

Abstract
Oxaliplatin‐based systemic chemotherapy has been proposed to have efficacy in hepatocellular carcinoma (HCC). We investigated the combination of vorinostat and oxaliplatin for possible synergism in HCC cells. SMMC7721, BEL7402, and HepG2 cells were treated with vorinostat and oxaliplatin. Cytotoxicity assay, tumorigenicity assay in vitro, cell cycle analysis, apoptosis analysis, western blot analysis, animal model study, immunohistochemistry, and quantitative PCR were performed. We found that vorinostat and oxaliplatin inhibited the proliferation of SMMC7721, BEL7402, and HepG2 cells. The combination index (CI) values were all <1, and the dose‐reduction index values were all greater than 1 in the three cell lines, indicating a synergistic effect of combination of the two agents. Coadministration of vorinostat and oxaliplatin induced G2/M phase arrest, triggered caspase‐dependent apoptosis, and decreased tumorigenicity both in vitro and in vivo. Vorinostat suppressed the expression of BRCA1 induced by oxaliplatin. In conclusion, cotreatment with vorinostat and oxaliplatin exhibited synergism in HCC cells. The combination inhibited cell proliferation and tumorigenicity both in vitro and in vivo through induction of cell cycle arrest and apoptosis. Our results predict that a combination of vorinostat and oxaliplatin may be useful in the treatment of advanced HCC.
Keywords: BRCA1, combination, hepatocellular carcinoma, oxaliplatin, vorinostat
Introduction
Hepatocellular carcinoma (HCC) remains a major health conundrum worldwide 1, 2. Although it is considered resistant to conventional chemotherapeutic agents 3, 4, 5, many reports have demonstrated that systemic chemotherapy containing oxaliplatin exerted significant anticancer effect on HCC 6, 7, 8, 9, 10, 11.
Oxaliplatin was first discovered in Japan in 1976 12, 13 and approved for clinical treatment in 1994 14. As a second‐generation platinum drug, it was initially used in metastatic colorectal cancer and afterward found to be effective in combination with other drugs for alimentary canal cancers 15. Its full chemical name is oxalato(trans‐L‐1,2‐diaminocyclohexane)platinum because of the presence of the 1,2‐diaminocyclohexane (DACH) carrier ligand and oxalate “leaving group” 16. The DACH carrier ligand renders it more effective than its analogs cisplatin and carboplatin. Its main mechanism of action is via the formation of DNA adducts 17. Once inside the body, DACH ligand is transformed into a DACH platinum compound that binds to the nitrogen atom (N7) of guanine, followed by formation of transient monoadducts and stable diadducts 15. In addition to DNA intrastrand crosslinks, oxaliplatin can induce DNA interstrand crosslinks and DNA‐protein crosslinks that cause DNA lesions, inhibition of DNA synthesis and repair, cell cycle arrest, and ultimately apoptosis 15, 18.
Epigenetic alterations occur in a variety of cancers including HCC. Among them, histone acetylation plays a crucial role in tumor initiation and development 19, 20, 21, and the use of histone deacetylase (HDAC) inhibitors has been considered as a novel approach for the treatment of numerous malignancies 22, 23, 24, 25, 26. A number of HDAC inhibitors are currently being used in experimental studies and clinical trials targeted to hematological malignancies and solid tumors. For solid tumors, HDAC inhibitors have to be combined with other chemotherapy agents to fully exert their antitumor activity 27.
The combination of HDAC inhibitors and oxaliplatin has been reported in gastric, pancreatic, and colorectal cancers 28, 29, 30, 31, 32, 33, but the effect on HCC remains unknown. The HDAC inhibitor vorinostat was first approved by the FDA for the therapy of refractory cutaneous T‐cell lymphoma 34, 35 and has been widely used in the treatment of many solid tumors.
In our study, we investigated the combination of vorinostat and oxaliplatin in HCC cell lines in vitro and in vivo and found that the combination of these two drugs had a synergistic effect on HCC.
Materials and Methods
Cell culture and agents
Human hepatocellular carcinoma cell lines SMMC7721 and BEL7402 and the human hepatoma cell line HepG2 were purchased from China Center for Type Culture Collection (CCTCC, Wuhan, China). All cell lines were cultured in high‐glucose Dulbecco's modified Eagle's medium (DMEM, Gibco, USA) containing 10% fetal bovine serum (FBS, Gibco, USA). Vorinostat, oxaliplatin, and DMSO were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Vorinostat and oxaliplatin were dissolved in DMSO.
Cell viability assay, CI analysis, and dose‐reduction index analysis
BEL7402 (2 × 103 cells per well), SMMC7721 (1.5 × 103 cells per well), and HepG2 (1 × 103 cells per well) were plated in 96‐well plates and treated with increasing concentrations of vorinostat or oxaliplatin for 48 h to generate a cell viability curve. Cell Counting Kit‐8 (CCK‐8, Dojindo, Japan) reagent was added to plates for 2 h and the optical density (OD) values were measured at 450 nm. The half maximal inhibitory concentration (IC50) was calculated by SPSS software. Next, the cells were treated with increasing concentrations of vorinostat and oxaliplatin in a fixed ratio of IC50 of the two agents for 48 h to form a new cell viability curve. Chou previously explained the CI model 36, 37. Based on his theory, the CI value in our study was calculated as: CI = a/A + a′/A′, where A and A′ are the concentrations of single drug required to inhibit x% of cells, and a and a′ are the respective drug concentrations of the combination that inhibits x% of cells. A CI < 1 indicates synergism of the combination. In his papers, Chou mentioned the dose‐reduction index (DRI) determined by the fold reduction in the dose of the agents compared with the dose of agent used in isolation at a fixed effect level 36, 37. DRI > 1 indicates a reduced dose and reduced toxicity after combination treatment. DRI for each corresponding drug was calculated as: DRI (A) = A/a or DRI (A′) = A′/a′.
Western blotting
Immunoblotting was performed as described previously 38. The primary antibodies against acetylated histone H3 and β‐actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and antibodies against cleaved‐caspase 9, cleaved‐caspase 7, PARP, and BRCA1 were purchased from Cell Signaling Technology (Beverly, MA, USA). All horseradish peroxidase (HRP) secondary antibodies were purchased from Jackson Immuno Research Laboratories (West Grove, PA, USA).
