

Abstract
Background and Purpose
The gut–liver axis is associated with the progression of non‐alcoholic fatty liver disease (NAFLD). Targeting the gut–liver axis and bile acid‐based pharmaceuticals are potential therapies for NAFLD. The effect of tauroursodeoxycholic acid (TUDCA), a candidate drug for NAFLD, on intestinal barrier function, intestinal inflammation, gut lipid transport and microbiota composition was analysed in a murine model of NAFLD.
Experimental Approach
The NAFLD mouse model was established by feeding mice a high‐fat diet (HFD) for 16 weeks. TUDCA was administered p.o. during the last 4 weeks. The expression levels of intestinal tight junction genes, lipid metabolic and inflammatory genes were determined by quantitative PCR. Tissue inflammation was evaluated by haematoxylin and eosin staining. The gut microbiota was analysed by 16S rRNA gene sequencing.
Key Results
TUDCA administration attenuated HFD‐induced hepatic steatosis, inflammatory responses, obesity and insulin resistance in mice. Moreover, TUDCA attenuated gut inflammatory responses as manifested by decreased intestinal histopathology scores and inflammatory cytokine levels. In addition, TUDCA improved intestinal barrier function by increasing levels of tight junction molecules and the solid chemical barrier. The components involved in ileum lipid transport were also reduced by TUDCA administration in HFD‐fed mice. Finally, the TUDCA‐treated mice showed a different gut microbiota composition compared with that in HFD‐fed mice but similar to that in normal chow diet‐fed mice.
Conclusions and Implications
TUDCA attenuates the progression of HFD‐induced NAFLD in mice by ameliorating gut inflammation, improving intestinal barrier function, decreasing intestinal fat transport and modulating intestinal microbiota composition.
Abbreviations
- ACOX1
peroxisomal acyl‐CoA oxidase 1
- ANOSIM
analysis of similarities
- C3GNT
core 3β1,3‐N‐acetyl glucosaminyltransferase
- CYP7a
cholesterol 7α‐hydroxylase
- ER
endoplasmic reticulum
- FABP
fatty acid‐binding protein
- FATP4
fatty acid transport protein 4
- FAR3
fatty acid receptor 3
- H&E
haematoxylin and eosin
- HFD
high‐fat diet
- HOMA‐IR
homeostasis model assessment of the insulin resistance index
- Iap
intestinal alkaline phosphatase
- ICAM1
intercellular cell adhesion molecule‐1
- IPGTT
i.p. glucose tolerance test
- IPITT
i.p. insulin tolerance test
- Irak4
IL‐1 receptor‐associated kinase 4
- JAM
junctional adhesion molecule
- Lcad
long‐chain acyl‐CoA dehydrogenase
- NAFLD
non‐alcoholic fatty liver disease
- NAS
non‐alcoholic fatty liver disease activity score
- NASH
non‐alcoholic steatohepatitis
- NCD
normal chow diet
- OTU
operational taxonomic unit
- PCoA
principal coordinates analysis
- Tab1
TGF‐β activated kinase 1 mitogen‐activated protein kinase kinase kinase 7‐binding protein 1
- TC
total cholesterol
- TEERs
transepithelial electrical resistances
- TGs
triglycerides
- TLR
toll‐like receptor
- Tram
toll or IL‐1 receptor domain‐containing adaptor inducing IFN‐β‐related adaptor molecule
- TUDCA
tauroursodeoxycholic acid
- UDCA
ursodeoxycholic acid
- ZO‐1
zonula occludens‐1
Introduction
Non‐alcoholic fatty liver disease (NAFLD) is a part of metabolic syndrome that has become a worldwide health concern. Cardiovascular diseases and type II diabetes are closely associated with the progression of NAFLD (Targher et al., 2016). The clinical spectrum of NAFLD includes isolated steatosis, steatosis with inflammation and fibrosis. Ten to twenty percent of NAFLD cases will develop non‐alcoholic steatohepatitis (NASH) (Hyysalo et al., 2014), which increases the risk of cardiovascular diseases, malignancy and liver‐related death. Fibrosis also aggravates the prognosis of NAFLD (Ratziu et al., 2015). Approximately 10–15% of NASH will progress to cirrhosis, and the latter increases the incidence of hepatocellular carcinoma. Lifestyle intervention, as the main therapeutic option, could control only 2.8% of NASH development (Ratziu et al., 2015). Therefore, the development of effective therapies for NAFLD is a crucial clinical goal.
Studies have revealed that the increased intestinal mucosal inflammation and intestinal epithelial barrier disruption, which increase the likelihood of the translocation of microbial products, are closely involved in the progression of NAFLD (Rahman et al., 2016). Mice with defects in intestinal epithelial permeability developed more severe steatohepatitis after consuming a diet containing 0.2% cholesterol, 20% protein, 43% carbohydrates and 23% fat than their counterparts fed a normal diet containing 16% protein, 61% carbohydrates and 7.2% fat (Rahman et al., 2016). Additionally, a human study revealed that patients with NAFLD have increased gut epithelial permeability, decreased levels of tight junction proteins, zonula occludens‐1 (ZO‐1), claudin 1 and occludin and higher levels of inflammation; these changes are closely associated with the occurrence and progression of NAFLD (Xin et al., 2014). A meta‐analysis indicated that, compared with healthy volunteers, patients with NAFLD and NASH were more likely to have enhanced intestinal permeability (Luther et al., 2015). The increased gut permeability increases liver exposure to intestine‐derived bacterial products (such as LPS, short‐chain fatty acids, bile acids, cytokines and ethanol), which increase hepatic inflammation and dyslipidaemia by activating toll‐like receptor (TLR) signalling and the inflammasome (Csak et al., 2011; Henao‐Mejia et al., 2012). Additionally, studies have shown that NAFLD is closely associated with the changed composition of intestinal microbiota. Many species in human gut microbiota are thought to be associated with the progression of NAFLD such as Bifidobacterium, Roseburia and Ruminococcus (Le Roy et al., 2013; Boursier et al., 2016). Moreover, various distinct mechanisms have been suggested for the microbiome in NAFLD and complications of dysbiosis: dysfunctional intestinal barrier with small intestinal bacterial overgrowth (Miele et al., 2009), inflammatory responses and metabolites produced or modified by the microbiota such as bile acids and LPS (Abu‐Shanab and Quigley, 2010). This complicated crosstalk among gut microbiota, intestinal permeability and the immune system collectively modulate the progression of NAFLD to NASH.
Bile acids have been reported to induce multiple effects on the intestinal lumen and intestinal wall, including pro‐intestinal or anti‐intestinal inflammatory responses (Martinez‐Moya et al., 2013; Renga et al., 2013), resolution of endoplasmic reticulum (ER) stress in intestinal epithelial cells underlying the pathology of inflammatory bowel disease (Berger and Haller, 2011), improvement of gut barrier dysfunction (Stenman et al., 2013) and regulation of gut microbes (Arab et al., 2017). These findings suggest that there is a complex relationship between bile acids and the intestine. Tauroursodeoxycholic acid (TUDCA), as a conjugated bile acid derivative, has been demonstrated to treat NAFLD via acting as an endogenous chemical chaperone to protect cells against ER stress (Xie et al., 2002; Choi et al., 2014; Itoh et al., 2016). Moreover, TUDCA can also decrease the inflammation in the intestine of mice with dextran sulfate sodium‐induced colitis (Cao et al., 2013a). As the intestinal micro‐environment (including inflammation status, function of the epithelial tight junction and gut microbiota) has an essential role in the progression of NAFLD and there is a close relationship between bile acids and the intestinal state, we hypothesized that TUDCA ameliorates NAFLD via gut–liver crosstalk: the compound reduces gut inflammation, augments intestinal barrier function, decreases intestinal fat transport and modulates gut microbiota.
Methods
Drug and diet
TUDCA (purity ≥98%) was obtained from Bruschettini S.r.l. (Genoa, Italy) and was dissolved in 0.9% saline for use. Both the normal chow diet (NCD) (containing 10% fat by energy) and high‐fat diet (HFD) (containing 60% fat by energy) were purchased from Beijing HFK Bio‐Technology Co., Ltd. (Beijing, China).
