

Abstract
EBV transforms small resting primary B cells into large lymphoblastoid cells which are able to grow and survive in vitro indefinitely. These cells represent a model for oncogenesis. In this unit, variants of conventional Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR), namely the activation (CRISPRa) and interference (CRISPRi) methods, are discussed in the context of gene regulation at promoter and enhancer elements on genomic DNA. LCLs stably expressing dCas9-VP64 (Cas9 associated with CRISPRa) or dCas9-KRAB (Cas9 associated with CRISPRi) are transduced with lentivirus that encode a single guide RNA (sgRNA) that targets a specific gene locus. The ribonucleoprotein complex formed by the dCas9 molecule and its cognate sgRNA enables sequence-specific binding at a promoter or enhancer of interest to drive epigenetic changes in its vicinity, which in turn affects the expression of genes regulated by the targeted promoter or enhancer.
Keywords: CRISPR, transcription regulation, Epstein-Barr virus, enhancer, promoter
Introduction
EBV infects primary human B cells to produce transformed lymphoblastoid B cells (McFadden et al., 2016; Schneider & zur Hausen, 1975). EBV-positive lymphoblastoid B cells can be established as separate clonal populations termed lymphoblastoid B cell lines (LCLs) that can be used for a number of purposes, such as maintaining renewable sources of nucleic acids from healthy and diseased human individuals and allowing the study of virus-host interactions in the context of oncogenesis.
Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR) technologies are becoming increasingly popular for genome editing and manipulation. With conventional CRISPR, the programmable endonuclease Cas9 and target-specific single-guide RNAs (sgRNAs) associate and engage target DNA to achieve gene-specific knockout (Jinek et al., 2012).
CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) are extensions of the conventional CRISPR knockout system. Instead of wild-type Cas9, engineered variants of a nuclease-deficient Cas9 (dCas9) carrying the D10A and H840A mutations are used (Gilbert et al., 2013; Qi et al., 2013). In CRISPRa, dCas9 is fused to a general transcription activation protein domain, such as herpes simplex virus (HSV) viral protein 16 (VP16) activation domain (known as VP64) or the nuclear factor of kappa-B (NF-κB)-p65 activating domain (p65AD) (Maeder et al., 2013; Perez-Pinera et al., 2013). Because dCas9-VP64 and dCas9-p65AD are incapable of inducing DNA cleavage, when programmed with an sgRNA, they bind to cognate DNA sequences to recruit factors that promote gene transcription. On the other hand, CRISPRi involves dCas9 that is fused to Krüppel-associated box (KRAB), a repressor domain that silences transcription. When dCas9-KRAB is in complex with sgRNA and engages cognate sequences on DNA, transcription repression happens through steric hindrance and exclusion of transcription activators and via the interaction between the KRAB domain and KRAB-associated protein 1 (KAP1) that results in the recruitment of histone deacetylases, histome lysine-methyltransferases and chromatin remodeling proteins (Lupo et al., 2013). CRISPRa and CRISPRi are particularly useful in instances where knockout of a certain gene results in rapid onset of death that precludes extended experimental analysis. They are also useful for precise manipulation of gene dosage. One other application for both CRISPRa and CRISPRi is that they can be used to modulate non-coding RNA levels, a lacking aspect of wild type Cas9 (Ghosh, Tibbit, & Liu, 2016; Joung et al., 2017). Finally, CRISPRa also has utility as a tool for gain-of-function experiments (Konermann et al., 2015).
This Unit provides detailed procedures for CRISPRa and CRISPRi in LCLs, complementary to wild-type Cas9 methods described in Jiang et al. (CPMB Unit 31.12). In general, pooled lymphoblastoid B cells stably expressing chimeric dCas9 proteins can be used for experimentation. However, if the user should desire to establish dCas9 clones for reduced experimental variability, Basic Protocol 1 describes the process for doing so. Basic Protocol 2 describes the procedure for designing and cloning sgRNAs that are suitable for use with various dCas9 variants, as well as the design process for sgRNAs targeting specific regulatory elements.
Basic Protocol 1 – Generation and Validation of CRISPRa- and CRISPRi-Competent B Cell Lines
Both dCas9 and the sgRNA of interest can be cloned into a single vector. However, dCas9 is encoded by a gene that is approximately 4.5 kilobases in length; lentiviral packaging of the dCas9-encoding RNA becomes inefficient and titers are consequently lowered by several magnitudes. To improve overall dCas9 activity, we recommend first obtaining pools of lymphoblastoid B cells that stably express dCas9 at high levels before performing a second round of transduction with lentiviruses encoding the sgRNAs of interest [see Jiang et al. (CPMB, 2017, Chapter 33)]. Blasticidin may be used to maintain dCas9 expression in LCL pools but we suggest against it for two reasons – first, the continual presence of blasticidin in culture media causes a certain level of toxicity and death to a number of LCLs that we have tested, and second, dCas9 expression appears to be maintained even in the absence of continual blasticidin treatment. To obtain single-cell dCas9-expressing clones, dCas9-expressing pools will have to be generated first, followed by dilution and expansion of single-cell clones. Empirical confirmation of dCas9 expression should be performed on the clones obtained. This Protocol describes the steps to be taken to establish dCas9-expressing LCLs, the process of validating dCas9 activity, as well as the optional procedure for single-cell cloning.
Materials
-
▪
GM12878 LCL. These are ENCODE Tier 1 EBV-positive lymphoblastoid B cells obtained from Coriell (GM12878). GM12878 LCLs should ideally be maintained at 1 × 105 cells/mL of R10 media at 37°C in a humidified incubator supplemented with 5% CO2. Cells should be passaged every two to three days to maintain at the ideal cell density and culture media pH.
-
▪
R10 media (See Reagents and Solutions)
-
▪
R10-blasticidin (See Reagents and Solutions)
-
▪
R10-puromycin (See Reagents and Solutions)
-
▪
Phosphate-buffered saline (PBS) (Gibco)
-
▪
Freezing media (See Reagents and Solutions)
-
▪
Lentiviral supernatants (pXPR_109 for dCas9-VP64 and pXPR_121 for KRAB-dCas9). For details on how to package lentiviruses using these plasmids, please refer to Support Protocol 1 of Jiang et al. (CPMB Unit 31.12).
