

Abstract
Background and Purpose
Following inflammatory stimuli, neutrophils are recruited to sites of inflammation and exert effector functions that often have deleterious effects on tissue integrity, which can lead to organ failure. Bruton's tyrosine kinase (Btk) is expressed in neutrophils and constitutes a promising pharmacological target for neutrophil‐mediated tissue damage. Here, we evaluate a selective reversible inhibitor of Btk, PRN473, for its ability to dampen neutrophil influx via inhibition of adhesion receptor signalling pathways.
Experimental Approach
In vitro assays were used to assess fMLP receptor 1 (Fpr‐1)‐mediated binding of ligands to the adhesion receptors macrophage antigen‐1 (Mac‐1) and lymphocyte function antigen‐1. Intravital microscopy of the murine cremaster was used to evaluate post‐adhesion strengthening and endoluminal crawling. Finally, neutrophil influx was visualized in a clinically relevant model of sterile liver injury in vivo. Btk knockout animals were used as points of reference for Btk functions.
Key Results
Pharmacological inhibition of Btk by PRN473 reduced fMLP‐induced phosphorylation of Btk and Mac‐1 activation. Biochemical experiments demonstrated the specificity of the inhibitor. PRN473 (20 mg·kg−1) significantly reduced intravascular crawling and neutrophil recruitment into inflamed tissue in a model of sterile liver injury, down to levels seen in Btk‐deficient animals. A higher dose did not provide additional reduction of intravascular crawling and neutrophil recruitment.
Conclusions and Implications
PRN473, a highly selective inhibitor of Btk, potently attenuates sterile liver injury by inhibiting the activation of the β2‐integrin Mac‐1 and subsequently neutrophil recruitment into inflamed tissue.
Abbreviations
- Btk
Bruton's tyrosine kinase
- fMLP
N‐formylmethionine‐leucyl‐phenylalanine
- Fpr1
N‐formylmethionine‐leucyl‐phenylalanine receptor 1
- ICAM‐1
intercellular adhesion molecule 1
- LFA‐1
lymphocyte function antigen‐1
- Mac‐1
macrophage antigen‐1
Introduction
Blood leukocytes are recruited to sites of tissue inflammation where they execute host defence functions such as release of radical oxygen species and digestive enzymes (Nourshargh and Alon, 2014). However, especially in the context of sterile injury where a targetable pathogen is missing, neutrophil recruitment is often excessive, thereby causing significant collateral tissue damage, which at times can even affect remote organs. In these settings, such as following ischaemic injury, trauma or sterile liver injury, neutrophil‐driven innate immune functions constitute more of a poorly controlled autoimmune disease than beneficial host protection. Depending on clinical severity, this process can ultimately lead to multiorgan dysfunction and thus significantly contribute to patient mortality.
For this reason, preclinical studies have investigated the effects of inhibiting the molecular mechanisms of the leukocyte recruitment in autoimmunity and other sterile inflammatory diseases. Growing evidence demonstrates beneficial therapeutic potential of decreasing neutrophil influx, with reduced collateral tissue damage and preserved organ function (Etzioni, 1996; Singbartl and Ley, 2004; Herter et al., 2014; Volmering et al., 2016).
Over the last decade, our understanding of leukocyte recruitment has greatly increased, with the different steps of this process, named the ‘leukocyte recruitment cascade’, now identified (Herter and Zarbock, 2013). The interaction of endothelial E‐selectin and P‐selectin with neutrophil P‐selectin glycoprotein ligand‐1 (PSGL‐1) mediates the initial capture and rolling of white blood cells along the vessel wall (Herter and Zarbock, 2013). During leukocyte rolling, engagement of endothelial‐bound chemokines to G‐coupled receptors on the neutrophil triggers full arrest via activation of the β2‐integrin lymphocyte function antigen‐1 (LFA‐1) (Zarbock et al., 2007). Of note, the chemokine receptor CXCR2 seems to be the predominant chemokine receptor mediating neutrophil arrest. Following arrest, post‐adhesion strengthening and consecutive endoluminal crawling occur prior to extravasation to the tissue (Herter and Zarbock, 2013). Both of these processes require chemokine‐induced activation of the β2‐integrin macrophage antigen‐1 (Mac‐1) (Phillipson et al., 2006; Volmering et al., 2016).
We previously reported that Bruton's tyrosine kinase (Btk) is essential for the activation of the β2‐integrin Mac‐1 following engagement of N‐formylmethionine‐leucyl‐phenylalanine receptor 1 (Fpr1) (Volmering et al., 2016). This process is required for both the sustained adhesion at the vessel wall and endoluminal crawling prior to transmigration to the tissue and thus constitutes a prerequisite for neutrophil recruitment (Volmering et al., 2016). Accordingly, Btk constitutes a promising pharmacological target to inhibit overwhelming neutrophil influx to preserve organ function. Furthermore, Btk inhibitors have proven their clinical utility and are already clinically used with great success to treat B‐cell malignancies (Seiler and Dreyling, 2017).
Previous studies have suggested improved efficiency of inverted cyanoacrylamide inhibitors targeting a non‐catalytic cysteine in Btk to prolong on‐target residence and enhance selectivity (Bradshaw et al., 2015). However, the feasibility of this approach to inhibit neutrophil responses in vivo was unknown. Here, we demonstrate the therapeutic potential of PRN473, a reversible, selective inhibitor of Btk phosphorylation of the inverted cyanoacrylamide type (Bisconte et al., 2015), designed to achieve extended Btk pharmacodynamic effects. Treatment with PRN473 potently abolished N‐formylmethionine‐leucyl‐phenylalanine (fMLP)‐induced Mac‐1 activation both in vivo and in vitro and greatly reduced neutrophil recruitment in a model of sterile liver injury.
Methods
Animals
All animal care and experimental procedures were approved by the Animal Care and Use Committees of North Rhine Westphalia, Germany (LANUV). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The 8‐ to 15‐week‐old C57Bl/6 (Janvier, Le Genest‐Saint‐Isle, France), LysMGFP + (Faust et al., 2000), Btk −/− (The Jackson Laboratory, Bar Harbor, ME, USA) and LysMGFP +–Btk −/− mice were housed in the specified pathogen‐free (SPF) facility. PRN473 was dissolved in DMSO prior to usage and diluted from a 10 mM stock solution for in vitro experiments. DMSO of equal concentration was used as vehicle control. For in vivo experiments, PRN473 was dissolved in Captex355 NP/EF (supplied by Principia Biopharma) and diluted from a 4 mg·mL−1 stock solution, injected i.p. for 3 days daily, prior to the experiment, with the last dose on the day of the study. PRN473 was administered at 20 or 40 mg·kg−1 to target partial or complete occupancy of Btk respectively (Supporting Information Figure 2). Captex355 NP/EF at equal concentrations was used as vehicle control.
