

Abstract
Background and Purpose
Incretin‐based therapies based on glucagon‐like peptide‐1 (GLP‐1) receptor agonists are effective treatments of type 2 diabetes. Abundant research has focused on the development of long‐acting GLP‐1 receptor agonists. However, all GLP‐1 receptor agonists in clinical use or development are based on human or Gila GLP‐1. We have identified a potent GLP‐1 receptor agonist, xGLP‐1B, based on Xenopus GLP‐1.
Experimental Approach
To further modify the structure of xGLP‐1B, alanine scanning was performed to study the structure ‐activity relationship of xGLP‐1B. Two strategies were then employed to improve bioactivity. First, the C‐terminal tail of lixisenatide was appended to cysteine‐altered xGLP‐1B analogues. Second, polyethylene glycol (PEG) chains with different molecular weights were conjugated with the peptides, giving a series of PEGylated conjugates. Comprehensive bioactivity studies of these conjugates were performed in vitro and in vivo.
Results
From the in vitro receptor activation potency and in vivo acute hypoglycaemic activities of conjugates 25 ‐36, 33 was identified as the best candidate for further biological assessments. Conjugate 33 exhibited prominent hypoglycaemic and insulinotropic activities, as well as improved pharmacokinetic profiles in vivo. The prolonged antidiabetic duration of 33 was further confirmed by pre‐oral glucose tolerance tests (OGTT) and multiple OGTT. Furthermore, chronic treatment of db/db mice with 33 ameliorated non‐fasting blood glucose and insulin levels, reduced HbA1c values and normalized their impaired glucose tolerance. Importantly, no in vivo toxicity was observed in mice treated with 33.
Conclusions and Implications
Peptide 33 is a promising long‐acting type 2 diabetes therapeutic deserving further investigation.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- GLP‐1
glucagon‐like peptide‐1
- IPGTT
i.p. glucose tolerance test
- mPEG‐MAL
methoxy‐PEG‐maleimides
- OGTT
oral glucose tolerance tests
- RP‐HPLC
reversed phase HPLC
- SAR
structure–activity relationship
- TC
serum total cholesterol
- TG
triglycerides
- T2DM
type 2 diabetes mellitus
Introduction
Type 2 diabetes mellitus (T2DM) is a progressive disease with two major characteristics: (1) impaired insulin secretion caused by beta cell dysfunction and (2) reduced insulin sensitivity in association with obesity (Agyemang et al., 2016; Mishra et al., 2017). Many drugs which promote beta cell proliferation and/or neogenesis and restore beta cell function have side effects such as weight gain or hypoglycaemia (Nathan et al., 2009). Glucagon‐like peptide‐1 (GLP‐1), a GLP‐1 receptor agonist secreted from intestinal L‐cells in response to nutrient ingestion, has emerged as an attractive alternative for T2DM treatment without the undesirable side effects mentioned above (Meier, 2012; Manandhar and Ahn, 2015). GLP‐1 stimulates insulin release in a glucose‐dependent manner and plays an important physiological role in nutrient metabolism and glucose homeostasis (Evers et al., 2017). However, its inactivation by dipeptidyl peptidase IV and neutral endopeptidase 24.11, as well as rapid glomerular filtration, results in a short in vivo t 1/2 of GLP‐1, compromising its clinical application (Murage et al., 2010; Yang et al., 2014). Therefore, much research effort has focused on the development of novel long‐acting GLP‐1 analogues, represented by exenatide and liraglutide. However, all of the GLP‐1 receptor agonists used in the clinic or in clinical development are based on the peptide sequence of human GLP‐1 (hGLP‐1) or Gila GLP‐1, such as exendin‐4 (Mapelli et al., 2009; Tomlinson et al., 2016).
We have recently explored a non‐traditional approach to find novel GLP‐1 receptor agonists based on Xenopus GLP‐1 (Irwin et al., 1997; Han et al., 2017). Amino acid modification of Xenopus GLP‐1 generated a new GLP‐1 derivative xGLP‐1B (Table 1), which is a highly potent activator of GLP‐1 receptors and better at lowering glucose levels than hGLP‐1. In the present study, we describe the activities of the compounds obtained from further modification of xGLP‐1B. Firstly, alanine scanning was performed to investigate the structure–activity relationship of xGLP‐1B. The results suggested positions 12, 14, 17 and 26 as suitable sites for derivatization. Next, the C‐terminal sequence (PSSGA PPSKK KKKK) of lixisenatide was appended to xGLP‐1B and its four new cysteine‐altered analogues, generating xGLP‐1B‐Sen and peptides 21–24. The in vitro receptor activation potency and in vivo glucose‐lowering activity of these peptides and some of their polyethylene glycol (PEG) conjugates were screened. Several candidates were selected and subjected to further biological and pharmacokinetic evaluations, with the aim of identifying novel long‐acting hypoglycaemic and insulinotropic agents with prominent therapeutic potential.
Table 1.
The in vitro receptor activation potency of xGLP‐1B analogues and their in vivo AUCglucose 0–120 min values in KM mice
| Peptides | Sequence | EC50 (nM)a | EC50 (% of hGLP‐1) | AUC0–120 min b |
|---|---|---|---|---|
| Xenopus glucagon | HSQGT FTSDY SKYLD SRRAQ DFVQW WLMNT | – | – | – |
| hGLP‐1 | HAEGT FTSDV SSYLE GQAAK EFIAW LVKGR | 2.1 ± 0.2 | – | 670 ± 38 |
| xGLP‐1B | HGEGT YTNDV TEYLE EKAAK EFIEW LIKGK | 1.5 ± 0.5 | 71.4 | 648 ± 18 |
| 1 | HGEGT YTNDV AEYLE EKAAK EFIEW LIKGK | 3.2 ± 0.9 | 152.4 | 731 ± 32 |
| 2 | HGEGT YTNDV TAYLE EKAAK EFIEW LIKGK | 0.54 ± 0.09 | 25.7 | 579 ± 22 |
| 3 | HGEGT YTNDV TEALE EKAAK EFIEW LIKGK | 4.3 ± 1.1 | 204.8 | 714 ± 27 |
| 4 | HGEGT YTNDV TEYAE EKAAK EFIEW LIKGK | 0.9 ± 0.2 | 42.9 | 638 ± 14 |
| 5 | HGEGT YTNDV TEYLA EKAAK EFIEW LIKGK | 14.1 ± 1.9 | 671.4 | 942 ± 39 |
| 6 | HGEGT YTNDV TEYLE AKAAK EFIEW LIKGK | 21.2 ± 3.7 | 1009.5 | 1054 ± 19 |
| 7 | HGEGT YTNDV TEYLE EAAAK EFIEW LIKGK | 0.12 ± 0.02 | 5.71 | 515 ± 23 |
| 8 | HGEGT YTNDV TEYLE EKRAK EFIEW LIKGK | 2.4 ± 0.7 | 114.3 | 687 ± 12 |
| 10 | HGEGT YTNDV TEYLE EKAAA EFIEW LIKGK | 2.6 ± 0.4 | 123.8 | 681 ± 35 |
| 11 | HGEGT YTNDV TEYLE EKAAK AFIEW LIKGK | 13.3 ± 2.9 | 633.3 | 1086 ± 19 |
| 12 | HGEGT YTNDV TEYLE EKAAK EAIEW LIKGK | 11.9 ± 1.6 | 566.7 | 1232 ± 24 |
| 13 | HGEGT YTNDV TEYLE EKAAK EFAEW LIKGK | 22.1 ± 4.6 | 1052.4 | 1579 ± 63 |
| 14 | HGEGT YTNDV TEYLE EKAAK EFIAW LIKGK | 5.9 ± 1.2 | 281.0 | 814 ± 15 |
| 15 | HGEGT YTNDV TEYLE EKAAK EFIEA LIKGK | 2.9 ± 0.7 | 138.1 | 749 ± 13 |
| 16 | HGEGT YTNDV TEYLE EKAAK EFIEW AIKGK | 0.44 ± 0.06 | 21.0 | 622 ± 38 |
| 17 | HGEGT YTNDV TEYLE EKAAK EFIEW LAKGK | 7.5 ± 2.4 | 357.1 | 931 ± 19 |
| 18 | HGEGT YTNDV TEYLE EKAAK EFIEW LIAGK | 6.2 ± 1.9 | 295.2 | 875 ± 53 |
| 19 | HGEGT YTNDV TEYLE EKAAK EFIEW LIKAK | 9.2 ± 2.5 | 438.1 | 952 ± 68 |
| 20 | HGEGT YTNDV TEYLE EKAAK EFIEW LIKGA | 3.4 ± 0.4 | 161.9 | 782 ± 24 |
The receptor potency data are presented as EC50 (means ± SD). All experiments were performed in triplicate and repeated three times (n = 3).