Animal experiments
Juvenile male C57BL/6J mice (7 ± 1 weeks old, body weight 20 ± 1 g; Beijing HFK Bio‐Technology Co., Ltd.) were housed in individually ventilated cages (four animals per cage) at the SPF facility of Huazhong University of Science and Technology under controlled environmental conditions (temperature 22 ± 2°C; relative humidity 60–70%) with free access to standard laboratory chow and tap water and were maintained on a regular 12/12 h light/dark cycle. All animal care and experimental procedures were approved by the Animal Care Ministry of Health and were performed in accordance with national and EU guidelines for the handling and use of experimental animals. All animal studies were approved by the Animal Experimentation Ethics Committee of Huazhong University of Science and Technology. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Mice were acclimatized to their environment for 1 week before the experiments. Mice were randomly allocated into three groups (n = 9 for each group). A co‐worker blinded to the experimental protocol randomized animals into these groups. One group of animals was fed a NCD and the other two a HFD. The HFD‐induced NAFLD model has been used for years. After 12 weeks of HFD feeding, one group of HFD‐treated mice was administered TUDCA 1000 mg·kg−1 body weight, p.o., once daily (HFD + TUDCA group) for 4 weeks, whereas the NCD‐treated mice and other group of HFD‐treated mice were treated with an equal volume of saline (NCD or HFD group) for 4 weeks. During the experiment, four mice (one from the NCD group and three from the HFD + TUDCA group) were unexpectedly killed early due to technical problems during gavage. The whole study lasted 16 weeks, during which the body weight and food intake of each animal were measured every week.
At week 16, an i.p. glucose tolerance test (IPGTT) and i.p. insulin tolerance test (IPITT) were performed according to previously described methods (Cao et al., 2013b). Fresh stool samples were collected and stored immediately at −80°C for subsequent analysis. At the end of the trial, after overnight fasting for 12 h, blood was collected, and serum was isolated by centrifugation at 1006 × g for 15 min at 4°C. Fasting serum insulin was determined using elisa kits (EMD Millipore Corporation, Burlington, MA, USA). The serum levels of total cholesterol (TC) and triglycerides (TGs) were determined using corresponding assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China). After the collection of blood, all mice were killed promptly by cervical vertebra dislocation. Tissues, including the liver and ileum [The small intestine was divided into two pieces of equal length called the jejunum and ileum. The ileum is from the distal part of the intestine located 1 cm from the ileocaecal valve (Navarrete et al., 2015)], were weighed; one portion of the tissues was fixed with formalin for histological analysis, and the other portion was immediately frozen in liquid nitrogen for further analysis.
Gene expression analysis
Total mRNA was isolated from the livers and ileums using TRIzol reagent (TAKARA, Tokyo, Japan) and was reverse transcribed into cDNA using a high‐capacity cDNA reverse transcription kit (TAKARA, Tokyo, Japan) according to the manufacturer's protocol. The mRNA levels were quantified by quantitative PCR (qPCR) with SYBR Green (Qiagen, Hilden, Germany). The relative mRNA levels were normalized to Gapdh mRNA levels in the same samples. The primer pairs used in this study are listed in Supporting Information Table S1.
Histological examination
The intestinal and liver histopathologies were evaluated by haematoxylin and eosin (H&E) staining. The intestinal histopathology was scored from 0 to 4, and the detailed criteria were as described previously (Steck et al., 2011). The NAFLD activity score (NAS) ranged from 0 to 8. NAS includes three histological scores: lobular inflammation (0–3), steatosis (0–3) and ballooning degeneration (0–2) (Kleiner et al., 2005).
16S rRNA sequencing
Frozen stool samples were used to characterize the gut microbiota. After the samples had been slowly thawed, total DNA was isolated from 0.2 g of faeces suspended in 1.4 mL of buffer solution and vortexed using a FastPrep FP120 instrument (Yan et al., 2011) for 1 min at 4°C, followed by incubation at 70°C for 5 min. After centrifugation at 14 000 × g for 1 min, 1.2 mL of the upper phase was collected, and total DNA was obtained using the TIANamp Stool DNA Kit (Tiangen Biotech, Beijing, China), as per the manufacturer's protocol. The DNA obtained was used to amplify the V3–V4 region of 16S rRNA genes. After purification, the amplicons were equally combined and subjected to a sequencing library preparation for 454 GS FLX pyrosequencing. A total of 1 388 221 sequences were subjected to quality control standards. Sequences had to meet the following criteria (Huse et al., 2010): (i) no Ns in the trimmed sequence, (ii) an exact match to the 5′ primer and (iii) Lucy's identified region of poor quality at the 0.002 threshold did not extend beyond the 5′ primer. The 5′ primer was trimmed from the sequences before analysis. Any sequences that did not meet a length requirement from 180 to 280 bases after trimming were discarded. The remaining 653 569 sequences were subjected to the next analysis.
Using QIIME, 8530 operational taxonomic units (OTUs) at the similarity level of 97% were obtained (Huse et al., 2010). The most abundant sequence of each OTU was selected as the representative sequence and was assigned taxonomy using RDP classification software with a bootstrap cut‐off of 50% (Wang et al., 2007). The abundance data of representative sequences were normalized for each sample and were log transformed. The statistical significance of differences in bacterial composition among the different samples was assessed by analysis of similarities (ANOSIM) test.
Cell culture
Human epithelial colorectal adenocarcinoma (Caco‐2) cells were cultured in DMEM supplemented with 10% FBS, penicillin and streptomycin. Cells were maintained in a 5% CO2 incubator at 37°C. Cells were routinely tested to exclude mycoplasma contamination. During the experiment, the palmitic acid (PA) + LPS group was stimulated with palmitic acid (600 μM) and LPS (10 μg·mL−1), and the PA + LPS + TUDCA group was co‐stimulated with palmitic acid (600 μM), LPS (10 μg·mL−1) and TUDCA (500 mM).
Transepithelial electrical resistance measurement
Transepithelial electrical resistance (TEER) measurements were made using an EVOM metre (World Precision Instruments, Sarasota, FL, USA) and chopstick‐style electrodes as performed previously (Li et al., 2010). Briefly, Caco‐2 cells were seeded onto Corning Costar™ Transwell™ Permeable Supports (0.4 μm pore size) in 12‐well plates at 2×105cells.cm‐2. After 14 days, monolayer confluency was reached, and cells were used in further experiments. Cells were treated as indicated. All TEER measurements were performed at 37°C.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). In all studies, a co‐worker was blinded to the experimental protocol. All data in this study are presented as the means ± SEM. Differences between two groups were determined by Student's two‐tailed t‐test. When more than two groups was examined, one‐way ANOVA followed by Newman–Keuls post hoc test for multiple comparisons was used. The non‐parametric ANOSIM test was used for independent variables. Post hoc tests were run only if F achieved P < 0.05, and there was no significant variance inhomogeneity. A P value <0.05 was considered statistically significant. All statistical analyses were performed using SPSS software, version 19.0. All of the data are representative results from more than five biological replicates and three analytical replicates per biological replicate.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
TUDCA attenuates HFD‐induced hepatic steatosis and inflammation
We first examined whether TUDCA administration influences the liver metabolic phenotype and inflammation in HFD‐induced NAFLD mice. After 16 weeks of HFD treatment, the liver size, liver weight and the ratio of the liver weight to body weight were significantly increased compared with those in the NCD group, while increases in these indexes were significantly reversed by TUDCA (Figure 1A). Consistent with these findings, H&E and Oil Red O staining showed more lipid droplets in the liver of HFD‐fed mice than controls, whereas TUDCA administration reduced the lipid droplets in the liver (Figure 1B). To further examine the severity of NAFLD, the NAS was examined according to the criteria of Kleiner et al. (2005). The NAS of the HFD‐treated group was significantly higher than that of the control group and was reduced after TUDCA administration (Figure 1B). As shown in Figure 1C, the concentrations of TG and TC in the serum and livers were also remarkably decreased after TUDCA administration. Furthermore, quantitative real‐time PCR was performed to determine the expression levels of genes related to fatty acid synthesis, transport and β‐oxidation in liver tissue. The expression levels of genes related to cholesterol synthesis (3‐hydroxy‐3‐methylglutaryl CoA reductase) and fatty acid‐binding protein 1 (Fabp1) were significantly suppressed after TUDCA administration. Meanwhile, β‐oxidation‐related genes [peroxisomal acyl‐CoA oxidase 1 (Acox1) and long‐chain acyl‐CoA dehydrogenase (Lcad)] that were suppressed in the HFD group were restored after TUDCA treatment (Figure 1D). Cholesterol 7α‐hydroxylase (CYP7A1; Cyp7a1) is involved in bile acid and cholesterol metabolism. TUDCA also reversed the decrease in Cyp7a1 gene expression after HFD feeding (Figure 1D). Furthermore, the lipoprotein lipase mRNA level was significantly increased in the HFD group compared with that in the control group and was reversed by TUDCA treatment (Figure 1D). These data indicated that TUDCA administration attenuates liver steatosis induced by HFD. As NAFLD is closely linked to a chronic inflammatory response, the expression levels of inflammatory mediators in the liver samples were measured. The levels of pro‐inflammatory cytokines [including IL‐1β, IL‐6, CCL2, CCL4, intercellular cell adhesion molecule‐1 (ICAM1) and TNF‐α] (Figure 1E) and innate immunity components [TLR4, TLR2, Cd14, TRIF‐related adaptor molecule (TRAM), IL‐1 receptor‐associated kinase 4 (IRAK4) and TGF‐β activated kinase 1 (MAP3K7)‐binding protein 1 (TAB1)] (Wang et al., 2017) (Figure 1F) were significantly higher in HFD‐treated mice than those in the control group, but the gene expression levels of these pro‐inflammatory cytokines and signalling molecules were markedly decreased by TUDCA.
