

Abstract
Ormeloxifene (ORM), is a clinically approved selective estrogen receptor modulator, which has also shown excellent anti-cancer activity, thus it can be an ideal repurposing pharmacophore. Herein, we report therapeutic effects of ORM on prostate cancer (PrCa) and elucidate a novel molecular mechanism of its anti-cancer activity. ORM treatment inhibited epithelial to mesenchymal transition (EMT) process as evident by repression of N-cadherin, Slug, Snail, and vimentin, MMPs (MMP2 and MMP3), β-catenin/TCF-4 transcriptional activity, and induced the expression of pGSK3β. In molecular docking analysis, ORM showed proficient docking with β-catenin and GSK3β. In addition, ORM induced apoptosis, inhibited growth and metastatic potential of PrCa cells and arrested cell cycle in G0-G1 phase via modulation of cell cycle regulatory proteins (inhibition of Mcl-1, cyclin D1, and CDK4 and induction of p21 and p27). In functional assays, ORM remarkably reduced tumorigenic, migratory and invasive potential of PrCa cells. Additionally, ORM treatment significantly (P<0.01) regressed the prostate tumor growth in the xenograft mouse model while administered through intra-peritoneal route (250 μg/mouse; thrice weekly). These molecular effects of ORM were also observed in excised tumor tissues as shown by immunohistochemistry analysis. Our results, for the first time, demonstrate repurposing potential of ORM as an anti-cancer drug for the treatment of advanced stage metastatic PrCa through a novel molecular mechanism involving β-catenin and EMT pathway.
Keywords: Ormeloxifene, prostate cancer, β-catenin, EMT
Introduction
Prostate Cancer (PrCa) is the second leading cause of cancer-related death among American men. The American Cancer Society projected that a total of 161,360 new cases of PrCa would be diagnosed and approximately 26,730 men will die in the United States alone in the year of 2017(1). Despite the initial success of androgen-ablation therapy, resistance to anti-androgen therapy manifests by progression to androgen-independent PrCa, which is the end stage that accounts for the majority of cancer deaths (2). Current chemotherapeutic drugs such as docetaxel, cabazitaxel and mitoxantrone provide moderate treatment benefits for the management of advanced PrCa but all of them suffer from severely toxic side effects. Moreover, these drugs do not target major oncogenic signaling pathways such as β-catenin/EMT which lead to tumor growth and metastasis of PrCa (3).
Studies have shown that loss of E-cadherin and overexpression of N-cadherin is involved in EMT, leading to aggressive and metastatic PrCa phenotypes (4,5). Altered β-catenin expression/subcellular localization also plays a major role in EMT process in various malignancies including PrCa (6). Studies have suggested involvement of β‐catenin in the development, progression and therapy resistance of advanced PrCa (3). β-catenin is present in the cytoplasm as a heterodimeric protein complex that includes glycogen synthase kinase 3β (GSK3β), axin, and adenomatosis polyposis coli (APC) (7). GSK3β-dependent phosphorylation of β-catenin enhances its proteasomal degradation and inhibits its translocation into the nucleus, where it binds to T-cell Factor (Tcf) family of transcription factors, leading to transcriptional activation of various downstream target oncogenes. Various studies including our have reported increased nuclear β-catenin expression correlates with higher prostate tumor grades as compared to normal adjacent tissues (5,8). The stabilization of the transcriptional co-activator β‐catenin regulates expression of many genes which are involved in cell proliferation, differentiation and the EMT (9). These studies suggest that β-catenin appears to be a very important molecular target in cancer therapy, and its targeting may lead to successful therapeutic approach for the management of metastatic PrCa. Thus, there is an urgent need to develop non-toxic agents/pharmacological inhibitors that target aforementioned signaling pathways. These agent(s) could be used alone or in combination with conventional chemotherapy for the treatment of advanced PrCa.