The AUCglucose 0–120 min values were calculated by acute glucose‐lowering experiment in KM mice by IPGTT. Results are presented as means ± SD, n = 6.
Methods
Animals
Male Kunming (KM) mice (6–7 weeks old, 20–25 g) and Sprague–Dawley rats (9–10 weeks old, 200–250 g) were acquired from the Comparative Medical Centre of Yangzhou University (Jiangsu, China). Male C57BL/6 db/db mice (6–8 weeks old, 22–25 g) were acquired from the Model Animal Research Centre of Nanjing University (Jiangsu, China). KM mice are the species commonly used in preliminary antidiabetic activity research, and Sprague–Dawley rats are the species widely used in pharmacokinetic research. C57BL/6 db/db mice are mildly hyperglycaemic and commonly used as a model of type 2 diabetes in diabetes research. All the in vivo experimental data were collected and analysed by two independent observers, and treatment groups were blinded during result assessment. Animals were housed in specific pathogen‐free facilities in polypropylene cages, and corncob was used as the bedding material. Three rats or six mice were housed in a single cage. Animals were acclimatized for 1 week prior to the experiment under controlled temperature (25 ± 3°C) and a reverse 12 h light‐dark cycle. Animals were allowed free access to water and food (standard laboratory chow) throughout the study. All experimental animal procedures were performed in accordance with the Laboratory Animal Management Regulations in China, the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (revised 2011) and approved by Jiangsu Normal University ethical committee. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
General synthetic route of peptides and PEGylated conjugates
Peptides 1–24 were synthesized by a semi‐automatic PSI‐200 peptide synthesizer (Peptide Scientific Inc.) using a standard N‐Fmoc/tBu solid phase peptide synthesis protocol, as described elsewhere (Yang et al., 2015). Cysteine altered peptides (21–24) were conjugated with 1, 2 and 5 kDa methoxy‐PEG‐maleimides (mPEG‐MAL) in PBS (0.05 mol·L−1, pH 7.0). The reaction was stirred at 25°C under N2 until completion as confirmed by HPLC. The crude peptides and PEGylated conjugates were purified by reversed phase (RP)‐HPLC and characterized by Thermo LC‐MS.
In vitro GLP‐1 receptor activation potency test
The potency of the peptides and conjugates selected towards the GLP‐1 receptor was evaluated by functional assays using HEK‐293 cell lines stably overexpressing the human GLP‐1 receptor, as previously described (Han et al., 2014). Briefly, HEK‐293 cells were grown in DMEM‐31053 (Invitrogen, Carlsbad, CA, USA) containing 50 U·mL−1 penicillin, 0.5% FBS, 50 μg·mL−1 streptomycin, 2 mmol·L−1 L‐glutamine and 20 mmol·L−1 HEPES at 37°C in 5% CO2. Two hours before the experiment, cells were resuspended in the growth medium and plated in 96‐well plates. The test peptides and conjugates were dissolved in DMSO and diluted with the medium (0.5% FBS substituted by 0.1% BSA fraction V) over a range of concentrations. The test peptides, diluted in medium, were added to the cells, followed by incubation at room temperature for 20 min. The accumulation of cAMP was assayed by using the cAMP dynamic 2 kit (Cisbio, USA). Fluorescence was measured by using homogeneous time resolved fluorescence technology and was detected using an Envision 2104 Multilabel Reader (Perkin Elmer, UK). The concentration of cAMP was determined via the conversion of the fluorescence data to cAMP concentration using a cAMP standard curve. The in vitro potency of the test peptides was quantified by determining the EC50 using GraphPad Prism version 5.0 (San Diego, USA).
Acute glucose‐lowering activity in Kunming mice
The acute in vivo glucose‐lowering activities of peptides 1–36 were evaluated by the i.p. glucose tolerance test (IPGTT) in KM mice, using a previously described method (Cui et al., 2016). Details are shown in the Supporting Information (Table S1, Entry 1). The glucose‐lowering activities of the test peptides were quantified by measuring the AUC values using GraphPad Prism version 5.0 (San Diego, USA).
Glucose‐lowering and insulin secretion assay in db/db mice
The glucose‐lowering and insulinotropic activities of peptide 33 were measured by IPGTT on db/db mice using a previously described method with some modifications (Han et al., 2013a). Details of the blood glucose levels are shown in the Supporting Information (Table S1, Entry 2). The plasma insulin levels were measured at 0, 5, 10, 15, 30, 45, 60 and 120 min by collecting blood samples (approximately 50 μL) from the tail vein in microcentrifuge tubes containing EDTA. Plasma was separated by centrifugation (1250× g, 10 min) and the plasma insulin concentrations were determined using a mouse insulin ELISA kit (Lot. 201701, Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions.
Pharmacokinetics assay in Sprague–Dawley rats
The pharmacokinetic parameters of lixisenatide and 33 were measured in male Sprague–Dawley rats (n = 3, ~250 g) using a previously‐described method with some modifications (Han et al., 2013b). Briefly, Sprague–Dawley rats were randomly divided into two groups and fasted for 12 h before receiving a single s.c. injection of 200 nmol·kg−1 of test peptides. Blood samples (~0.1 mL) were collected from the lateral tail vein in EDTA‐coated Microtainer tubes at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4 and 6 h post s.c. injection. Blood samples were centrifuged at 4°C to obtain the plasma, and the plasma samples were stored at −20°C until tested. The in vitro cell‐based GLP‐1 receptor‐activation assay described above was used to determine the plasma concentration of the test peptides in each group of rats (Bianchi et al., 2013).
Acute effect on food intake
For acute food intake studies, male 8‐week‐old db/db mice were randomly distributed according to their body weight and individually housed in cages to which they were acclimatized to for 1 week before the experiment (van Witteloostuijn et al., 2016a). Mice were pricked five times to accustom them to the i.p. dosing procedure on separate days. Mice were fasted for 18 h with free access to water before the start of the experiment. On the day of the study, animals were i.p. treated with saline (control), lixisenatide (25 nmol·kg−1) and peptide 33 (25 or 100 nmol·kg−1). Following the i.p. injection, mice were returned to their cage and had free access to pre‐weighed chow that was placed in a Petri dish, and the accumulative food intake was determined at 0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 16, 18 and 24 h post‐injection.
In vivo long‐term hypoglycaemic activity tests
The long‐term glucose‐lowering activity of 33 was evaluated by pre‐oral glucose tolerance tests (OGTT) and further verified by multiple OGTT as previously described (Han et al., 2017). Details are shown in the Supporting Information (Table S1, Entries 3–5).
Chronic in vivo studies and histological analyses
Eight week male db/db mice were randomly divided into three groups (n = 6) based on non‐fasting glucose levels; after 1 week of acclimatization, blood samples were collected from the tail vein (~10 μL) of mice, and the HbA1c level (day 0) was determined by using a DCA 2000+ chemistry analyser (Bayer Diagnostics, USA), based on an immunoagglutination inhibition method. Based on the pharmacokinetics results and as mice have a relatively high metabolic rate, saline (control) and lixisenatide (25 nmol·kg−1) were administered i.p. three times a day (7:00, 15:00 and 23:00 h), and 33 (25 nmol·kg−1) was administered i.p. twice daily (7:00 and 19:00 h) for 5 weeks. Food consumption and body weight were monitored daily. Non‐fasting plasma insulin levels and blood glucose levels were determined at 8:00 h every 2 days according to the same procedure described in the glucose‐lowering and insulin secretion assay. At the end of the treatment period (day 36), the HbA1c values of each group mice were determined using the same method described above. Following the 5 week treatment, IPGTT were performed in 18 h‐fasted mice (Supporting Information Table S1, Entry 6). Plasma insulin was also measured at 0, 5, 10, 15, 30, 45, 60 and 120 min, using the same method described above.