Colony formation assay and soft agar assay
Colony formation assay was carried out as described previously 39. HepG2 (500 cells/well) and SMMC7721 (300 cells/well) were seeded in six‐well plates and treated with vorinostat and/or oxaliplatin for 48 h. Media were refreshed every other day. The wells were stained with crystal violet (Sigma‐Aldrich, USA) and their images were acquired at day 14. The numbers of colonies were counted and analyzed by Alpha Innotech Imaging system (Alphatron Asia Pte Ltd, Singapore). Soft agar assay was performed as previously reported 40. HepG2 (5000 cells/well) and SMMC7721 (5000 cells/well) were plated in six‐well plates and treated with culture media containing vorinostat and/or oxaliplatin, which was replaced every 2 days. At day 14, the colonies were counted and analyzed as described above.
Cell cycle and apoptosis analysis
The flow cytometry analysis was carried out as described previously 41. For cell cycle analysis, HepG2 and BEL7402 cells were treated with vorinostat and/or oxaliplatin for 48 h. A total of 1 × 106 cells per sample were analyzed using FACSAria Cell Cytometer (BD Biosciences, San Jose, CA, USA). For apoptosis analysis, 1 × 105 cells per well were tested. All data were analyzed using CellQuest software (BD Biosciences).
Xenograft tumorigenicity assay
The animal studies were performed as previously described 39, 40. All procedures performed in animal studies were approved by the Committee on the Ethics of Animal Experiments of Zhongnan Hospital, Wuhan University. HepG2 cells were subcutaneously injected into the mice. Drug treatment started when the tumors reached 100 mm3 in size. Vorinostat (25 mg·kg−1) was injected intraperitoneally everyday, and oxaliplatin (5 mg·kg−1) was injected intraperitoneally twice a week. Subcutaneous tumor xenografts were removed and conserved for subsequent analysis.
Immunohistochemistry analysis
Immunohistochemistry was performed as previously described 39, 40. Ki‐67 primary antibody was obtained from Dako (Golstrup, Denmark). The paraffin‐embedded sections of the xenografts were detected using the TUNEL assay kit (R&D Systems, Minneapolis, MN, USA) for apoptosis analysis.
Real‐time quantitative PCR analysis
PCR was performed as described previously 42. The primer sequences for BRCA1 were as follows: sense 5′‐GGCTATCCTCTCAGAGTGACATTT‐3′, anti‐sense 5′‐GCTTTATCAGGTTATGTTGCATGG‐3′. Expression of β‐actin mRNA was used as an internal control for normalization. Results were calculated as fold induction relative to β‐actin.
Transient RNA interference
Small interfering RNA (siRNA) duplexes targeting human BRCA1 sequences and a scrambled siRNA were designed as described previously 43, 44. All siRNAs were synthesized by Ribobio (Guangzhou, China). Transfection of the siRNA duplexes was performed using jetPRIME (Polyplus‐transfection SA, Illkirch, France) according to the manufacturer's instructions.
Statistical analyses
Data analyses were carried out using GraphPad Prism 5.0 (La Jolla, CA, USA) or SPSS 13.0 (Chicago, IL, USA). All of the experiments were performed at least three independent times. The results were presented as mean ± SEM. Comparisons between the different groups were analyzed by one‐way ANOVA with P < 0.05 considered statistically significant.
Results
Vorinostat and oxaliplatin attenuate the growth of HCC cells
We first investigated the effect of vorinostat or oxaliplatin alone on cell growth in three HCC cell lines. HepG2, SMMC7721, and BEL7402 cells were cultured with different concentrations of vorinostat or oxaliplatin for 48 h. Both vorinostat and oxaliplatin inhibited proliferation of the three cell lines. The IC50 values for vorinostat and oxaliplatin are shown in Figure 1A and Table 1.
Figure 1.
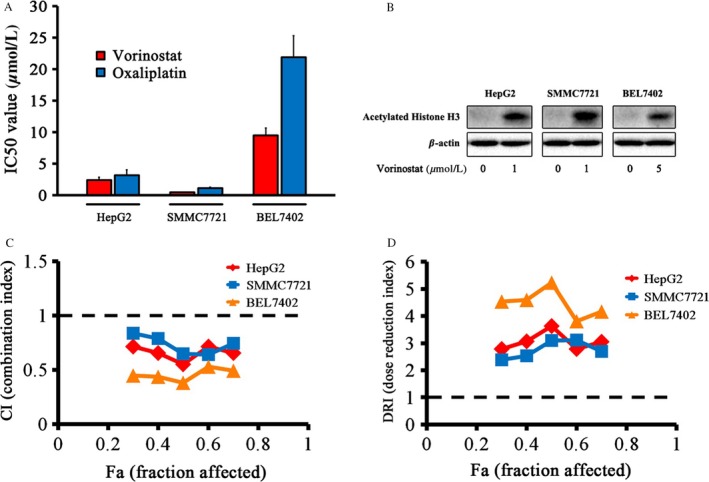
Vorinostat and oxaliplatin attenuated the proliferation of HCC cell lines. (A) Cytotoxicity assay. HepG2, SMMC7721, and BEL7402 cells in 96‐well plates were treated with different concentrations of vorinostat and oxaliplatin for 48 h and cell viability was detected by the Cell Counting Kit‐8 (CCK‐8) assay. The half maximal inhibitory concentration (IC50) was calculated by SPSS software. (B) HepG2 and SMMC7721 cells were treated with 1 μmol/L vorinostat and BEL7402 cells were treated with 5 μmol/L vorinostat. The expression of acetylated Histone H3 was detected by western blotting. (C) Combination index (CI) values of HepG2, SMMC7721, and BEL7402 cells at different fractions affected. (D) Dose‐reduction index (DRI) values of HepG2, SMMC7721, and BEL7402 cells at different fractions affected. All experiments were performed at least three independent times.
Table 1.
The IC50 values for vorinostat and oxaliplatin in HCC cell lines
| Cell line | IC50 values of drugs (μmol/L) | |
|---|---|---|
| Vorinostat | Oxaliplatin | |
| HepG2 | 2.39 ± 0.48 | 3.17 ± 0.87 |
| SMMC7721 | 0.47 ± 0.08 | 1.14 ± 0.14 |
| BEL7402 | 9.49 ± 1.18 | 21.92 ± 3.38 |
Vorinostat increases the acetylation of histone H3 in HCC cells
The effect of vorinostat on the acetylation of histone H3 was tested in HCC cells. The results demonstrated that vorinostat treatment for 48 h increased the acetylation level of histone H3 in HepG2, SMMC7721, and BEL7402 cell lines (Fig. 1B).
