

Abstract
Objectives:
To identify a novel, generalizable diagnostic for acute respiratory distress syndrome using whole-blood gene expression arrays from multiple acute respiratory distress syndrome cohorts of varying etiologies.
Data Sources:
We performed a systematic search for human whole-blood gene expression arrays of acute respiratory distress syndrome in National Institutes of Health Gene Expression Omnibus and ArrayExpress. We also included the Glue Grant gene expression cohorts.
Study Selection:
We included investigator-defined acute respiratory distress syndrome within 48 hours of diagnosis and compared these with relevant critically ill controls.
Data Extraction:
We used multicohort analysis of gene expression to identify genes significantly associated with acute respiratory distress syndrome, both with and without adjustment for clinical severity score. We performed gene ontology enrichment using Database for Annotation, Visualization and Integrated Discovery and cell type enrichment tests for both immune cells and pneumocyte gene expression. Finally, we selected a gene set optimized for diagnostic power across the datasets and used leave-one-dataset-out cross validation to assess robustness of the model.
Data Synthesis:
We identified datasets from three adult cohorts with sepsis, one pediatric cohort with acute respiratory failure, and two datasets of adult patients with trauma and burns, for a total of 148 acute respiratory distress syndrome cases and 268 critically ill controls. We identified 30 genes that were significantly associated with acute respiratory distress syndrome (false discovery rate < 20% and effect size >1.3), many of which had been previously associated with sepsis. When metaregression was used to adjust for clinical severity scores, none of these genes remained significant. Cell type enrichment was notable for bands and neutrophils, suggesting that the gene expression signature is one of acute inflammation rather than lung injury per se. Finally, an attempt to develop a generalizable diagnostic gene set for acute respiratory distress syndrome showed a mean area under the receiver-operating characteristic curve of only 0.63 on leave-one-dataset-out cross validation.
Conclusions:
The whole-blood gene expression signature across a wide clinical spectrum of acute respiratory distress syndrome is likely confounded by systemic inflammation, limiting the utility of whole-blood gene expression studies for uncovering a generalizable diagnostic gene signature.
Keywords: acute respiratory distress syndrome, diagnostic, gene expression, human, microarray
The acute respiratory distress syndrome (ARDS) is defined by the acute onset of hypoxic respiratory failure, with a Pao2/Fio2 ratio less than 300 and bilateral infiltrates on chest radiograph not related to volume overload (1, 2). This definition captures a broad array of patients; the LUNG-SAFE study estimated that 23% of ICU patients on mechanical ventilation meet ARDS criteria (3). As a result, there are diverse critical illness settings in pediatric and adult patients with multiple primary problems including sepsis, pneumonia, trauma, burns, and surgical insults. Whether these insults lead to a common underlying pathophysiology is unclear. A potential explanation for the lack of novel therapeutic targets in ARDS is that the broad syndrome may in fact be composed of several lung injury endotypes (4–6). There is thus a critical need 1) to establish whether all patients with “ARDS” really have the same disease and 2) to develop new molecular diagnostics either for the broad syndrome, or to instead use genomics to help define specific ARDS subgroups.
Many groups have hypothesized that the lung inflammation present in ARDS may be correlated with specific gene signatures of circulating leukocytes. These groups have used microarrays to study the transcriptome in patients with lung injury. This is perhaps partly because ARDS is a syndrome understood to be an inflammatory response and partly because blood is much easier to sample than lung tissue. These studies of ARDS have come from a wide variety of clinical settings, including adult sepsis (7–9), pediatric sepsis (10–12), and severe trauma and burns (13–15). There is low overlap of disease-related genes and pathways identified in each of these studies, and no unified ARDS-defining gene signature has emerged to date, which is possibly due to a number of reasons. First, the syndrome of ARDS may be caused by different underlying mechanisms. Second, insufficient data may be available to find a common signal despite the clinical heterogeneity. Third, gene expression in blood samples from ARDS patients may not reflect the disease state in lung tissue.
We have developed a multicohort analysis framework that combines heterogeneous gene expression datasets to identify robust parsimonious gene signatures in specific defined disease states such as pulmonary tuberculosis and influenza, and in clinically heterogeneous syndromes such as sepsis and organ transplant rejection (16–22). We thus hypothesized that a multicohort analysis of all publically available gene expression data in ARDS would identify robust gene expression signature capable of classifying this syndrome across diverse populations of critically ill patients.
