, Gunnar T, Lillsunde P, Kalso EA (2015). Opioid concentrations in oral fluid and plasma in cancer patients with pain. J Pain Symptom Manage 50: 524–532. [DOI] [PubMed] [Google Scholar]
- Hysek CM, Simmler LD, Nicola VG, Vischer N, Donzelli M, Krahenbuhl S et al (2012). Duloxetine inhibits effects of MDMA (‘ecstasy’) in vitro and in humans in a randomized placebo‐controlled laboratory study. PLoS One 7: e36476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS, Pollack GM (2007). Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p‐glycoprotein efflux ratios. Drug Metab Dispos 35: 660–666. [DOI] [PubMed] [Google Scholar]
- Kinoshita H, Ohkubo T, Yasuda M, Yakushiji F (2011). Serotonin syndrome induced by dextromethorphan (Medicon) administrated at the conventional dose. Geriatr Gerontol Int 11: 121–122. [DOI] [PubMed] [Google Scholar]
- Kitamura A, Higuchi K, Okura T, Deguchi Y (2014). Transport characteristics of tramadol in the blood‐brain barrier. J Pharm Sci 103: 3335–3341. [DOI] [PubMed] [Google Scholar]
- Larsen JJ, Hyttel J (1985). 5‐HT‐uptake inhibition potentiates antinociception induced by morphine, pethidine, methadone and ketobemidone in rats. Acta Pharmacol Toxicol (Copenh) 57: 214–218. [DOI] [PubMed] [Google Scholar]
- Luccarini P, Perrier L, Degoulange C, Gaydier AM, Dallel R (2004). Synergistic antinociceptive effect of amitriptyline and morphine in the rat orofacial formalin test. Anesthesiology 100: 690–696. [DOI] [PubMed] [Google Scholar]
- Luethi D, Trachsel D, Hoener MC, Liechti ME (2017a). Monoamine receptor interaction profiles of 4‐thio‐substituted phenethylamines (2C‐T drugs). Neuropharmacology . https://doi.org/10.1016/j.neuropharm.2017.07.012. [DOI] [PubMed] [Google Scholar]
- Luethi D, Kolaczynska KE, Docci L, Krahenbuhl S, Hoener MC, Liechti ME (2017b). Pharmacological profile of mephedrone analogs and related new psychoactive substances. Neuropharmacology . https://doi.org/10.1016/j.neuropharm.2017.07.026. [DOI] [PubMed] [Google Scholar]
- Marechal C, Honorat R, Claudet I (2011). Serotonin syndrome induced by tramadol intoxication in an 8‐month‐old infant. Pediatr Neurol 44: 72–74. [DOI] [PubMed] [Google Scholar]
- Martin DC, Introna RP, Aronstam RS (1991). Fentanyl and sufentanil inhibit agonist binding to 5‐HT1A receptors in membranes from the rat brain. Neuropharmacology 30: 323–327. [DOI] [PubMed] [Google Scholar]
- Meini M, Moncini M, Daini L, Giarratana T, Scaramelli D, Chericoni S et al (2015). Relationship between plasma concentrations of the l‐enantiomer of methadone and response to methadone maintenance treatment. Eur J Pharmacol 760: 1–6. [DOI] [PubMed] [Google Scholar]
- Monte AA, Chuang R, Bodmer M (2010). Dextromethorphan, chlorphenamine and serotonin toxicity: case report and systematic literature review. Br J Clin Pharmacol 70: 794–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moraes NV, Lauretti GR, Coelho EB, Godoy AL, Neves DV, Lanchote VL (2016). Impact of fraction unbound, CYP3A, and CYP2D6 in vivo activities, and other potential covariates to the clearance of tramadol enantiomers in patients with neuropathic pain. Fundam Clin Pharmacol 30: 153–161. [DOI] [PubMed] [Google Scholar]
- Pope LE, Khalil MH, Berg JE, Stiles M, Yakatan GJ, Sellers EM (2004). Pharmacokinetics of dextromethorphan after single or multiple dosing in combination with quinidine in extensive and poor metabolizers. J Clin Pharmacol 44: 1132–1142. [DOI] [PubMed] [Google Scholar]
- Rastogi R, Swarm RA, Patel TA (2011). Case scenario: opioid association with serotonin syndrome: implications to the practitioners. Anesthesiology 115: 1291–1298. [DOI] [PubMed] [Google Scholar]
- Rickli A, Luethi D, Reinisch J, Buchy D, Hoener MC, Liechti ME (2015). Receptor interaction profiles of novel N‐2‐methoxybenzyl (NBOMe) derivatives of 2,5‐dimethoxy‐substituted phenethylamines (2C drugs). Neuropharmacology 99: 546–553. [DOI] [PubMed] [Google Scholar]