Vorinostat increases the antiproliferative efficacy of oxaliplatin in HCC cells
Combination treatment with vorinostat and oxaliplatin attenuated cell growth more than either drug used in isolation in the three cell lines. The CI values in HepG2, SMMC7721, and BEL7402 cells were all less than 1, which indicated a synergistic effect after combination treatment with the two drugs (Fig. 1C). The DRI values of the two drugs were greater than 1 in all three cell lines (Fig. 1D). The CI and DRI values for coadministration of vorinostat and oxaliplatin are listed in Table 2.
Table 2.
CI and DRI values after combination treatment with vorinostat and oxaliplatin
| Cell line | ||||
|---|---|---|---|---|
| HepG2 | SMMC7721 | BEL7402 | ||
| CI values at % fraction affected | 30% | 0.72 ± 0.03 | 0.84 ± 0.04 | 0.45 ± 0.08 |
| 40% | 0.66 ± 0.06 | 0.79 ± 0.02 | 0.44 ± 0.03 | |
| 50% | 0.55 ± 0.06 | 0.65 ± 0.05 | 0.38 ± 0.03 | |
| 60% | 0.72 ± 0.03 | 0.64 ± 0.04 | 0.53 ± 0.07 | |
| 70% | 0.67 ± 0.05 | 0.75 ± 0.05 | 0.49 ± 0.10 | |
| DRI values at % fraction affected | 30% | 2.79 ± 0.13 | 2.40 ± 0.14 | 4.54 ± 0.79 |
| 40% | 3.06 ± 0.29 | 2.53 ± 0.06 | 4.59 ± 0.32 | |
| 50% | 3.64 ± 0.38 | 3.10 ± 0.31 | 5.24 ± 0.43 | |
| 60% | 2.79 ± 0.10 | 3.12 ± 0.23 | 3.81 ± 0.49 | |
| 70% | 2.98 ± 0.21 | 2.69 ± 0.25 | 4.17 ± 0.81 | |
CI, combination index; DRI, dose‐reduction index.
Vorinostat and oxaliplatin decrease the tumorigenicity of HCC cells in vitro
To investigate the effect of vorinostat and oxaliplatin on the tumorigenicity of HCC cells, colony formation assays and soft agar assays were performed in HepG2 and SMMC7721 cell lines. Vorinostat or oxaliplatin reduced both the number and size of colonies. Moreover, combination with the two agents further attenuated the tumorigenicity compared with single‐drug treatment (Fig. 2A and B).
Figure 2.
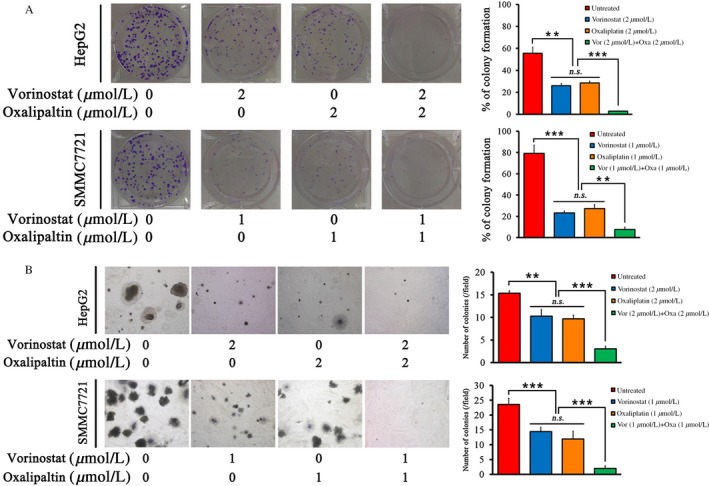
Combination of vorinostat and oxaliplatin inhibited tumorigenicity in vitro. (A) Colony formation assay. HepG2 cells (500 cells/well) were treated with 2 μmol/L vorinostat and/or 2 μmol/L oxaliplatin in six‐well plates for 48 h. SMMC7721 (300 cells/well) were treated with 1 μmol/L vorinostat and/or 1 μmol/L oxaliplatin in six‐well plates for 48 h. The wells were stained and images were acquired at day 14. The percentage of cells that formed colonies was calculated. (B) Soft agar assay. HepG2 cells (5000 cells/well) were treated with 2 μmol/L vorinostat and/or 2 μmol/L oxaliplatin in six‐well plates for 14 days. SMMC7721 (5000 cells/well) were treated with 1 μmol/L and/or 1 μmol/L oxaliplatin in six‐well plates for 14 days. The average number of colonies in a field was calculated. ***P < 0.001, **P < 0.01.
Vorinostat and oxaliplatin trigger cell cycle arrest and apoptosis in HCC cells
After vorinostat treatment, the number of cells in G0/G1 and G2/M phases was increased with an accompanying reduction in the number of cells in S phase compared with the control group. After oxaliplatin treatment, the number of cells in S and G2/M phases increased with a concomitant decrease in the numbers of cells in G0/G1 phase. Cotreatment with the two agents induced a greater proportion of cells in G2/M phase compared with either drug used in isolation (Fig. 3A). Both vorinostat and oxaliplatin induced apoptosis in HepG2 and BEL7402 cells and coadministration significantly augmented the apoptotic rate compared with either vorinostat or oxaliplatin used alone (Fig. 3B). Western blot analysis showed that the expression of apoptotic proteins including cleaved‐PARP, cleaved‐caspase 7, and cleaved‐caspase 9 was increased after combined treatment with the two agents (Fig. 3C).
Figure 3.