Biochemical assays
Bone marrow‐derived wild‐type (WT) or Btk −/− neutrophils were isolated using a 62% single‐layer Percoll gradient and resuspended in PBS (containing 1 mM each CaCl2 and MgCl2). Neutrophils were stimulated with fMLP (10 μM) or were left unstimulated. Neutrophils were then lysed in RIPA buffer and boiled with sample buffer (10 min, 95°C) or, in some experiments, incubated with Sepharose A/G beads (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and anti‐Btk (Santa Cruz Biotechnology, Inc.) antibody for 4 h at 4°C. Beads were washed four times, and bound proteins were eluted by adding boiling sample buffer. Cell lysates and immunoprecipitates were run on 10% SDS‐PAGE and immunoblotted using antibodies against phosphotyrosine (4G10, EMD Millipore, Burlington, MA, USA), Btk (Santa Cruz Biotechnology, Inc.), phospho‐Src (Tyr416), phospho‐p38 MAPK, p38 MAPK, phospho‐Akt and Akt (Cell Signalling, Danvers, MA, USA). Immunoblots were developed using an enhanced chemiluminescence system (GE Healthcare, Little Chalfont, UK). Densitometric quantifications were conducted using ImageJ software.
Soluble ICAM‐1‐binding and fibrinogen‐binding assay
The soluble intercellular adhesion molecule 1 (ICAM‐1)‐binding and fibrinogen‐binding assays were performed as previously described (Lefort et al., 2012). In brief, to assess LFA‐1‐specific ICAM‐1 binding, isolated murine neutrophils were purified using Histopaque, preincubated with a functional blocking anti‐CD11b (clone M1/70, 10 μg·mL−1) antibody to prevent Mac‐1‐dependent ICAM‐1. Afterwards, neutrophils were stimulated with fMLP (10 μM) or CXCL1 (KC, 100 ng·mL−1) for 3 min at 37°C or left unstimulated in the presence of ICAM‐1/Fc (20 μg·mL−1, R&D Systems Inc., Minneapolis, MN, USA) or IgG control and allophycocyanin (APC)‐conjugated anti‐human IgG1 (Fc specific, Southern Biotechnology, Birmingham, AL, USA). Neutrophils were fixed on ice and stained with FITC‐conjugated anti‐Ly6G (Biolegend, San Diego, CA, USA). LFA‐1‐specific binding to ICAM‐1/Fc was measured by flow cytometry. To investigate Mac‐1 affinity to fibrinogen, isolated murine neutrophils were incubated for 10 min at 37°C with 150 μg·mL−1 Alexa 647‐conjugated fibrinogen (Invitrogen, Carlsbad, CA, USA) and stimulated with fMLP or were left unstimulated. Neutrophils were treated with EDTA (2 mM) as negative controls. Fluorescence intensity was measured by flow cytometry. The percentage of neutrophils positive for fibrinogen binding was calculated by defining a threshold of the fluorescence intensity where 95% of neutrophils in the EDTA control were considered as negative.
Intravital microscopy of the murine cremaster muscle
Mice were anaesthetized with injection of 125 mg·kg−1 ketamine hydrochloride (Sanofi Winthrop Pharmaceuticals, Bridgewater, NJ, USA) and 12.5 mg·kg−1 xylazine (Tranqui Ved, Phonix Scientific, St. Joseph, MO, USA) i.p., and the cremaster muscle was prepared for intravital imaging as previously described (Herter et al., 2013). Intravital microscopy was performed on an upright microscope (Axioskop, Carl Zeiss Microscopy, Jena, Germany) with a 40× 0.75 numerical aperture saline immersion objective. Neutrophil adhesion was determined by transillumination intravital microscopy. Recorded images were analysed offline with ImageJ (NIH, Rockville, MD, USA) software. The microcirculation was recorded with a digital camera (Sensicam QE, The Cooke Corporation, Romulus, MI, USA). For GPCR‐induced arrest, the carotid artery was cannulated for injection of fMLP (16 μg). In a representative vessel, the initial number of adherent neutrophils was determined, fMLP was injected and the vessel was recorded for 15 min. Movies were analysed with SlideBook software (Intelligent Imaging Innovations, Göttingen, Germany). Blood flow centreline velocity was measured using a dual photodiode sensor system (Circusoft Instrumentation, Hockessin, DE, USA) and was used to calculate blood flow velocity (Zarbock et al., 2007) and estimated Newtonian wall shear rates (Ley and Gaehtgens, 1991) as described previously.
Intravascular in vivo crawling assay
Using intravital microscopy, intravascular crawling of neutrophils was examined as described previously (Phillipson et al., 2006). Briefly, anti‐Gr‐1 antibody (clone RB6‐8C5), labelled with Alexa Fluor 488 (Molecular Probes, Eugene, OR, USA), was injected via the cannulated carotid artery prior to the experiment. Following preparation and exteriorization, the cremaster was superfused with fMLP (10 μM) and time‐lapse microscopy was performed for 2 h. The number of adherent cells was determined, and the neutrophil crawling velocity and crawled distance were analysed using SlideBook software (version 5, Intelligent Imaging Innovations).
Spinning disc confocal intravital microscopy of the murine liver after focal hepatic necrosis
Mice were anaesthetized i.p. with a mixture of ketamine hydrochloride (125 mg·kg−1) and xylazine (12.5 mg·kg−1). After anaesthesia, mice were placed on a heating pad set to 37°C to maintain the body temperature for the duration of all performed experiments. The left carotid artery was cannulated for administration of additional anaesthetics.
