

ABSTRACT
Recently, we reported that homozygous deletion of alternative exon 33 of CaV1.2 calcium channel in the mouse resulted in ventricular arrhythmias arising from increased CaV1.2Δ33 ICaL current density in the cardiomyocytes. We wondered whether heterozygous deletion of exon 33 might produce cardiac phenotype in a dose-dependent manner, and whether the expression levels of RNA splicing factors known to regulate alternative splicing of exon 33 might change in human heart failure. Unexpectedly, we found that exon 33+/− cardiomyocytes showed similar CaV1.2 channel properties as wild-type cardiomyocyte, even though CaV1.2Δ33 channels exhibit a gain-in-function. In human hearts, we found that the mRNA level of splicing factor Rbfox1, but not Rbfox2, was downregulated in dilated cardiomyopathy, and CACNA1C mRNA level was dramatically decreased in the both of dilated and ischemic cardiomyopathy. These data imply Rbfox1 may be involved in the development of cardiomyopathies via regulating the alternative splicing of CaV1.2 exon 33. (149 words)
KEYWORDS: alternative splicing, cardiomyopathy, CaV1.2 calcium channel, heterozygous knockout, Rbfox
Introduction
CaV1.2 L-type calcium channels have essential roles in the cardiac excitation-contraction coupling and development. Alternative splicing in CACNA1C, encoding the CaV1.2 pore-forming subunit α1C, modulates the function of CaV1.2 calcium channels. Exon 33 has been identified in human and rodent CaV1.2 α1C subunit by transcript-scanning;1,2 this alternative exon has been reported to affect the biochemical and biophysical functions of heterologously expressed CaV1.2 channels.1–6 Recently, we generated CaV1.2 exon 33 specific knockout mice (exon 33−/−) and these exon 33-null mice were found to develop cardiac arrhythmia and dysfunction, owing to increased CaV1.2 channel currents arising from a leftward shift of voltage-dependent activation and inactivation potentials as compared to wild-type (WT) (exon 33+/+) cardiomyocytes.7 Thus, we asked whether cardiomyocytes isolated from heterozygous exon 33-knockout mice might produce intermediate changes in biophysical properties of the CaV1.2 channels and how this might affect excitability of the cardiomyocytes.
In our previous work, we found that the expression of CaV1.2 alternative exon 33 was significantly increased in human failing hearts,7 and these data raise a potential relevance to clinical management of heart failure. A second question we asked was the mechanism by which alternative splicing of exon 33 of CaV1.2 channels might be regulated in the heart. The (U)GCAUG elements, which can be recognized and bound by RNA binding protein Rbfox1/2, were identified in the intronic sequence surrounding CaV1.2 exon 33.8 Functionally, Rbfox1/2 could enhance the inclusion of alternative exon 33 of CaV1.2 calcium channels.8,9 To date, Rbfox1/2 was reported to be crucial in cardiac development and different cardiomyopathies via regulation of serial splicing events,10–14 indicating a plausible role in the regulation of alternative exon 33 of CaV1.2 calcium channels in the heart. In this follow-up study of our previous work, we measured Rbfox1/2 mRNA levels in human failing and non-failing heart samples, in order to explore the possible association between the expressions of Rbfox and exon 33 of CaV1.2 in cardiomyopathies.
Results
Exon 33+/− cardiomyocyte has similar CaV1.2 channel properties with WT cardiomyocyte
In the exon 33+/− ventricular tissue, the upper band density of CaV1.2 channels with inclusion of exon 33 (CaV1.233) is similar with the lower band, which are the CaV1.2 channels with exon 33 skipping (CaV1.2Δ33),7 indicating ∼50% CaV1.2Δ33 channels in exon 33+/− hearts, compared with ∼7.6% CaV1.2Δ33 channels in WT hearts, and 100% CaV1.2Δ33 channels in exon 33−/− hearts.7 To investigate the CaV1.2 current properties of the exon 33+/− cardiomyocytes, we performed voltage-clamp recordings in isolated ventricular myocytes. Unexpectedly, the current density of exon 33+/− cardiomyocyte is almost same with WT cardiomyocyte, but much smaller than exon 33−/− cardiomyocyte (Fig. 1A–C). Moreover, the current-voltage (I-V) relationship curve (Fig. 1D) and steady-state inactivation potential (Fig. 1E) of exon 33+/− cardiomyocyte were also similar with WT cardiomyocyte, but rightward shifted compared with exon 33−/− cardiomyocyte. Taken together, our data indicated that heterozygous deletion of alternative exon 33 produced half the Δexon33-containing CaV1.2 transcripts but functionally the biophysical properties of the CaV1.2 Ca2+ currents recorded in CaV1.2 exon 33+/− cardiomyocytes were similar to WT. As we could not raise an antibody to specifically detect exon 33, we are unable to determine the levels of surface expressions of CaV1.233 or CaV1.2Δ33 channels.