At the end of the study, mice were bled by retroorbital puncture to collect blood for biochemical analysis. Then mice were killed by cervical dislocation, and the pancreas was immediately removed. For biochemical analysis, blood samples were centrifuged for separating the serum, serum total cholesterol (TC), triglycerides (TG), HDL cholesterol, LDL cholesterol, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) that were analysed by using Hitachi 7060 Automatic Analyser (Tokyo, Japan) (Sun et al., 2016). TC and TG values were determined by an enzymatic colorimetric method, while HDL‐cholesterol and LDL‐cholesterol were assayed by a direct method. AST and ALT were determined using a kinetic rate method. The commercially available kits for testing these values were purchased from Ningbo Medical System Biotechnology Co., Ltd. (Ningbo, China). For histochemical analysis, the methodology was as described in detail elsewhere (Han et al., 2017). In brief, pancreas samples were fixed in 4% paraformaldehyde for 24 h at 4°C and subsequently embedded in paraffin blocks, then sliced into 3‐μm‐thick sections. Then the tissue sections were stained with haematoxylin–eosin so that islet morphology could be assessed. The area and number of islets, six islets per mouse pancreas and six mice per group, were analysed by using an Olympus DP2‐BSW digital camera software (Olympus, Center Valley, PA, USA).
Data analysis
The data are presented as means ± SD. Data analysis was performed by one‐way ANOVA for multiple comparisons with post hoc Tukey's tests if F achieved statistical significance (P < 0.05) and there was no significant variance in homogeneity. P values of <0.05 were considered as significant. No data were excluded from any of the studies. All pharmacological data analyses were performed using GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA). Pharmacokinetic parameters of peptide 33 were determined using the Bioavailability Programme Package software (BAPP, Version 2.2, Centre of Drug Metabolism and Pharmacokinetics, China Pharmaceutical University). The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
mPEG‐MAL (1, 2 and 5 kDa) was obtained from Xi'an Ruixi Biological Technology Co., Ltd (Xi'an, China). GLP‐1, lixisenatide, Fmoc‐protected amino acids and Fmoc Rink amide‐MBHA resin were purchased from GL Biochem (Shanghai, China). The mouse insulin ELISA kit and cAMP dynamic kit were acquired from Nanjing Jiancheng Bioengineering Institute (Jiangsu, China) and Cisbio (Bedford, MA, USA) respectively. All other reagents, unless otherwise indicated, were obtained from Sigma‐Aldrich Co. (St. Louis, MO, USA) and used as received.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in The Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Structure–activity relationship (SAR) studies of xGLP‐1B by alanine scanning
We initially conducted alanine scanning to investigate the SAR of xGLP‐1B. Considering that a conservative N‐terminal sequence is critical for the ability of GLP‐1 analogues to activate the GLP‐1 receptor (Hager et al., 2017), alanine scanning was started from position 11. Amino acids at positions 11–17 and 20–30 were serially replaced by L‐alanine, giving peptides 1–7 and 10–20. Peptide 8 was obtained by substituting the Ala18 of xGLP‐1B with arginine, which is at position 18 of Xenopus glucagon (Table 1) (Adelhorst et al., 1994). Since alanine is found at position 19 for both Xenopus glucagon and xGLP‐1B, mutagenesis at this site was not performed. Hence, a total number of 19 xGLP‐1B analogues (1–8 and 10–20, Table 1) were synthesized using the N‐Fmoc/tBu solid‐phase strategy. They were all characterized by LC‐MS after purification by RP‐HPLC (see Supporting Information).
The potency of the xGLP‐1B analogues at activating the GLP‐1 receptor was tested, and the results are listed in Table 1. L‐Alanine substitutions made at positions 15, 16, 21, 22 and 23 caused a great loss of receptor activation potency, as shown from the EC50 values (>10 nM) of peptide 5, 6, 11, 12 and 13. Substituting Thr11, Tyr13, Lys20, Trp25 and Lys30 with L‐alanine almost had no effect on the receptor activation potency of peptides 1, 3, 10, 15 and 20, suggesting that these positions may not be essential for receptor activation. Similarly, introducing the Arg18 of Xenopus glucagon into xGLP‐1B did not affect the receptor activation activity of 8. Importantly, when Glu12, Leu14, Lys17 and Leu26 were mutated into alanine, improved receptor activation potency was observed for 2, 4, 7 and 16, indicating that these positions might hinder the agonist–receptor interaction and could be suitable sites for modification.
In addition to the in vitro agonist potency study, the in vivo hypoglycaemic abilities of peptides 1–20 were also evaluated by IPGTT in KM mice. As shown in Figure 1A, B, the administration of peptides 1–20 resulted in enhanced glucose tolerance patterns, particularly for peptides 2, 4, 7, and 16. This is in good accordance with the receptor activation results. Furthermore, glucose AUC values calculated also revealed that the hypoglycaemic effects of peptides 2, 4, 7 and 16 were better than those of xGLP‐1B, hGLP‐1 and other analogues. Taken together, our alanine scanning results suggested positions 12, 14, 17, and 26 in xGLP‐1B are suitable sites for modification.
Figure 1.
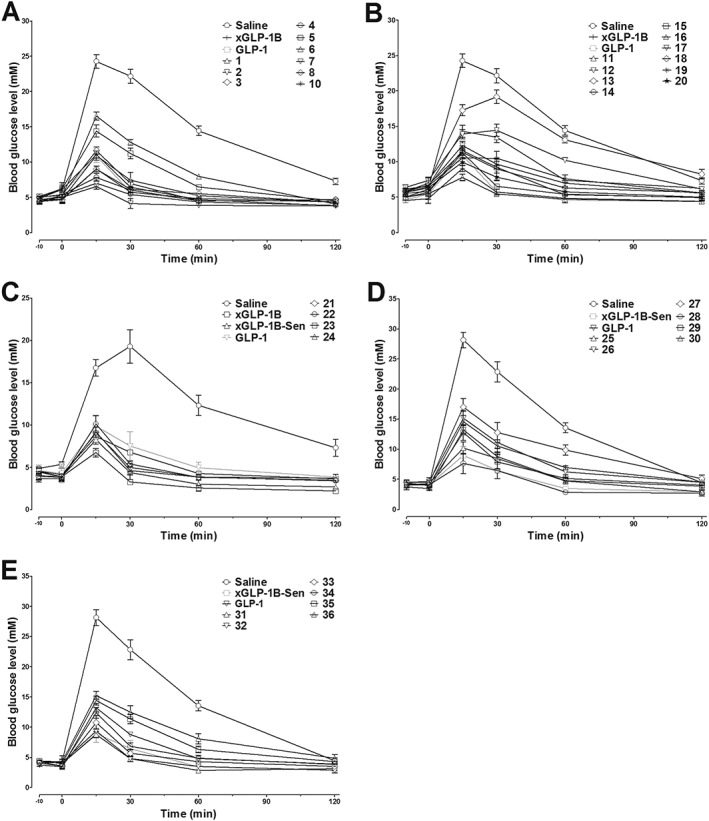
The acute hypoglycaemic activity of GLP‐1, xGLP‐1B, xGLP‐1B‐Sen and peptides 1–36 in KM mice as assessed by IGPTT. Saline (control), GLP‐1, xGLP‐1B, xGLP‐1B‐Sen and peptides 1–36 were i.p. administered 10 min prior to the glucose load (i.p. 2 g·kg−1). The blood glucose curves during the IPGTT are shown in panels A–E. Results are presented as means ± SD, n = 6.