For intravital microscopy of the liver, mice were prepared as previously described (McDonald et al., 2010). In brief, a midline laparotomy was performed followed by removal of the skin and abdominal muscle to expose the liver lobe. A thin platinic wire was heated and briefly pressed on the liver to induce a focal injury on the surface of the liver. Necrotic cells were labelled with a single application of 2 μL of a propidium iodide solution (1.0 mg·mL−1) diluted 1:25 in PBS to the surface of the liver. To avoid dehydration, all exposed tissues were moistened with PBS‐soaked tissues. After preparation, the exposed liver was visualized with an upright spinning disc confocal microscope (CellObserver SD, Carl Zeiss Microscopy, Jena, Germany) equipped with a 5×/0.25 FLUAR objective, and time‐lapse Z‐stacks were recorded for up to 4 h after focal injury. The number of adherent neutrophils was determined for different time points per field of view, or after 4 h after injury within specific regions (within injury, proximal 150 μm around injury and beyond 150 μm from injury border). Duration of adhesion and neutrophil crawling was determined within the necrotaxis zone 2.5 h after injury. Thirty neutrophils per field of view were randomly selected for tracking, and chemotaxis parameters (migration plots, forward migration index and crawling velocities) were set. All investigated parameters were analysed with ImageJ or Chemotaxis and Migration tool (Ibidi, Martinsried, Germany).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as mean ± SEM and were analysed using SPSS (version 22, IBM, Armonk, NY, USA). Differences between the groups were evaluated by two‐way ANOVA with post hoc Bonferroni correction or Student's t‐test where appropriate. In the case of repeated measurements (chemokine‐induced arrest adhesion of time), ANOVA for repeated measurements was used. The number of experiments is indicated by the n values, and a P value of <0.05 was considered statistically significant.
Materials
PRN473 was provided by Principia Biopharma (San Francisco, CA, USA). The chemokine KC was obtained from Peprotech, Rocky Hill, NJ, USA and fMLP from Sigma‐Aldrich, Taufkirchen, Germany.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
PRN473 is a potent inhibitor of neutrophil Fpr1‐induced Btk phosphorylation
PRN473 is a reversible covalent inhibitor targeting a non‐catalytic cysteine in Btk (Bradshaw et al., 2015), designed to selectively bind Btk with a slow off‐rate to provide long duration of inhibition of the target. To investigate the effects of PRN473 on neutrophil signalling, we stimulated WT and Btk‐deficient neutrophils isolated using a single layer 62% Percoll gradient with fMLP in the presence or absence of PRN473 and performed an immunoprecipitation of Btk to analyse phosphorylation of the molecule with a phosphotyrosine‐specific antibody. Stimulation of neutrophils with fMLP triggers Btk phosphorylation through Fpr1 in an Hck‐dependent manner (Volmering et al., 2016). We found that even low concentrations of PRN473 significantly decreased Btk phosphorylation to levels seen in Btk‐deficient neutrophils (Figure 1A, C and Supporting Information Figure 1A–C). By contrast, upstream activation of Src ‐family kinases was unaffected by PRN473 treatment, while downstream signalling molecules Akt and p38 were inhibited to the level seen in Btk −/− neutrophils (Figure 1B, D–F). These data demonstrate that PRN473 is capable of inhibiting Btk activation following Fpr1 engagement in neutrophils.
Figure 1.
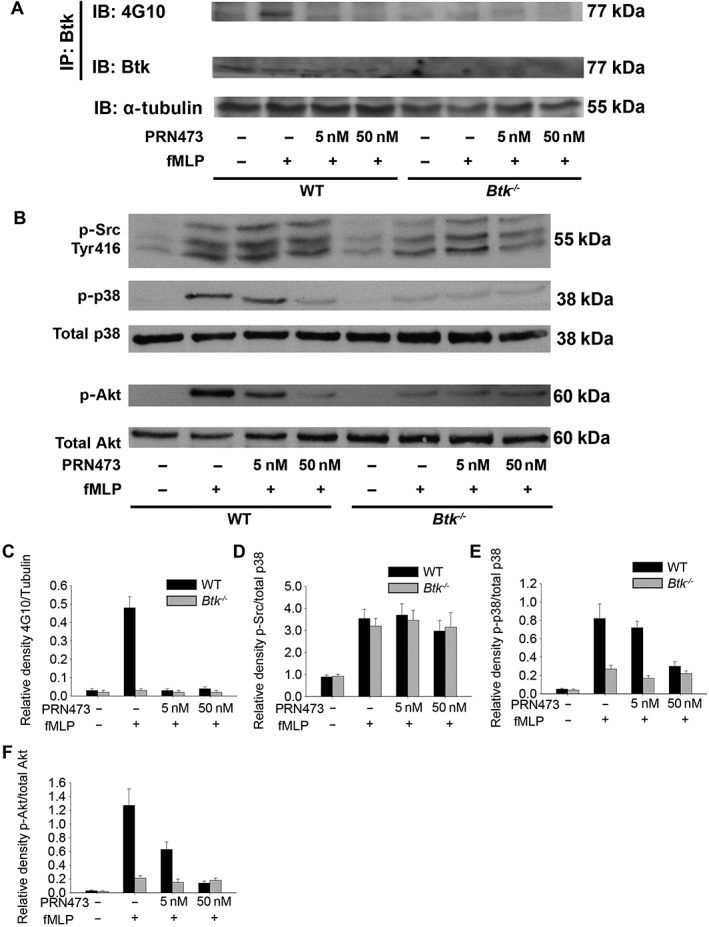
PRN473 is a potent inhibitor of Fpr1‐induced Btk phosphorylation in neutrophils. (A) Neutrophils were stimulated with fMLP, in the presence or absence of PRN473 at the indicated concentrations, for 1 min, after which lysates were prepared, and Btk was immunoprecipitated and probed on Western blots with the phosphotyrosine‐specific antibody (4G10). Total Btk is absent in Btk‐deficient animals. Tubulin was used as a loading control. (B) Representative Western blots of neutrophils stimulated with fMLP probed for phospho‐Src (Tyr416), phospho‐p38 MAPK, total p38 MAPK, phospho‐Akt (p‐Akt) and total Akt, following treatment with different concentrations of PRN473 as indicated. (C, D) Densities of phospho‐proteins relative to total protein [4G10 to tubulin (C), p‐Src to p38 (D), p‐p38 to p38 (E) and p‐Akt to Akt (F)]; data shown are means ± SEM; n = 5.