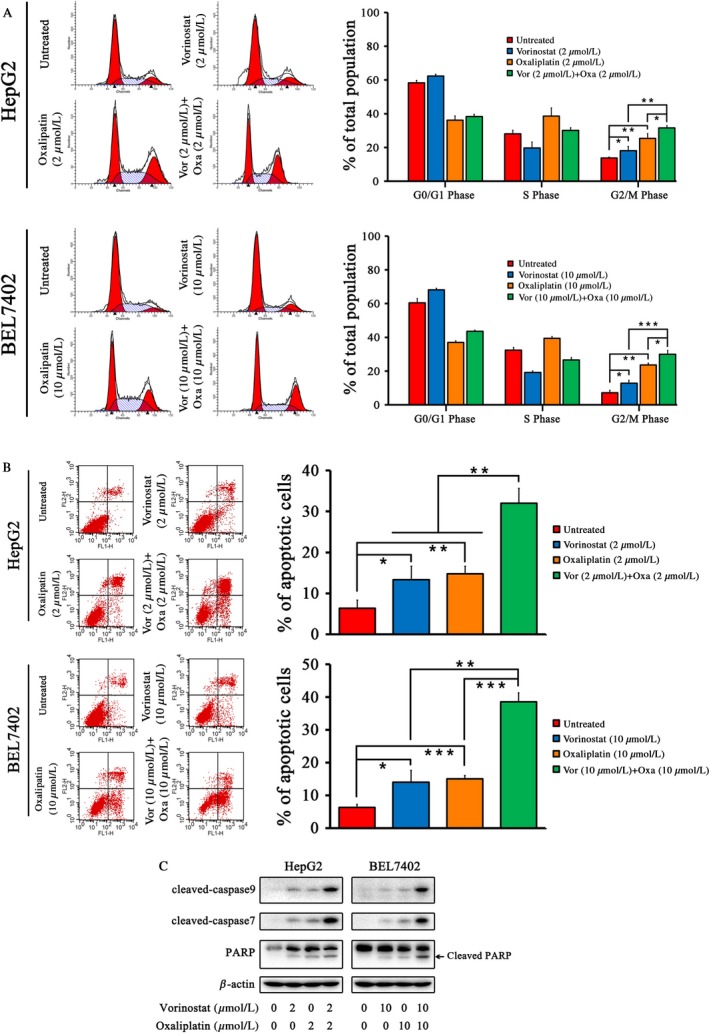
Combination of vorinostat and oxaliplatin induced cell cycle arrest and caspase‐dependent apoptosis in HCC cells. (A) Cell cycle analysis after 48‐h treatment with vorinostat and/or oxaliplatin in HepG2 and BEL7402 cells and the proportion of cells in each cell cycle phase. (B) Apoptosis analysis after 48‐h treatment with vorinostat and/or oxaliplatin and the apoptotic rate of cells in HepG2 and BEL7402 cells. (C) Western blot analysis of the expression of cleaved‐caspase 9, cleaved‐caspase 7, and PARP including cleaved‐PARP after 48‐h drug treatment. HepG2 cells were treated with 2 μmol/L vorinostat and/or 2 μmol/L oxaliplatin and BEL7402 cells were treated with 10 μmol/L vorinostat and/or 10 μmol/L oxaliplatin. ***P < 0.001, **P < 0.01, *P < 0.05.
Vorinostat enhances the anticancer effect of oxaliplatin in an animal model
We next investigated the effect of vorinostat and oxaliplatin on HCC cell growth in vivo. In this study, animal body weight was determined as an indicator of toxicity. The results of body weight curve showed no significant loss in body weight in all experimental groups (Fig. 4A). Drug treatment started when the tumor size grew to approximately 100 mm3 at day 9. Mice were sacrificed at day 27. Neither vorinostat (25 mg·kg−1 everyday) nor oxaliplatin (5 mg·kg−1 twice a week) alone significantly decreased HCC cell proliferation compared with the control group. However, the combination of the two drugs induced a significant decrease in HCC tumor weight and size (Fig. 4B–D). The proportion of Ki‐67‐positive cells, representing the proliferation index, was significantly reduced and the proportion of TUNEL‐positive cells, representing the apoptotic index, was significantly elevated after cotreatment with vorinostat and oxaliplatin (Fig. 4E–G).
Figure 4.
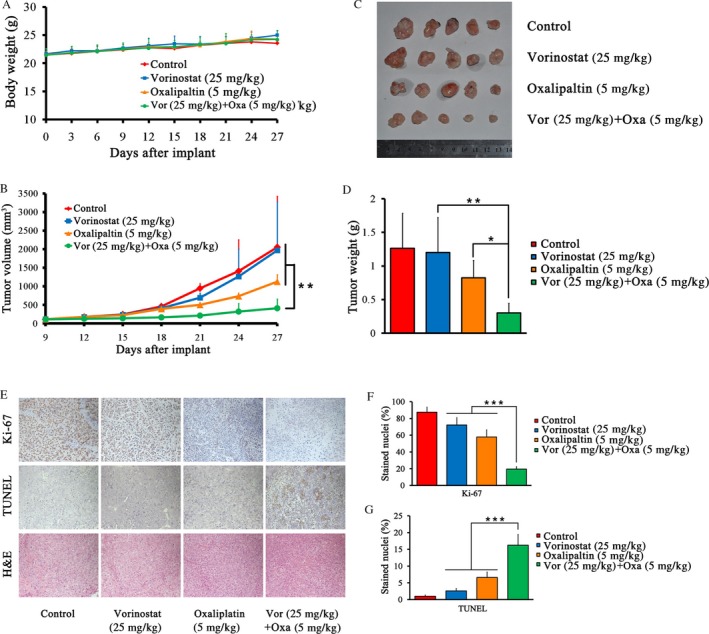
Combination of vorinostat and oxaliplatin inhibited tumorigenicity in animal model. (A) Body weight curve for different groups. (B) Tumor growth curve during the animal model study. At day 9 after injection of cancer cells, drug treatment was initiated and the tumor volume was calculated every third day. Vorinostat (25 mg·kg−1) was injected intraperitoneally everyday and oxaliplatin (5 mg·kg−1) was injected intraperitoneally twice a week. (C) Photographs of HCC xenografts of all groups on the day of sacrifice. (D) Tumor weights of each group. (E) Representative images of Ki‐67, TUNEL, and hematoxylin and eosin (H&E) staining for growth and apoptosis analysis (200×). (F, G) Proliferation index as determined by the proportion of Ki‐67‐stained nuclei and the apoptotic index as determined by the proportion of TUNEL‐stained nuclei in different groups. ***P < 0.001, **P < 0.01, *P < 0.05.