METHODS
We carried out a systematic search for clinical studies of ARDS in two public gene expression repositories (National Institutes of Health [NIH] GEO, ArrayExpress) using the search terms: ARDS, respiratory distress syndrome, ALI, lung injury, ICU, mechanical ventilation. We excluded studies done in animals and of experimental lung injury in humans. ARDS was defined according to each included study; if a study included multiple gradations of lung injury, we used the Berlin criteria (23) definition. One study, GSE66099 (17), is from a cohort of pediatric sepsis (10, 11, 24, 25). In this study, lung injury cases were defined as children at ICU day 1 with a P/F ratio less than 200 but excluded those for whom the likely diagnosis was cyanotic heart disease. No chest radiographic findings were available for this dataset. Controls for GSE66099 were defined as patients with sepsis with concomitant respiratory failure, but not meeting the P/F ratio criterion for lung injury. In each study, cases were defined as the lung injury phenotypes described above, and controls were taken to match cases on the basis of major diagnosis. For instance, if the ARDS class only included septic patients, then only septic controls were used, but healthy or noninfected systemic inflammatory response syndrome was excluded. Only patients with gene expression data taken within 48 hours of the diagnosis of ARDS were included.
In addition to the publicly available clinical whole-blood datasets, we used the Inflammation and Host Response to Injury Program (Glue Grant) trauma and burn blood datasets (13–15). The Glue Grant datasets are drawn from three longitudinal cohorts, of which two profiled either buffy coat or sorted cells from patients with posttraumatic injury, and one profiled patients with postburn injury. As previously described (17), the samples were divided into subcohorts of time since injury (e.g., 1–3 or 3–6 d post injury), and cases and controls were compared only within these subcohorts. ARDS cases were defined according to Berlin criteria, and controls were defined as non-ARDS mechanically ventilated patients. Criteria were assumed to apply to a patient if they were noted within ±1 day of the sample being drawn. Infection was defined in these cohorts as previously described (17). Use of the Glue Grant was approved by both the Glue Grant Consortium and the Stanford University Institutional Review Board (protocol 29798).
Gene expression datasets that used Affymetrix arrays were gcRMA normalized using R package affy, and other arrays were quantile normalized using R package limma (26) if not already normalized. All microarray data were log-2 transformed, and probes were summarized to genes within datasets using a fixed-effect inverse variance model.
Multicohort analysis of gene expression was done as previously described (16–18, 21). Briefly, we applied two meta-analysis methods: one combining effect sizes using Hedges’ g, the other using Fisher’s sum-of-logs method combining p values. Genes set as “significant” passed a minimum false discovery rate (FDR) and a minimum effect size. We chose effect size thresholds that were relatively low so as to reduce false positives while still maintaining a high number of true positives (19).
Gene ontology enrichment was tested using the DAVID online tool (http://david.ncifcrf.gov/) with default variables. Bonferroni-corrected p value of less than 0.01 was set as the threshold of significance.
In addition to testing for differential gene expression in ARDS, we also performed metaregression analysis via synthesis-of-slopes to correct for clinical severity. For each cohort, the model was a regression on ARDS status (dependent) as a function of clinical severity and gene expression. We modeled each gene independently by running a separate regression for every gene present on the microarrays. To keep the scales between datasets similar, 1) the available clinical severity scores (Acute Physiology and Chronic Health Evaluation (APACHE) II, APACHE III, Pediatric Risk of Mortality, Denver score, and Multi-Organ Dysfunction Score) were converted to log-odds mortality based on models in their describing papers and 2) all datasets were ComBat-normalized together prior to meta-analysis and then analyzed separately. ComBat resets the location and scale of each gene but preserves within-cohort differences. The metaregression was performed using the closed-form method-of-moments random-effects model variation (27) of the synthesis-of-slopes regression method described by Becker and Wu (28). Thus, a gene was considered significant overall if it had statistically significant regression coefficients (betas) across cohorts for the prediction of mortality independent of clinical severity. The final list of p values was converted to FDR using Benjamini-Hochberg correction (29).
Cell type enrichment tests were performed as previously described (17, 22). Briefly, gene expression profiles of relevant immune and lung cell types in vitro were downloaded and conormalized. A gene signature of interest can then be tested for expression level in each in vitro cell type. The score is standardized across cell types, and a p value of the resulting Z score is calculated based on a normal assumption.
To determine a set of genes optimized for diagnostic power across the cohorts for any given analysis, we used a greedy forward search algorithm, as previously described (17, 18, 21). Briefly, the expression values of the derived gene set were converted to a single score by taking the difference of the geometric means of the positive genes and the negative genes. The resulting score was then evaluated for diagnostic power using AUC. The forward search starts with no genes and selects the single gene which best improves the AUC at each step, until no improvement in AUC is possible. Leave-one-dataset-out cross validation (LODOCV) was performed by leaving out one cohort from an analysis, performing meta-analysis on the remaining k–1 cohorts, followed by a forward search, and then testing the obtained forward search gene set in the left-out cohort. The LODOCV analysis is then repeated k times in a round-robin fashion. The mean LODOCV AUCs reported are thus the mean-of-means of k rounds of analysis.