-
▪
Polybrene reagent (optional) (EMD Millipore, TR-1003-G)
-
▪
T25 cell culture flasks
-
▪
T75 cell culture flasks
-
▪
Clear 6-well plates, sterile (Sigma-Aldrich, CLS3516-10EA)
-
▪
Clear 96-well plates, sterile (Sigma-Aldrich, CLS3300-50EA)
-
▪
15 mL Falcon tubes, sterile
-
▪
1.5 mL microcentrifuge tubes, sterile
-
▪
Centrifuge suitable for 15 mL Falcon tubes
-
▪
10% bleach in H2O
-
▪
37°C tissue culture incubator supplemented with 5% CO2
-
▪
BSL2/BSL2+ certified tissue culture hood
-
▪
Flow cytometer
-
▪
Alexa Fluor 488 anti-human CD19 antibody (eBioscience, 53-0199-41)
-
▪
FITC anti-human CD86 monoclonal antibody (BD Biosciences, 557343)
-
▪
PE anti-human CD95 (FAS) antibody (Biolegend, 305607)
-
▪
Optional: mouse anti-Cas9 monoclonal antibody (clone 7A9-3A3, Active Motif 61578)
CAUTION: Ensure that cells are mycoplasma-free by testing regularly and practicing good aseptic techniques.
CAUTION: Although lentiviruses used in this Protocol is replication-defective, handle with care. Always bleach all pipettes, tips etc. that have come into contact with lentiviral supernatants before disposal.
TIP Pre-warm media at 37°C before addition to LCLs.
Protocol
A. Establishment of LCLs stably expressing dCas9-VP64 or KRAB-dCas9
NOTE: Unless otherwise stated, all steps in this Basic Protocol that mention the use of lentiviral supernatants refer to lentiviruses that can encode either dCas9-VP64 (also known as lenti dCas9-VP64_Blast; Addgene #61425) or KRAB-dCas9 (also known as pLX_311-KRAB-dCas9; Addgene #96918). The procedure of establishing stable cell lines and evaluating their dCas9 activities is identical, regardless of the mechanism of CRISPR gene regulation.
Day 1
-
1
Seed 6 × 105 to 1.2 × 106 cells at 3 × 105 cells/mL in a T25 flask for growth overnight. Ensure that cells are mycoplasma-free and in the log phase of growth.
Day 2
-
2
Shake the T25 flask vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
3
If using frozen aliquots of lentiviral supernatant, thaw the lentiviral supernatants in a 37°C water bath.
-
4
Add an equal volume of lentiviral supernatant to the cell suspension in the T25 flask. To ensure that cells are adequately exposed to the lentivirus, resuspend thoroughly by pipetting up and down several times. Avoid excessive bubbling.
-
5
Place the T25 flask in a 37°C incubator for 16 to 24 hours.
Day 3
-
6
Check visually or using a microscope that the transduced cells are healthy. Healthy lymphoblastoid cells will form clumps when left overnight.
-
7
Shake the T25 flask vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
8
Add a third volume of lentiviral supernatant to the cell suspension. Example: if 6 × 105 cells were seeded in 2 mL of R10 media on Day 1, add 2 mL of lentiviral supernatant on Day 3. The final volume will be 2 mL (Day 1) + 2 mL (Day 2) + 2 mL (Day 3) = 6 mL.
-
9
Resuspend thoroughly by pipetting up and down several times, taking care to avoid the introduction of bubbles.
-
10
Place the T25 flask in a 37°C incubator for 16 to 24 hours.
Day 4
-
11
Check by eye or using a microscope that the transduced cells are healthy. Healthy lymphoblastoid cells will form clumps when left overnight.
-
12
Shake the T25 flask vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
13
Transfer the suspension into a sterile 15 mL Falcon tube.
-
14
Centrifuge the cell suspension for 5 minutes at 300 × g at room temperature.
-
15
Use a glass Pasteur pipette to remove the supernatant.
-
16
Tap the bottom of the Falcon tube gently to disperse the cells in the remaining fluid.
-
17
Resuspend cells in the same volume of pre-warmed R10-blasticidin. Example: if the volume at the end of Day 3 was 6 mL, resuspend cells with 6 mL of R10-blasticidin.
-
18
Place the T25 flask in the incubator. Selection with blasticidin requires at least a week. During this period, passage or expand the transduced cells every two to three days with R10-blasticidin. Transfer to a T75 flask if necessary. Cells that have been successfully transduced will form clumps and be able to grow and survive in R10-blasticidin.
-
19
After one to two weeks of blasticidin selection, centrifuge cells for 5 minutes at 300 × g at room temperature and resuspend with antibiotic-free R10 media to a density of 3 × 105 cells/mL.
B. Validation of dCas9 expression and activity
Day 1
-
20
Shake the T25 flask vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
21
Count cells. In each of two 15 mL Falcon tubes, centrifuge a volume of cell suspension containing 6 × 105 cells for 5 minutes at 300 × g at room temperature.
-
22
Discard the supernatant from both tubes.
-
23
To one tube, add 100 μL of 1X Laemmli sample buffer to the cell pellet and boil the sample at 95°C for 5 minutes. Sonicate the sample briefly and centrifuge for 5 minutes at 300 × g at room temperature to clarify the solution. Perform SDS-PAGE and immunoblotting for dCas9 with the sample, using non-transduced GM12878 cells as a negative control. dCas9 should produce a band that migrates at 150-160 kDa.
-
24
To the other tube, resuspend cells with 2 mL of pre-warmed R10 media by pipetting up and down several times. Avoid excessive bubbling.
-
25
Add 2 mL of supernatant containing sgRNA-encoding lentiviruses to the cell suspension. Resuspend well by pipetting up and down several times, taking care not to introduce bubbles.
-
26
Transfer the cell-virus suspension into a T25 flask.
-
27
Place the T25 flask in the incubator for 16 to 24 hours.