Figure 1.
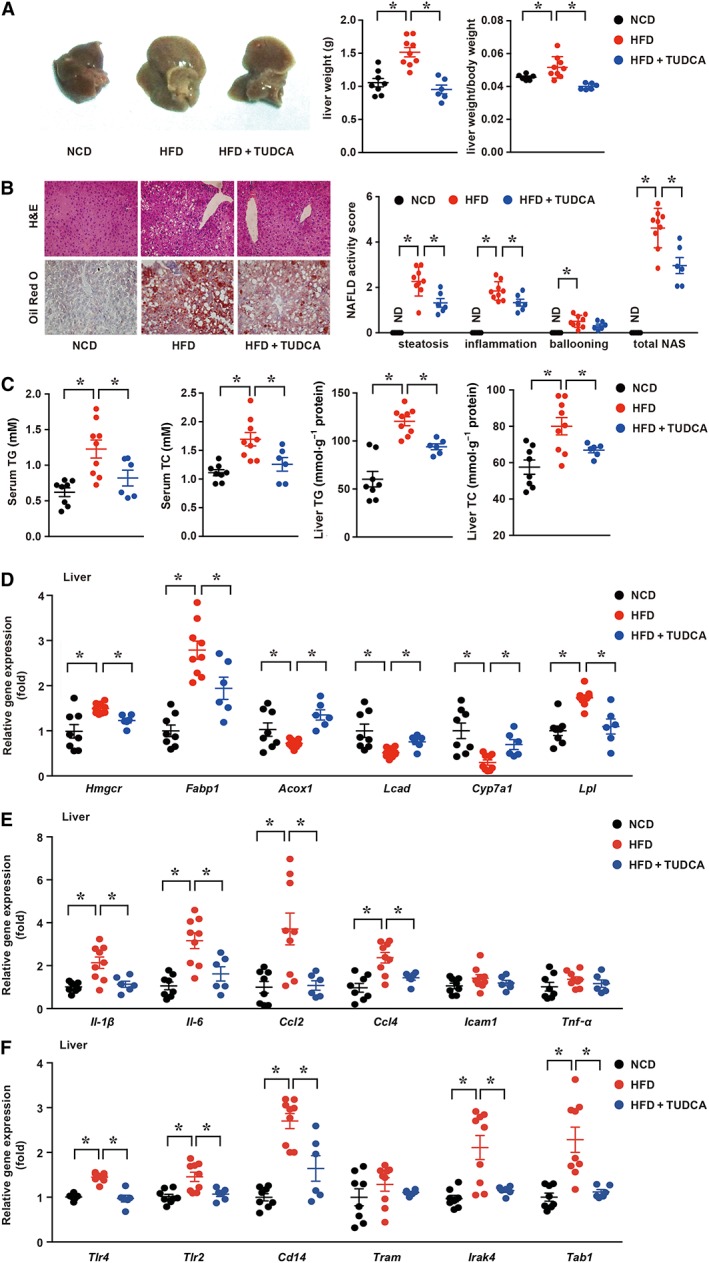
TUDCA attenuates HFD‐induced hepatic steatosis and inflammatory responses in NAFLD mice. (A, left) Representative macroscopic images of the livers of NCD, HFD and TUDCA mice, (A, middle) Liver weight and (A, right) the ratio of the liver weight and body weight of the three groups. (B, left) Representative H&E‐treated and Oil Red O‐stained sections in liver samples and (B, right) NAS of three groups. (C) The serum and hepatic TG and TC contents of mice in the indicated groups were measured using the elisa. (D) The mRNA expression levels of genes associated with fatty acid synthesis, transport and β‐oxidation, (E) inflammatory cytokines and (F) innate immunity components in the liver were detected by qPCR. The data are presented as the means ± SEM. One‐way ANOVA followed by Newman–Keuls post hoc test for multiple comparison. NCD group, n = 8; HFD group, n = 9; and HFD + TUDCA, n = 6. *P < 0.05. Hmgcr, 3‐hydroxy‐3‐methylglutaryl CoA reductase. ND, not detected.
TUDCA ameliorates HFD‐induced obesity and insulin resistance
As NAFLD is tightly associated with systemic metabolic disorders such as obesity and insulin resistance (Gaggini et al., 2017), apart from the impact of TUDCA on the liver, we examined whether TUDCA might affect the systemic metabolic status, such as body weight and glucose metabolic indexes. As shown in Figure 2A, despite a similar food intake (Supporting Information Figure S2), the gain in body weight of TUDCA‐treated mice was less than that of HFD‐fed mice. Indeed, the body weight was 76% lower in TUDCA‐treated mice than in mice without TUDCA treatment after HFD feeding for 16 weeks and was merely 7% higher in NCD‐fed mice. These data indicate that TUDCA treatment also reduced body weight gain in HFD‐fed mice. We also found that TUDCA treatment decreased fasting hyperglycaemia and hyperinsulinaemia as well as increasing the homeostasis model assessment of the insulin resistance index (HOMA‐IR) (Figure 2B–D). The IPGTT revealed that TUDCA administration significantly improved HFD‐induced glucose intolerance (Figure 2E). The IPGTT curves from TUDCA‐ and HFD‐treated mice revealed that plasma glucose levels at 15, 30 and 60 min after glucose injection were more elevated in HFD‐fed mice, suggesting that TUDCA could improve insulin resistance. Additionally, the IPITT of TUDCA‐treated mice demonstrated significantly better insulin sensitivity (Figure 2F). TUDCA‐treated mice had a higher response to insulin injection. Collectively, these data suggest that TUDCA administration ameliorates HFD‐induced obesity and insulin resistance.
Figure 2.
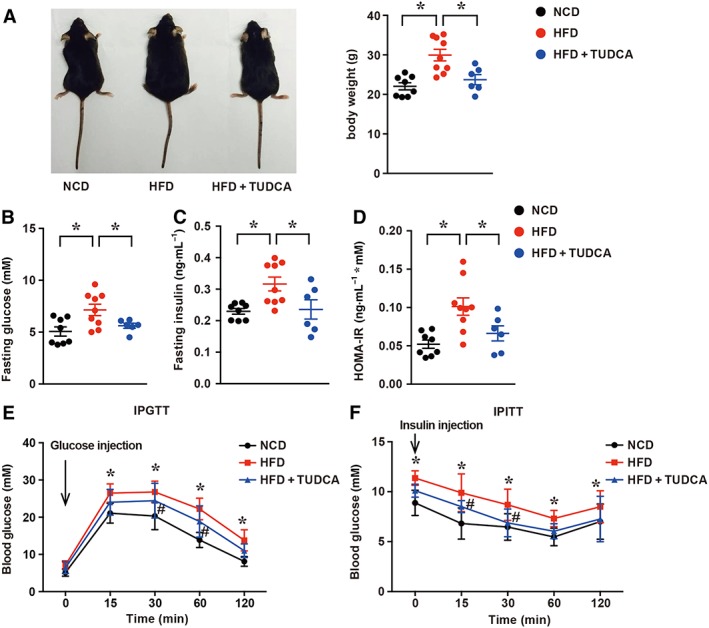
TUDCA ameliorates HFD‐induced obesity and insulin resistance in NAFLD mice. (A, left) Representative macroscopic pictures and (A, right) body weight of the NCD, HFD and HFD + TUDCA groups. (B) Fasting glucose, (C) fasting insulin and (D) HOMA‐IR in mice treated with NCD, HFD or HFD + TUDCA. Fasting glucose and fasting insulin levels were measured at the endpoint of this experiment. HOMA‐IR was calculated as HOMA‐IR = (FBG (mM) × FINS (ng·mL−1))/22.5. (E) The IPGTT and (F) IPITT assays were performed to evaluate the insulin sensitivity of mice in the indicated groups treated with NCD, HFD or HFD + TUDCA. The data are presented as the mean ± SEM for (A–D) and mean ± SD for (E–F). One‐way ANOVA followed by Newman–Keuls post hoc test for multiple comparison. NCD group, n = 8; HFD group, n = 9; and HFD + TUDCA, n = 6. *P < 0.05 versus the NCD group; # P < 0.05 versus the HFD group.