RESULTS
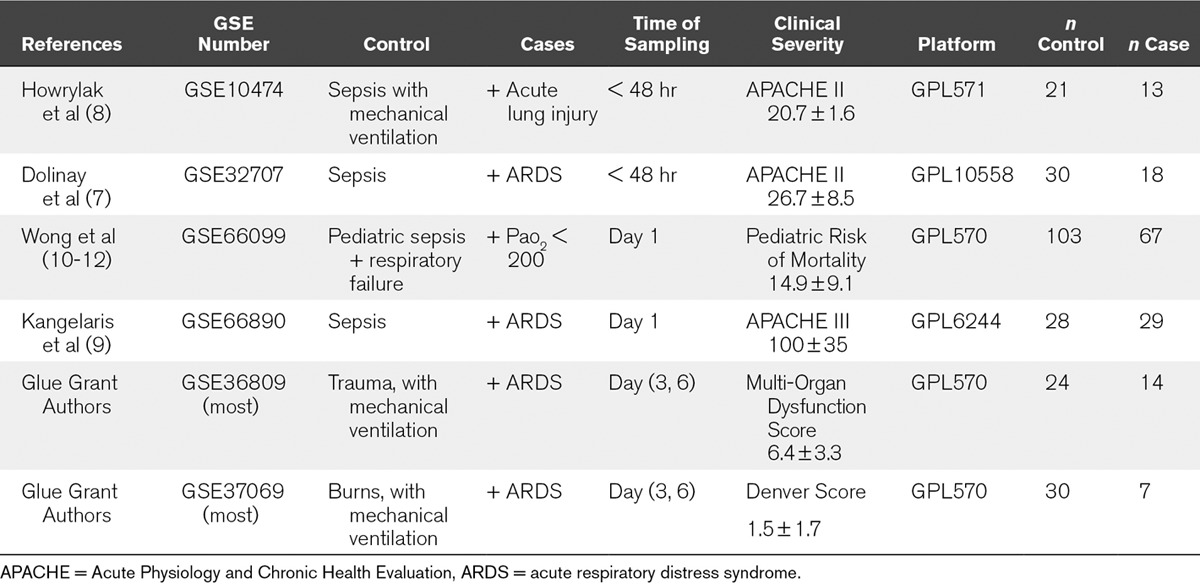
We first performed a systematic search of NIH GEO and ArrayExpress for clinical studies of ARDS or acute lung injury (ALI). We found three clinical whole-blood datasets which compared septic ARDS/ALI patients to septic patients without ARDS/ALI (GSE10474 [8], GSE32707 [7], GSE66890 [9]). In addition, we analyzed one large dataset of septic pediatric patients (GSE66099) for hypoxemia (P/F ratio < 200) not due to cyanotic heart disease. No chest radiographic data were available in this study, so the severe hypoxemia was assumed due to lung injury. Finally, the Glue Grant trauma and burn cohorts were sorted into patients with ARDS versus time-matched mechanically ventilated non-ARDS controls. In the Glue Grant cohorts, only patients at more than 72 hours after initial injury were included to avoid including patients with traumatic or inhalational lung injury. In combination, this yielded six whole-blood ARDS cohorts spanning a broad clinical spectrum (n controls = 236, n cases = 148) (Table 1).
TABLE 1.
Clinical Whole Blood Acute Respiratory Distress Syndrome/Acute Lung Injury Datasets Included in the Multicohort Analysis
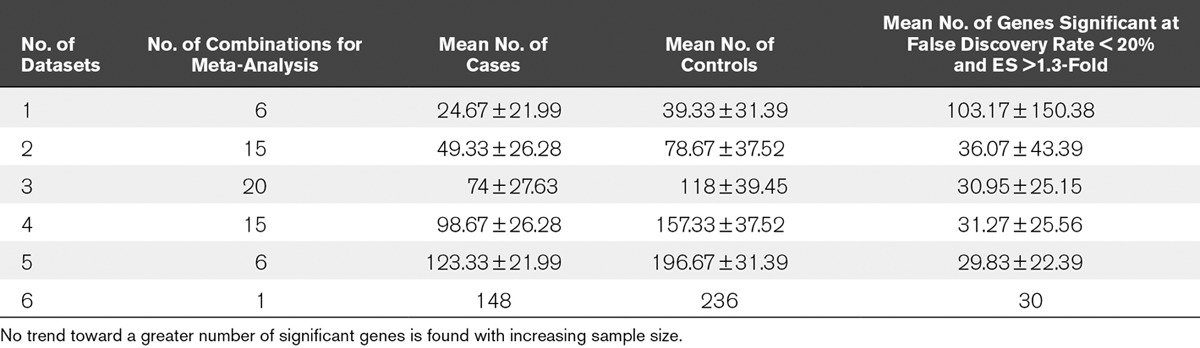
To find genes significantly differentially expressed in lung injury, we used all six datasets for a multicohort analysis as previously described (16–18, 21). We initially set the significance threshold at FDR less than 1% and effect size greater than 1.3-fold, but only nine genes were significant at this threshold. We thus relaxed the criteria to FDR less than 20% and effect size greater than 1.3-fold. This yielded significant 30 genes in the clinical studies using whole-blood samples (Table 2). When we restricted the analysis to only patients with sepsis by removing the Glue Grant trauma and burns datasets, the number of genes identified as significant at the same thresholds was reduced to 18. The number of significant genes reduced to 21 when we removed the pediatric dataset (GSE66099), and to 29 when we removed both trauma and pediatric datasets (leaving only adult sepsis datasets). We further ran meta-analyses for every possible subcombination of the six datasets (Table 3). We found that there was no increase in the average number of significant genes as more samples were included, indicating that adding further data is unlikely to significantly boost the number of significant genes identified. Out of the 30 differentially regulated genes in the clinical whole-blood studies, several genes have been shown to be differentially expressed in sepsis and septic shock, including MMP8, MPO, RETN, ELANE, DEFA1, TGFBI, LCN2, and TREM1 (17, 30–32). Only two Gene Ontology terms were found to be enriched at a Bonferroni p value of less than 0.01: “defense response to fungus”, and “killing of cells of other organism”.