Vorinostat suppresses the expression of BRCA1 in HCC cells
We examined the effect of 48‐h treatment with vorinostat and oxaliplatin on the expression of BRCA1 in HCC cells in vitro. Western blotting showed that vorinostat downregulated the protein expression of BRCA1 in a dose‐dependent manner (Fig. 5A). The mRNA level of BRCA1 was also decreased by vorinostat using quantitative PCR (qPCR) (Fig. 5B). Oxaliplatin increased the protein and mRNA expression of BRCA1, as determined by western blotting and qPCR (Fig. 5C and D). This effect was partially reversed by the addition of vorinostat (Fig. 5C and D). BRCA1 expression was specifically inhibited by siRNA, as verified by western blotting (Fig. 5E). The relative cell viability of HCC cells was much lower after BRCA1 inhibition and exposure to oxaliplatin than after treatment with oxaliplatin alone (Fig. 5F).
Figure 5.
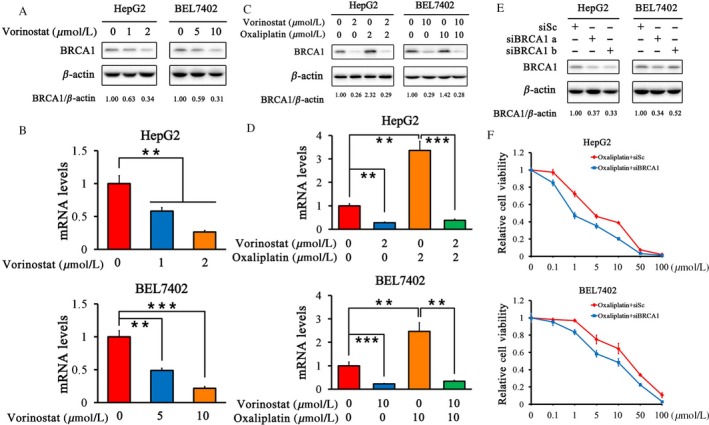
Vorinostat suppressed the expression of BRCA1 induced by oxaliplatin in HCC cells. (A and B) Protein and mRNA expression of BRCA1 by western blotting and qPCR after treatment with vorinostat. HepG2 cells were treated with vorinostat (1 μmol/L and 2 μmol/L). BEL7402 cells were treated with vorinostat (5 μmol/L and 10 μmol/L). (C and D) Protein and mRNA expression of BRCA1 by western blotting and qPCR after treatment with vorinostat and/or oxaliplatin. HepG2 cells were treated with 2 μmol/L vorinostat and/or 2 μmol/L oxaliplatin. BEL7402 cells were treated with 10 μmol/L vorinostat and/or 10 μmol/L oxaliplatin. (E) Inhibition of BRCA1 expression by siRNA was confirmed by western blotting. (F) Relative viability curves of HCC cells after treatment with oxaliplatin alone or with BRCA1 inhibition. Quantification of each band by densitometry was performed and the BRCA1/β‐actin ratio was indicated. ***P < 0.001, **P < 0.01.
Discussion
Our study evaluated the combined effect of vorinostat and oxaliplatin on HCC. Both vorinostat and oxaliplatin alone attenuated the proliferation of HepG2, SMMC7721, and BEL7402 cells. When the two agents were combined, the CI values were less than 1 in all three cell lines, indicating a synergistic effect of the two agents, and all DRI values were greater than 1, demonstrating reduced dose and toxicity for the combination. Cotreatment with vorinostat and oxaliplatin induced G2/M phase arrest as shown by flow cytometry. Vorinostat or oxaliplatin used in isolation triggered apoptosis as evidenced by flow cytometry and western blotting and coadministration had a synergistic effect. The combination of vorinostat and oxaliplatin significantly inhibited HCC tumorigenicity in vitro and in vivo.
Sorafenib is a noncurative therapy that improves the survival of patients with advanced HCC 45, 46, 47. However, it is not widely used in Asia because of the vast expense and potentially increased adverse events in the Asian population 48, 49, 50. Therefore it is necessary to identify novel agents for testing in clinical trials.
Histone acetylation is a key factor in the regulation of chromatin accessibility as determined by the nuclear distribution of microinjected fluorescein‐labeled dextrans assessed by measuring the fluorescein‐dextran sizes combined with image correlated spectroscopy 19, 51. After treatment with HDAC inhibitors, acetylation of the core histones is increased, the N‐terminal tails of histones are removed, the interplay between histone and chromatin is loosened, and the DNA configuration becomes open and much more accessible to transcriptional factors and DNA‐targeting drugs 52, 53, 54.
Chemoresistance to oxaliplatin is a very challenging problem to circumvent. DNA damage repair is the most important mechanism underlying tumor resistance to oxaliplatin and occurs through four basic mechanisms, including DNA double‐strand break (DSB) repair. Homologous recombination (HR) repair is the most important pathway of DSB repair 55, 56 and breast cancer susceptibility gene 1 (BRCA1) is a key member of the HR pathway 57, 58. BRCA1 was identified in 1990 59. It is frequently mutated in ovarian and breast carcinomas and is involved in multiple cellular functions such as DNA repair, transcription, and recombination 60, 61. BRCA1 deficiency is associated with increased sensitivity to oxaliplatin 62. In this study, we found that BRCA1 expression correlated with resistance to oxaliplatin (Fig. 6). The expression of BRCA1 was elevated after oxaliplatin treatment and BRCA1 inhibition increased chemosensitivity to oxaliplatin.
Figure 6.
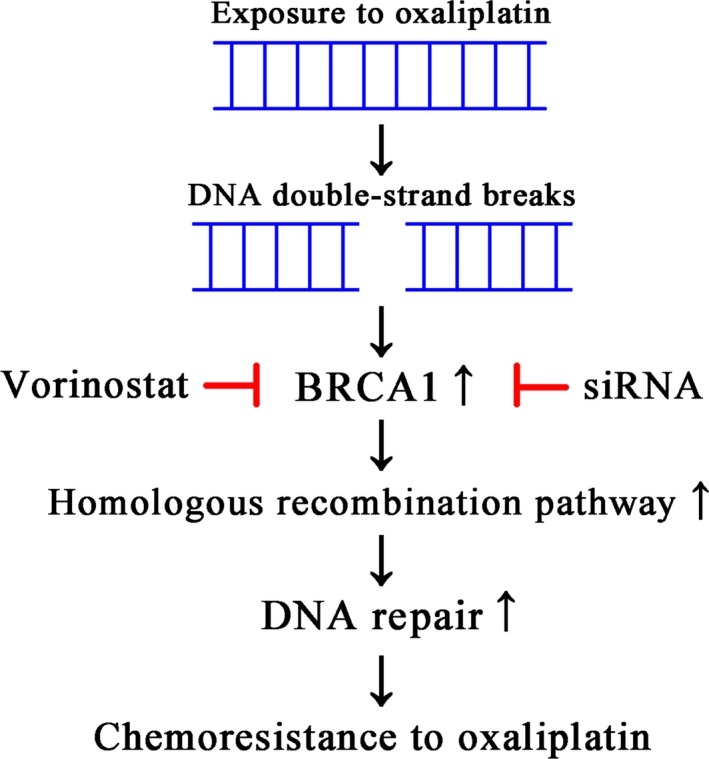
Schematic representation of the mechanism underlying chemoresistance to oxaliplatin.