Rational design of 21, 22, 23 and 24 as lead peptides with enhanced bioactivity
The C‐terminal sequence of lixisenatide contributes to its higher affinity for the GLP‐1 receptor compared with exendin‐4 and GLP‐1 (Fonseca et al., 2012). To further improve the biological activity of xGLP‐1B, the C‐terminal sequence (PSSGA PPSKK KKKK) of lixisenatide was attached to Lys30 of xGLP‐1B, giving xGLP‐1B‐Sen. Furthermore, based on the above SAR study of xGLP‐1B, the Glu12, Leu14, Lys17 and Leu26 of xGLP‐1B‐Sen were individually replaced by cysteine, giving four cysteine‐altered xGLP‐1B‐Sen analogues 21–24 (Table 2). The structures of both xGLP‐1B‐Sen and 21–24 were unambiguously confirmed by LC‐MS after purification by RP‐HPLC (see Supporting Information). As shown in Table 2 and Figure 1C, both the in vitro receptor activation potency and the in vivo glucose‐lowering activity of xGLP‐1B‐Sen were superior to those of xGLP‐1B, demonstrating that the introduction of C‐terminal PSSGA PPSKK KKKK sequence effectively improved the bioactivity of xGLP‐1B. Cysteine replacement had no negative impacts on the receptor activation potency and hypoglycaemic activity, further confirming the reliability of our SAR results. Therefore, 21–24 were selected as lead peptides for the following PEG conjugation.
Table 2.
The in vitro receptor activation potency of xGLP‐1B‐Sen analogues and their in vivo AUCglucose 0–120 min values in KM mice
| Peptides | Sequence | EC50 (nM)a | EC50 (% of hGLP‐1) | AUC0–120 min b |
|---|---|---|---|---|
| hGLP‐1 | HAEGT FTSDV SSYLE GQAAK EFIAW LVKGR | 3.8 ± 1.1 | – | 686 ± 80 |
| xGLP‐1B | HGEGT YTNDV TEYLE EKAAK EFIEW LIKGK | 2.4 ± 0.9 | 63.2 | 614 ± 94 |
| xGLP‐1B‐Sen |
HGEGT YTNDV TEYLE EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.29 ± 0.07 | 7.6 | 466 ± 35 |
| 21 |
HGEGT YTNDV TCYLE EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.49 ± 0.15 | 12.9 | 552 ± 14 |
| 22 |
HGEGT YTNDV TEYCE EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.58 ± 0.14 | 15.3 | 575 ± 26 |
| 23 |
HGEGT YTNDV TEYLE ECAAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.12 ± 0.05 | 3.2 | 384 ± 13 |
| 24 |
HGEGT YTNDV TEYLE EKAAK EFIEW CIKGK PSSGA PPSKK KKKK |
0.34 ± 0.11 | 8.9 | 553 ± 24 |
The receptor potency data are presented as EC50 (means ± SD). All experiments were performed in triplicate and repeated three times (n = 3).
The AUCglucose 0–120 min values were calculated by an acute glucose‐lowering experiment in KM mice by IPGTT. Results are presented as means ± SD, n = 6.
Synthesis, characterization and biological activity of PEGylated conjugates
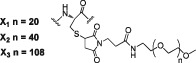
To study the impacts of PEG size on the bioactivity of PEGylated conjugates, three maleimide‐labelled PEG chains (mPEG‐MAL) with different molecular weights (PEG1000, PEG2000 and PEG5000) were conjugated with the cysteine residue in peptides 21, 22, 23 and 24. A total of 12 PEGylated conjugates (25–36, Table 3) were synthesized through thiol‐Michael addition reaction. The crude conjugates were purified by semi‐preparative RP‐HPLC and subsequently characterized by electrospray ionization‐MS (see Supporting Information). As summarized in Table 3, when PEG1000 was attached, GLP‐1 receptor activation potency was not affected, as observed from the sub‐nmol EC50 values of 25, 28, 31 and 34. However, an increase in the size of the PEG chain led to decreased receptor activation potency, particularly for 26, 27, 29, 30, 35 and 36. Interestingly, PEGylation was well tolerated by 23, regardless of the PEG size. Conjugates 31–33, PEGylated products from 23, continued as potent GLP‐1 receptor agonists with sub‐nmol EC50 values (Representative concentration–response curves of hGLP‐1 and PEGylated conjugates are shown in Supporting Information Figure S1). Consistent with the in vitro results, the in vivo hypoglycaemic test showed a similar trend (Figure 1D, E). All the conjugates decreased the blood glucose levels as compared with the saline. In particular, the glucose‐lowering activity of 31–33 was better than GLP‐1 as demonstrated from both the blood glucose curves (Figure 1E) and the glucose AUC values calculated (Table 3). In summary, 31–33 were found to have similar antidiabetic activity, superior to other conjugates. From previous reports and our own experience, PEGylated peptides with longer PEG chains usually display better pharmacokinetic behaviours. As a result, the PEG5000‐modified conjugate 33 was finally selected for the following bioactivity studies.
Table 3.
The in vitro receptor activation potency of PEGylated conjugates and their in vivo AUCglucose 0–120 min values in KM mice
| Peptides | Sequencea | EC50 (nM)b | EC50 (% of hGLP‐1) | AUC0–120 min c |
|---|---|---|---|---|
| hGLP‐1 | HAEGT FTSDV SSYLE GQAAK EFIAW LVKGR | 2.5 ± 0.9 | – | 756 ± 21 |
| xGLP‐1B‐Sen |
HGEGT YTNDV TEYLE EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.15 ± 0.08 | 6.0 | 553 ± 67 |
| 25 |
HGEGT YTNDV TX1YLE EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.25 ± 0.05 | 10.0 | 674 ± 53 |
| 26 |
HGEGT YTNDV TX2YLE EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
3.28 ± 1.38 | 131.2 | 927 ± 111 |
| 27 |
HGEGT YTNDV TX3YLE EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
7.41 ± 1.59 | 296.4 | 1173 ± 61 |
| 28 |
HGEGT YTNDV TEYX1E EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.78 ± 0.2 | 31.2 | 499 ± 61 |
| 29 |
HGEGT YTNDV TEYX2E EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
2.21 ± 0.49 | 88.4 | 752 ± 69 |
| 30 |
HGEGT YTNDV TEYX3E EKAAK EFIEW LIKGK PSSGA PPSKK KKKK |
3.98 ± 1.58 | 159.2 | 935 ± 50 |
| 31 |
HGEGT YTNDV TEYLE EX1AAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.13 ± 0.04 | 5.2 | 488 ± 22 |
| 32 |
HGEGT YTNDV TEYLE EX2AAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.29 ± 0.08 | 11.6 | 522 ± 21 |
| 33 |
HGEGT YTNDV TEYLE EX3AAK EFIEW LIKGK PSSGA PPSKK KKKK |
0.41 ± 0.15 | 16.4 | 613 ± 15 |
| 34 |
HGEGT YTNDV TEYLE EKAAK EFIEW X1IKGK PSSGA PPSKK KKKK |
0.37 ± 0.04 | 14.8 | 699 ± 28 |
| 35 |
HGEGT YTNDV TEYLE EKAAK EFIEW X2IKGK PSSGA PPSKK KKKK |
1.69 ± 0.28 | 67.6 | 913 ± 29 |
| 36 |
HGEGT YTNDV TEYLE EKAAK EFIEW X3IKGK PSSGA PPSKK KKKK |
2.91 ± 0.92 | 116.4 | 1050 ± 41 |
|
The average molecular mass of PEGylated xGLP‐1B‐Sen conjugates were calculated by M + 44n (for PEG1000, n = 20; for PEG2000, n = 40; for PEG5000, n = 108).
The receptor potency data are presented as EC50 (means ± SD). All experiments were performed in triplicate and repeated three times (n = 3).
The AUCglucose 0–120 min values were calculated by an acute glucose‐lowering experiment in KM mice by IPGTT. Results are presented as means ± SD, n = 6.
Hypoglycaemic and insulinotropic activities of 33 in db/db mice
The in vivo hypoglycaemic and insulinotropic activities of 33 were examined by IPGTT in db/db mice, using lixisenatide as a positive control. As shown in Figure 2A, blood glucose levels in lixisenatide‐ or 33‐treated groups were significantly lower than those in the saline group after the glucose challenge. The time courses of blood glucose levels following the administration of lixisenatide and 33 were similar. Importantly, the decreased blood glucose levels were accompanied by increased plasma insulin levels (Figure 2C), demonstrating a GLP‐1‐dependent mechanism of action of 33. The plasma insulin levels in the 33‐treated group were similar to those in the lixisenatide‐treated animals and were significantly higher than those in the saline group during 10–60 min. The AUCglucose and AUCinsulin values (Figure 2B, D) further quantitatively confirmed that the hypoglycaemic and insulinotropic activities of 33 were slightly better than those of lixisenatide.