TUDCA attenuates gut inflammatory responses
It has been recently reported that increased gut inflammation and compromised intestinal epithelial permeability could permit the enhanced translocation of a multitude of gut microbial products (pathogen‐associated molecular patterns) involved in the progression of NAFLD (Chen et al., 2015; Rahman et al., 2016). Therefore, the intestinal histopathology was evaluated by H&E staining (Figure 3A). HFD induced low‐grade inflammatory cell infiltration with an increased histopathological score and mRNA levels of inflammatory cytokines (e.g. Il‐1β, Ccl2, Ccl4 and Icam1; Figure 3B). However, TUDCA administration significantly decreased the elevated mRNA level of Il‐1β (threefold), Ccl2 (twofold) and Icam1 (1.5‐fold) (Figure 3B). The mRNA levels of innate immunity components (Tlr4, Tlr2, Cd14, Tram, Irak4 and Tab1) were also significantly down‐regulated by TUDCA administration (Figure 3C). To further demonstrate the impact of TUDCA on intestinal inflammation, we performed in vitro experiments. Human epithelial colorectal adenocarcinoma cells (Caco‐2 cell line) were co‐stimulated with palmitic acid (600 μM) and LPS (10 μg·mL−1) for 6 h, so that they mimicked the NAFLD intestinal condition, including the intake of fatty acid and a burst of microbiota production. We found that the co‐stimulation with palmitic acid and LPS significantly increased the expression of inflammation‐related factors and innate immunity components (Il‐6, Il‐1β, Ccl2, Cd14, Irak4 and Tab1). As expected, TUDCA (500 mM) administration, along with palmitic acid and LPS treatment, attenuated the increased levels of inflammation‐related factors, findings similar to the in vivo observations. Taken together, these findings indicate intestinal inflammatory responses were significantly alleviated after TUDCA treatment.
Figure 3.
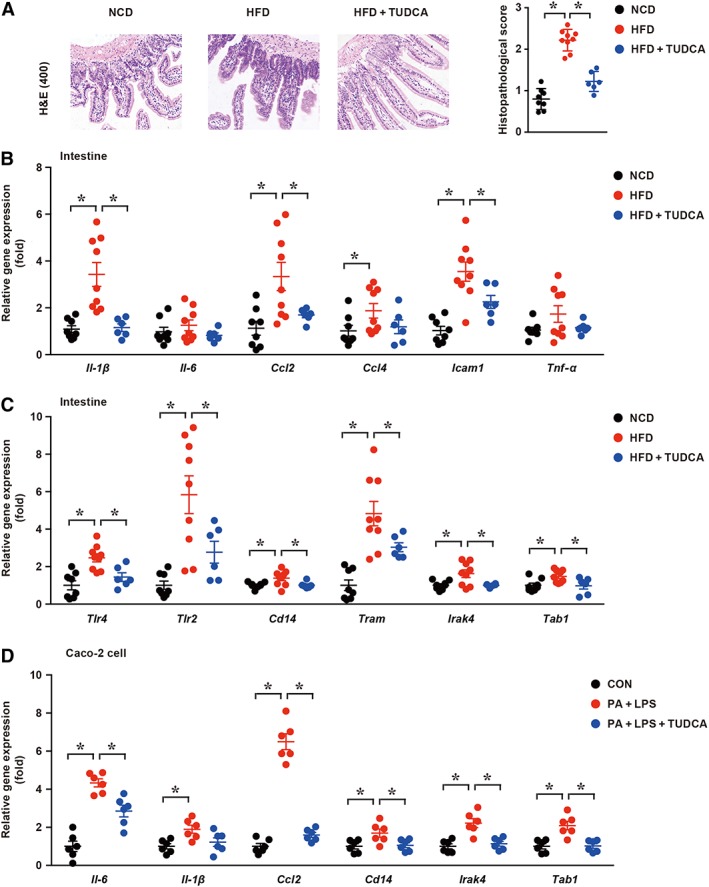
TUDCA attenuates gut inflammation. (A) Representative ileum H&E staining sections (left panel) and corresponding histopathological score (right panel) in the NCD, HFD and HFD + TUDCA groups; (B, C) the mRNA expression levels of inflammatory cytokines and components of innate immune signalling were measured in the indicated groups; (D) the mRNA expression levels of inflammatory cytokines and components of innate immune signalling were measured in the indicated groups in Caco‐2 cells. CON, control group treated with vehicle; PA + LPS group, Caco‐2 cells co‐stimulated with 600 μM palmitate and 10 μg·mL−1 LPS for 6 h; PA + LPS + TUDCA group, Caco‐2 cells treated with the combination of palmitate (600 mΜ), LPS (10 μg·mL−1) and TUDCA (500 mM) for 6 h. The data are presented as the means ± SEM. One‐way ANOVA followed by the Newman–Keuls post hoc test for multiple comparisons. NCD group, n = 8; HFD group, n = 9; and HFD + TUDCA, n = 6. In vitro experiment, n = 5. *P < 0.05.
TUDCA improves intestinal barrier function
Apart from the gut inflammation status, we also measured intestinal barrier function. Epithelial tight junction molecules, such as ZO‐1, junctional adhesion molecule (JAM), occludin and claudin 4, are markers of the epithelium integrity. As expected, the mRNA levels of Zo‐1, Jam, Occludin and Claudin4 were significantly down‐regulated in HFD‐fed mice compared with those in NCD‐treated mice. In contrast, TUDCA administration largely reversed the decrease in these tight junction molecules in HFD‐fed mice (Figure 4A).
Figure 4.
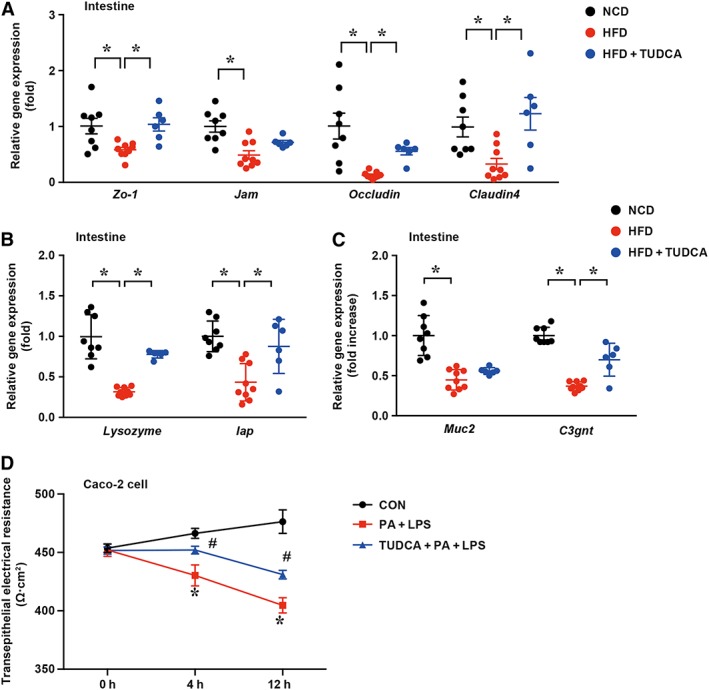
TUDCA improves intestinal barrier function. (A) The mRNA levels of ileum tight junction molecules were measured by real‐time PCR. (B, C) The mRNA levels of gut mucin lysozyme, Iap, Muc2 and C3gnt in the mice of different groups were detected by qPCR. (D) The TEERs of the CON, PA + LPS and PA + LPS + TUDCA groups were detected in Caco‐2 cells. CON, control group treated with vehicle; PA + LPS group, Caco‐2 cells co‐stimulated with 600 μM palmitate and 10 μg·mL−1 LPS for the indicated times; PA + LPS + TUDCA group, Caco‐2 cells treated with the combination of palmitate (600 mΜ), LPS (10 μg·mL−1) and TUDCA (500 mM) for the indicated times. The data are presented as the means ± SEM. One‐way ANOVA followed by the Newman–Keuls post hoc test for multiple comparisons. NCD group, n = 8; HFD group, n = 9; and HFD + TUDCA, n = 6. *P < 0.05; in vitro experiment, n = 5. *P < 0.05 versus the CON group; # P < 0.05 versus the PA + LPS group.