HDAC inhibitors can attenuate DNA damage repair and suppress the expression of BRCA1 in many tumors including cervical, head and neck cancer, prostate, ovarian, and breast cancers 63, 64, 65, 66, 67, 68. In this study, we showed that BRCA1 expression was upregulated by oxaliplatin exposure alone, whereas vorinostat downregulated BRCA1 expression in the presence or absence of oxaliplatin in HCC cells (Fig. 6).
Vorinostat, a broad‐spectrum HDAC inhibitor, can inhibit many histone deacetylases including HDAC1 and HDAC2. HDAC1 and HDAC2 have been proven to cooperate in regulating BRCA1 transcript and protein expression in acute myeloid leukemia (AML) cells 69. It has been reported that BRCA1 associates with some components of the histone deacetylase complex and that the carboxylterminal domain (BRCT) of BRCA1 interacts with the histone deacetylases HDAC1 and HDAC2 61. We presume that vorinostat suppresses HDAC1 and HDAC2, which interact with and inhibit BRCA1. This might be one of the mechanisms underlying the regulation of BRCA1 expression by vorinostat, and requires further research.
Taken together, the combination of vorinostat and oxaliplatin has a synergistic effect in HCC cells. Their coadministration attenuates the growth of cells, induces G2/M phase arrest, causes apoptosis, and decreases tumorigenicity both in vitro and in vivo. Vorinostat enhances the anticancer effect of oxaliplatin on HCC cells by suppressing the expression of BRCA1 that is induced by oxaliplatin. Vorinostat is a potent chemosensitizer of oxaliplatin, which is a DNA‐targeting agent. Cotreatment with vorinostat and oxaliplatin may be a promising novel strategy against advanced HCC.
Conflict of Interest
All the authors declared no competing interests.
Acknowledgments
This work was supported by the Fundamental Research Funds for the Central Universities of Ministry of Education of China (Grant No. 2042017kf0094).
Cancer Medicine 2018; 7(1):196–207
References
- 1. Torre, L. A. , Bray F., Siegel R. L., Ferlay J., Lortet‐Tieulent J., and Jemal A.. 2015. Global cancer statistics, 2012. CA Cancer J. Clin. 65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. Chen, W. , Zheng R., Baade P. D., Zhang S., Zeng H., Bray F., et al. 2016. Cancer statistics in China, 2015. CA Cancer J. Clin. 66:115–132. [DOI] [PubMed] [Google Scholar]
- 3. Forner, A. , Llovet J. M., and Bruix J.. 2012. Hepatocellular carcinoma. Lancet 379:1245–1255. [DOI] [PubMed] [Google Scholar]
- 4. Bruix, J. , and Sherman M.. 2011. Management of hepatocellular carcinoma: an update. Hepatology 53:1020–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yau, T. , Chan P., Epstein R., and Poon R. T.. 2009. Management of advanced hepatocellular carcinoma in the era of targeted therapy. Liver Int. 29:10–17. [DOI] [PubMed] [Google Scholar]
- 6. Qin, S. , Bai Y., Lim H. Y., Thongprasert S., Chao Y., Fan J., et al. 2013. Randomized, multicenter, open‐label study of oxaliplatin plus fluorouracil/leucovorin versus doxorubicin as palliative chemotherapy in patients with advanced hepatocellular carcinoma from Asia. J. Clin. Oncol. 31:3501–3508. [DOI] [PubMed] [Google Scholar]
- 7. Qin, S. , Cheng Y., Liang J., Shen L., Bai Y., Li J., et al. 2014. Efficacy and safety of the FOLFOX4 regimen versus doxorubicin in Chinese patients with advanced hepatocellular carcinoma: a subgroup analysis of the EACH study. Oncologist 19:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun, W. , Sohal D., Haller D. G., Mykulowycz K., Rosen M., Soulen M. C., et al. 2011. Phase 2 trial of bevacizumab, capecitabine, and oxaliplatin in treatment of advanced hepatocellular carcinoma. Cancer 117:3187–3192. [DOI] [PubMed] [Google Scholar]
- 9. Zaanan, A. , Williet N., Hebbar M., Dabakuyo T. S., Fartoux L., Mansourbakht T., et al. 2013. Gemcitabine plus oxaliplatin in advanced hepatocellular carcinoma: a large multicenter AGEO study. J. Hepatol. 58:81–88. [DOI] [PubMed] [Google Scholar]
- 10. Petrelli, F. , Coinu A., Borgonovo K., Cabiddu M., Ghilardi M., Lonati V., et al. 2014. Oxaliplatin‐based chemotherapy: a new option in advanced hepatocellular carcinoma. a systematic review and pooled analysis. Clin. Oncol. (R. Coll. Radiol.) 26:488–496. [DOI] [PubMed] [Google Scholar]
- 11. Zhu, A. X. 2006. Systemic therapy of advanced hepatocellular carcinoma: how hopeful should we be? Oncologist 11:790–800. [DOI] [PubMed] [Google Scholar]
- 12. Kidani, Y. , Noji M., and Tashiro T.. 1980. Antitumor activity of platinum(II) complexes of 1,2‐diamino‐cyclohexane isomers. Gan 71:637–643. [PubMed] [Google Scholar]
- 13. Gustavsson, B. , Carlsson G., Machover D., Petrelli N., Roth A., Schmoll H. J., et al. 2015. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin. Colorectal Cancer 14:1–10. [DOI] [PubMed] [Google Scholar]
- 14. Monneret, C. 2011. Platinum anticancer drugs. From serendipity to rational design. Ann. Pharm. Fr. 69:286–295. [DOI] [PubMed] [Google Scholar]
- 15. Alcindor, T. , and Beauger N.. 2011. Oxaliplatin: a review in the era of molecularly targeted therapy. Curr. Oncol. 18:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piccart, M. J. , Lamb H., and Vermorken J. B.. 2001. Current and future potential roles of the platinum drugs in the treatment of ovarian cancer. Ann. Oncol. 12:1195–1203. [DOI] [PubMed] [Google Scholar]
- 17. Todd, R. C. , and Lippard S. J.. 2009. Inhibition of transcription by platinum antitumor compounds. Metallomics 1:280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carrato, A. , Gallego J., and Diaz‐Rubio E.. 2002. Oxaliplatin: results in colorectal carcinoma. Crit. Rev. Oncol. Hematol. 44:29–44. [DOI] [PubMed] [Google Scholar]