Figure 1.
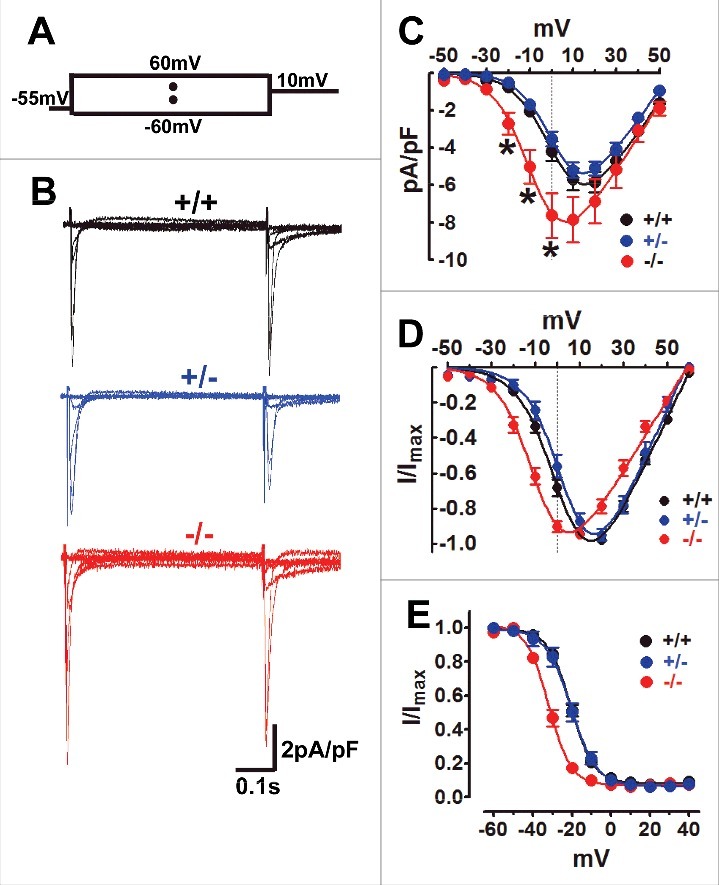
Exon 33+/− cardiomyocyte has similar CaV1.2 channel properties with WT cardiomyocyte. (A) The stimulus waveform was used to induce ICa of cardiomyocyte, briefly ICa was recorded under the different testing potentials, increased from −60 mV to 60 mV (10 mV increase each step) followed by 10 mV depolarizing-pulse to induce steady-state inactivation in cardiomyocytes when using the 1.8 mmol/L Ca2+ as charger carrier. (B) Exemplary current traces were recorded from WT (black), exon 33+/− (blue) or exon 33−/− (red) cardiomyocytes. (C) Current densities of CaV1.2 channels in WT (n = 11 cells, V0.5 = 2.82±2.3 mV), exon 33+/− (n = 20 cells, V0.5 = 3.18±1.7 mV) and exon 33−/− (n = 18 cells, V0.5 = −7.28±3.3 mV) cardiomyocytes (*P<0.05 vs. WT, unpaired t test). (D) Normalized current-voltage relationships of CaV1.2 channels in WT, exon 33+/− and exon 33−/− cardiomyocytes. (E) Steady-state inactivation of CaV1.2 channels in WT (n = 16 cells, V0.5,inact = −20.63±0.44 mV), exon 33+/− (n = 14 cells, V0.5,inact = −20.46±0.85 mV) and exon 33−/− (n = 12 cells, V0.5,inact = −31.94±0.64 mV) cardiomyocytes.