Treatment of neutrophils with PRN473 abolishes Mac‐1 but not LFA‐1 activation in vitro
To determine if inhibition of Btk by PRN473 decreases fMLP‐induced activation of the β2‐integrin Mac‐1, we performed a binding assay of the Mac‐1 ligand fibrinogen. Inhibition of Btk by PRN473 inhibited neutrophil binding to fibrinogen in a Mac‐1‐dependent manner to levels seen in neutrophils from Btk −/− mice (Figure 2A, B), thus illustrating impaired Fpr1‐mediated activation of the integrin Mac‐1. To test whether these effects are Mac‐1 specific, we performed binding assays for the integrin LFA‐1. Treatment of neutrophils with PRN473 did not compromise binding of LFA‐1 to its ligand ICAM‐1 (Figure 2C, D). These results are in line with our previous studies (Volmering et al., 2016), indicating that Btk is dispensable for LFA‐1 activation following Fpr1 engagement and thereby confirming specificity of PRN473 treatment to Fpr1‐mediated activation of Mac‐1 but not LFA‐1. To further examine the specificity of Btk inhibition for Fpr1‐mediated Mac‐1 activation, we performed both fibrinogen‐binding and ICAM‐1‐binding assays using CXCR2 stimulation by CXCL1 (KC, Figure 2E, F). LFA‐1 and Mac‐1 activation were unaffected in neutrophils treated with PRN473 or in cells derived from Btk −/− mice, confirming that Btk is only required for the Fpr1‐triggered pathway of Mac‐1 activation.
Figure 2.
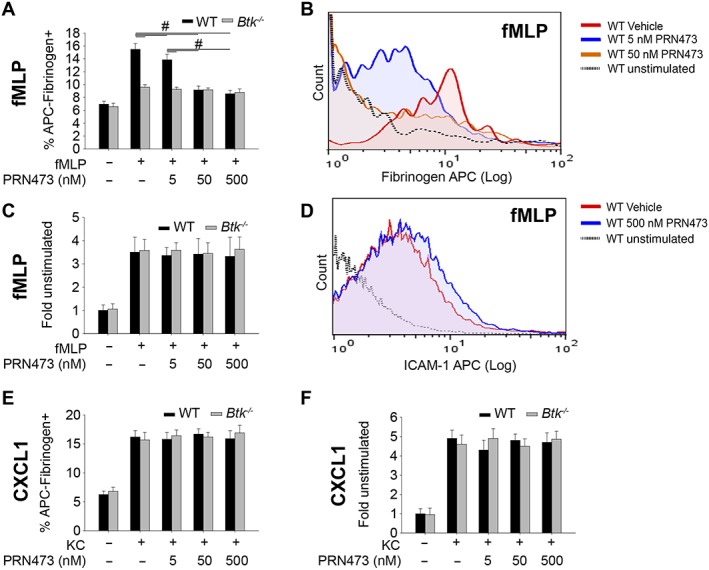
Treatment of neutrophils with PRN473 abolishes Mac‐1 activation in vitro. (A) Fibrinogen binding to isolated neutrophils from WT or Btk −/− animals, following stimulation with fMLP, in the presence or absence of PRN473 at the indicated concentrations, presented as mean percentage of allophycocyanin (APC)–fibrinogen positive cells. (B) Representative FACS histogram of (A). (C) Binding of the LFA‐1 ligand ICAM‐1 in the presence of anti‐Mac‐1 as in (A) presented as mean fold fluorescence of unstimulated samples. (D) Representative FACS histogram of (C). (E) Experiment as in (A) but with CXCL1 stimulation. (F) Experiment as in (C) but with CXCL1 stimulation. Data shown are means ± SEM, n = 5. # P < 0.05, significantly different as indicated.
Post‐adhesion strengthening is reduced following PRN473 treatment in vivo
During neutrophil recruitment, one important function of the integrin Mac‐1 is the sustained adhesion at the vessel wall following cell arrest (Volmering et al., 2016). While the initial arrest on the endothelium is mediated by chemokine‐induced LFA‐1 activation, Mac‐1 is required for sustained adhesion over time. Without proper functioning of Mac‐1, leukocytes lose contact with the vessel wall and detach (Volmering et al., 2016). We therefore investigated if this step is affected by PRN473 treatment in vivo, at two dose levels targeting partial or complete Btk occupancy (Supporting Information Figure 2). Chemokine‐induced arrest was similar in treated and untreated animals following i.a. injection of fMLP (Figure 3A, B), confirming our in vitro data that LFA‐1‐dependent adhesion was not impaired by PRN473 treatment in vivo. However, neutrophils in mice treated with PRN473 detached from the vessel wall more rapidly than in mice treated with vehicle control (Figure 3A, B), thereby indicating reduced Mac‐1‐dependent post‐adhesion strengthening in PRN473‐treated mice. Again, we performed similar experiments using CXCL1 stimulation and found no difference in adhesion or post‐adhesion strengthening (Figure 3C, D), confirming the specificity of PRN473 inhibition for Fpr1‐mediated Mac‐1 activation in vivo.
Figure 3.
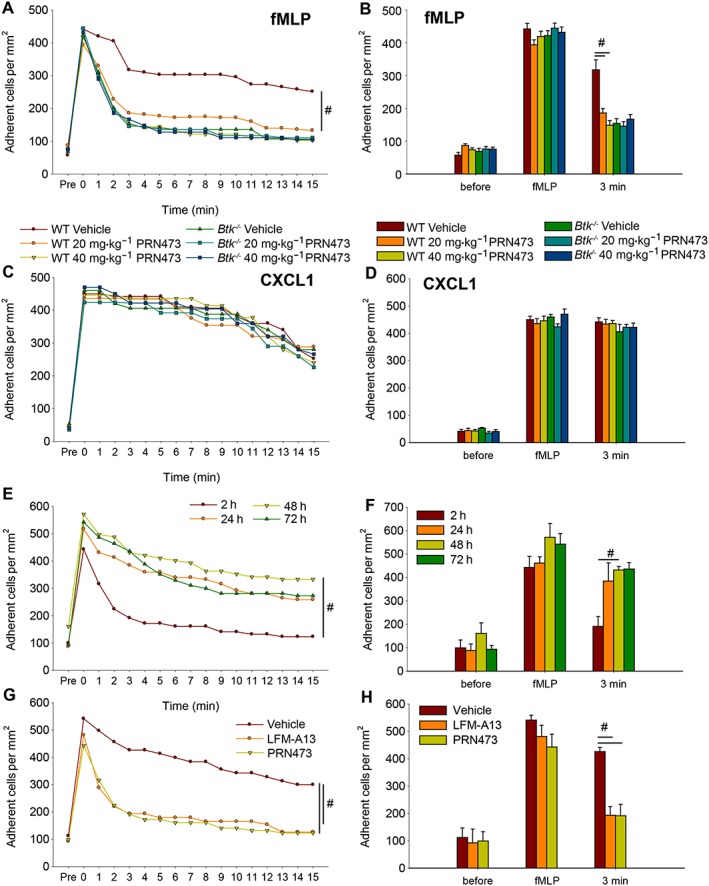
Post‐adhesion strengthening is reduced following PRN473 treatment in vivo. (A) Timeline of the mean number of adherent cells before and after injection of fMLP (time point 0) of WT and Btk −/− mice pretreated with PRN473, n = 5. (B) Quantification of time points before and after stimulation with fMLP and 3 min of (A). (C, D) Corresponding figures to (A) and (B) using CXCL1 as a stimulus. (E) Timeline as in (A) for different time points after last dose of PRN473 administration as indicated. (F) Corresponding numbers of mean number of adherent cells 3 min after fMLP injection of (E). (G) Timeline as in (A) and corresponding numbers of mean number of adherent cells at 3 min after fMLP injection (H) following pretreatment with LFM‐A13, PRN473 or vehicle. Data shown are means ± SEM. # P < 0.05, significantly different as indicated.