- 19. Ozaki, K. , Kishikawa F., Tanaka M., Sakamoto T., Tanimura S., and Kohno M.. 2008. Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross‐resistance to a wide range of DNA‐damaging drugs. Cancer Sci. 99:376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Esteller, M. 2007. Cancer epigenomics: DNA methylomes and histone‐modification maps. Nat. Rev. Genet. 8:286–298. [DOI] [PubMed] [Google Scholar]
- 21. Jones, P. A. , and Baylin S. B.. 2007. The epigenomics of cancer. Cell 128:683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anestopoulos, I. , Voulgaridou G. P., Georgakilas A. G., Franco R., Pappa A., and Panayiotidis M. I.. 2015. Epigenetic therapy as a novel approach in hepatocellular carcinoma. Pharmacol. Ther. 145:103–119. [DOI] [PubMed] [Google Scholar]
- 23. Bose, P. , Dai Y., and Grant S.. 2014. Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol. Ther. 143:323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lakshmaiah, K. C. , Jacob L. A., Aparna S., Lokanatha D., and Saldanha S. C.. 2014. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer. Res. Ther. 10:469–478. [DOI] [PubMed] [Google Scholar]
- 25. Lane, A. A. , and Chabner B. A.. 2009. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 27:5459–5468. [DOI] [PubMed] [Google Scholar]
- 26. Lee, M. J. , Kim Y. S., Kummar S., Giaccone G., and Trepel J. B.. 2008. Histone deacetylase inhibitors in cancer therapy. Curr. Opin. Oncol. 20:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bots, M. , and Johnstone R. W.. 2009. Rational combinations using HDAC inhibitors. Clin. Cancer Res. 15:3970–3977. [DOI] [PubMed] [Google Scholar]
- 28. Zhou, C. , Ji J., Shi M., Yang L., Yu Y., Liu B., et al. 2014. Suberoylanilide hydroxamic acid enhances the antitumor activity of oxaliplatin by reversing the oxaliplatininduced Src activation in gastric cancer cells. Mol. Med. Rep. 10:2729–2735. [DOI] [PubMed] [Google Scholar]
- 29. Piacentini, P. , Donadelli M., Costanzo C., Moore P. S., Palmieri M., and Scarpa A.. 2006. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Arch. 448:797–804. [DOI] [PubMed] [Google Scholar]
- 30. Kim, J. C. , Kim D. D., Lee Y. M., Kim T. W., Cho D. H., Kim M. B., et al. 2009. Evaluation of novel histone deacetylase inhibitors as therapeutic agents for colorectal adenocarcinomas compared to established regimens with the histoculture drug response assay. Int. J. Colorectal Dis. 24:209–218. [DOI] [PubMed] [Google Scholar]
- 31. Flis, S. , Gnyszka A., and Splawinski J.. 2009. HDAC inhibitors, MS275 and SBHA, enhances cytotoxicity induced by oxaliplatin in the colorectal cancer cell lines. Biochem. Biophys. Res. Commun. 387:336–341. [DOI] [PubMed] [Google Scholar]
- 32. Fakih, M. G. , Pendyala L., Fetterly G., Toth K., Zwiebel J. A., Espinoza‐Delgado I., et al. 2009. A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5‐fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin. Cancer Res. 15:3189–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Na, Y. S. , Kim S. M., Jung K. A., Yang S. J., Hong Y. S., Ryu M. H., et al. 2010. Effects of the HDAC inhibitor CG2 in combination with irinotecan, 5‐fluorouracil, or oxaliplatin on HCT116 colon cancer cells and xenografts. Oncol. Rep. 24:1509–1514. [DOI] [PubMed] [Google Scholar]
- 34. Duvic, M. , Talpur R., Ni X., Zhang C., Hazarika P., Kelly C., et al. 2007. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T‐cell lymphoma (CTCL). Blood 109:31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olsen, E. A. , Kim Y. H., Kuzel T. M., Pacheco T. R., Foss F. M., Parker S., et al. 2007. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T‐cell lymphoma. J. Clin. Oncol. 25:3109–3115. [DOI] [PubMed] [Google Scholar]
- 36. Chou, T. C. 2006. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58:621–681. [DOI] [PubMed] [Google Scholar]
- 37. Chou, T. C. 2010. Drug combination studies and their synergy quantification using the Chou‐Talalay method. Cancer Res. 70:440–446. [DOI] [PubMed] [Google Scholar]
- 38. Chen, L. , Zhang W., Zhou Q. D., Yang H. Q., Liang H. F., Zhang B. X., et al. 2012. HSCs play a distinct role in different phases of oval cell‐mediated liver regeneration. Cell Biochem. Funct. 30:588–596. [DOI] [PubMed] [Google Scholar]
- 39. Ding, Z. Y. , Jin G. N., Wang W., Chen W. X., Wu Y. H., Ai X., et al. 2014. Reduced expression of transcriptional intermediary factor 1 gamma promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology 60:1620–1636. [DOI] [PubMed] [Google Scholar]
- 40. Zhang, B. , Halder S. K., Kashikar N. D., Cho Y. J., Datta A., Gorden D. L., et al. 2010. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 138:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei, S. , Xiong M., Zhan D. Q., Liang B. Y., Wang Y. Y., Gutmann D. H., et al. 2012. Ku80 functions as a tumor suppressor in hepatocellular carcinoma by inducing S‐phase arrest through a p53‐dependent pathway. Carcinogenesis 33:538–547. [DOI] [PubMed] [Google Scholar]
- 42. Chen, L. , Zhang W., Liang H. F., Zhou Q. F., Ding Z. Y., Yang H. Q., et al. 2014. Activin A induces growth arrest through a SMAD‐ dependent pathway in hepatic progenitor cells. Cell Commun. Signal. 12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang, S. , Liu F., Zhu J., Chen P., Liu H., Liu Q., et al. 2016. DNA repair genes ERCC1 and BRCA1 expression in non‐small cell lung cancer chemotherapy drug resistance. Med. Sci. Monit. 22:1999–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu, X. , Lv Y. G., Yan C. Y., Yi J., and Ling R.. 2016. Enforced expression of hsa‐miR‐125a‐3p in breast cancer cells potentiates docetaxel sensitivity via modulation of BRCA1 signaling. Biochem. Biophys. Res. Commun. 479:893–900. [DOI] [PubMed] [Google Scholar]