TABLE 2.
Genes Found to be Significant at False Discovery Rate Less Than 20%, Effect Size Greater Than 1.3-Fold Comparing Acute Respiratory Distress Syndrome Versus Non–Acute Respiratory Distress Syndrome Critically Ill Controls Using Transcriptomics From Whole Blood
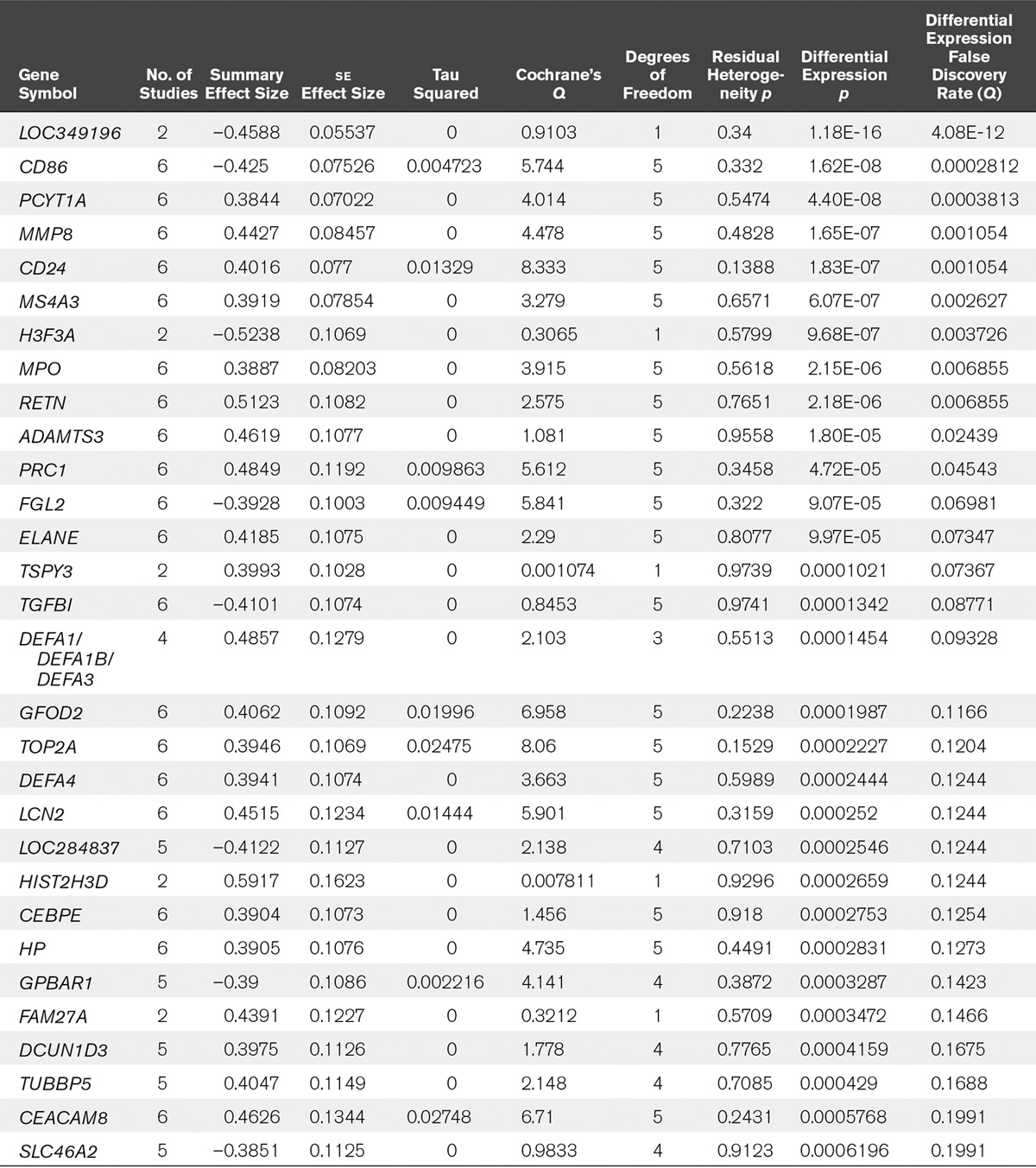
TABLE 3.