Figure 2.
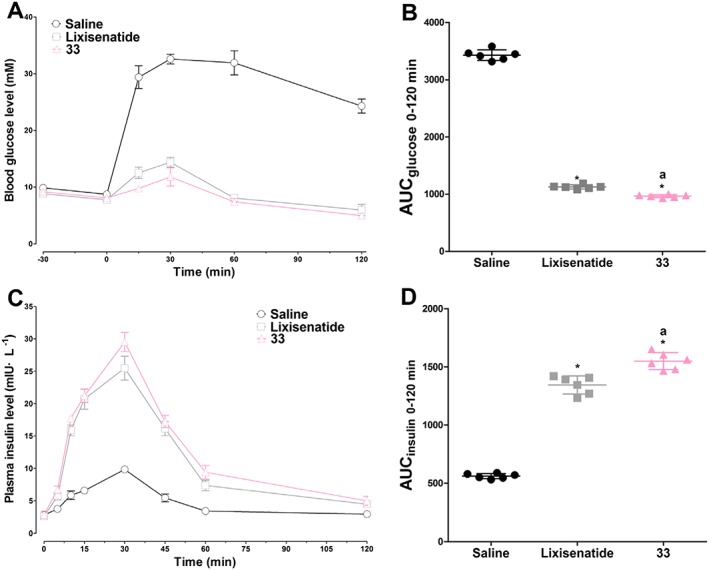
In vivo hypoglycaemic and insulinotropic activities of 33 in db/db mice as assessed by IPGTT. Saline, lixisenatide and 33 (25 nmol·kg−1) were i.p. injected 30 min prior to 1 g·kg−1 i.p. glucose load. (A) The time course of blood glucose levels in each group. (B) The calculated AUCglucose 0–120 min values of each group. (C) The time course of plasma insulin levels in each group. (D) The calculated AUCinsulin 0–120 min values of each group. Results are presented as means ± SD, n = 6. * P < 0.05 versus control (saline), a P < 0.05 versus lixisenatide.
Pharmacokinetics of peptide 33 in Sprague–Dawley rats
The pharmacokinetics of 33 and lixisenatide were measured in the Sprague–Dawley rats, and the results are shown in Figure 3. After s.c. administration of lixisenatide, the plasma concentrations increased rapidly with a Tmax of 0.5 h and then rapidly decreased to baseline within 3 h. The elimination half‐life (t 1/2) of lixisenatide was 0.8 h. Conjugate 33 showed a different pharmacokinetic profile. Following s.c. administration, 33 showed a delayed absorption pattern with a Tmax of 1.1 h. Furthermore, the t 1/2 of 33 (t 1/2 = 2.0 h) was much longer than that of lixisenatide. Detailed pharmacokinetic analysis also revealed that PEG5000 conjugation not only extended the elimination t 1/2 but also significantly increased the drug utilization. The AUCinf value of 33 was approximately 2.7‐fold greater than that of lixisenatide (Figure 3).
Figure 3.
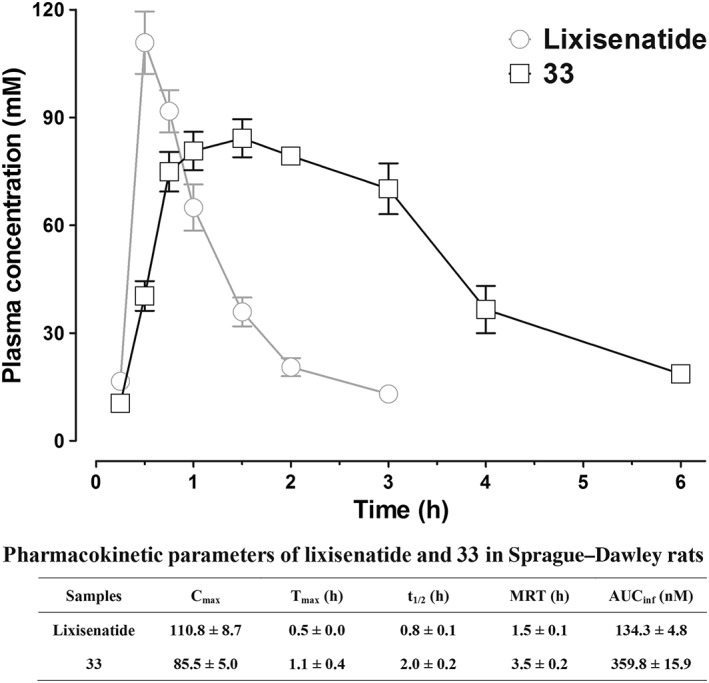
Pharmacokinetic characterizations and detailed pharmacokinetic parameters of lixisenatide and 33 in Sprague–Dawley rats after s.c. administration (200 nmol·kg−1). Results are presented as means ± SD, n = 3.
Effect of peptide 33 on acute food intake
In addition to their glucose‐stabilization ability, GLP‐1 receptor agonists are also known to exert an anorectic effect on rodents and humans (Kerr et al., 2010). Therefore, we next investigated the anorectic effect of 33 in db/db mice at two doses (25 and 100 nmol·kg−1). As shown in Figure 4, at a dose of 25 nmol·kg−1, both lixisenatide and 33 potently reduced the food intake by 37–45% versus saline, and the anorectic effect of 33 was slightly better than that of lixisenatide. When administered at a higher dose (100 nmol·kg−1), 33 reduced the cumulative food intake by 65%, exhibiting a dose‐dependent pattern.
Figure 4.
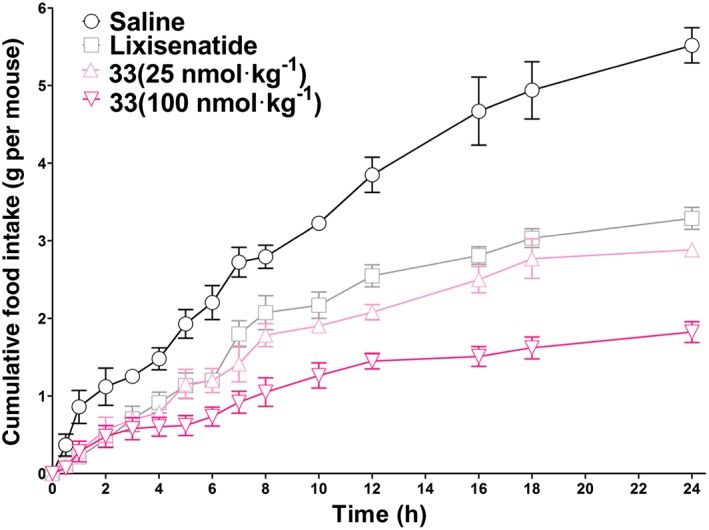
Cumulative food intake following acute i.p. administration of lixisenatide (25 nmol·kg−1) or 33 (25 or 100 nmol·kg−1) to db/db mice. Results are presented as means ± SD, n = 6.
Long‐term hypoglycaemic activity of 33 in db/db mice
The glucose stabilization effect of 33 was examined at two doses (25 or 100 nmol·kg−1) by assessing two types of OGTT in db/db mice. In the pre‐OGTT test, pretreatment with 25 nmol·kg−1 lixisenatide and 25 or 100 nmol·kg−1 33 120 min prior to the oral glucose load exhibited similar glucose‐stabilizing abilities. The blood glucose values in the lixisenatide‐ or 33‐treated groups were significantly lower than that in the saline group (Figure 5A). When mice were pretreated with lixisenatide (25 nmol·kg−1) 360 min before an oral glucose load, the glucose‐lowering activity was significantly reduced. However, the hypoglycaemic effect of 33 during the same condition was only slightly compromised (Figure 5B). Moreover, at a dose of 100 nmol·kg−1, 33 showed a constant glucose stabilization activity regardless of the pretreatment time.