Exon 33+/− cardiomyocyte shows normal membrane excitation
Further, we explored the properties of cardiomyocyte excitation under current-clamp recording. Unlike exon 33−/− cardiomyocyte, the action potential duration (APD) after 90% repolarization (APD90%) of exon 33+/− cardiomyocyte was not increased but was similar to WT cardiomyocyte (Fig. 2A–B). This result could be explained by the similar characterized current properties between exon 33+/− and WT cardiomyocyte (Fig. 1C–E). Following that, we also measured the early after-depolarization (EAD) and autonomous action potentials (APs), two hallmarks of cardiac arrhythmias.15,16 Our data showed the occurrence of EAD in exon 33+/− cardiomyocytes (∼3%) had no differences as compared with WT cardiomyocytes (∼5%), while exon 33−/− cardiomyocytes had a significant increase of EAD occurrence (∼18%) (Fig. 2C–D). Another indicator of cardiac arrhythmia is the generation of autonomous APs after the cessation of electrical stimulation, exon 33−/− cardiomyocytes produced much higher frequency of autonomous APs (∼36%) as previously indicated,7 but exon 33+/− cardiomyocytes showed the similar low frequency of autonomous APs as compared with WT cardiomyocytes (Fig. 2E–F). In sum, exon 33+/− cardiomyocytes did not show any signs of abnormal membrane excitations, such as EAD and autonomous APs, indicating heterozygous knockout of alternative exon 33 of CaV1.2 calcium channels might be not contribute to cardiac arrhythmia.
Figure 2.
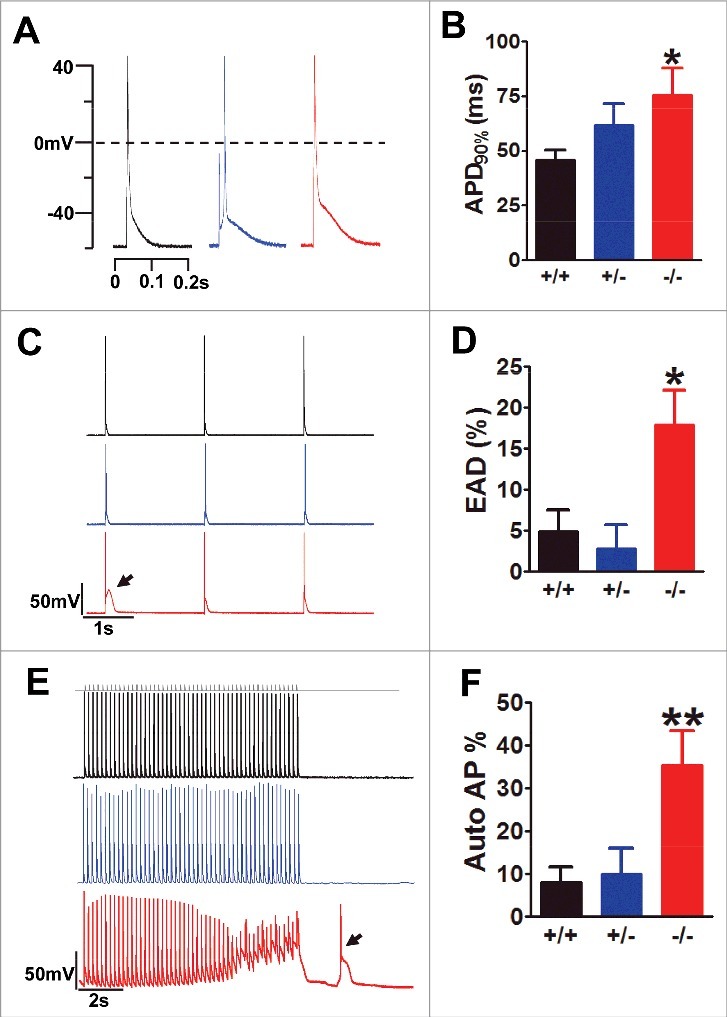
Exon 33+/− cardiomyocyte did not show any signs of abnormal excitations. (A) AP waveforms recorded from WT (black), exon 33+/− (blue) and exon 33−/− (red) cardiomyocytes. (B) APD after 90% of repolarization (APD90%) from WT (n = 11 cells), exon 33+/− (n = 14 cells) and exon 33−/− (n = 16 cells) (*P<0.05 vs. WT, unpaired t test). (C and D) Detection of EADs in cardiomyocytes of WT (n = 73 cells, 17 mice), exon 33+/− (n = 24 cells, 5 mice) or exon 33−/− mice (n = 73 cells, 16 mice) at a 0.5-Hz pacing rate. (P = 0.0139 one-way ANOVA; *P<0.05 vs. WT, Bonferroni post hoc test). (E and F) Detection of autonomous APs in cardiomyocytes from WT (n = 73 cells, 17 mice), exon 33+/− (n = 24 cells, 5 mice) and exon 33−/− (n = 73 cells, 16 mice) a 5-Hz pacing rate (P = 0.0052, one-way ANOVA; **P<0.01 vs. WT, Bonferroni post hoc test).