Meta-Analyses of All Possible Combinations of the Six Datasets Comparing Acute Respiratory Distress Syndrome Versus Non–Acute Respiratory Distress Syndrome Critically Ill Controls
In addition, we performed a metaregression via random-effects synthesis-of-slopes, assessing for differential expression related to ARDS status while correcting for the clinical severity scores associated with each cohort listed in Table 1. No genes were found to be significant at FDR less than 20%. Together, this suggests that any ARDS gene expression signature in whole blood is likely confounded by the generalized severe inflammation from sepsis and shock.
Changes in gene expression in a complex tissue such as whole blood can be due either to intrinsic changes in gene expression within cells, or to cell type shifts, or both. We thus tested the 30 significant genes for enrichment in several in vitro cell types from both the immune system and the lung (Fig. 1). The theory is that enrichment in a given in vitro cell type may indicate an overexpression of that cell type in the blood. The 30-gene ARDS signature was enriched in immature granulocytes (bands and metamyelocytes), which likely reflects the severity of systemic inflammation present in these patients. Notably, the clinical whole-blood signature showed a trend toward decreased inferred abundance in monocytes and M2-polarized macrophages, and as expected, no strong representation of pneumocytes.
Figure 1.
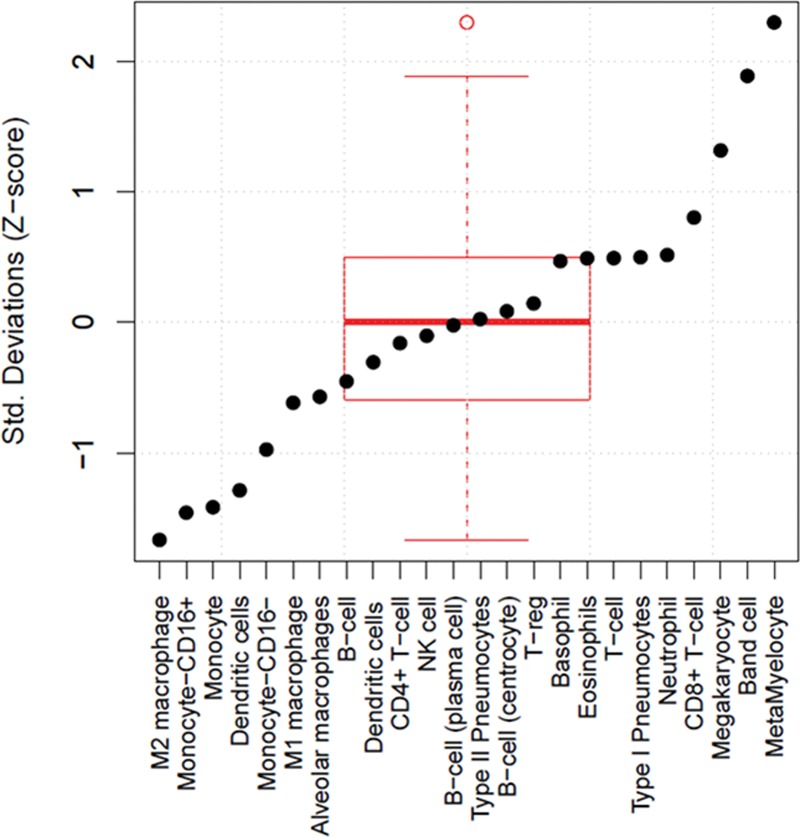
The 30 genes found to be significantly differentially expressed in acute respiratory distress syndrome (ARDS) were tested for enrichment in several in vitro cell catheters (both immune cell types and pneumocytes) to assess for the possibility of cell type enrichment in whole blood in ARDS NK = Natural Killer.
A blood diagnostic signature for ARDS would be a useful clinical tool. Therefore, we studied whether gene expression classifiers could robustly separate lung injury profiles. We thus used a greedy forward search algorithm (17, 18, 21) to search for a subset of the 30 significant genes that was maximized for diagnostic power. Generalizability was assessed with LODOCV application of the same method, since no independent validation datasets were available. The forward search conducted using all datasets produced a seven-gene set (up-regulated: ADAMTS3, MPO, HIST2H3D, TUBBP5, FAM27A, PRC1; down-regulated: LOC284837). However, the performance was poor even in the datasets across which it was discovered and in which is assumed to be overfit (mean AUC, 0.74; range, 0.60–0.85) (Fig. 2). Using the Youden method to select an optimal cutoff for each curve yielded a mean sensitivity of 63% and a mean specificity of 74%. Furthermore, the same procedure had a mean (± sd) LODOCV AUC of 0.63 ± 0.07, indicating that the seven-gene signature likely has poor generalizability for diagnosing ARDS. Finally, we performed LODOCV analyses for several subgroups of datasets. The subgroups and mean (± sd) LODOCV AUCs were 1) pediatric patients removed; mean LODOCV 0.54 ± 0.04, 2) trauma/burn datasets removed; mean LODOCV 0.59 ± 0.09, and 3) both pediatric and trauma/burn datasets removed, LODOCV 0.55 ± 0.09. Thus, choosing more homogeneous subgroups, albeit with smaller sample size, did not lead to improved discriminatory power either.