It is important to note that PRN473 treatment had no effect on centreline velocity (vehicle: 3.60 ± 0.09 mm·s−1, 20 mg·kg−1: 3.48 ± 0.16 mm·s−1, 40 mg·kg−1: 3.46 ± 0.10 mm·s−1, n = 5), blood flow velocity (vehicle: 2.25 ± 0.06 mm·s−1, 20 mg·kg−1: 2.18 ± 0.10 mm·s−1, 40 mg·kg−1: 2.16 ± 0.06 mm·s−1) or wall shear stress (vehicle: 925 ± 44 per s, 20 mg·kg−1: 957 ± 82 per s, 40 mg·kg−1: 946 ± 60 per s) with similar vessel wall diameters (vehicle: 31.4 ± 1.3 μm, 20 mg·kg−1: 30.2 ± 2.8 μm, 40 mg·kg−1: 29.6 ± 1.1 μm).
Furthermore, to ensure reversibility of the observed effects caused by PRN473 administration, we performed chemokine‐induced arrest experiments following 2, 24, 48 and 72 h after the last dosing of PRN473 with 40 mg·kg−1. We observed reversal of PRN473‐mediated impairment of post‐adhesion strengthening as soon as 24 to 48 h after the last dose (Figure 3E, F). These results are compatible with previously published results (Bisconte et al., 2015) (Supporting Information Figure 3).
Finally, we compared pretreatment with PRN473 to a pretreatment with a known Btk inhibitor, LFM‐A13, and observed similar results following similar dosing (40 mg·kg−1) in its ability to prohibit post‐adhesion strengthening (Figure 3G, H).
Pretreatment with PRN473 impairs intravascular crawling
Following post‐adhesion strengthening, neutrophils undertake Mac‐1‐dependent migratory activity on the vascular side of the vessel wall prior to transmigration (Herter and Zarbock, 2013). Previous studies have highlighted the importance of this step for successful neutrophil recruitment (Phillipson et al., 2006). To investigate the effects of Btk inhibition on this step of the leukocyte recruitment cascade, we evaluated intraluminal crawling following fMLP‐mediated arrest in vivo. Migratory activity was severely decreased in animals treated with PRN473 compared to vehicle control in terms of arrested cells that crawled (Figure 4A) migration distance (Figure 4B) and velocity (Figure 4C) to levels seen in mice lacking Btk (Figure 4A–C). These data demonstrate that Btk inhibition with PRN473 potently prohibits intraluminal crawling of adherent neutrophils.
Figure 4.
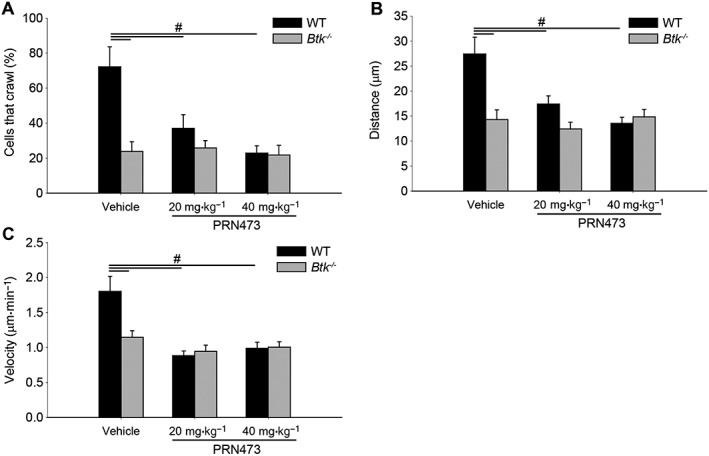
Pretreatment with PRN473 impairs intravascular crawling. Intravascular crawling of neutrophils in the cremaster in WT and Btk −/− animals following PRN473 pretreatment as indicated. (A) Quantification of adherent cells that crawled presented as the mean percentage of cells that translocated throughout the observation period. (B) Average crawling distance. (C) Average crawling velocity. Data shown are means ± SEM, n = 5. # P < 0.05, significantly different as indicated.
PRN473 abolishes neutrophil recruitment following sterile liver injury
Finally, we tested the hypothesis that pretreatment with PRN473 would dampen neutrophil recruitment to the liver following sterile liver injury. While the recruitment of neutrophils outside the necrotic focus is dependent on CXCR2 and thus does not require Btk, neutrophil accumulation to the necrotic inflammatory core is dependent on an fMLP gradient guiding Mac‐1‐dependent migration to the point of injury (McDonald and Kubes, 2011, 2012; McDonald et al., 2010; Volmering et al., 2016). Even though we observed a similar recruitment of neutrophils outside the injury zone and within the peri‐necrotic zone (Figure 5A left and middle panel), the number of neutrophils arriving in the necrotic zone was severely diminished in animals treated with PRN473 (Figure 5A right panel, B). These results demonstrate intact CXCR2‐dependent recruitment of cells to the outer regions of the necrotic focus but impaired Mac‐1‐mediated recruitment of cells to the necrotic zone in PRN473‐treated mice. The decreased accumulation in the lesion was due to impaired migratory activity of recruited neutrophils as evidenced by the reduced migration velocity and distance of cells in PRN473‐treated mice (Figure 5C–E) and not due to a difference in baseline neutrophil adherence as demonstrated in treated animals without heat injury (outside zone vehicle 0.84 ± 0.11 vs. PRN473 0.76 ± 0.05, peri‐necrotic zone 1.98 ± 0.08 vs. 2.07 ± 0.13 and necrotic zone 0.85 ± 0.08 vs. 1.08 ± 0.24 adherent cells per 10 000 μm2). To examine whether this effect translates into a significant effect on liver function, we obtained blood samples 8 h after injury and observed a marked decrease of serum glutamic oxaloacetic transaminase and glutamic pyruvic transaminase in injured animals pretreated with 40 mg·kg−1 PRN473 (Figure 5F) despite similar levels of lactate (2.83 ± 0.54 mmol·L−1 vehicle vs. 2.61 ± 0.81 mmol·L−1 PRN473). Taken together, these data demonstrate that PRN473‐mediated Btk inhibition abolishes Mac‐1‐dependent accumulation of neutrophils at the site of sterile injury in the liver in vivo while preserving CXCR2‐mediated recruitment.