Rbfox1 expression is correlated with exon 33 and CACNA1C mRNA levels in human hearts
To address the possible pathological relevance, we collected human non-failing and failing heart samples7 to measure RBFOX1/2 and CACNA1C mRNA expression. Specifically, Rbfox1 expression in dilated cardiomyopathy (DCM) failing hearts was significantly lower than non-failing hearts (Fig. 3A). However, Rbfox2 expression had no significant differences among non-failing, dilated and ischemic cardiomyopathy (ICM) failing hearts (Fig. 3B). Moreover, CACNA1C mRNA expression was dramatically downregulated in both dilated and ischemic cardiomyopathy failing hearts (Fig. 3C), implying that decreased CaV1.2 calcium channel may also be involved in the induction of hypertrophy and/or heart failure.17 More interestingly, we found that Rbfox1 expression was negatively correlated with exon 33 inclusion (Fig. 3D), but positively correlated with CACNA1C mRNA expression in human hearts (Fig. 3E), these suggest dysregulated Rbfox1 might play some roles in the regulation of human CaV1.2 expression. Therefore, it is reasonable to believe that altered Rbfox1 expression may contribute to the changes in expression and alternative splicing of CaV1.2 calcium channels that led to lower channel activity in failing hearts.
Figure 3.
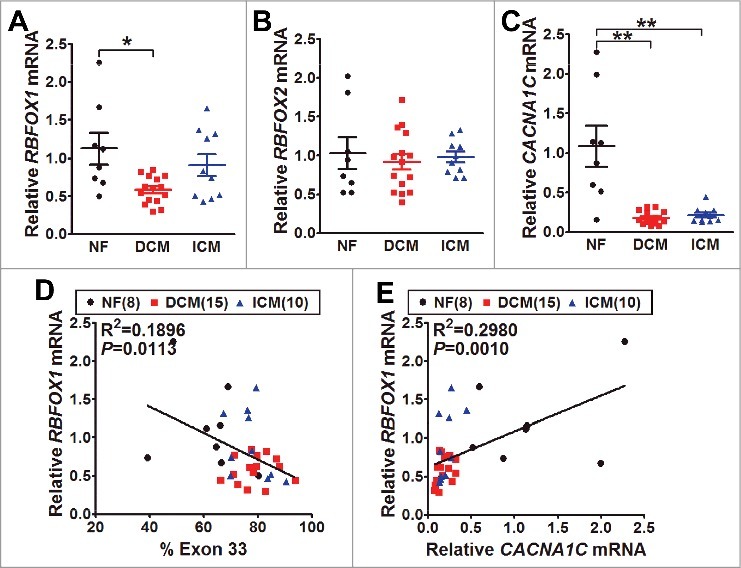
The mRNA expression of Rbfox1/2 and CaV1.2 α1C in human hearts. (A) The mRNA levels of Rbfox1 were measured by real-time RT-PCR from the samples of human normal (NF, black, n = 8), dilated cardiomyopathy (DCM, red, n = 15) and ischemic cardiomyopathy (ICM, blue, n = 10) hearts. The relative expression was normalized by internal expression of human GAPDH and RPLPO mRNA (P = 0.0103, one-way ANOVA; *P<0.05 vs. WT, Bonferroni post hoc test). (B) The mRNA levels of Rbfox2 were also measured by real-time RT-PCR from NF (n = 8), DCM (n = 15) and ICM (n = 10) hearts. (C) CaV1.2 α1C mRNA expression was measured by real-time RT-PCR from NF (n = 8), DCM (n = 15) and ICM (n = 10) hearts (**P<0.01, one-way ANOVA; **P<0.01 vs. WT, Bonferroni post hoc test). The correlations between alternative exon 33 expression and Rbfox1 (D) or CaV1.2 α1C (E) mRNA level in human hearts were analyzed by linear regression, R2 refers to the coefficient of determination.