Figure 2.
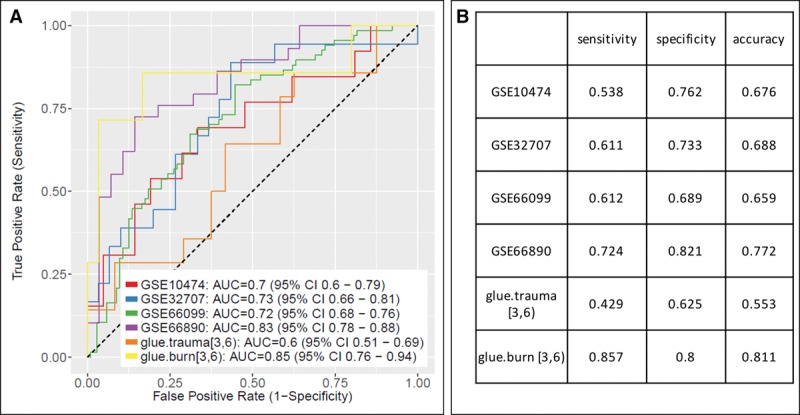
Discriminatory power for prediction of acute respiratory distress syndrome (ARDS) versus non-ARDS critically ill controls using transcriptomics from whole blood. A single seven-gene set was chosen via forward search. A, Receiver-operating characteristic (ROC) curves. B Test characteristics of each curve at its Youden cutoff. AUC = area under the ROC curve.
DISCUSSION
Better diagnostics and prognostics are needed for ARDS. While the Berlin and Pediatric Acute Lung Injury Consensus Conference criteria are reasonably clear, they do not necessarily reflect underlying pathophysiology (2, 23), and whether ARDS is best “lumped” as one disease with shared underlying pathophysiology, or “split” into multiple distinct endotypes, is unclear (4–6). We here used our established multicohort analytic framework on a comprehensive set of peripheral blood ARDS gene expression datasets across a very wide clinical spectrum to test whether we could identify shared ARDS genes and a diagnostic gene set. While we identified 30 differentially expressed genes at a relaxed significance threshold (FDR < 20%), many of these genes are known to be associated with sepsis severity and may not relate to ARDS pathophysiology per se. Further, when we adjusted for clinical severity score, none of these remained significant. When we paired down to the seven genes that best separated ARDS from non-ARDS across all datasets, this performed poorly when applied to any single population in cross validation, with a mean AUC of 0.63, little better than flipping a coin.
Why did our methods, which worked well in multiple other disease states, fail to identify a gene signature in ARDS? In this study, we purposely included studies from the broad clinical spectrum of disease, incorporating both children and adults (17, 18, 21, 33) and underlying ARDS risk factors including sepsis and trauma/burns. Broad inclusion allows the biggest question to be posed, namely, whether any whole-blood gene expression signature is generally true across patients with lung injury. While focused studies of (for instance) just adults with sepsis might yield a better classifier, restricting our analysis to just those more homogeneous cohorts did not yield a greater number of significant genes or a better classifier. In fact, we tested every possible subgrouping of datasets (thus encompassing every possible combination of “more similar” clinical circumstances) and never found more significant genes or a better classifier. Thus, our failure to find a robust classifier across all populations may represent evidence that there is not a unified pathophysiology across the wide clinical spectrum of ARDS, or at least none that can be captured from whole-blood gene expression. However, this does not preclude the possibility that focused studies on either individual cell types in the blood or narrowed clinical phenotypes could yield more accurate, albeit narrower, diagnostics.
A second possible reason for these negative results is the possibility that whole-blood gene expression, which largely reflects changes in leukocytes (which in the acute phase is often primarily neutrophils), may not carry diagnostic information about lung pathology. As anticipated, we did not find a strong signal for pneumocyte expression in whole blood, since whole-blood gene expression mostly reflects circulating leukocytes. This does not preclude the use of other techniques (such as metabolomics or proteomics) to study ARDS in peripheral blood, as many molecules or proteins may “leak” into the bloodstream from the lung but would not be detected via gene expression microarray (which measures mostly gene expression in leukocytes). Similarly, it is possible that gene expression studies of ARDS-relevant tissues (e.g., bronchoalveolar fluid) may be revealing (22).