Day 2
-
28
Check by eye or using a microscope that the transduced cells are healthy. Healthy lymphoblastoid cells will form clumps when left overnight.
-
29
Shake the T25 flask vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
30
Add 2 mL of supernatant containing sgRNA-encoding lentiviruses to the cell suspension. Resuspend thoroughly by pipetting up and down several times, taking care to avoid the introduction of bubbles.
-
31
Place the T25 flask in a 37°C incubator for 16 to 24 hours.
Day 3
-
32
Check visually or using a microscope that the transduced cells are healthy. Healthy lymphoblastoid cells will form clumps when left overnight.
-
33
Shake the T25 flask vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
34
Transfer the suspension into a sterile 15 mL Falcon tube.
-
35
Centrifuge the cell suspension for 5 minutes at 300 × g at room temperature.
-
36
Use a glass Pasteur pipette to remove the supernatant.
-
37
Tap the bottom of the Falcon tube gently to disperse the cells in the remaining fluid.
-
38
Resuspend cells in 6 mL of pre-warmed R10-puromycin.
-
39
Place the T25 flask in the incubator. Selection with puromycin at 3 μg/mL typically requires at least two days and should ideally be done over three days. After selection, passage or expand the transduced cells every two to three days with antibiotic-free R10 media. Transfer to a T75 flask if necessary. Cells that have been successfully transduced will form clumps and be able to grow and survive in R10-puromycin.
Day 6
-
40
Check visually or using a microscope that the transduced cells are healthy. Healthy lymphoblastoid cells will form clumps when left overnight.
-
41
Shake the T25 flask vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
42
Count cells and centrifuge a volume of cell suspension containing 1 × 105 to 5 × 105 cells for 5 minutes at 300 × g at room temperature.
-
43
Resuspend cells in 400 μL of PBS to obtain a suspension of dispersed cells.
-
44
Perform flow cytometric analysis using antibodies against CD19. Use non-transduced cells for baseline comparison.
TIP: We find that a good positive control for CRISPRa and CRISPRi in LCLs is a CD19-targeting sgRNA (see Table 1 for sequences). Cell surface availability of CD19 epitopes will be perturbed with CRISPRa and CRISPRi, which in turn affects the fluorescent signal for flow cytometric analysis. Reproducible enhancement (Figure 1) and repression (Figure 2) of surface CD19 expression in B cell lines such as GM12878 LCL and P3HR-1 Burkitt lymphoma cells can be achieved.
Table 1.
Single guide RNA sequences used.
| Gene Name | 20-mer sequence (5′ to 3′) |
|---|---|
| CD19_ss CD19_as |
ACACACAAGATCATTTCCCG CGGGAAATGATCTTGTGTGT |
| CD86_ss CD86_as |
TCAAAATCTGTAGAGAAAAG CTTTTCTCTACAGATTTTGA |
| FAS-1_ss FAS-1_as FAS-2_ss FAS-2_as |
GGTGTTCAAAGACGCTTCTG CAGAAGCGTCTTTGAACACC CTCGCGCAAGAGTGACACAC GTGTGTCACTCTTGCGCGAG |
| POU3F1-1_ss POU3F1-1_as POU3F1-2_ss POU3F1-2_as POU3F1-3_ss POU3F1-3_as |
AGATCGGGTTAATGAAACTG CAGTTTCATTAACCCGATCT TCAGAGGTGACAAATGTAGG CCTACATTTGTCACCTCTGA CATGTTTTCACATTACTGGG CCCAGTAATGTGAAAACATG |
ss – oligonucleotide sequence is identical to the sense strand; as – oligonucleotide sequence is identical to the anti-sense strand. Each ss and as oligo pair is used for sgRNA cloning as described in Basic Protocol 2.
Figure 1.
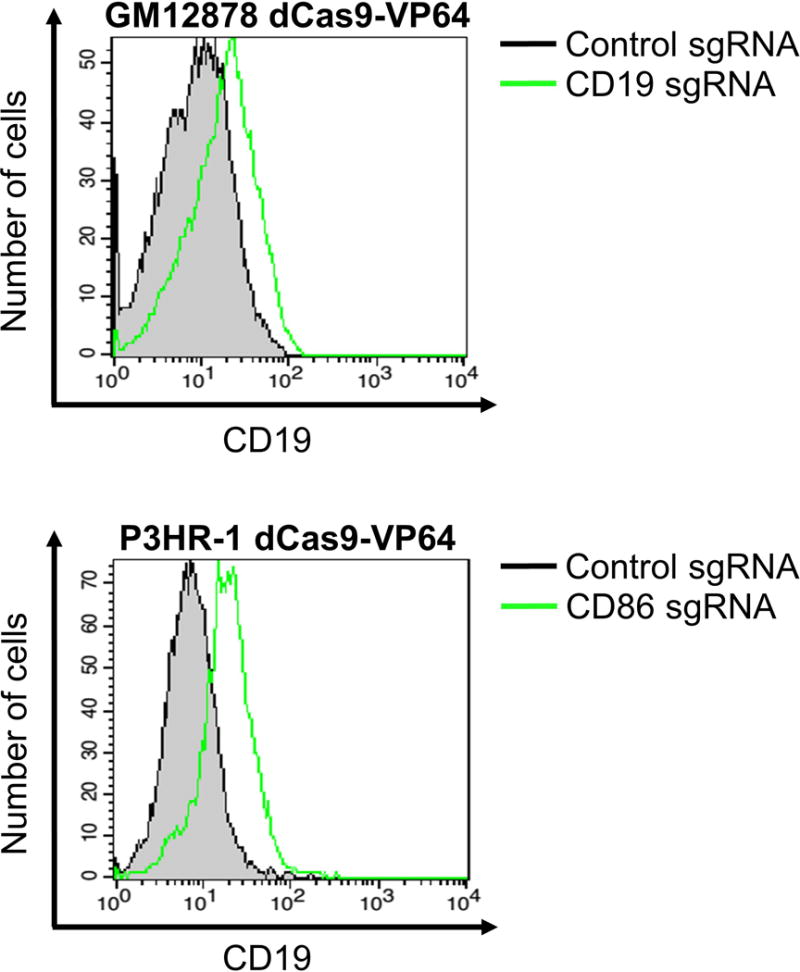
GM12878 LCL and P3HR-1 Burkitt lymphoma cells stably expressing activating dCas9-VP64 were transduced with either control sgRNA or sgRNAs targeting the indicated genes (CD19 or CD86). Two days after puromycin selection for cells that had been successfully transduced with sgRNA-encoding lentiviruses, cells were collected for flow cytometric analysis using antibodies against CD19 and CD86.