Discussion
Alternative splicing of CaV1.2 calcium channel is recognized as an important post-transcriptional modification that provides modulation of channel function. Our previous work has directly addressed the in vivo significance of altered CaV1.2 calcium channel property arising from alternative splicing of exon 33 that showed the deletion of exon 33 induced enhanced channel activity and abnormal cardiomyocyte excitation. This process in turn resulted in ventricular arrhythmia and cardiac dysfunction in mice.7 In normal rodent hearts, the proportion of CaV1.2 calcium channels with inclusion of exon 33 is more than 90%.2-4,7 As such, what could the phenotype of the heart be if this proportion decreases to ∼50%? Here, we investigated that 50% expression at the transcript level of exon 33 in CaV1.2 channel in heterozygous knockout mice did not produce any significant changes in channel properties and therefore not surprisingly no gross differences in cardiac electrical properties in cardiomyocytes. Our data raised the question on how the presence of CaV1.233 could completely override the contribution of CaV1.2Δ33 channels in cardiomyocytes. There are several possibilities to explain this phenotype. First, CaV1.2 channels with exon 33 (CaV1.233) make a chief role in the excitation-contraction coupling in the normal heart, and a small portion of CaV1.233 channels is sufficient to maintain the regular activities of CaV1.2 channels. Second, CaV1.2 α1C subunits are known to be bound with CaVβ subunits to traffic them to cell membrane;18 though CaV1.2Δ33 channels are transcribed in cardiomyocytes, the trafficking of these CaV1.2Δ33 channels might be disturbed when competing with CaV1.233 channels, thus reducing the contribution of CaV1.2Δ33 channels on the cell surface in heterozygous knockout cardiomyocytes. Third, as oligomerization of CaV1.2 channels can form a “coupled gating” function in excitable cells,19,20 CaV1.2 channels might have a “selective ability” to form the oligomerization with neighboring CaV1.233 channels, but not CaV1.2Δ33 channels in cardiomyocyte; thus the exclusion of alternative exon 33 may affect the oligomerization of CaV1.2 channels. Nevertheless, the detailed mechanisms of CaV1.233-CaV1.2Δ33 channel interaction are warranted for further investigation.
Another issue is which mechanism(s) regulates the specific expression pattern of alternative exon 33 of CaV1.2 calcium channels in human hearts. The possible candidate is Rbfox1/2, which belong to the RNA-binding proteins,21 it has been known to directly enhance the inclusion of alternative exon 33 of CaV1.2 channel during neuronal development.8 Therefore, what is the relevance of Rbfox1/2 in the pathology of cardiac diseases? In this work, we found the expression of Rbfox1 was dramatically decreased in human DCM when compared with non-failing hearts, which is consistent with Gao's report in which they indicated the expression of Rbfox1 was markedly diminished in human DCM and in transverse aortic restriction-induced murine heart failure.12 These imply Rbfox1 indeed takes part in the alternative splicing regulation of diseased hearts. Although Rbfox1 was reported to enhance the inclusion of CaV1.2 alternative exon 33 in rodent8, in contrast we found the expression of Rbfox1 was negatively correlated with the expression of exon 33 in human hearts. This result could be considered as the increased expression of CaV1.233 channels could be a compensatory response to heart failure in humans, but we cannot exclude other potential splicing factors that might directly or indirectly regulate the alternative splicing of CaV1.2 exon 33 in human heart. Here, we also found the dramatic decrease of CaV1.2 α1C mRNA in human DCM and ICM hearts, in line with the reduced activities of CaV1.2 channels in human failing hearts22. Moreover, the expression of Rbfox1 was positively correlated with the expression of CaV1.2 calcium channels in human hearts by our study. Therefore, induction of Rbfox1 expression might have some beneficial effects on the cardiomyopathies.12
In conclusion, 50% loss of alternative exon 33 in CaV1.2 calcium channel did not affect the electrophysiological properties of CaV1.2 channels recorded in mouse cardiomyocytes, and Rbfox1 might be a modulator of exon 33 alternative splicing in human heart. Nevertheless, further studies are required to clarify the molecular mechanisms how the presence of exon 33 affect the structure and function of CaV1.2 channels and what upstream signals may be important to regulate Rbfox expression.