Besides diagnostic signatures or the lack thereof, we found several important insights in these data. First, there was a significant signal in these gene expression datasets for immature granulocytes; this was true in ARDS patients of all etiologies (sepsis, burns, and trauma). The importance of neutrophil-related gene expression was noted in the Kangelaris et al study (GSE66890) (9), which specifically tested and found that this neutrophil-dominance is not related to differences in neutrophil count in patients with ARDS. We also tested for a signal of alveolar cells but could not find any; this confirmed our hypothesis that gene expression signal from the lung may be overwhelmed by the large inflammatory response in peripheral blood neutrophils.
One limitation of our study is that although we included all gene expression datasets published to date, the overall n remains moderately low (a total of 148 ARDS cases and 236 critically ill controls). However, we have shown in methodologic work that typically three to five datasets with a total n of 300 or more are typically enough to find reproducible differential gene expression signal if one exists (19). We previously showed that multicohort analyses of an equivalent sample size and number of datasets should yield between 200 and 2,000 significantly expressed genes, substantially more than the 30 we identified here, and were able to successfully build classifiers using similar numbers of samples when signal was present in myriad other diseases (16–18, 21). Another weakness is that we had access to only one pediatric dataset, so our multicohort strategy could not be applied to identify a gene signature specifically within pediatric ARDS.
Overall, the present study has several important findings. Despite using methods and sample sizes that have been used to build robust classifiers in other disease states including sepsis, we could not find a robust ARDS whole-blood gene signature across either a wide clinical spectrum or in narrower clinical subgroups. We show that whole-blood gene expression profiling in ARDS may be confounded by systemic inflammation, and that this likely contributes to the inability to find a generalizable diagnostic signature. Given the prior success of the multicohort analysis framework for discovering generalizable biomarkers (16–18, 21, 34), and the fact that including more clinical cohorts did not increase the number of significant genes found in common, our failure to find a robust discriminatory gene set here may be evidence of absence of a common gene signature in ARDS, instead of absence of evidence. This, in turn, may suggest that the syndrome of ARDS does not represent a single disease state. Overall, however, we see this as a positive step forward for the field; with this evidence of absence, the community can choose to study more specific subgroups of ARDS (e.g., pediatric ARDS alone or endotype-based studies), alternate tissues (e.g., specific circulating cell types, bronchoscopy washings, or lung biopsies), or using different methodologies (e.g., proteomics and metabolomics) in future work.
ACKNOWLEDGMENTS
We thank many authors who contributed the gene expression data reanalyzed here, without whom this would not have been possible. We thank Dr. Jake Hughey for independent statistical review. We thank the Glue Grant investigators for sharing their data publicly; they are supported in this by National Institute of General Medical Sciences Glue Grant Legacy Award R24GM102656.
Footnotes
Drs. Rogers and Khatri are cosenior authors.
Since the time of the article, Dr. Sweeney has moved to Inflammatix.
Drs. Sweeney, Rogers, and Khatri conceived the study. Dr. Sweeney carried out the analyses. Drs. Thomas, Howrylak, and Wong annotated the clinical pediatric data. All members revised the article and approved its final version.
Drs. Sweeney and Khatri received funding from Inflammatix (co-founders and stock). Dr. Thomas’ institution received funding from the Food and Drug Administration, and he received funding from Therabron and CareFusion. Dr. Wong’s institution received funding from the National Institutes of Health (NIH). Drs. Wong and Rogers received support for article research from the NIH. Dr. Rogers received funding from K23 HL125663. Dr. Khatri received funding from the National Institute of Allergy and Infectious Disease (grants 1U19AI109662, U19AI057229, and U54I117925) and the Bill and Melinda Gates Foundation, and he received support for article research from the NIH and the Bill and Melinda Gates Foundation. Dr. Howrylak disclosed that she does not have any potential conflicts of interest.
Data and Materials Availability: The Gene Expression Omnibus data can be found online using their accession numbers listed here. The Glue Grant data are publicly available pending institutional review board approval per the instructions found at https://www.gluegrant.org/. The data and code necessary to recreate the multicohort analysis have been deposited online at http://khatrilab.stanford.edu/sepsis; access to the data will be granted after approval by the Glue Grant consortium.