In addition to tight junction molecules serving as mechanical barriers, chemical barriers, including various digestive enzymes, lysozyme, mucopolysaccharide, antimicrobial peptides and other components secreted by the digestive tract, are also components of the intestinal mucosal barrier (Yan et al., 2013). Therefore, the intestinal mRNA levels of antibacterial peptides, including those of Reg3b, Reg3g, Defb1 and angiogerin‐1, were measured by qPCR. However, these antibacterial peptides exhibited no significant differences between the HFD group and HFD + TUDCA group (Supporting Information Figure S1). Lysozyme was demonstrated to bind to bacterial LPS with a high affinity to produce a complex and inhibit the biological activities of LPS (Takada et al., 1994). Additionally, intestinal alkaline phosphatase is a gut mucosal defence factor known to dephosphorylate LPS (Estaki et al., 2014). As expected, the mRNA levels of lysozyme and intestinal alkaline phosphatase were significantly lower in the ileum from HFD‐fed mice than those in the NCD group (0.316‐fold and 0.438‐fold, respectively); by comparison, these lower mRNA levels were significantly increased by 2.5‐fold and 2.1‐fold in TUDCA‐treated HFD‐fed mice respectively (Figure 4B). Moreover, qPCR demonstrated that the expression of core 3β1,3‐N‐acetyl glucosaminyltransferase (C3gnt) was reduced by 2.7‐fold in HFD‐fed mice but was increased in TUDCA‐treated HFD‐fed mice, although muc2 displayed no difference between these groups (Figure 4C). C3GnT is responsible for the glycosylation of intestinal mucins, providing an important source of growth substrates for intestinal bacteria, and is a component of the intestinal mucosal barrier (Xia, 2010). To further determine the effect of TUDCA on the integrity of cellular barriers, the TEER of three groups – the control group, PA + LPS group and PA + LPS + TUDCA group – were measured in Caco‐2 cells at different time points (0, 4 and 12 h). At the beginning of the experiment, no differences were detected among the three groups. However, after 4 and 12 h treatments, the TEER of the PA + LPS group was significantly decreased compared with that of the control group and this effect was reversed by the combination treatment with TUDCA (Figure 4D), demonstrating that TUDCA treatment could protect the integrity of cellular barriers. The data above suggest that intestinal barrier function is restored by TUDCA administration.
TUDCA suppresses gut lipid transport
With regard to the gut–liver axis, abnormal lipid metabolism in the intestine may lead to excessive lipid flow into the portal circulation, resulting in more lipid accumulated in the liver. To determine whether TUDCA alters lipid metabolism in the intestine, we further explored gut lipid transport. As expected, TUDCA administration significantly reduced the abundance of ileum lipid transport‐related genes induced by HFD, such as fatty acid translocase (Cd36) (7.7‐fold reduction), Fabp (2.1‐fold reduction), fatty acid transport protein 4 (Fatp4) (2.2‐fold reduction) and fatty acid receptor 3 (Ffar3) (3.4‐fold reduction) (Figure 5A). In vitro experiments showed a similar result (Figure 5B). Taken together, from these we concluded that TUDCA suppresses intestinal lipid absorption.
Figure 5.
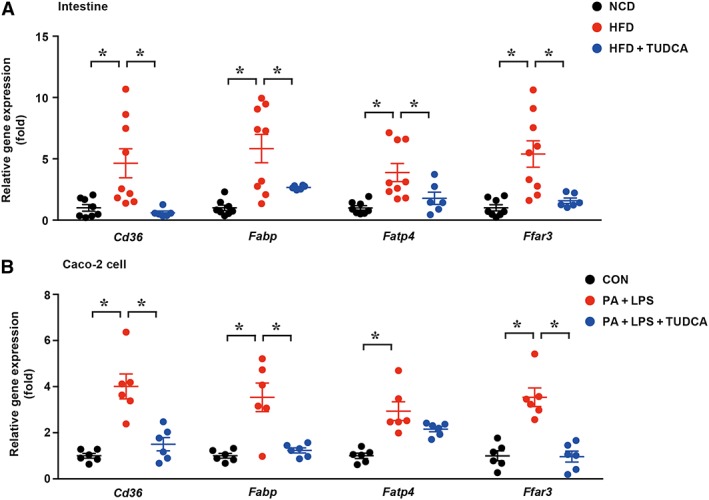
TUDCA suppresses gut lipid transport genes expression. (A) The mRNA levels of ileum lipid transport‐related genes (Cd36, Fabp, Fatp4 and Ffar3) in the NCD, HFD and HFD + TUDCA groups were measured. NCD group, n = 8; HFD group, n = 9; and HFD + TUDCA, n = 6. *P < 0.05. (B) The expression levels of ileum lipid transport‐related genes were detected in Caco‐2 cells (n = 5). CON, control group treated with vehicle; PA + LPS group, Caco‐2 cells co‐stimulated with 600 μM palmitate and 10 μg·mL−1 LPS for 6 h; PA + LPS + TUDCA group, Caco‐2 cells treated with the combination of palmitate (600 mΜ), LPS (10 μg·mL−1) and TUDCA (500 mM) for 6 h. The data are presented as the means ± SEM. One‐way ANOVA followed by the Newman–Keuls post hoc test for multiple comparisons. *P < 0.05.
TUDCA‐treated mice showed different gut microbiota composition compared with HFD‐fed mice
There is considerable evidence that dysbiosis contributes to the process of NAFLD; this has been provided by animal studies in which the gut microbiota is manipulated and by observational studies in patients with NAFLD (Leung et al., 2016). To explore whether gut microbiota showed differences among the three groups, we investigated the composition of the microbiota population in mice with in‐depth genetic sequencing. The phylogenetically informative 16S rRNA gene (hypervariable region V3–V4) was amplified from total DNA extracted from murine faecal samples. The overall structural changes of the gut microbiota were then analysed using the unsupervised multivariate statistical method UniFrac distance‐based principal coordinates analysis (PCoA). The PCoA of the weighted and unweighted UniFrac distance from the data set showed apparent separation into colour‐coded clusters according to HFD without or with TUDCA treatment (Figure 6A). ANOSIM of PCoA matrix scores indicated a significant separation among the microbiota of the NCD, HFD and HFD + TUDCA groups (P < 0.05). The calculated relative abundance of bacteria at the phylum level in the three groups is presented in Figure 6B. HFD‐fed mice displayed an increased abundance of Firmicutes (64 vs. 51%) and decreased abundance of Bacteroidetes (20 vs. 43%), a gut microbial composition associated with obesity and NAFLD (Turnbaugh et al., 2006; De Minicis et al., 2014). In contrast, the TUDCA treated group showed a tendency to have a gut microbial composition close to that of NCD mice. The abundance of Proteobacteria was increased (14 vs. 4%) in HFD‐fed mice compared with that in control mice, while TUDCA treatment ameliorated this change to a small extent. Cox et al. (2014) showed that following low‐dose penicillin treatment there was a strong increase in Proteobacteria that was concomitant with increased adiposity, a finding that was in accordance with our result. However, the abundance of Actinobacteria was too low to be detected. Further in‐depth analysis indicated that, among the changed microbiota, two anti‐inflammatory bacterial taxa, Faecalibacterium and Akkermansia (Everard et al., 2013; Quevrain et al., 2016), were markedly increased in the TUDCA‐treated group, with an increase of 370‐fold and 68‐fold respectively (Figure 6C). However, the TUDCA‐treated group showed a significantly decreased abundance of Mucispirillum and Ruminococcusgnavus by 11‐fold and threefold respectively. Mucispirillum and Ruminococcusgnavus were reported to play important roles in pro‐inflammatory responses (El Aidy et al., 2014; Titecat et al., 2014). Additionally, Paraprevotella, YS2 and Dehalobacterium (Figure 6D) were initially found to be changed; in contrast, in the TUDCA‐treated group the changes in these bacteria tended to be reversed and similar to those in the NCD group. These data indicate that the TUDCA‐treated mice had a different gut microbiota composition compared with HFD‐fed mice but a similar composition to that in the NCD‐fed mice. Therefore, TUDCA may play an important role in modulating the composition of gut microbiota.