Materials and methods
Human samples and animals
Human heart samples and knockout mice of alternative exon 33 of CaV1.2 (exon 33−/−) were obtained or generated as described previously.7 Exon 33−/− mice were crossed with C57BL/6J mice to generate heterozygous knockout mice (exon 33+/−). The genotype was determined by PCR method. Isolation of cardiomyocytes was also described in our previous report.7 All animals were treated ethically in accordance with approved institutional IACUC protocol of the National University of Singapore.
RT-PCR
Total RNA was extracted using Trizol (Invitrogen) as indicated in manufacturer's protocol. Reverse transcription was performed using Superscript III (Invitrogen). Real-time PCR was used in quantitative analysis of the Rbfox1/2 and CaV1.2 channel mRNA expression level. FAM labeled CACNA1C probe located at the junction of constitutive exon 3 and exon 4 (Assay ID Hs00167681_m1, Applied Biosystems), and RBFOX1/2 (Assay ID Hs01125659_m1 and Hs00204814_m1, Applied Biosystems) were used to measure the standard mRNA expressions. Human GAPDH (Assay ID 4333764T, Applied Biosystems) and RPLPO (large ribosomal protein) (Assay ID 4333761T, Applied Biosystems) were used as endogenous control (FAM™ Dye/MGB Probe, Applied Biosystems).
Electrophysiology
Whole-cell L-type calcium current and action potentials of cardiomyocytes were recorded using the patch-clamp technique as previously described.7
Statistical analysis
Data is reported as mean ± S.E.M. Statistical significance was analyzed using a student t test, or one-way ANOVA followed by post hoc multiple comparisons test. The relationships between Rbfox1 expression and exon 33 or CaV1.2 expression were analyzed by linear regression. A value of P < 0.05 was considered as statistical significance.
Funding Statement
The research was supported by funding from the Singapore National Medical Research Council and Biomedical Research Council to TWS.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Tang ZZ, Liang MC, Lu S, Yu D, Yu CY, Yue DT, Soong TW. Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J Biol Chem. 2004;279:44335–43. doi: 10.1074/jbc.M407023200. PMID:15299022. [DOI] [PubMed] [Google Scholar]
- 2.Tang ZZ, Hong X, Wang J, Soong TW. Signature combinatorial splicing profiles of rat cardiac- and smooth-muscle Cav1.2 channels with distinct biophysical properties. Cell Calcium. 2007;41:417–28. doi: 10.1016/j.ceca.2006.08.002. PMID:16979758. [DOI] [PubMed] [Google Scholar]
- 3.Tang ZZ, Liao P, Li G, Jiang FL, Yu D, Hong X, Yong TF, Tan G, Lu S, Wang J, et al.. Differential splicing patterns of L-type calcium channel Cav1.2 subunit in hearts of Spontaneously Hypertensive Rats and Wistar Kyoto Rats. Biochim Biophys Acta. 2008;1783:118–30. doi: 10.1016/j.bbamcr.2007.11.003. PMID:18070605. [DOI] [PubMed] [Google Scholar]
- 4.Liao P, Li G, Yu DJ, Yong TF, Wang JJ, Wang J, Soong TW. Molecular alteration of Ca(v)1.2 calcium channel in chronic myocardial infarction. Pflugers Arch. 2009;458:701–11. doi: 10.1007/s00424-009-0652-4. PMID:19263075. [DOI] [PubMed] [Google Scholar]
- 5.Liao P, Yu D, Li G, Yong TF, Soon JL, Chua YL, Soong TW. A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J Biol Chem. 2007;282:35133–42. doi: 10.1074/jbc.M705478200. PMID:17916557. [DOI] [PubMed] [Google Scholar]