Figure 2.
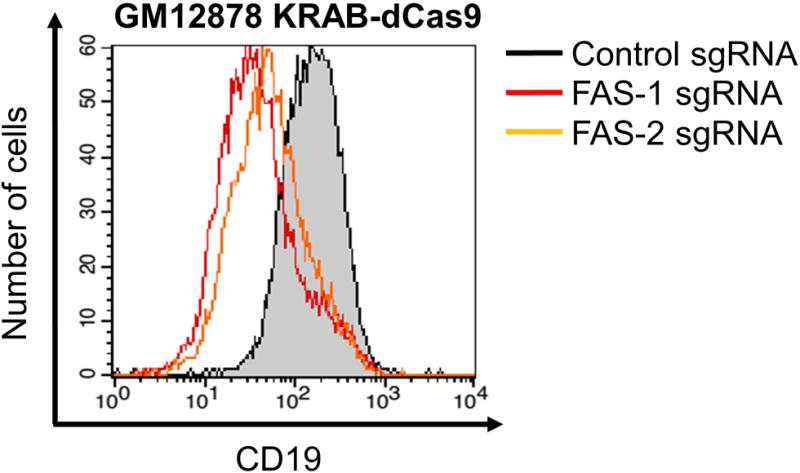
GM12878 LCL stably expressing activating KRAB-dCas9 was transduced with either control sgRNA or distinct sgRNAs targeting CD19 (CD19 sgRNA 1 and CD19 sgRNA 2). Two days after puromycin selection for cells that had been successfully transduced with sgRNA-encoding lentiviruses, cells were collected for flow cytometric analysis using antibodies against CD19.
-
▪
For CRISPRa: the CD19 fluorescent signal should be higher than baseline.
-
▪
For CRISPRi: the CD19 fluorescent should be lower than baseline.
-
45
If dCas9 activity is functional in the pooled LCLs at this point and single-cell clones are desired, proceed with the next section.
TIP: Spinoculation of lentiviruses in the presence of polybrene at a final concentration of 8 μg/mL enhances transduction efficiency. Briefly, instead of simple addition of lentiviral supernatants to the cell suspension, lentiviral supernatant is added to the cell suspension, followed by addition of polybrene at 8 μg/mL and gentle centrifugation for two hours at 300 × g at room temperature. The volumetric ratio of lentiviral supernatant to cell suspension can range between 1:1 and 2:1. The cell-virus suspension is then incubated at 37°C in a 5% CO2 atmosphere for four to six hours before centrifugation and resuspension in polybrene-free R10 media.
C. Isolation of dCas9-expressing single-cell clones through serial dilution
-
46
Prepare conditioned R10 media by centrifuging LCL culture media prior to passage or expansion and passing the clarified supernatant through a 0.45 μm filter. Store the conditioned R10 media at 4°C when not in use. Conditioned R10 media contains growth factors and cytokines secreted by the lymphoblastoid cells which aid in outgrowth at low cell densities.
-
47
Shake the flask of cells obtained from Step 38 vigorously or use a pipette to draw the cell suspension up and down a few times to disperse the cells.
-
48
Count cells and centrifuge a volume of cell suspension containing 1 × 106 cells for 5 minutes at 300 × g at room temperature.
-
49
Decant the supernatant and resuspend cells in 10 mL of pre-warmed conditioned R10 media.
-
50
Dilute the cell suspension further with conditioned R10 media to a cell density of 200 cells/mL.
-
51
Using a multi-channel pipette, aliquot 50 μL of conditioned R10 media into each well of a sterile 96-well plate.
-
52
Briefly and gently vortex the tube of cell suspension obtained from Step 49.
-
53
Using a single-channel pipette, aliquot 50 μL of cell suspension into each well of the top row (Row A) of the 96-well plate.
-
54
Using the multi-channel pipette, perform a two-fold serial dilution series from the top row down to the bottom row (Row H).
-
55
Place the 96-well plate in a 37°C incubator to allow growth.
-
56
Check the 96-well plate every other day to ensure that wells do not dry out. If the liquid level is low, add 25 μL of conditioned R10 media to each well.
-
57
Outgrowth of single cells takes approximately one to two weeks. One week after setting up the plate, check the plate every three days for single clumps.
-
58
To reduce the likelihood of heterogeneous clones, ensure that wells containing a higher dilution do not show cellular outgrowth/clumps of cells.
-
59
Once single-cell clones are observed, transfer the cells into a well of a sterile 6-well plate. Top up with 1 mL of conditioned R10 media.
-
60
Place the 6-well plate in a 37°C incubator for expansion.
-
61
When confluent (approximately 6 × 105 cells/mL), collect 6 × 105 cells for validation of dCas9 activity (see earlier section entitled, “B. Validation of dCas9 expression and activity”).
-
62
Expand cells further, growing them in fresh (non-conditioned) R10 media.
-
63
Test cells for mycoplasma before freezing in aliquots for future use.
Basic Protocol 2 – Designing and Cloning of Single-Guide RNAs for CRISPRa and CRISPRi
The design principles for CRISPRa and CRISPRi sgRNAs are similar to that for conventional CRISPR. A 20-mer sequence in the sgRNA dictates binding specificity of the dCas9-sgRNA complex. Binding of dCas9 is also dependent on the presence of a PAM site on the target DNA.
However, transcription factors are often pleiotropic in nature; they do not usually bind to a single site on the entire genome. Co-evolution of transcription factors and their binding sites on DNA likely has occurred over time (Yang et al., 2011) such that sequences across the genome that bind a given transcription factor usually appear similar or identical. Given that the specificity of CRISPRa and CRISPRi sgRNAs is largely attributed to the short 20-mer, which is approximately double the size of the average transcription factor-binding site (Stewart, Hannenhalli, & Plotkin, 2012), it is expected that any given dCas9-sgRNA complex will bind to several different locations on genomic DNA.