Figure 6.
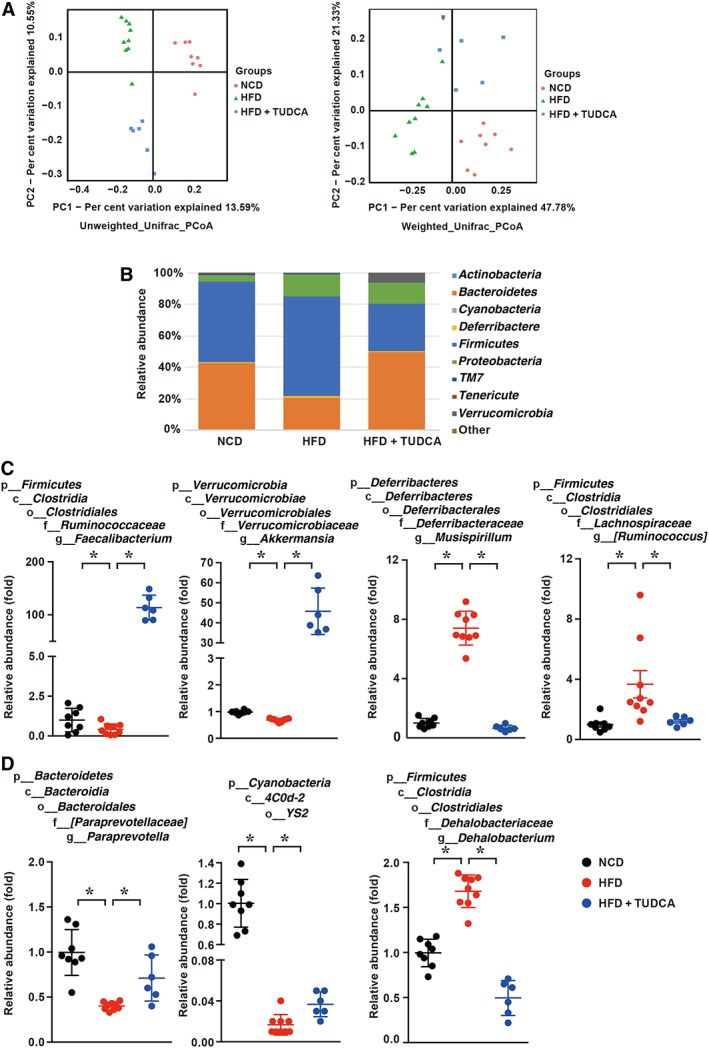
TUDCA treated mice show different gut microbiota composition compared with HFD‐fed mice. (A) PCoA score plot; PCoA score plot based on unweighted (left panel) and weighted (right panel) UniFrac metrics; (B) average phylum distribution of gut microbiomes in the NCD, HFD and HFD + TUDCA groups; and (C, D) comparison of the taxonomic abundance among the indicated groups. The data are presented as the means ± SEM. The statistical significance in bacterial composition among the different samples was assessed by the ANOSIM test. NCD group, n = 8; HFD group, n = 9; and HFD + TUDCA, n = 6. *P < 0.05.
Discussion
In the present study, we demonstrated that TUDCA treatment markedly ameliorated HFD‐induced NAFLD in mice. In addition to its commonly accepted protective effect on ER stress, we found that the mechanisms through which TUDCA alleviated NAFLD included (i) attenuation of gut inflammation, (ii) improvement of intestinal barrier function, (iii) decrease of gut fat transport and (iv) partial modulation of intestinal microbiota composition.
Obesity and NAFLD are rising health problems worldwide with an estimated prevalence of 20–30%, which increases the risk of diabetes, cardiovascular diseases and cancer (Hannah and Harrison, 2016). Therefore, it is important to develop therapeutic approaches to prevent an adverse outcome. TUDCA, an amphiphilic bile acid, is the taurine conjugate form of ursodeoxycholic acid (UDCA). It is known as a chemical chaperone against ER stress (Ozcan et al., 2006) and has been recently reported to reduce liver steatosis in several studies (Choi et al., 2014; Legry et al., 2014). In our present study, we demonstrated the effectiveness of TUDCA in alleviating hepatic steatosis and inflammation, as well as improving obesity and insulin resistance. We observed that mice fed a HFD developed NAFLD compared with control mice, while TUDCA improved the condition. The ability of TUDCA to redress the glucose metabolism disorder in NAFLD mice has also been reported (Cho et al., 2014). We also assessed the effects of TUDCA on lipid droplets in the liver, NAS, liver injury indexes and genes related to fatty acid synthesis and elimination. Our results revealed that lipid metabolism homeostasis was disrupted after HFD administration, and TUDCA restored this metabolic disorder to some extent. This result is in accord with that reported by Choi et al. (2014). Meanwhile, because inflammatory responses can result in the development of a metabolic disorder, we tested the levels of inflammatory mediators in different groups. The levels of inflammatory mediators were increased in HFD‐treated mice compared with the control levels, while TUDCA administration reduced the expression of inflammatory cytokines. In a previous study, TUDCA was also demonstrated to alleviate the inflammatory responses in acute pancreatitis (Seyhun et al., 2011). Collectively, the efficacy of TUDCA on HFD‐induced NAFLD involves effects on insulin resistance, hepatic steatosis and inflammatory responses.
Regarding the mechanism by which TUDCA ameliorates NAFLD, major studies have unanimously considered that TUDCA functions as an ER stress protectant (Cho et al., 2014; Choi et al., 2014). Yang et al. (2010) found that oral TUDCA treatment mainly decreased the expression of genes involved in lipogenesis and fatty acid uptake in ob/ob mice. Similarly, we also confirmed that TUDCA could prevent hepatosteatosis induced by the HFD through increasing β‐oxidation genes (Acox1 and Lcad) in the liver and suppressing fatty acid uptake genes (such as Cd36) in the intestine. Cho et al. (2014) found that TUDCA attenuated the progression of methionine–choline‐deficient (MCD) diet‐induced steatohepatitis by reducing ER stress. In their study, ER stress was the key mechanism of MCD‐induced liver inflammation and the key therapeutic target for TUDCA. Likewise, we also demonstrated that TUDCA could alleviate liver inflammation. However, apart from the traditionally reported mechanism, our study primarily found that TUDCA could alleviate NAFLD in mice by improving the intestinal micro‐environment (including the inflammatory state, function of the epithelial tight junction, lipid transport and gut microbiota). We found that gut inflammation and the TLR2/4 signalling pathways were markedly suppressed by TUDCA treatment. Moreover, TUDCA treatment dramatically improved the expression of molecules associated with intestinal epithelial tight junctions, including Zo‐1, Jam, Occludin and Claudin4. Additionally, the expression levels of lysozyme (responsible for protection from bacterial infections) and IAP (detoxification of bacterial LPS), serving as the chemical barrier of the intestine, were increased after TUDCA treatment. Unexpectedly, no significant difference in the levels of antibacterial peptides was observed, a finding that was inconsistent with that obtained after obeticholic acid treatment (Ubeda et al., 2016). Obeticholic acid was reported to improve ileum expression of antimicrobial peptides to reduce bacterial translocation and inhibit intestinal inflammation in cirrhotic rats. The difference in these findings is most likely due to the different affinity of the two bile acids to farnesoid X receptor. Additionally, the levels of muc2 and C3GnT were decreased in the HFD group and were up‐regulated after TUDCA treatment. Muc2 is a gene encoding a mucin protein that can disassociate pathogenic and commensal bacteria (Bergstrom et al., 2010). C3GnT is a core enzyme in the synthesis of core 3‐derived O‐glycans, the predominant components of intestinal mucus. Mice lacking glycans display a thinner intestinal mucus barrier (Zarepour et al., 2013), which represents an important source of growth substrates for intestinal bacteria. Therefore, the changes in the gut chemical barrier, including those of lysozyme, Iap, Muc2 and C3gnt, influence gut permeability. The study by Kim et al. (2012) demonstrated that, in coordination with the increased permeability via activation of the TLR4 signalling pathway, intestinal inflammation by altering the gut microbiota accelerated obesity in HFD mice. These improvements in the ileum might delay and alleviate the development of NAFLD.