- 6.Cheng X, Pachuau J, Blaskova E, Asuncion-Chin M, Liu J, Dopico AM, Jaggar JH. Alternative splicing of Cav1.2 channel exons in smooth muscle cells of resistance-size arteries generates currents with unique electrophysiological properties. Am J Physiol Heart Circ Physiol. 2009;297:H680–8. doi: 10.1152/ajpheart.00109.2009. PMID:19502562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li G, Wang J, Liao P, Bartels P, Zhang H, Yu D, Liang MC, Poh KK, Yu CY, Jiang F, et al.. Exclusion of alternative exon 33 of CaV1.2 calcium channels in heart is proarrhythmogenic. Proc Natl Acad Sci U S A. 2017;114:E4288–E95. doi: 10.1073/pnas.1617205114. PMID:28490495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang ZZ, Zheng S, Nikolic J, Black DL. Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol Cell Biol. 2009;29:4757–65. doi: 10.1128/MCB.00608-09. PMID:19564422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damianov A, Black DL. Autoregulation of Fox protein expression to produce dominant negative splicing factors. RNA. 2010;16:405–16. doi: 10.1261/rna.1838210. PMID:20042473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blech-Hermoni Y, Ladd AN. RNA binding proteins in the regulation of heart development. Int J Biochem Cell Biol. 2013;45:2467–78. doi: 10.1016/j.biocel.2013.08.008. PMID:23973289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frese KS, Meder B, Keller A, Just S, Haas J, Vogel B, Fischer S, Backes C, Matzas M, Köhler D, et al.. RNA splicing regulated by RBFOX1 is essential for cardiac function in zebrafish. J Cell Sci. 2015;128:3030–40. doi: 10.1242/jcs.166850. PMID:26116573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao C, Ren S, Lee JH, Qiu J, Chapski DJ, Rau CD, Zhou Y, Abdellatif M, Nakano A, Vondriska TM, et al.. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J Clin Invest. 2016;126:195–206. doi: 10.1172/JCI84015. PMID:26619120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, Lozano MJ, Abass IJ, Ijaz T, Brasier AR, et al.. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep. 2016;15:2200–13. doi: 10.1016/j.celrep.2016.05.002. PMID:27239029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei C, Qiu J, Zhou Y, Xue Y, Hu J, Ouyang K, Banerjee I, Zhang C, Chen B, Li H, et al.. Repression of the Central Splicing Regulator RBFox2 Is Functionally Linked to Pressure Overload-Induced Heart Failure. Cell Rep. 2015. pii: S2211-1247(15)00141-2. doi: 10.1016/j.celrep.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogeholz N, Pauls P, Bauer BK, Schulte JS, Dechering DG, Frommeyer G, Kirchhefer U, Goldhaber JI, Müller FU, Eckardt L, et al.. Suppression of Early and Late Afterdepolarizations by Heterozygous Knockout of the Na+/Ca2+ Exchanger in a Murine Model. Circ Arrhythm Electrophysiol. 2015;8:1210–8. doi: 10.1161/CIRCEP.115.002927. PMID:26338832. [DOI] [PubMed] [Google Scholar]
- 16.Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–9. doi: 10.1016/j.hrthm.2010.09.017. PMID:20868774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, et al.. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122:280–90. doi: 10.1172/JCI58227. PMID:22133878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel beta-subunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature 1994;368:67–70. doi: 10.1038/368067a0. PMID:7509046. [DOI] [PubMed] [Google Scholar]
- 19.Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010;106:748–56. doi: 10.1161/CIRCRESAHA.109.213363. PMID:20110531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc Natl Acad Sci U S A. 2012;109:1749–54. doi: 10.1073/pnas.1116731109. PMID:22307641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuroyanagi H. Fox-1 family of RNA-binding proteins. Cell Mol Life Sci. 2009;66:3895–907. doi: 10.1007/s00018-009-0120-5. PMID:19688295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Piacentino V 3rd, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91:517–24. doi: 10.1161/01.RES.0000033988.13062.7C. PMID:12242270. [DOI] [PubMed] [Google Scholar]