Like conventional CRISPR, CRISPRa and CRISPRi sgRNA scores are important determinants for choosing which sgRNA to use in experiments. The “On-Target Efficacy” score is calculated to indicate how specific the sgRNA is in a given CRISPR application (Doench et al., 2016), with a score of one meaning that every regulatory element recognized by the sgRNA is bound. Generally, we find most scores to be in the range of 0.5 to 0.6.
Successful CRISPRa and CRISPRi can be assessed by immunoblotting or quantitative RT-PCR for the target protein and transcript, or by flow cytometric analysis if the protein of interest has an extracellular epitope that can be engaged by fluorophore-conjugated antibodies.
In this Basic Protocol 2, the details of sgRNA design and cloning will be described in the contexts of promoter (Sections A and C) and enhancer (Sections B and C) targeting.
Materials
-
▪
Oligonucleotides (see Sections A and B for procedures to design sgRNAs against promoters and enhancers, respectively), purified by standard desalting and diluted to 100 μM with nuclease-free water
-
▪
FastDigest BsmBI/Esp3I (Fermentas)
-
▪
10X FastDigest Buffer (Fermentas)
-
▪
T4 DNA ligase (New England BioLabs)
-
▪
10X T4 Ligase Buffer (New England BioLabs)
-
▪
One Shot Stbl3 Chemically Competent E. coli (Invitrogen)
-
▪
Sterile S.O.C. media (Invitrogen)
-
▪
LB-ampicillin (100 μg/mL) plates
Protocol
A. In silico CRISPRa and CRISPRi sgRNA design using the Broad GPP Web Portal
This sgRNA design tool is best suited to generating sequences that target gene promoters.
-
1
Go to https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design-crisprai.
-
2
For CRISPR Enzyme, select “S. pyogenes (NGG)”.
-
3
For Target Taxon, select “human”.
-
4
For Input Gene IDs or Symbols, type the gene of interest in capital letters. For example, type ‘CD19’, not ‘Cd19’ or ‘cd19’.
-
5
Select the appropriate CRISPR mechanism, which is either “CRISPRa” or “CRISPRi”.
-
6
Leave Quota as “5” (default). This returns five sgRNAs that target the promoter of interest.
-
7
Check the “Report Unpicked Sequences?” box.
-
8
Check the “I’m not a robot” box and click “Submit”.
-
9
Click on the hyperlink that says, “sgRNA Picking Results”, to download a tab-delimited text file containing the best candidate sgRNAs.
-
10
Open the tab-delimited text file. Select all, copy and paste into an Excel spreadsheet to enable easy visualization.
-
11
Sort the candidate guide RNA sequences according to On-Target Efficacy Scores (the higher, the better) and Off-Target Rank (the lower, the better). In this example, the best guide RNA 20-mer is ACACACAAGATCATTTCCCG (On-Target Efficacy Score of 0.7232 and Off-Target Rank of 6).
-
12
Determine the reverse complement and add the sequences (shown in bold) for ligation into BsmBI/Esp3I cut sites. The PAM site is denoted in red. Using the first candidate sgRNA found in Step 11 as an example:
CD19 genomic sequence: 5′……. ATAAACACACAAGATCATTTCCCGTGGTAG ...... 3′ 3′……. TATTTGTGTGTTCTAGTAAAGGGCACCATC ……5′ CD19 sgRNA sequence: 5′-CACCGACACACAAGATCATTTCCCG -3′ 3′-CTGTGTGTTCTAGTAAAGGGCCAAA-5′
B. In silico sgRNA design using chromatin interaction analysis by paired-end tag (ChIA-PET) sequencing data and Benchling
This section looks at the use of publicly accessible ChIA-PET data to inform the design of CRISPRi sgRNAs that target enhancers. Enhancers are typically several kilobases in length, which is impractical to target with a tiling strategy. ChIA-PET enables the visualization of highly frequent associations between two distant DNA elements e.g. a particular Transcription Factor-Binding Site (TFBS) within the enhancer with a target gene promoter. Knowledge of specific TFBSs within the enhancer that regulates a gene promoter facilitates sgRNA design. To this end, we have compiled a WashU Epigenome Browser session in EBV-transformed LCLs (Jiang et al., 2017), which includes the long-range chromatin interactions mediated by viral and cellular transcription factors in LCLs.
-
13
Open the WashU Epigenome Browser (http://epigenomegateway.wustl.edu/browser/?genome=hg19&session=AuL8qiK9Bf)
-
14
Select “Jiang et al 2017 Cell Host Microbe”, which contains tracks pertaining to all ChIP-seq and long-range interactions (CTCF and RNA Polymerase II ChIA-PET) experiments. These tracks are based on the hg19 human genome.
-
15
In the top-left corner of the window, put in the coordinates or name of the gene of interest. Here, POU3F1 will be used as an example. Type ‘POU3F1’ into the gene name field and click ‘Go’.
-
16
A selection of mRNA sequences will appear. Click on the upper-most option, which is the refGene entry.
-
17
The purple arcs will disappear. Zoom out by clicking ‘−1’ near the top-left corner of the window until the purple arcs re-appear.
-
18
In the RNA Polymerase II (RNAPII) track, two arcs should appear to link the intergenic enhancer between UTP11L and POU3F1, to the POU3F1 promoter. Right-click on the title of the RNAPII track and select ‘Configure’. Set the filter threshold score for positive values to 5. The result should be a single arc that links the intergenic enhancer to the POU3F1 promoter (Figure 3A).
-
19
Double-click on the arc in the RNAPII track to see the coordinates of the interacting regions. The enhancer stretch should read chr1: 38493863-38494431, while the promoter stretch should read chr1: 38510537-38511406. For this guided example, we are interested in chr1: 38493750-38493961 as it coincides with a ChIP-seq peak in the EBNA-LP (EBV-encoded nuclear antigen-leader protein) track.