- 45. Llovet, J. M. , Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J. F., et al. 2008. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359:378–390. [DOI] [PubMed] [Google Scholar]
- 46. Cheng, A. L. , Kang Y. K., Chen Z., Tsao C. J., Qin S., Kim J. S., et al. 2009. Efficacy and safety of Sorafenib in patients in the Asia‐Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double‐blind, placebo‐controlled trial. Lancet Oncol. 10:25–34. [DOI] [PubMed] [Google Scholar]
- 47. Cheng, A. L. , Guan Z., Chen Z., Tsao C. J., Qin S., Kim J. S., et al. 2012. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: subset analyses of the phase III Sorafenib Asia‐Pacific trial. Eur. J. Cancer 48:1452–1465. [DOI] [PubMed] [Google Scholar]
- 48. Chen, P. J. , Furuse J., Han K. H., Hsu C., Lim H. Y., Moon H., et al. 2010. Issues and controversies of hepatocellular carcinoma‐targeted therapy clinical trials in Asia: experts' opinion. Liver Int. 30:1427–1438. [DOI] [PubMed] [Google Scholar]
- 49. Poon, D. , Anderson B. O., Chen L. T., Tanaka K., Lau W. Y., Van Cutsem E., et al. 2009. Management of hepatocellular carcinoma in Asia: consensus statement from the Asian Oncology Summit 2009. Lancet Oncol. 10:1111–1118. [DOI] [PubMed] [Google Scholar]
- 50. Lee, H. C. 2008. Systemic chemotherapy of hepatocellular carcinoma–Korean experience. Oncology 75(Suppl 1):114–118. [DOI] [PubMed] [Google Scholar]
- 51. Gorisch, S. M. , Wachsmuth M., Toth K. F., Lichter P., and Rippe K.. 2005. Histone acetylation increases chromatin accessibility. J. Cell Sci. 118:5825–5834. [DOI] [PubMed] [Google Scholar]
- 52. Lee, D. Y. , Hayes J. J., Pruss D., and Wolffe A. P.. 1993. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72:73–84. [DOI] [PubMed] [Google Scholar]
- 53. Kim, M. S. , Blake M., Baek J. H., Kohlhagen G., Pommier Y., and Carrier F.. 2003. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 63:7291–7300. [PubMed] [Google Scholar]
- 54. Diyabalanage, H. V. , Granda M. L., and Hooker J. M.. 2013. Combination therapy: histone deacetylase inhibitors and platinum‐based chemotherapeutics for cancer. Cancer Lett. 329:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thompson, L. H. , and Schild D.. 2001. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 477:131–153. [DOI] [PubMed] [Google Scholar]
- 56. Khanna, K. K. , and Jackson S. P.. 2001. DNA double‐strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27:247–254. [DOI] [PubMed] [Google Scholar]
- 57. Wu, J. , Lu L. Y., and Yu X.. 2010. The role of BRCA1 in DNA damage response. Protein Cell. 1:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dever, S. M. , White E. R., Hartman M. C., and Valerie K.. 2012. BRCA1‐directed, enhanced and aberrant homologous recombination: mechanism and potential treatment strategies. Cell Cycle 11:687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hall, J. M. , Lee M. K., Newman B., Morrow J. E., Anderson L. A., Huey B., et al. 1990. Linkage of early‐onset familial breast cancer to chromosome 17q21. Science 250:1684–1689. [DOI] [PubMed] [Google Scholar]
- 60. Powell, S. N. , and Kachnic L. A.. 2003. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 22:5784–5791. [DOI] [PubMed] [Google Scholar]
- 61. Yarden, R. I. , and Brody L. C.. 1999. BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl Acad. Sci. USA 96:4983–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fedier, A. , Steiner R. A., Schwarz V. A., Lenherr L., Haller U., and Fink D.. 2003. The effect of loss of Brca1 on the sensitivity to anticancer agents in p53‐deficient cells. Int. J. Oncol. 22:1169–1173. [PubMed] [Google Scholar]
- 63. Fukuda, T. , Wu W., Okada M., Maeda I., Kojima Y., Hayami R., et al. 2015. Class I histone deacetylase inhibitors inhibit the retention of BRCA1 and 53BP1 at the site of DNA damage. Cancer Sci. 106:1050–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang, Y. , Carr T., Dimtchev A., Zaer N., Dritschilo A., and Jung M.. 2007. Attenuated DNA damage repair by trichostatin A through BRCA1 suppression. Radiat. Res. 168:115–124. [DOI] [PubMed] [Google Scholar]
- 65. Kachhap, S. K. , Rosmus N., Collis S. J., Kortenhorst M. S., Wissing M. D., Hedayati M., et al. 2010. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS ONE 5:e11208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chao, O. S. , Goodman O. B. Jr. 2014. Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol. Cancer Res. 12:1755–1766. [DOI] [PubMed] [Google Scholar]
- 67. Konstantinopoulos, P. A. , Wilson A. J., Saskowski J., Wass E., and Khabele D.. 2014. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol. Oncol. 133:599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Weberpals, J. I. , O'Brien A. M., Niknejad N., Garbuio K. D., Clark‐Knowles K. V., and Dimitroulakos J.. 2011. The effect of the histone deacetylase inhibitor M344 on BRCA1 expression in breast and ovarian cancer cells. Cancer Cell Int. 11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhao, J. , Xie C., Edwards H., Wang G., Taub J. W., and Ge Y.. 2017. Histone deacetylases 1 and 2 cooperate in regulating BRCA1, CHK1, and RAD51 expression in acute myeloid leukemia cells. Oncotarget 8:6319–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]