Figure 5.
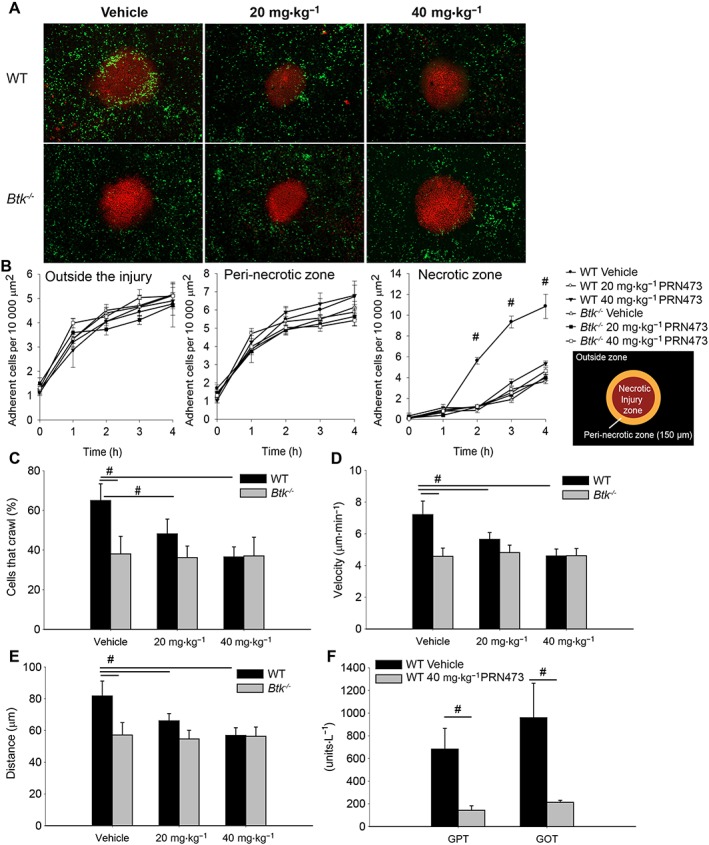
PRN473 abolishes neutrophil recruitment following sterile liver injury. (A) Representative micrographs of neutrophil (eGFP, green) recruitment to the necrotic zone (propidium iodide, red) 4 h after heat injury as obtained using spinning disc time‐lapse microscopy. (B) Timeline of adherent cells outside the injury zone (left panel), in the peri‐necrotic zone (middle panel) and within the necrotic injury zone (right panel) presented as mean number of adherent cells per time point ± SEM. (C) Average percentage of cells that crawled. (D) Average crawling velocity of cells. (E) Average crawling distance. (F) Liver enzymes (glutamic oxaloacetic transaminase, GOT; glutamic pyruvic transaminase, GPT) 8 h post injury in mice treated with vehicle control vs. 40 mg·kg−1 PRN473. Data shown are means ± SEM, n = 5. # P < 0.05, significantly different as indicated.
Discussion
Here, we report that inhibition of Btk with the reversible covalent inhibitor PRN473 can effectively prevent neutrophil recruitment via blockade of Fpr1‐induced activation of Mac‐1. We found PRN473 to be a potent inhibitor of Btk phosphorylation. Even at small doses, the level of Btk phosphorylation observed was almost absent, as evidenced by comparison to Btk‐deficient neutrophils. This potent effect was also evident in the in vitro ligand‐binding assays where a strong reduction of fibrinogen binding was found. It is a remarkable finding that PRN473 decreases downstream effects of Btk signalling to levels seen in the absence of Btk.
The kinase Btk fulfils a unique role in neutrophils comprising the positive regulation of integrin activation (Mueller et al., 2010; Volmering et al., 2016) as well as the negative regulation of ROS production and stimulation‐induced apoptosis (Honda et al., 2012). Increased ROS production and prolonged survival following functional stimulation, as seen in Btk‐deficient neutrophils, usually leads to exacerbated inflammation and increased tissue damage (Milot and Filep, 2011; El Kebir and Filep, 2013; Mittal et al., 2014; Leliefeld et al., 2016). Remarkably, We have described protective effects of Btk deficiency in previous studies, in particular, in following the sterile liver injury model used in this study (Volmering et al., 2016). This observation indicates that Btk‐mediated neutrophil recruitment and not ROS production or prolonged survival is the determining factor for organ damage following inhibition of Btk. Our observations support previous results using an siRNA‐based approach to silence Btk following acute lung injury (Krupa et al., 2014) and a recently published paper showing amelioration of liver damage following reperfusion injury after treatment with an irreversible Btk inhibitor (Palumbo et al., 2017).
However, it is worth mentioning that Btk expression is not limited to neutrophils. Yet, for instance, inflammatory states with significant involvement of B‐cell immunity, such as viral infections, are less likely to benefit from Btk inhibition (Bermejo et al., 2013), while recent work has implicated Btk in, for example, the production of IL‐17 in response to Trypanosoma cruzi infection. The sterile liver injury model employed here, similar to acute lung injury and some arthritis models, is independent of B‐cell‐mediated adaptive immunity (Tsuboi et al., 2008; McDonald and Kubes, 2012; Rosetti et al., 2012). However, the flipside of this coin is that Btk is not expressed in T‐cells and Btk inhibition thus preserves T‐cell functions, which have quite recently been shown to contribute to the orchestration of innate immunity (Day et al., 2006; Kasten et al., 2010).