-
20
Create and log in to a Benchling account (https://www.benchling.com).
-
21
To the left side of the window, click ‘Create’ and mouse over to select ‘CRISPR’ and then ‘CRISPR Guides’.
-
22
In the pop-up window, import data from ‘Chromosomal Coordinates’, select ‘hg19’ as the source genome, and put in ‘chr1 38493750-38493961’. Click ‘Next’.
-
23
A new pop-up window showing various design parameters will appear. Leave the default selections as they are and click ‘Finish’.
-
24
The sgRNAs will not be generated yet. In the split window on the right, put in the coordinates again and click ‘Create’.
-
25
A list of sgRNAs will appear. Sort them by ‘On-Target Score’ – the higher the score, the greater the efficiency of the sgRNA. For this example, the sequences of the top three guide RNAs and their reverse complement can be found in Table 1. Click the ‘Save’ button situated above the list of sgRNAs and select ‘Export All (.tsv)’ to copy and paste all sgRNA sequences into an Excel spreadsheet.
-
26
Proceed with Step 12.
Figure 3.
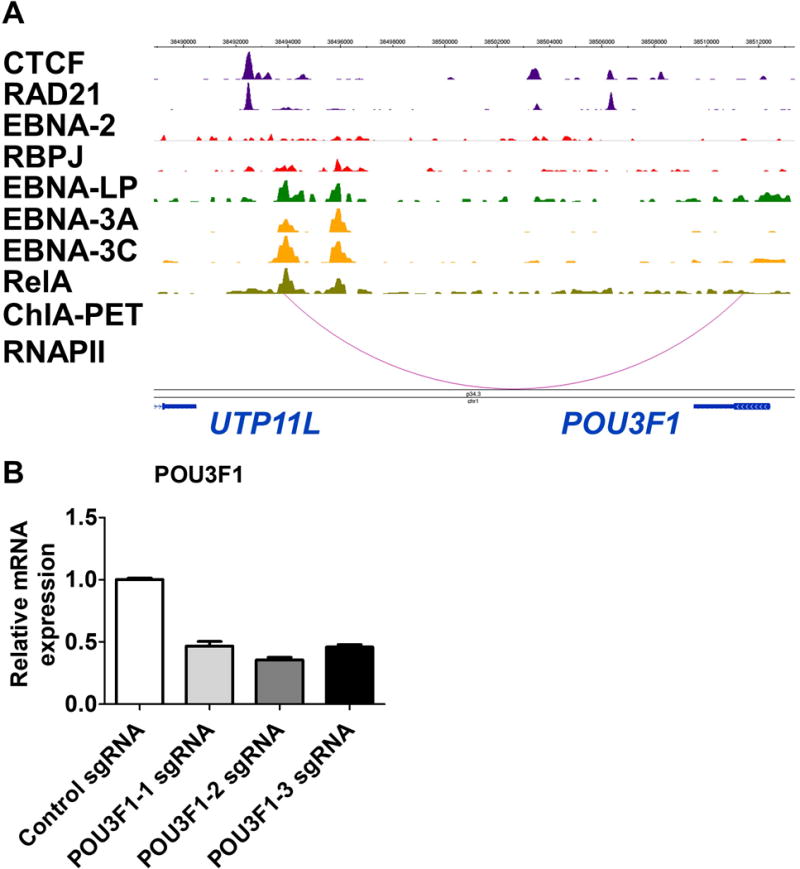
(A) Genome snapshot displaying ChIP-seq and ChIA-PET tracks for various viral and host transcription factors at chr1: 38488884-38513387 (Consortium, 2012; Landt et al., 2012). The ChIA-PET RNAPII track indicates physical linkage between the intergenic enhancer and the POU3F1 promoter. (B) Quantitative RT-PCR of the POU3F1 transcript after CRISPRi treatment. GM12878 cells stably expressing KRAB-dCas9 were transduced with either sgCtrl (control non-targeting sgRNA) or one of three distinct sgRNAs targeting the intergenic enhancer between UTP11L and POU3F1 (sgPOU3F1). Total RNA was harvested and subject to quantitative RT-PCR. POU3F1 transcript levels were normalized to that of β-actin.
C. Cloning of sgRNA into pXPR_501 (CRISPRa) or pLentiGuide-Puro (CRISPRi)
Section C is adapted from Feng Zhang’s protocol (Sanjana, Shalem, & Zhang, 2014; Shalem et al., 2014) with minor modifications (see Internet Resources).
-
27Prepare digestion mix as follows:
-
▪pXPR_501 or pLentiGuide-Puro, 5 μg
-
▪FastDigest BsmBI or Esp3I (Fermentas), 3 μL
-
▪10X FastDigest Buffer (Fermentas), 3 μL
-
▪ddH2O to bring final volume to 30 μL
-
▪
-
28
Incubate digestion mix at 37°C for 1 hour.
-
29
Load mix directly onto a 0.8% agarose gel to separate the cut vector from the filler DNA. Run 1 μg of uncut vector as negative control.
-
30
Gel extract and purify the cut vector using a standard kit e.g. QIAquick Gel Extraction Kit or Macherey-Nagel Nucleospin Gel and PCR Clean Up Kit. For the elution step, perform it twice, each time using 10 μL of the kit’s elution buffer, to obtain a final eluate of volume 20 μL.
-
31
Measure the DNA concentration of the eluate using a Nanodrop device. Dilute to 50 ng/μL for use. Optional: transform 1 μL of the 50 ng/μL stock into bacteria to see if colonies form. If the vector has been successfully cut, no colonies should be produced.
-
32Prepare the following 1X reaction for annealing of sgRNA oligos (designed using the steps described in either Section A or Section B) in a PCR tube:
-
▪Oligo 1 (100 μM), 1 μL
-
▪Oligo 2 (100 μM), 1 μL
-
▪10X T4 Ligation Buffer (New England BioLabs), 1 μL
-
▪ddH2O, 7 μL
-
▪
TIP: Prepare a master mix of 10X T4 Ligation Buffer and ddH2O before addition to the oligo pairs.