Figure 5.
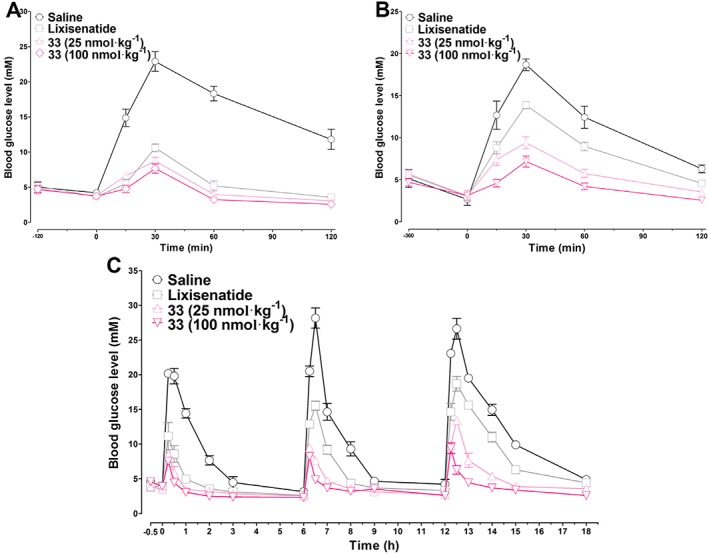
Long‐term hypoglycaemic effects of lixisenatide and 33 tested by pre‐OGTT and multiple OGTT in db/db mice. Each group of mice were i.p. injected with saline (control), lixisenatide (25 nmol·kg−1) or 33 (25 or 100 nmol·kg−1) 120 min (A) or 360 min (B) prior to oral glucose loads (1.5 g·kg−1). For multiple OGTT, mice were i.p. injected with saline (control), lixisenatide (25 nmol·kg−1), or 33 (25 or 100 nmol·kg−1) 0.5 h prior to the first glucose load (1.5 g·kg−1), and glucose loads were repeated at 6 and 12 h. The blood glucose levels were detected at the times indicated. Results are presented as means ± SD, n = 6.
To better evaluate the long‐acting hypoglycaemic activity of 33, a multiple OGTT was performed. As shown in Figure 5C, the blood glucose levels in saline‐treated mice rapidly increased over 18 mmol·L−1 30 min after each oral glucose load. At a dose of 25 nmol·kg−1, lixisenatide and 33 showed similar glucose‐lowering effects in the first OGTT. The hypoglycaemic efficacy of lixisenatide was compromised in the second OGTT and dramatically reduced in the third OGTT. The glucose‐lowering activity of 33 was similar in the first and second OGTT and was moderately decreased in the third OGTT. When administered at a dose of 100 nmol·kg−1, the glucose‐stabilizing effect of 33 was stable and relatively unchanged during the whole experiment. Taken together, the OGTT tests revealed that 33 increased the glucometabolic ability of db/db mice in a dose‐dependent manner, and its long‐acting antidiabetic effect was superior to that of lixisenatide.
Chronic in vivo studies
To further evaluate the potential therapeutic utility of 33, db/db mice were treated chronically (for 5 weeks) with 33 or lixisenatide. Lixisenatide was administered three times a day (i.p. 25 nmol·kg−1), and 33 was given twice daily (i.p. 25 nmol·kg−1), based on their pharmacokinetic profiles. The HbA1c value in the saline group was significantly enhanced (P < 0.05) after 35 days. In contrast, treatment with 33 and lixisenatide reduced the glycaemic control marker HbA1c after 35 days, and 33 was more effective at lowering HbA1c than lixisenatide (P < 0.05, Figure 6A). As shown in Figure 6B, C, the body weight in the saline group continually increased over the 5 week treatment period. In contrast, body weight changed little in 33‐ and lixisenatide‐treated mice, and 33 was better at suppressing the weight increase than lixisenatide. Similarly, administration of 33 reduced the food intake during the whole treatment period, to a greater extent than lixisenatide. Moreover, the non‐fasting blood glucose levels and plasma insulin levels were monitored every 2 days. Both 33 and lixisenatide significantly reduced hyperglycaemia in db/db mice from day 2 to day 36 (Figure 6D), accompanied by significantly increased non‐fasting plasma insulin levels (Figure 6E). At the end of the treatment (day 37), an IPGTT test was performed to evaluate the chronic treatment effects of 33 and lixisenatide. As shown in Figure 6F, the glucose excursions in the 33‐ and lixisenatide‐treated groups were smaller than in the saline group. Thus, chronic 33 and lixisenatide treatment improved the glucose tolerance of db/db mice. The plasma insulin levels in the 33‐ and lixisenatide‐treated groups were markedly higher on day 37 than in the saline group, indicating improved beta cell function (Figure 6G).
Figure 6.
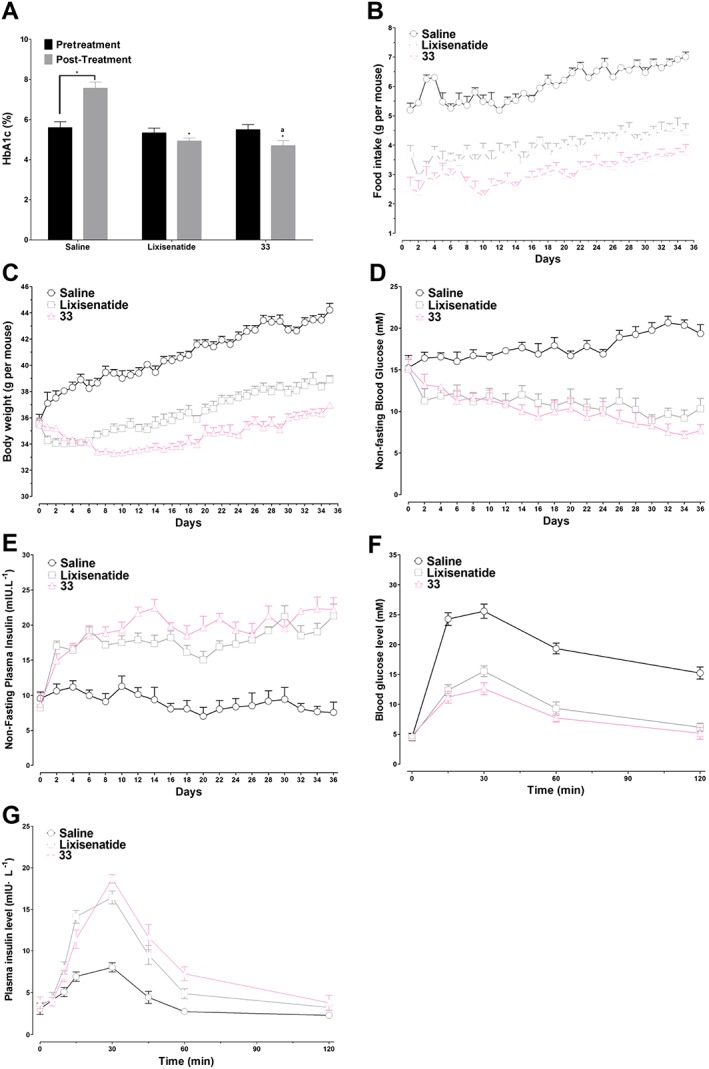
Effects of long‐term treatment, for 35 days, with 33 or lixisenatide on db/db mice. (A) HbA1c (%) values on day 0 compared with on day 36. * P < 0.05 versus control, a P < 0.05 versus lixisenatide. (B) Food intake. (C) Body weight change. (D) Non‐fasting blood glucose levels. (E) Non‐fasting plasma insulin levels. (F) The time course of blood glucose levels in each group in IPGTT test on day 37. (G) The time course of plasma insulin levels in each group in IPGTT test on day 37. Results are presented as means ± SD, n = 6.