- Saarikoski T, Saari TI, Hagelberg NM, Neuvonen M, Neuvonen PJ, Scheinin M et al (2013). Rifampicin markedly decreases the exposure to oral and intravenous tramadol. Eur J Clin Pharmacol 69: 1293–1301. [DOI] [PubMed] [Google Scholar]
- Saarikoski T, Saari TI, Hagelberg NM, Backman JT, Neuvonen PJ, Scheinin M et al (2015). Effects of terbinafine and itraconazole on the pharmacokinetics of orally administered tramadol. Eur J Clin Pharmacol 71: 321–327. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Reid A (1987). Effect of 6‐hydroxydopamine‐induced lesions to ascending and descending noradrenergic pathways on morphine analgesia. Brain Res 419: 156–165. [DOI] [PubMed] [Google Scholar]
- Scholze P, Zwach J, Kattinger A, Pifl C, Singer EA, Sitte HH (2000). Transporter‐mediated release: a superfusion study on human embryonic kidney cells stably expressing the human serotonin transporter. J Pharmacol Exp Ther 293: 870–878. [PubMed] [Google Scholar]
- Schou M, Varnas K, Lundquist S, Nakao R, Amini N, Takano A et al (2015). Large variation in brain exposure of reference CNS drugs: a PET study in nonhuman primates. Int J Neuropsychopharmacol 18: pyv036: pyv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder W, Vry JD, Tzschentke TM, Jahnel U, Christoph T (2010). Differential contribution of opioid and noradrenergic mechanisms of tapentadol in rat models of nociceptive and neuropathic pain. Eur J Pain 14: 814–821. [DOI] [PubMed] [Google Scholar]
- Schroder W, Tzschentke TM, Terlinden R, De Vry J, Jahnel U, Christoph T et al (2011). Synergistic interaction between the two mechanisms of action of tapentadol in analgesia. J Pharmacol Exp Ther 337: 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz AR, Pizon AF, Brooks DE (2008). Dextromethorphan‐induced serotonin syndrome. Clin Toxicol (Phila) 46: 771–773. [DOI] [PubMed] [Google Scholar]
- Shakoor M, Ayub S, Ahad A, Ayub Z (2014). Transient serotonin syndrome caused by concurrent use of tramadol and selective serotonin reuptake inhibitor. Am J Case Rep 15: 562–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikholeslami B, Gholami M, Lavasani H, Rouini M (2016). Evaluation of the route dependency of the pharmacokinetics and neuro‐pharmacokinetics of tramadol and its main metabolites in rats. Eur J Pharm Sci 92: 55–63. [DOI] [PubMed] [Google Scholar]
- Siebenhuener K, Eschmann E, Kienast A, Schneider D, Minder CE, Saller R et al (2017). Chronic pain: how challenging are DDIs in the analgesic treatment of inpatients with multiple chronic conditions? PLoS One 12: e0168987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J et al (2013). Pharmacological characterization of designer cathinones in vitro . Br J Pharmacol 168: 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmler LD, Rickli A, Hoener MC, Liechti ME (2014). Monoamine transporter and receptor interaction profiles of a new series of designer cathinones. Neuropharmacology 79: 152–160. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GK, Bell TE, Yenari MA (1996). Dose escalation safety and tolerance study of the N‐methyl‐D‐aspartate antagonist dextromethorphan in neurosurgery patients. J Neurosurg 84: 860–866. [DOI] [PubMed] [Google Scholar]
- Sternbach H (1991). The serotonin syndrome. Am J Psychiatry 148: 705–713. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Naito T, Mino Y, Kawakami J (2016). Validated determination method of tramadol and its desmethylates in human plasma using an isocratic LC‐MS/MS and its clinical application to patients with cancer pain or non‐cancer pain. J Pharm Health Care Sci 2: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Auerbach SB (1995). Involvement of the dorsal raphe but not median raphe nucleus in morphine‐induced increases in serotonin release in the rat forebrain. Neuroscience 68: 553–561. [DOI] [PubMed] [Google Scholar]