-
33Run the following thermal cycler program:
-
▪37°C, 30 minutes
-
▪95°C, 5 minutes
-
▪Decrease temperature to 25°C at 5°C/minute
-
▪
-
34
Dilute annealed oligos 1:100 with nuclease-free water. Invert several times to mix thoroughly.
-
35Prepare the following reaction for DNA ligation:
-
▪Dilution of annealed oligos, 1 μL
-
▪BsmBI/Esp3I-digested plasmid (50 ng/μL), 1 μL
-
▪10X T4 Ligation Buffer, 1 μL
-
▪ddH2O, 7 μL
-
▪
NOTE: If the solution containing digested plasmid has a DNA concentration lower than 50 ng/μL, it can still be used as long as 50 ng of input plasmid DNA is used, with the appropriate adjustment made to the volume of ddH2O put into the reaction.
-
36
Add 1 μL of T4 DNA ligase and incubate at room temperature for 1 hour or 16°C overnight.
-
37Transform 50 μL of Stbl3 bacterial suspension with 10 μL of the ligation reaction.
-
▪Thaw the Stbl3 bacteria on ice.
-
▪Incubate bacteria with the ligation reaction for 30 minutes on ice.
-
▪Heat shock the bacteria for 30 seconds at 42°C. NOTE: To ensure maximal transformation efficiency, it is crucial that the heat shock step is at least 30 seconds and does not exceed 45 seconds.
-
▪Place bacteria back on ice for 2 minutes.
-
▪Add 250 μL of pre-warmed SOC media to the bacteria. Pipette up and down several times gently to mix thoroughly.
-
▪Shake the tube at 300 to 500 rpm at 30°C for an hour. At the same time, warm an LB-ampicillin plate at 30°C.
-
▪Plate the contents of the entire tube on the LB-ampicillin plate.
-
▪Incubate the LB-ampicillin plate at 30°C for at least 16 hours and till a maximum of 24 hours.
-
▪
-
38
Select colonies for miniprep. Optional: sequence the miniprep DNA using the standard hU6 forward primer.
-
39
Miniprep DNA can be used for transfection of 293FT cells to generate lentiviral supernatants. The supernatants can then be used as per Basic Protocol 1 Section B. Assaying gene expression changes can be done via quantitative RT-PCR of the target transcript or, less preferably, via immunoblotting or flow cytometry with antibodies specific to the target protein.
Reagents and Solutions
All media should be stored at 4°C until use. Minimize prolonged warming at 37°C to prevent degradation of L-glutamine. For R10 media, they can be used for approximately one month or until the phenol red indicator starts to turn pink, whichever comes earlier.
R10 culture media
-
▪
RPMI-1640 (+) L-glutamine (Gibco, 11875-085)
-
▪
10% fetal bovine serum (FBS) (Gibco)
-
▪
10X U/mL penicillin-streptomycin (Gibco)
-
▪
2 mM L-glutamine (Gibco) (optional)
R10-blasticidin
-
▪
R10 culture media
-
▪
10 μg/mL blasticidin (Invivogen, ant-bl-5)
R10-puromycin
-
▪
R10 culture media
-
▪
3 μg/mL puromycin dihydrochloride (Life Technologies, A11138-03)
Freezing media
-
▪
90% FBS (Gibco)
-
▪
10% sterile DMSO (Fisher, bp231-1)
Commentary
Background Information
For an overview of CRISPRa and CRISPRi, several recent publications provide an excellent starting point (Dominguez, Lim, & Qi, 2016; La Russa & Qi, 2015).
Critical Parameters
To increase the efficiency of lentiviral transduction of LCLs, spinoculation in the presence of 8 μg/mL polybrene is recommended.
To monitor the extent of CRISPR activation or interference, quantitative RT-PCR is the preferred method. Immunoblotting and flow cytometry may also be performed. However, the success of the latter two methods depends on several factors, such as the target protein’s half-life and the specificity of the detection antibody.
Troubleshooting
Please refer to Table 2.
Table 2.
Common problems, possible causes, and solutions to address the problems.
| Problem | Possible Cause | Solution |
|---|---|---|
| No lymphoblastoid cells alive after selection | Transfection is unsuccessful | Check that 293FT cells produce green fluorescence signal 24h after transfection of pXPR_011. |
| Check the concentrations of the packaging (at least 1 μg/μL) and lentiviral expression constructs (at least 100 ng/μL). Perform maxiprep if necessary. | ||
| Packaging of lentiviruses is unsuccessful | Use low-passage (<20 passages) 293FT cells for transfection. | |
| No colonies present after sgRNA cloning | Oligo annealing is unsuccessful | Ensure that the sgRNA oligos have been diluted to the correct concentration. Make sure that the correct pairs of oligos have been used in the reaction, especially when multiple sgRNAs are being cloned at the same time. |
| Ligation is unsuccessful | Keep T4 DNA ligase at -20°C and minimize prolonged exposure to higher temperatures. Perform ligation reaction longer than 16 hours. | |
| Too many colonies (> 100) present after sgRNA cloning | Linearized vector is contaminated with uncut vector | Perform vector digestion again, incubating for longer periods or using more restriction enzyme. Ensure that cut and uncut forms of the vector are sufficiently separated on the agarose gel. |
| Quantitative RT-PCR shows no change to gene expression levels in sgRNA-transduced CRISPRi-competent cells | Alternative promoter usage with the use of a qPCR primer pair that amplifies a region common to the transcript isoforms | Use a primer pair that amplifies as close as possible to the target promoter’s transcription start site. |
| Quantitative RT-PCR shows no change in gene expression levels in sgRNA-transduced cells | sgRNA sequence is incorrect | Sequence the sgRNA expression construct to ensure that the targeting sequence is correct. |
| Immunoblotting and/or flow cytometry shows no change to protein expression levels in sgRNA-transduced cells | Antibody does not detect the target protein | Perform conventional CRISPR, followed by immunoblotting to see if the band corresponding to the detected protein disappears. If the band persists, purchase a new antibody and test it against the same whole-cell lysate. |
Understanding Results
In our experience, CRISPRi targeting of an enhancer can give up to 50% reduction of target gene transcript levels (Figure 3B; see Table 3 for sequences of validation primers). However, the effects can vary widely between different genomic loci for several reasons. First, CRISPRa and CRISPRi sgRNAs are targeted towards non-protein-coding regions which have not been under the same evolutionary pressures as protein-coding gene bodies. Hence, CRISPRa and CRISPRi sgRNAs are expected to be more promiscuous in their binding of regulatory elements on DNA, yielding substantial variation in gene expression modulation. Second, nucleosome positioning may influence Cas9 activity; if the PAM site is obscured within the nucleosome core, Cas9-sgRNA RNPs can become restricted in their ability to recognize cognate sequences (Hinz, Laughery, & Wyrick, 2015; Isaac et al., 2016).