Abnormal lipid metabolism in the intestine may lead to excessive lipid flow into the portal circulation, resulting in more lipid accumulated in the liver. Thus, we further explored gut lipid transport. We found that gut lipid transport‐related genes (Cd36, Fabp, Fatp4 and Ffar3) were markedly increased in HFD mice, whereas they were significantly decreased after TUDCA administration. Previous studies also demonstrated that these lipid transport receptors may have potential relevance to the regulation of intestinal lipid metabolism (Abumrad and Davidson, 2012). Therefore, TUDCA may alleviate NAFLD progression through suppressing gut lipid transport.
Furthermore, animal studies in which the gut microbiota is manipulated, as well as observational studies in patients with NAFLD, have provided considerable evidence that dysbiosis contributes to the process of NAFLD (Leung et al., 2016). In this study, we found that the ratio of Bacteroidetes/Firmicutes in the HFD group was decreased compared with that in the control group, while TUDCA restored the ratio. In previous studies, Ley et al. (2005) reported that the intestinal microbiota of ob/ob mice showed a reduction in the proportion of Bacteroidetes and an increased level of Firmicutes compared with that of wild‐type mice. It was reported that obese individuals had a lower abundance of Bacteroidetes and a higher abundance of Firmicutes than lean individuals (Manco et al., 2010). These data suggest a relationship between the ratio of Firmicutes to Bacteroidetes and metabolic disorders (Semova et al., 2012). In addition, the TUDCA treated group showed increased abundance of two anti‐inflammatory bacterial taxa, Faecalibacterium and Akkermansia. Levels of Faecalibacterium (Munukka et al., 2014) and Akkermansia (Dao et al., 2016) have been shown to be decreased in NAFLD patients. Additionally, for the first time, we found that the abundance of some taxa was changed dramatically in HFD‐fed mice and was reversed in TUDCA‐treated mice. These bacteria might be new candidate phylotypes for predicting and treating metabolic disorders.
The present study has a few limitations: (i) although the HFD‐induced NAFLD in the mouse model shares many common characteristics with higher mammalian diseases and is commonly used to explore cellular and molecular mechanisms, potential limitations still exist regarding extrapolating data from mice to humans. (ii) The equivalent dose of TUDCA corresponding to 1000 mg·kg−1 in mice is approximately 81 mg·kg−1 in humans (Reagan‐Shaw et al., 2008). However, this dose is much higher than the usual dose of UDCA used to treat primary biliary cirrhosis (12–15 mg·kg−1) (Carey et al., 2015). Therefore, the safety and efficacy of high‐dose TUDCA in humans should be further evaluated.
In conclusion, our present study, for the first time, demonstrated that TUDCA treatment ameliorates HFD‐induced murine NAFLD by improving gut inflammation and intestinal barrier function, decreasing intestinal fat transport and modulating intestinal dysbiosis. Our study uncovered possible new mechanisms for TUDCA as a potential therapy for NAFLD. These data provide direction for future studies designed to address the use of TUDCA in clinical settings of NAFLD.
Author contributions
W.W. and J.Z. contributed in the study concept and design, acquisition of data, analysis and interpretation of data and drafting of the manuscript; W.G. to the acquisition of data; D.S., H.D., L.X. and Q.Z. to the acquisition and analysis of data; B.S. to the analysis and interpretation of data and critical revision of the manuscript for important intellectual content; and K.H., L.Y. and X.H. in the study supervision, study concept and design, analysis and interpretation of data, writing of the manuscript and obtaining funding.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Sequences of Primers Used for Real‐Time Quantitative PCR.
Figure S1 TUDCA does not increase the expression of intestinal antibacterial peptides in NAFLD mice. The mRNA levels of several antibacterial peptides (Reg3b, Reg3g, Defb1 and Angiogerin‐1) were measured by qPCR. The data are reported as the means ± SEM. One‐way ANOVA (ANOVA) followed by the Newman–Keuls post hoc test for multiple comparisons. NCD: normal chow diet group, n = 8; HFD: high‐fat diet group, n = 9; HFD + TUDCA: high‐fat diet with TUDCA treatment group, n = 6. *P < 0.05.
Figure S2 TUDCA has no influence on food intake. The weekly food intake of the NCD, HFD, HFD + TUDCA groups was calculated. The data are reported as the means ± SEM. One‐way analysis of variance (ANOVA) followed by the Newman–Keuls post hoc test for multiple comparisons. NCD: normal chow diet group, n = 8; HFD: high‐fat diet group, n = 9; HFD + TUDCA: high‐fat diet with TUDCA treatment group, n = 6. *P < 0.05.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (nos. 81370550, 81570530 and 30500658 to L.Y. and 81570486 and 81330014 to X.H.).
Wang, W. , Zhao, J. , Gui, W. , Sun, D. , Dai, H. , Xiao, L. , Chu, H. , Du, F. , Zhu, Q. , Schnabl, B. , Huang, K. , Yang, L. , and Hou, X. (2018) Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non‐alcoholic fatty liver disease. British Journal of Pharmacology, 175: 469–484. doi: 10.1111/bph.14095.
Contributor Information
Kai Huang, Email: huangkai1@hust.edu.cn.
Ling Yang, Email: hepayang@163.com, Email: lingyang70@gmail.com.
Xiaohua Hou, Email: houxh@medmail.com.cn.