Biochemical analysis revealed that TC and TG were significantly reduced in the 33‐ and lixisenatide‐treated groups after the 5 week treatment, as compared with the saline group (Figure 7A, B). Meanwhile, 33 and lixisenatide led to large decreases in LDL cholesterol levels (Figure 7D) but did not affect the HDL cholesterol (Figure 7C). Furthermore, the AST and ALT values in the 33‐treated mice were slightly lower than in the saline group and were comparable to those in the lixisenatide group (Figure 7E, F). These results indicate the safety profile of the chronic treatment with 33 in vivo. Finally, to examine whether long‐term 33 administration was accompanied by changes in the area and number of pancreatic islets, a histological examination was performed on pancreata from db/db mice at the end of the 5 week treatment period. As shown in Figure 7G, H, the islet area and number were significantly increased in mice treated with 33 as compared with saline.
Figure 7.
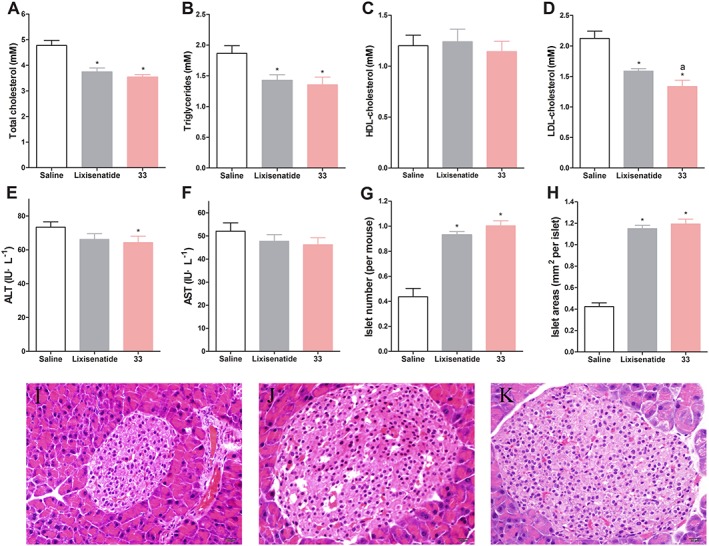
Biochemical analysis and histological examination of each group of db/db mice following the 5 week treatment. (A) Total cholesterol. (B) Triglyceride. (C) HDL cholesterol. (D) LDL cholesterol. (E) ALT. (F) AST. (G) Number of pancreatic islets. (H) Area of pancreatic islets. Representative images of HE staining (I) saline (control), (J) lixisenatide and (K) 33 histological samples. Results are presented as means ± SD, n = 6. * P < 0.05 versus control, a P < 0.05 versus lixisenatide.
Discussion
Unlike the GLP‐1 receptor agonists currently in clinical use or in clinical development, which are based on the amino acid sequence of hGLP‐1 or Gila GLP‐1 (Tomlinson et al., 2016), our previous research identified a new GLP‐1 derivative xGLP‐1B from Xenopus glucagon. xGLP‐1B was more potent as an activator of the GLP‐1 receptor than hGLP‐1, suggesting its potential role as a novel amphibian‐derived GLP‐1 analogue. The improved receptor activation potency of xGLP‐1B may be attributed to its unique amino acid sequence, which differs from hGLP‐1 at positions 2, 6, 8, 11, 12, 16, 17, 24, 27 and 30 (Table 1). Before performing further modifications on xGLP‐1B, its SAR was studied by alanine scanning. Because alanine is the smallest optically‐active amino acid with high α‐helix propensity, alanine scanning is commonly used for study the SAR of peptides (Kompella et al., 2015). We found that when Glu12, Leu14, Lys17 and Leu26 of xGLP‐1B were replaced by L‐alanine, the corresponding peptides 2, 4, 7 and 17 showed improved bioactivity, indicating that these positions were suitable for chemical modification.
The C‐terminal tail of lixisenatide contributes to its high GLP‐1 receptor affinity. We therefore hypothesized that the introduction of the C‐terminal tail of lixisenatide (PSSGA PPSKK KKKK) could improve the biological activities of xGLP‐1B. To this end, we synthesized xGLP‐1B‐Sen, which showed an eightfold enhancement in receptor activation potency and increased in vivo glucose‐lowering activity compared with xGLP‐1B (Table 2 and Figure 1). By substituting the Glu12, Leu14, Lys17 and Leu26 of xGLP‐1B‐Sen with cysteine, four analogues 21–24 were also synthesized, which exhibited potent receptor activation and hypoglycaemic activity similar to xGLP‐1B‐Sen (Table 2 and Figure 1), further confirming the conclusion from our SAR study.
Next, PEGylation of peptides 21–24 was carried out to further improve their pharmacokinetic behaviours (van Witteloostuijn et al., 2016b). mPEG‐MAL with different molecular weights (1, 2 and 5 kDa) were conjugated with 21–24 through the specific Machel addition between maleimide and cysteine, giving conjugates 25–36 (Table 3). The molecular weight of PEG obviously affected the bioactivity of the conjugates derived from 21, 22 and 24. Both the in vitro receptor activation potency and in vivo hypoglycaemic activity reduced as the PEG size increased. Interestingly, the PEG size had a minimal impact on the three conjugates derived from 23, as indicated by the similar bioactivity among 31–33. These results suggested that position 17 of xGLP‐1B‐Sen was the best site for derivatization. Based on our previous experience, peptide 33 with a PEG5000 chain was selected for further studies, since a longer PEG chain is more beneficial for achieving long‐acting effects.
In vivo hypoglycaemic and insulinotropic tests provided direct evidence that the insulinotropic activity of 33 was glucose‐dependent (Figure 2). Detailed pharmacokinetic analysis of 33 revealed that PEG conjugation not only extended its t 1/2 but also significantly increased drug utilization (Figure 3). The t 1/2 and AUC values of 33 were 2.5‐fold and 2.7‐fold that of lixisenatide respectively. Importantly, the Cmax of 33 was lower than that of lixisenatide. Since a high Cmax value probably leads to side effects like vomiting and nausea, as reported for lixisenatide and exendin‐4, 33 may avoid these adverse effects in humans. The longer in vivo t 1/2 of 33 was also reflected by the acute food intake test, in which 33 exhibited a better anorectic effect than lixisenatide at the same dose (Figure 4). The anorectic effect of 33 was dose‐dependent, similar to that of liraglutide. The duration of hypoglycaemic activity of 33 was further evaluated by pre‐OGTT and multiple OGTT in vivo. In accordance with the pharmacokinetic profiles, 33 exhibited better extended glucose‐lowering effects than lixisenatide in both pre‐OGTT and multiple OGTT (Figure 5). At a dose of 100 nmol·kg−1 in multiple OGTT, 33 sustained normal glycaemia for 18 h, which might be even longer in humans since the drug clearance is faster in rodents than in man (Kim et al., 2011).
Chronic treatment of 33 on db/db mice decreased the HbA1c value, body weight gain, food intake and blood glucose levels but increased the non‐fasting plasma insulin levels (Figure 6). Furthermore, 33 improved the glucose tolerance of db/db mice after the long‐term treatment. Blood biochemical analysis proved that the total cholesterol and triglycerides in mice treated with 33 were significantly reduced, but the ALT and AST values were unaffected (Figure 7). Thus, no toxicity was observed in mice treated with 33, indicating that it is safe for clinical use. Finally, histological examination revealed that the enhanced glucose tolerance of 33‐treated mice was presumably due to increased beta cell neogenesis and/or proliferation.
In conclusion, through structure optimization and chemical modification of xGLP‐1B, we discovered a novel GLP‐1 receptor agonist, peptide 33. Conjugate 33 was shown to possess improved pharmacokinetic profiles, prominent glucose‐lowering and insulinotropic potency, excellent long‐acting hypoglycaemic activity and marked long‐term treatment effects, making it a promising candidate for T2DM treatment.
Author contributions
J.H. and J.F. designed the research study and wrote the paper. Moreover, J.H., Y.F., F.Z., X.C. and J.F. performed the research and analysed the data. Y.Z. and L.L. contributed essential reagents and performed some of the research.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 The representative concentration response curves of hGLP‐1 and PEGylated conjugates. The data for the test peptides were normalized and plotted as the percentage of the response stimulated by saturating GLP‐1 (1000 nM).