Clinically, a number of trials investigating the efficiency of blocking leukocyte adhesion receptors have demonstrated that the preclinical success of blocking neutrophil influx can be translated into the clinic (Panes et al., 2007; Wun et al., 2014). Interestingly, limiting neutrophil influx is even discussed as a therapeutic strategy in sepsis, the very disease for which neutrophils were once regarded our strongest allies (Brown and Treacher, 2006). However, the disadvantage of directly blocking adhesion receptors, especially of the integrin family, is their overall abundance and thus lack of specificity for neutrophil recruitment (Nicolls and Gill, 2006). For instance, while essential for neutrophil recruitment, blocking of LFA‐1 compromises both effector and regulatory T‐cell function as LFA‐1 is also required for the formation of the immune synapse (Reisman et al., 2011). In this context, a FDA‐approved LFA‐1 antibody has been withdrawn as it was shown to increase risk for progressive multifocal leukoencephalopathy (Kothary et al., 2011). Inhibiting specific molecular pathways in leukocyte recruitment offers a more targeted approach and our present data indicate that pharmacological inhibition of Btk is a potent strategy to inhibit Fpr1‐dependent neutrophil influx. Such selectivity could overcome drawbacks of earlier, less targeted, attempts.
Author contributions
J.M.H. performed the experiments, analysed the data and wrote and revised the manuscript; A.M. performed the experiments, analysed the data and revised the manuscript; S.V. performed the experiments; B.E.C. helped with the cell culture and animal experiments; J.M.B., A.B., R.J.H. and C.L.L. characterized PRN473, provided materials and advised on utility in the studies; A.Z. and C.L. designed the study, provided overall supervision and wrote the manuscript.
Conflict of interest
J.M.B., A.B., R.J.H. and C.L.L. are current or former employees of Principia Biopharma, a biotechnology company developing therapeutics for autoimmune diseases and oncology.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Western blots corresponding to Figure 1. A. Representative complete Western blot of Btk IP probed with 4G10 phospho‐antibody (corresponding to Figure 1A). B. Western blot of WT and Btk deficient neutrophils probed with anti‐Btk antibody used for the Btk IP of Figure 1A. C. Western blot of the membrane of B, probed with anti‐vinculin.
{kind=link}
Figure S2 Extended Btk inhibition in vivo. Btk occupancy was assessed in mice following a single i.p. dose with 20 mg/kg PRN473 to assess the level of BTK inhibition achieved in vivo. BTK occupancy was measured over time in splenic immune cells utilizing a fluorescently labelled probe for BTK, normalized to total BTK and calculated as % BTK occupancy compared to vehicle control (Bradshaw et al., 2015). Mean ± SD, n = 3 per timepoint.
{kind=link}
Figure S3 In vitro characterization of PRN473 selectivity. (A) Kinase selectivity profile of PRN473. PRN473 was profiled at a concentration of 1 μM against a diverse panel of 250 kinases. The kinases inhibited by greater than 90% are indicated by small red dots while the kinases also inhibited greater than 98% are indicated by the large red dots. Kinases containing a conserved Cys with BTK are indicated by the yellow dots. Illustration reproduced courtesy of Cell Signalling Technology, Inc. (www.cellsignal.com) (B) PRN473 kinase selectivity, n = 2, and cell based potency, n ≥ 3, shows potency and selectivity of PRN473 to BTK. (C) Biochemical off rate of BTK. Prolonged BTK residence time exceeding 200 h (D) PRN473 reversibility of binding to BTK was determined by measuring the release of 83% of the compound following trypsin digestion, n = 2.
{kind=link}
Acknowledgements
The study was supported by the German Research Foundation (ZA428/12‐1 and INST‐211/604‐2_A05 to A.Z. as well as HE 6810/3‐1 to J.M.H.) and the Else Kröner Fresenius Stiftung (2014_A80 to J.M.H.).
The authors would like to thank Barbara Prystaj for expert technical assistance and Professor Dr Jezry Nofer for serum analysis.
Herter, J. M. , Margraf, A. , Volmering, S. , Correia, B. E. , Bradshaw, J. M. , Bisconte, A. , Hill, R. J. , Langrish, C. L. , Lowell, C. A. , and Zarbock, A. (2018) PRN473, an inhibitor of Bruton's tyrosine kinase, inhibits neutrophil recruitment via inhibition of macrophage antigen‐1 signalling. British Journal of Pharmacology, 175: 429–439. doi: 10.1111/bph.14090.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermejo DA, Jackson SW, Gorosito‐Serran M, Acosta‐Rodriguez EV, Amezcua‐Vesely MC, Sather BD et al (2013). Trypanosoma cruzi trans‐sialidase initiates a program independent of the transcription factors RORγt and Ahr that leads to IL‐17 production by activated B cells. Nat Immunol 14: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisconte A, Hill RJ, Bradshaw JM, Verner E, Karr D, Finkle D, et al (2015). Efficacy in collagen induced arthritis models with a selective, reversible covalent Bruton's tyrosine kinase (BTK) inhibitor PRN473 is driven by durable target occupancy rather than extended plasma exposure. In: American Association of Immunologist Annual Meeting, Vol. May 8–12, 2015. New Orleans, LA.
- Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT et al (2015). Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat Chem Biol 11: 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KA, Treacher DF (2006). Neutrophils as potential therapeutic targets in sepsis. Discov Med 6: 118–122. [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD (2006). Renal ischemia–reperfusion injury and adenosine 2A receptor‐mediated tissue protection: the role of CD4+ T cells and IFN‐γ. J Immunol 176: 3108–3114. [DOI] [PubMed] [Google Scholar]
- El Kebir D, Filep JG (2013). Targeting neutrophil apoptosis for enhancing the resolution of inflammation. Cell 2: 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etzioni A (1996). Adhesion molecules – their role in health and disease. Pediatr Res 39: 191–198. [DOI] [PubMed] [Google Scholar]
- Faust N, Varas F, Kelly LM, Heck S, Graf T (2000). Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96: 719–726. [PubMed] [Google Scholar]
- Herter J, Zarbock A (2013). Integrin regulation during leukocyte recruitment. J Immunol 190: 4451–4457. [DOI] [PubMed] [Google Scholar]
- Herter JM, Rossaint J, Block H, Welch H, Zarbock A (2013). Integrin activation by P‐Rex1 is required for selectin‐mediated slow leukocyte rolling and intravascular crawling. Blood 121: 2301–2310. [DOI] [PubMed] [Google Scholar]
- Herter JM, Rossaint J, Spieker T, Zarbock A (2014). Adhesion molecules involved in neutrophil recruitment during sepsis‐induced acute kidney injury. J Innate Immun 6: 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK et al (2012). The kinase Btk negatively regulates the production of reactive oxygen species and stimulation‐induced apoptosis in human neutrophils. Nat Immunol 13: 369–378. [DOI] [PubMed] [Google Scholar]
- Kasten KR, Tschop J, Adediran SG, Hildeman DA, Caldwell CC (2010). T cells are potent early mediators of the host response to sepsis. Shock 34: 327–336. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G (2011). Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol 65: 546–551. [DOI] [PubMed] [Google Scholar]
- Krupa A, Fol M, Rahman M, Stokes KY, Florence JM, Leskov IL et al (2014). Silencing Bruton's tyrosine kinase in alveolar neutrophils protects mice from LPS/immune complex‐induced acute lung injury. Am J Physiol Lung Cell Mol Physiol 307: L435–L448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ et al (2012). Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 119: 4275–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leliefeld PH, Wessels CM, Leenen LP, Koenderman L, Pillay J (2016). The role of neutrophils in immune dysfunction during severe inflammation. Crit Care 20: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Gaehtgens P (1991). Endothelial, not hemodynamic, differences are responsible for preferential leukocyte rolling in rat mesenteric venules. Circ Res 69: 1034–1041. [DOI] [PubMed] [Google Scholar]
- McDonald B, Kubes P (2011). Cellular and molecular choreography of neutrophil recruitment to sites of sterile inflammation. J Mol Med (Berl) 89: 1079–1088. [DOI] [PubMed] [Google Scholar]
- McDonald B, Kubes P (2012). Neutrophils and intravascular immunity in the liver during infection and sterile inflammation. Toxicol Pathol 40: 157–165. [DOI] [PubMed] [Google Scholar]
- McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC et al (2010). Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330: 362–366. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milot E, Filep JG (2011). Regulation of neutrophil survival/apoptosis by Mcl‐1. ScientificWorldJournal 11: 1948–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB (2014). Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20: 1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K et al (2010). Tyrosine kinase Btk regulates E‐selectin‐mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) γ2 and PI3Kγ pathways. Blood 115: 3118–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolls MR, Gill RG (2006). LFA‐1 (CD11a) as a therapeutic target. Am J Transplant 6: 27–36. [DOI] [PubMed] [Google Scholar]
- Nourshargh S, Alon R (2014). Leukocyte migration into inflamed tissues. Immunity 41: 694–707. [DOI] [PubMed] [Google Scholar]
- Palumbo T, Nakamura K, Lassman C, Kidani Y, Bensinger SJ, Busuttil R et al (2017). Bruton tyrosine kinase inhibition attenuates liver damage in a mouse warm ischemia and reperfusion model. Transplantation 101: 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panes J, Aceituno M, Gil F, Miquel R, Pique JM, Salas A et al (2007). Efficacy of an inhibitor of adhesion molecule expression (GI270384X) in the treatment of experimental colitis. Am J Physiol Gastrointest Liver Physiol 293: G739–G748. [DOI] [PubMed] [Google Scholar]
- Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P (2006). Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 203: 2569–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman NM, Floyd TL, Wagener ME, Kirk AD, Larsen CP, Ford ML (2011). LFA‐1 blockade induces effector and regulatory T‐cell enrichment in lymph nodes and synergizes with CTLA‐4Ig to inhibit effector function. Blood 118: 5851–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosetti F, Tsuboi N, Chen K, Nishi H, Ernandez T, Sethi S et al (2012). Human lupus serum induces neutrophil‐mediated organ damage in mice that is enabled by Mac‐1 deficiency. J Immunol 189: 3714–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler T, Dreyling M (2017). Bruton's tyrosine kinase inhibitors in B‐cell lymphoma: current experience and future perspectives. Expert Opin Investig Drugs 26: 909–915. [DOI] [PubMed] [Google Scholar]
- Singbartl K, Ley K (2004). Leukocyte recruitment and acute renal failure. J Mol Med (Berl) 82: 91–101. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi N, Asano K, Lauterbach M, Mayadas TN (2008). Human neutrophil Fcγ receptors initiate and play specialized nonredundant roles in antibody‐mediated inflammatory diseases. Immunity 28: 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volmering S, Block H, Boras M, Lowell CA, Zarbock A (2016). The neutrophil Btk signalosome regulates integrin activation during sterile inflammation. Immunity 44: 73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wun T, Styles L, DeCastro L, Telen MJ, Kuypers F, Cheung A et al (2014). Phase 1 study of the E‐selectin inhibitor GMI 1070 in patients with sickle cell anemia. PLoS One 9: e101301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarbock A, Deem TL, Burcin TL, Ley K (2007). Gαi2 is required for chemokine‐induced neutrophil arrest. Blood 110: 3773–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Western blots corresponding to Figure 1. A. Representative complete Western blot of Btk IP probed with 4G10 phospho‐antibody (corresponding to Figure 1A). B. Western blot of WT and Btk deficient neutrophils probed with anti‐Btk antibody used for the Btk IP of Figure 1A. C. Western blot of the membrane of B, probed with anti‐vinculin.
Figure S2 Extended Btk inhibition in vivo. Btk occupancy was assessed in mice following a single i.p. dose with 20 mg/kg PRN473 to assess the level of BTK inhibition achieved in vivo. BTK occupancy was measured over time in splenic immune cells utilizing a fluorescently labelled probe for BTK, normalized to total BTK and calculated as % BTK occupancy compared to vehicle control (Bradshaw et al., 2015). Mean ± SD, n = 3 per timepoint.
Figure S3 In vitro characterization of PRN473 selectivity. (A) Kinase selectivity profile of PRN473. PRN473 was profiled at a concentration of 1 μM against a diverse panel of 250 kinases. The kinases inhibited by greater than 90% are indicated by small red dots while the kinases also inhibited greater than 98% are indicated by the large red dots. Kinases containing a conserved Cys with BTK are indicated by the yellow dots. Illustration reproduced courtesy of Cell Signalling Technology, Inc. (www.cellsignal.com) (B) PRN473 kinase selectivity, n = 2, and cell based potency, n ≥ 3, shows potency and selectivity of PRN473 to BTK. (C) Biochemical off rate of BTK. Prolonged BTK residence time exceeding 200 h (D) PRN473 reversibility of binding to BTK was determined by measuring the release of 83% of the compound following trypsin digestion, n = 2.