References
- Abumrad NA, Davidson NO (2012). Role of the gut in lipid homeostasis. Physiol Rev 92: 1061–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu‐Shanab A, Quigley EM (2010). The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 7: 691–701. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M (2017). Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 65: 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger E, Haller D (2011). Structure‐function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem Biophys Res Commun 409: 610–615. [DOI] [PubMed] [Google Scholar]
- Bergstrom KS, Kissoon‐Singh V, Gibson DL, Ma C, Montero M, Sham HP et al (2010). Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 6: e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo‐Perez F et al (2016). The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63: 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE et al (2013a). The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 144: 989–1000 e1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Xue Y, Xue L, Jiang X, Wang X, Zhang Z et al (2013b). Hepatic menin recruits SIRT1 to control liver steatosis through histone deacetylation. J Hepatol 59: 1299–1306. [DOI] [PubMed] [Google Scholar]
- Carey EJ, Ali AH, Lindor KD (2015). Primary biliary cirrhosis. Lancet 386: 1565–1575. [DOI] [PubMed] [Google Scholar]
- Chen P, Starkel P, Turner JR, Ho SB, Schnabl B (2015). Dysbiosis‐induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology (Baltimore, Md) 61: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Yoon JH, Kwak MS, Jang ES, Lee JH, Yu SJ et al (2014). Tauroursodeoxycholic acid attenuates progression of steatohepatitis in mice fed a methionine–choline‐deficient diet. Dig Dis Sci 59: 1461–1474. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Shin HS, Choi HS, Park JW, Jo I, Oh ES et al (2014). Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP‐1c activation in hepatocytes. Lab Invest 94: 1114–1125. [DOI] [PubMed] [Google Scholar]
- Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I et al (2014). Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158: 705–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G (2011). Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology (Baltimore, Md) 54: 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao MC, Everard A, Aron‐Wisnewsky J, Sokolovska N, Prifti E, Verger EO et al (2016). Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65: 426–436. [DOI] [PubMed] [Google Scholar]
- De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L et al (2014). Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 59: 1738–1749. [DOI] [PubMed] [Google Scholar]
- El Aidy S, Derrien M, Aardema R, Hooiveld G, Richards SE, Dane A et al (2014). Transient inflammatory‐like state and microbial dysbiosis are pivotal in establishment of mucosal homeostasis during colonisation of germ‐free mice. Benef Microbes 5: 67–77. [DOI] [PubMed] [Google Scholar]
- Estaki M, DeCoffe D, Gibson DL (2014). Interplay between intestinal alkaline phosphatase, diet, gut microbes and immunity. World J Gastroenterol 20: 15650–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB et al (2013). Cross‐talk between Akkermansia muciniphila and intestinal epithelium controls diet‐induced obesity. Proc Natl Acad Sci U S A 110: 9066–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V et al (2017). Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology . [DOI] [PubMed] [Google Scholar]
- Hannah WN Jr, Harrison SA (2016). Noninvasive imaging methods to determine severity of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 64: 2234–2243. [DOI] [PubMed] [Google Scholar]
- Henao‐Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T et al (2012). Inflammasome‐mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482: 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Welch DM, Morrison HG, Sogin ML (2010). Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12: 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyysalo J, Mannisto VT, Zhou Y, Arola J, Karja V, Leivonen M et al (2014). A population‐based study on the prevalence of NASH using scores validated against liver histology. J Hepatol 60: 839–846. [DOI] [PubMed] [Google Scholar]
- Itoh H, Muramatsu‐Kato K, Ferdous UJ, Kohmura‐Kobayashi Y, Kanayama N (2016). Undernourishment in utero and hepatic steatosis in later life: a potential issue in Japanese people. Congenit Anom (Kyoto) . [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KA, Gu W, Lee IA, Joh EH, Kim DH (2012). High fat diet‐induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One 7: e47713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW et al (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (Baltimore, Md) 41: 1313–1321. [DOI] [PubMed] [Google Scholar]
- Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C et al (2013). Intestinal microbiota determines development of non‐alcoholic fatty liver disease in mice. Gut 62: 1787–1794. [DOI] [PubMed] [Google Scholar]
- Legry V, Van Rooyen DM, Lambert B, Sempoux C, Poekes L, Espanol‐Suner R et al (2014). Endoplasmic reticulum stress does not contribute to steatohepatitis in obese and insulin‐resistant high‐fat‐diet‐fed foz/foz mice. Clin Sci (Lond) 127: 507–518. [DOI] [PubMed] [Google Scholar]
- Leung C, Rivera L, Furness JB, Angus PW (2016). The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 13: 412–425. [DOI] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005). Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 102: 11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Guo D, Zhu H, Hering‐Smith KS, Hamm LL, Ouyang J et al (2010). Interleukin‐6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol 299: R590–R595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R et al (2015). Hepatic injury in nonalcoholic steatohepatitis contributes to altered intestinal permeability. Cell Mol Gastroenterol Hepatol 1: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manco M, Putignani L, Bottazzo GF (2010). Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr Rev 31: 817–844. [DOI] [PubMed] [Google Scholar]
- Martinez‐Moya P, Romero‐Calvo I, Requena P, Hernandez‐Chirlaque C, Aranda CJ, Gonzalez R et al (2013). Dose‐dependent antiinflammatory effect of ursodeoxycholic acid in experimental colitis. Int Immunopharmacol 15: 372–380. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R et al (2009). Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49: 1877–1887. [DOI] [PubMed] [Google Scholar]
- Munukka E, Pekkala S, Wiklund P, Rasool O, Borra R, Kong L et al (2014). Gut–adipose tissue axis in hepatic fat accumulation in humans. J Hepatol 61: 132–138. [DOI] [PubMed] [Google Scholar]
- Navarrete J, Vasquez B, Del Sol M (2015). Morphoquantitative analysis of the ileum of C57BL/6 mice (Mus musculus) fed with a high‐fat diet. Int J Clin Exp Pathol 8: 14649–14657. [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO et al (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313: 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J et al (2016). Identification of an anti‐inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn's disease. Gut 65: 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y et al (2016). Loss of junctional adhesion molecule A promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 151: 733–746 e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, Goodman Z, Sanyal A (2015). Current efforts and trends in the treatment of NASH. J Hepatol 62: S65–S75. [DOI] [PubMed] [Google Scholar]
- Reagan‐Shaw S, Nihal M, Ahmad N (2008). Dose translation from animal to human studies revisited. FASEB J 22: 659–661. [DOI] [PubMed] [Google Scholar]
- Renga B, Mencarelli A, Cipriani S, D'Amore C, Carino A, Bruno A et al (2013). The bile acid sensor FXR is required for immune‐regulatory activities of TLR‐9 in intestinal inflammation. PLoS One 8: e54472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semova I, Carten JD, Stombaugh J, Mackey LC, Knight R, Farber SA et al (2012). Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12: 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyhun E, Malo A, Schafer C, Moskaluk CA, Hoffmann RT, Goke B et al (2011). Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol 301: G773–G782. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck N, Hoffmann M, Sava IG, Kim SC, Hahne H, Tonkonogy SL et al (2011). Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 141: 959–971. [DOI] [PubMed] [Google Scholar]
- Stenman LK, Holma R, Gylling H, Korpela R. (2013). Genetically obese mice do not show increased gut permeability or faecal bile acid hydrophobicity. Br J Nutr 110: 1157–1164. [DOI] [PubMed] [Google Scholar]
- Takada K, Ohno N, Yadomae T (1994). Detoxification of lipopolysaccharide (LPS) by egg white lysozyme. FEMS Immunol Med Microbiol 9: 255–263. [DOI] [PubMed] [Google Scholar]
- Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C (2016). Non‐alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta‐analysis. J Hepatol 65: 589–600. [DOI] [PubMed] [Google Scholar]
- Titecat M, Wallet F, Vieillard MH, Courcol RJ, Loiez C (2014). Ruminococcus gnavus: an unusual pathogen in septic arthritis. Anaerobe 30: 159–160. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006). An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031. [DOI] [PubMed] [Google Scholar]
- Ubeda M, Lario M, Munoz L, Borrero MJ, Rodriguez‐Serrano M, Sanchez‐Diaz AM et al (2016). Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol 64: 1049–1057. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhang Y, Yang L, Li H (2017). The innate immune signaling in cancer and cardiometabolic diseases: friends or foes? Cancer Lett 387: 46–60. [DOI] [PubMed] [Google Scholar]
- Xia L (2010). Core 3‐derived O‐glycans are essential for intestinal mucus barrier function. Methods Enzymol 479: 123–141. [DOI] [PubMed] [Google Scholar]
- Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE et al (2002). Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress‐induced caspase‐12 activation. Hepatology 36: 592–601. [DOI] [PubMed] [Google Scholar]
- Xin D, Zong‐Shun L, Bang‐Mao W, Lu Z (2014). Expression of intestinal tight junction proteins in patients with non‐alcoholic fatty liver disease. Hepatogastroenterology 61: 136–140. [PubMed] [Google Scholar]
- Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E et al (2011). Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology (Baltimore, Md) 53: 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Yang C, Tang J (2013). Disruption of the intestinal mucosal barrier in Candida albicans infections. Microbiol Res 168: 389–395. [DOI] [PubMed] [Google Scholar]
- Yang JS, Kim JT, Jeon J, Park HS, Kang GH, Park KS et al (2010). Changes in hepatic gene expression upon oral administration of taurine‐conjugated ursodeoxycholic acid in ob/ob mice. PLoS One 5: e13858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A et al (2013). The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect Immun 81: 3672–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Sequences of Primers Used for Real‐Time Quantitative PCR.
Figure S1 TUDCA does not increase the expression of intestinal antibacterial peptides in NAFLD mice. The mRNA levels of several antibacterial peptides (Reg3b, Reg3g, Defb1 and Angiogerin‐1) were measured by qPCR. The data are reported as the means ± SEM. One‐way ANOVA (ANOVA) followed by the Newman–Keuls post hoc test for multiple comparisons. NCD: normal chow diet group, n = 8; HFD: high‐fat diet group, n = 9; HFD + TUDCA: high‐fat diet with TUDCA treatment group, n = 6. *P < 0.05.
Figure S2 TUDCA has no influence on food intake. The weekly food intake of the NCD, HFD, HFD + TUDCA groups was calculated. The data are reported as the means ± SEM. One‐way analysis of variance (ANOVA) followed by the Newman–Keuls post hoc test for multiple comparisons. NCD: normal chow diet group, n = 8; HFD: high‐fat diet group, n = 9; HFD + TUDCA: high‐fat diet with TUDCA treatment group, n = 6. *P < 0.05.