Table 3.
Sequences of quantitative PCR primers used.
| Gene Name | Primer Sequence (5′ to 3′) |
|---|---|
| POU3F1 Set 1 | Forward: GGAGAGTTCTCCATCCCCTC Reverse: GAGAGAAGAGGCGTGTAGCG |
| POU3F1 Set 2 | Forward: TCGCTACACGCCTCTTCTCT Reverse: GGAGTTAGAAGGACCCCAGG |
Time Consideration
Basic Protocol 1: approximately 3 weeks (if single-cell cloning is not performed) or 5 weeks (if single-cell cloning is performed)
-
▪
Section A: 11 days
-
▪
Section B: 7 days
-
▪
Section C: 2 to 3 weeks
Basic Protocol 2: three days
-
▪
Section A: one hour
-
▪
Section B: one hour
-
▪
Section C: 3 days (from sgRNA cloning to obtaining miniprep DNA for transfection)
Acknowledgments
L.W.W. is a recipient of Singapore’s Agency for Science, Technology and Research (A*STAR) National Science Scholarship (Ph.D.). S.J. is a recipient of the Howard Hughes Medical Institute (HHMI) International Student Research Fellowship. B.Z. is funded by the National Institutes of Health (R01AI123420). B.E.G. is a recipient of the Burroughs Wellcome Fund Career Award in Medical Sciences.
Footnotes
-
The Broad Institute’s Genetic Perturbation Platform CRISPRa and CRISPRi sgRNA design tool: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design-crispraiThis free online tool allows users to design CRISPRa and CRISPRi sgRNAs for use with either SpCas9 or Staphylococcus aureus Cas9 (SaCas9) in human and murine cells, and also returns on- and off-target information for the candidate sgRNAs.
-
WashU Epigenome Browser LCL ChIP-seq and ChIA-PET Tracks: http://epigenomegateway.wustl.edu/browser/?genome=hg19&session=AuL8qiK9BfThis session enables visualization of epigenomic data (Zhou et al., 2011) used in Jiang et al.
-
Benchling: https://benchling.com/This free online portal enables the user to put in defined coordinates of the human genome and obtain a list of candidate sgRNAs with associated on- and off-target scores (Doench et al., 2016; Hsu et al., 2013).
-
Zhang Lab’s sgRNA cloning protocol: https://media.addgene.org/data/plasmids/52/52961/52961-attachment_YrytFfyNLHp5.pdfThis document outlines the steps needed to generate sgRNA constructs.
Literature Cited
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Root DE. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17(1):5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Tibbit C, Liu JL. Effective knockdown of Drosophila long non-coding RNAs by CRISPR interference. Nucleic Acids Res. 2016;44(9):e84. doi: 10.1093/nar/gkw063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Qi LS. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz JM, Laughery MF, Wyrick JJ. Nucleosomes Inhibit Cas9 Endonuclease Activity in Vitro. Biochemistry. 2015;54(48):7063–7066. doi: 10.1021/acs.biochem.5b01108. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac RS, Jiang F, Doudna JA, Lim WA, Narlikar GJ, Almeida R. Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. Elife. 2016;5 doi: 10.7554/eLife.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Zhou H, Liang J, Gerdt C, Wang C, Ke L, Zhao B. 3D genome landscape of Epstein-Barr Virus oncoproteins and virus activated NF-kB in lymphoblastoid cells. Cell Host & Microbe. 2017 Oct; doi: 10.1016/j.chom.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Engreitz JM, Konermann S, Abudayyeh OO, Verdine VK, Aguet F, Zhang F. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017;548(7667):343–346. doi: 10.1038/nature23451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Russa MF, Qi LS. The New State of the Art: Cas9 for Gene Activation and Repression. Mol Cell Biol. 2015;35(22):3800–3809. doi: 10.1128/MCB.00512-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Snyder M. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupo A, Cesaro E, Montano G, Zurlo D, Izzo P, Costanzo P. KRAB-Zinc Finger Proteins: A Repressor Family Displaying Multiple Biological Functions. Curr Genomics. 2013;14(4):268–278. doi: 10.2174/13892029113149990002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nat Methods. 2013;10(10):977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden K, Hafez AY, Kishton R, Messinger JE, Nikitin PA, Rathmell JC, Luftig MA. Metabolic stress is a barrier to Epstein-Barr virus-mediated B-cell immortalization. Proc Natl Acad Sci U S A. 2016;113(6):E782–790. doi: 10.1073/pnas.1517141113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Gersbach CA. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods. 2013;10(10):973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider U, zur Hausen H. Epstein-Barr virus-induced transformation of human leukocytes after cell fractionation. Int J Cancer. 1975;15(1):59–66. doi: 10.1002/ijc.2910150108. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AJ, Hannenhalli S, Plotkin JB. Why transcription factor binding sites are ten nucleotides long. Genetics. 2012;192(3):973–985. doi: 10.1534/genetics.112.143370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Yalamanchili HK, Li X, Yao KM, Sham PC, Zhang MQ, Wang J. Correlated evolution of transcription factors and their binding sites. Bioinformatics. 2011;27(21):2972–2978. doi: 10.1093/bioinformatics/btr503. [DOI] [PubMed] [Google Scholar]
- Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, Wang T. The Human Epigenome Browser at Washington University. Nat Methods. 2011;8(12):989–990. doi: 10.1038/nmeth.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]