REFERENCES
- 1.Ferguson ND, Fan E, Camporota L, et al. The Berlin definition of ARDS: An expanded rationale, justification, and supplementary material. Intensive Care Med 2012; 38:1573–1582. [DOI] [PubMed] [Google Scholar]
- 2.Pediatric Acute Lung Injury Consensus Conference Grou: Pediatric acute respiratory distress syndrome: Consensus recommendations from the Pediatric Acute Lung Injury Consensus Conference. Pediatr Crit Care Med 2015;16:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellani G, Laffey JG, Pham T, et al. LUNG SAFE Investigators; ESICM Trials Group: Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016; 315:788–800. [DOI] [PubMed] [Google Scholar]
- 4.Prescott HC, Calfee CS, Thompson BT, et al. Toward smarter lumping and smarter splitting: Rethinking strategies for sepsis and acute respiratory distress syndrome clinical trial design. Am J Respir Crit Care Med 2016; 194:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calfee CS, Delucchi K, Parsons PE, et al. NHLBI ARDS Network: Subphenotypes in acute respiratory distress syndrome: Latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2014; 2:611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Famous KR, Delucchi K, Ware LB, et al. ARDS Network: Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med 2017; 195:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolinay T, Kim YS, Howrylak J, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med 2012; 185:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howrylak JA, Dolinay T, Lucht L, et al. Discovery of the gene signature for acute lung injury in patients with sepsis. Physiol Genomics 2009; 37:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kangelaris KN, Prakash A, Liu KD, et al. Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol Physiol 2015; 308:L1102–L1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong HR, Shanley TP, Sakthivel B, et al. Genomics of Pediatric SIRS/Septic Shock Investigators: Genome-level expression profiles in pediatric septic shock indicate a role for altered zinc homeostasis in poor outcome. Physiol Genomics 2007; 30:146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong HR, Cvijanovich N, Allen GL, et al. Genomics of Pediatric SIRS/Septic Shock Investigators: Genomic expression profiling across the pediatric systemic inflammatory response syndrome, sepsis, and septic shock spectrum. Crit Care Med 2009; 37:1558–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong HR, Cvijanovich NZ, Hall M, et al. Interleukin-27 is a novel candidate diagnostic biomarker for bacterial infection in critically ill children. Crit Care 2012; 16:R213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seok J, Warren HS, Cuenca AG, et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program: Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2013; 110:3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren HS, Elson CM, Hayden DL, et al. Inflammation and Host Response to Injury Large Scale Collaborative Research Program: A genomic score prognostic of outcome in trauma patients. Mol Med 2009; 15:220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao W, Mindrinos MN, Seok J, et al. Inflammation and Host Response to Injury Large-Scale Collaborative Research Program: A genomic storm in critically injured humans. J Exp Med 2011; 208:2581–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khatri P, Roedder S, Kimura N, et al. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J Exp Med 2013; 210:2205–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sweeney TE, Shidham A, Wong HR, et al. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci Transl Med 2015; 7:287ra71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sweeney TE, Braviak L, Tato CM, et al. Genome-wide expression for diagnosis of pulmonary tuberculosis: A multicohort analysis. Lancet Respir Med 2016; 4:213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sweeney TE, Haynes WA, Vallania F, et al. Methods to increase reproducibility in differential gene expression via meta-analysis. Nucleic Acids Res 2017; 45:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sweeney TE, Perumal TM, Henao R, et al. Mortality prediction in sepsis via gene expression analysis: A community approach. bioRxiv. 2016 doi: 10.1038/s41467-018-03078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sweeney TE, Wong HR, Khatri P.Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci Transl Med 2016; 8:346ra91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sweeney TE, Lofgren S, Khatri P, et al. Gene expression analysis to assess the relevance of rodent models to human lung injury. Am J Respir Cell Mol Biol 2017; 57:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012; 307:2526–2533. [DOI] [PubMed] [Google Scholar]
- 24.Wong HR, Cvijanovich N, Lin R, et al. Identification of pediatric septic shock subclasses based on genome-wide expression profiling. BMC Med 2009; 7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong HR, Freishtat RJ, Monaco M, et al. Leukocyte subset-derived genomewide expression profiles in pediatric septic shock. Pediatr Crit Care Med 2010; 11:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smyth G.Gentleman R CV, Dudoit S, Irizarry R, Huber W.Limma: Linear models for microarray data. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. 2005, pp New York, NY, Springer, 397–420. [Google Scholar]
- 27.Chen H, Manning AK, Dupuis J.A method of moments estimator for random effect multivariate meta-analysis. Biometrics 2012; 68:1278–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker B, Wu M.The synthesis of regression slopes in meta-analysis. Statistical Science 2007; 22:414–429. [Google Scholar]
- 29.Benjamini Y, Hochberg Y.Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B 1995; 57:289–300. [Google Scholar]
- 30.Almansa R, Heredia-Rodríguez M, Gomez-Sanchez E, et al. Transcriptomic correlates of organ failure extent in sepsis. J Infect 2015; 70:445–456. [DOI] [PubMed] [Google Scholar]
- 31.Tsalik EL, Langley RJ, Dinwiddie DL, et al. An integrated transcriptome and expressed variant analysis of sepsis survival and death. Genome Med 2014; 6:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sweeney TE, Wong HR.Risk stratification and prognosis in sepsis: What have we learned from microarrays? Clin Chest Med 2016; 37:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andres-Terre M, McGuire HM, Pouliot Y, et al. Integrated, multi-cohort analysis identifies conserved transcriptional signatures across multiple respiratory viruses. Immunity 2015; 43:1199–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen R, Khatri P, Mazur PK, et al. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res 2014; 74:2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]