- Tao R, Auerbach SB (2002). GABAergic and glutamatergic afferents in the dorsal raphe nucleus mediate morphine‐induced increases in serotonin efflux in the rat central nervous system. J Pharmacol Exp Ther 303: 704–710. [DOI] [PubMed] [Google Scholar]
- Tao R, Karnik M, Ma Z, Auerbach SB (2003). Effect of fentanyl on 5‐HT efflux involves both opioid and 5‐HT1A receptors. Br J Pharmacol 139: 1498–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi M, Groshan K, Blakely RD, Richelson E (1997). Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 340: 249–258. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM, Christoph T, Kogel B, Schiene K, Hennies HH, Englberger W et al (2007). (−)‐(1R,2R)‐3‐(3‐dimethylamino‐1‐ethyl‐2‐methyl‐propyl)‐phenol hydrochloride (tapentadol HCl): a novel mu‐opioid receptor agonist/norepinephrine reuptake inhibitor with broad‐spectrum analgesic properties. J Pharmacol Exp Ther 323: 265–276. [DOI] [PubMed] [Google Scholar]
- Van Oekelen D, Megens A, Meert T, Luyten WH, Leysen JE (2002). Role of 5‐HT(2) receptors in the tryptamine‐induced 5‐HT syndrome in rats. Behav Pharmacol 13: 313–318. [DOI] [PubMed] [Google Scholar]
- Walter C, Ball D, Duffy M, Mellor JD (2012). An unusual case of serotonin syndrome with oxycodone and citalopram. Case Rep Oncol Med 2012: 261787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff T, Samuelsson H, Hedner T (1995). Morphine and morphine metabolite concentrations in cerebrospinal fluid and plasma in cancer pain patients after slow‐release oral morphine administration. Pain 62: 147–154. [DOI] [PubMed] [Google Scholar]
- Xu XS, Smit JW, Lin R, Stuyckens K, Terlinden R, Nandy P (2010). Population pharmacokinetics of tapentadol immediate release (IR) in healthy subjects and patients with moderate or severe pain. Clin Pharmacokinet 49: 671–682. [DOI] [PubMed] [Google Scholar]
- Zannikos PN, Smit JW, Stahlberg HJ, Wenge B, Hillewaert VM, Etropolski MS (2013). Pharmacokinetic evaluation of tapentadol extended‐release tablets in healthy subjects. J Opioid Manag 9: 291–300. [DOI] [PubMed] [Google Scholar]
- Zawertailo LA, Tyndale RF, Busto U, Sellers EM (2010). Effect of metabolic blockade on the psychoactive effects of dextromethorphan. Hum Psychopharmacol 25: 71–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Monoamine uptake inhibition in stably transfected HEK 293 cells that expressed the human NET, DAT, or SERT. The data are presented as the mean ± SEM of three independent experiments. Curves were fitted by non‐linear regression, and corresponding IC50 values are shown in Table 1. DAT inhibition curves were not performed for substances that did not inhibit the DAT at 100 μM.
Figure S2 None of the opioids released serotonin (5‐HT) or norepinephrine (NE). Monoamine release was induced by 100 μM of the compounds after preloading HEK 293 cells that expressed the human NET or SERT with radiolabeled monoamine. The dashed line marks nonspecific ‘pseudo‐efflux’ that arises from monoamine diffusion and subsequent reuptake inhibition. Substances that caused significantly more monoamine efflux (***P < 0.001) than non‐releasing uptake inhibitors (open bars) were determined to be monoamine releasers. 3,4‐Methylenedioxymethamphetamine (MDMA) served as positive control known to release 5‐HT and NE. There was a significant main effect of 5‐HT and NE release (F9,64 = 89.13, P < 0.001 and F10,78 = 21.46, P < 0.001, respectively) but only the positive control MDMA induced significantly greater 5‐HT and NE release compared with citalopram and nisoxetine (both P < 0.001) respectively. The data are presented as the mean and SEM of five independent experiments.
Table S1 Cases of opioid‐associated serotonin syndrome reported by PubMed.