Table S1 Time table of in vivo IPGTT and OGTT tests.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (nos 81602964 and 81602960), the Natural Science Foundation of Jiangsu Province (grant nos BK20150243 and BK20161028), PAPD of Jiangsu Higher Education Institutions, the Open Project of State Key Laboratory of Natural Medicines (SKLNMKF201711) and Startup Funding for Introduced Talents of Nanjing Medical University (KY109RC1602).
Han, J. , Fei, Y. , Zhou, F. , Chen, X. , Zhang, Y. , Liu, L. , and Fu, J. (2018) Xenopus‐derived glucagon‐like peptide‐1 and polyethylene‐glycosylated glucagon‐like peptide‐1 receptor agonists: long‐acting hypoglycaemic and insulinotropic activities with potential therapeutic utilities. British Journal of Pharmacology, 175: 544–557. doi: 10.1111/bph.14107.
Contributor Information
Jing Han, Email: hj1986424@jsnu.edu.cn.
Junjie Fu, Email: jfu@njmu.edu.cn.
References
- Adelhorst K, Hedegaard B, Knudsen LB, Kirk O (1994). Structure‐activity studies of glucagon‐like peptide‐1. J Biol Chem 269: 6275–6278. [PubMed] [Google Scholar]
- Agyemang C, Meeks K, Beune E, Owusu‐Dabo E, Mockenhaupt FP, Addo J et al (2016). Obesity and type 2 diabetes in sub‐Saharan Africans – is the burden in today's Africa similar to African migrants in Europe? The RODAM study. BMC Med 14: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The concise guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi E, Carrington PE, Ingallinella P, Finotto M, Santoprete A, Petrov A et al (2013). A PEGylated analog of the gut hormone oxyntomodulin with long‐lasting antihyperglycemic, insulinotropic and anorexigenic activity. Bioorgan Med Chem 21: 7064–7073. [DOI] [PubMed] [Google Scholar]
- Cui X, Meng Q, Chu Y, Gu X, Tang Y, Zhou F et al (2016). Glucagon‐like peptide‐1 loaded phospholipid micelles for the treatment of type 2 diabetes: improved pharmacokinetic behaviours and prolonged glucose‐lowering effects. RSC Adv 6: 94408–94416. [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander S, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers A, Haack T, Lorenz M, Bossart M, Elvert R, Henkel B et al (2017). Design of novel exendin‐based dual glucagon‐like peptide 1 (GLP‐1)/glucagon receptor agonists. J Med Chem 60: 4293–4303. [DOI] [PubMed] [Google Scholar]
- Fonseca VA, Alvarado‐Ruiz R, Raccah D, Boka G, Miossec P, Gerich JE et al (2012). Efficacy and safety of the once‐daily GLP‐1 receptor agonist lixisenatide in monotherapy. Diabetes Care 35: 1225–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager MV, Clydesdale L, Gellman SH, Sexton PM, Wootten D (2017). Characterization of signal bias at the GLP‐1 receptor induced by backbone modification of GLP‐1. Biochem Pharmacol 136: 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Huang X, Sun L, Li Z, Qian H, Huang W (2013a). Novel fatty chain‐modified glucagon‐like peptide‐1 conjugates with enhanced stability and prolonged in vivo activity. Biochem Pharmacol 86: 297–308. [DOI] [PubMed] [Google Scholar]
- Han J, Sun L, Chu Y, Li Z, Huang D, Zhu X et al (2013b). Design, synthesis, and biological activity of novel dicoumarol glucagon‐like peptide 1 conjugates. J Med Chem 56: 9955–9968. [DOI] [PubMed] [Google Scholar]
- Han J, Sun L, Huang X, Li Z, Zhang C, Qian H et al (2014). Novel coumarin modified GLP‐1 derivatives with enhanced plasma stability and prolonged in vivo glucose‐lowering ability. Br J Pharmacol 171: 5252–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Wang Y, Meng Q, Li G, Huang F, Wu S et al (2017). Design, synthesis and biological evaluation of PEGylated Xenopus glucagon‐like peptide‐1 derivatives as long‐acting hypoglycemic agents. Eur J Med Chem 132: 81–89. [DOI] [PubMed] [Google Scholar]
- Irwin DM, Satkunarajah M, Wen Y, Brubaker PL, Pederson RA, Wheeler MB (1997). The Xenopus proglucagon gene encodes novel GLP‐1‐like peptides with insulinotropic properties. P Natl Acad Sci USA 94: 7915–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BD, Flatt PR, Gault VA (2010). Effects of γ‐glutamyl linker on DPP‐IV resistance, duration of action and biological efficacy of acylated glucagon‐like peptide‐1. Biochem Pharmacol 80: 396–401. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Jiang HH, Lee S, Youn YS, Park CW, Byun Y et al (2011). Mono‐PEGylated dimeric exendin‐4 as high receptor binding and long‐acting conjugates for type 2 anti‐diabetes therapeutics. Bioconjug Chem 22: 625–632. [DOI] [PubMed] [Google Scholar]
- Kompella SN, Hung A, Clark RJ, Marí F, Adams DJ (2015). Alanine scan of α‐conotoxin RegIIA reveals a selective α3β4 nicotinic acetylcholine receptor antagonist. J Biol Chem 290: 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manandhar B, Ahn J‐M (2015). Glucagon‐like peptide‐1 (GLP‐1) analogs: recent advances, new possibilities, and therapeutic implications. J Med Chem 58: 1020–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli C, Natarajan SI, Meyer J‐P, Bastos MM, Bernatowicz MS, Lee VG et al (2009). Eleven amino acid glucagon‐like peptide‐1 receptor agonists with antidiabetic activity. J Med Chem 52: 7788–7799. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ (2012). GLP‐1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol 8: 728–742. [DOI] [PubMed] [Google Scholar]
- Mishra R, Chesi A, Cousminer DL, Hawa MI, Bradfield JP, Hodge KM et al (2017). Relative contribution of type 1 and type 2 diabetes loci to the genetic etiology of adult‐onset, non‐insulin‐requiring autoimmune diabetes. BMC Med 15: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murage EN, Gao G, Bisello A, Ahn J‐M (2010). Development of potent glucagon‐like peptide‐1 agonists with high enzyme stability via introduction of multiple lactam bridges. J Med Chem 53: 6412–6420. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R et al (2009). Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. Diabetes Care 32: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Huang X, Han J, Cai X, Dai Y, Chu Y et al (2016). Site‐specific fatty chain‐modified exenatide analogs with balanced glucoregulatory activity and prolonged in vivo activity. Biochem Pharmacol 110: 80–91. [DOI] [PubMed] [Google Scholar]
- Tomlinson B, Hu M, Zhang Y, Chan P, Liu Z‐M (2016). An overview of new GLP‐1 receptor agonists for type 2 diabetes. Expert Opin Inv Drug 25: 145–158. [DOI] [PubMed] [Google Scholar]
- van Witteloostuijn SB, Mannerstedt K, Wismann P, Bech EM, Thygesen MB, Vrang N et al (2016a). Neoglycolipids for prolonging the effects of peptides: self‐assembling glucagon‐like peptide 1 analogues with albumin binding properties and potent in vivo efficacy. Mol Pharm 14: 193–205. [DOI] [PubMed] [Google Scholar]
- van Witteloostuijn SB, Pedersen SL, Jensen KJ (2016b). Half‐life extension of biopharmaceuticals using chemical methods: alternatives to PEGylation. ChemMedChem 11: 2474–2495. [DOI] [PubMed] [Google Scholar]
- Yang B, Li X, Zhang C, Yan S, Wei W, Wang X et al (2015). Design, synthesis and biological evaluation of novel peptide MC2 analogues from Momordica charantia as potential anti‐diabetic agents. Org Biomol Chem 13: 4551–4561. [DOI] [PubMed] [Google Scholar]
- Yang X, Li Y, Wang Y, Zheng X, Kong W, Meng F et al (2014). Long‐acting GLP‐1 analogue in V‐shaped conformation by terminal polylysine modifications. Mol Pharm 11: 4092–4099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The representative concentration response curves of hGLP‐1 and PEGylated conjugates. The data for the test peptides were normalized and plotted as the percentage of the response stimulated by saturating GLP‐1 (1000 nM).
Table S1 Time table of in vivo IPGTT and OGTT tests.