Ormeloxifene (ORM) has demonstrated excellent anti-cancer activity in many different tumor types such as breast cancer (10), head and neck squamous cell carcinoma (HNSCC) (11), and ovarian cancer (12). We have recently demonstrated potent therapeutic efficacy of ORM in pancreatic cancer via inhibiting sonic hedgehog (SHH) signaling pathway, and modulation of tumor microenvironment (13). However, its effects on EMT processes and Wnt/β-catenin signaling are not investigated thus far. Herein, we have shown that ORM effectively inhibits molecular signatures of EMT, β-catenin/TCF-4 transcriptional activity, and induces phosphorylation of GSK3β, and degrades β-catenin leading to the suppression of prostate tumor growth in xenograft mouse model. Since, ORM is reported to have an excellent therapeutic index and is safe for human use for anti-fertility (contraception) purpose (14), ORM appears to be an ideal pharmacological agent for its repurposing as an anti-cancer agent against metastatic PrCa.
Materials and Methods
Cell lines
The human PrCa cells (PC3 and DU145) were the kind gift of Dr. Rajesh Singh, Assistant Professor, Morehouse School of Medicine, Atlanta, GA. They purchased these cells from ATCC (Manassas, Virginia) in January, 2016. Upon receipt cells were expanded and frozen aliquots (passage < 6) were stored in liquid nitrogen. When needed, cells were thawed and grown for less than 6 months. These cell lines were propagated in RPMI-1647 media supplemented with 10% fetal bovine serum (FBS) and 1× antibiotic and antimycotic solution. The media components were purchased from Lonza (Lonza, Walkersville, MD).
Chemicals and antibodies
Specific monoclonal and polyclonal antibodies of β-actin (cat. # 3700), cyclin D1 (cat. # 2922), CDK4 (cat. # 12790), p21 (cat. # 2947), p27 (cat. # 3686), Mcl-1 (cat. # 5453), pGSK3β (cat. # 5558), Histone H3 (cat. #4499), GAPDH (cat # 5174), N-Cadherin (cat. # 4061), Slug (cat. 9585), Snail (cat. # 3879), and Vimentin (cat. # 5741), PARP (cat. #9532S) and MMP2 (cat. # 4022) were obtained from Cell Signaling Technology Inc. β-catenin (cat # SC-7199), E-cadherin (cat. # SC-7870) and MTA1 (cat. # SC-17773) antibody was obtained from Santa Cruz Biotechnology. MMP3 (cat. # IM36) antibody was procured from Calbiochem, Merck Biosciences. HRP conjugated anti-mouse and anti-rabbit antibodies were acquired from Promega, Madison. Anti-mouse cy3 secondary antibody was purchased from Thermo Fisher Scientific, Carlsbad, CA. Ormeloxifene (ORM) was synthesized and characterized in Dr. Fathi Halaweish laboratory at South Dakota State University, Brookings, SD. The detail procedure for synthesis and characterization is described in our previous published manuscript (12).
MTT assay
Cell proliferation was determined by using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, 5×103 cells of PC3 or DU145 were plated in 96-well plates and incubated for 24 hrs in incubator at 37°C containing 5% CO2. Cells were treated with ORM (5-40 μM) for 24 hrs. Twenty microliter of 5 mg/ml MTT was added in each well containing 100 μl of cell media. The cells were then further incubate for 6 hrs in incubator and media was replaced with 150 μl of DMSO. Plates was vigorously shaked for 15 min and absorbance was taken at 570 nm on microplate reader (Cytation 3, BioTek, Winooski, VT, USA).
Colony forming assay
To investigate the effects of ORM on clonogenic potential of PC3 and DU145 cells, colony formation assay was performed. In brief, 500 cells were seeded per well in 6-well plate and allowed to stand for next three days. The cells were treated with ORM (2.5–7.5 μM) for seven days. Control cells were treated with DMSO (0.1%) as a vehicle control. The cells were maintained under standard cell culture conditions at 37°C and 5% CO2 in a humid environment. Colonies were fixed in methanol, stained with haematoxylin, and counted using UVP 810 software.
Western blot analysis
Western blot analysis was performed to investigate the effect of ORM on protein levels of various oncogenes link to prostate carcinogenesis. Briefly, PrCa cells (70–80% confluent) were treated with ORM (10–20 μM) concentrations for 24 hrs. Control cells were treated with vehicle (0.1% DMSO). Total cell lysates were prepared as described (15). Cytoplasmic and nuclear lysates were prepared using nuclear extract kit (Active Motif). Forty microgram of protein lysates were subjected for Western blot analysis using 4–20% SDS-PAGE gels, blotted onto PVDF membrane (Bio-Rad), and blocked with 10% bovine serum albumin (BSA) one hr at room temperature. The membranes were then incubated with the indicated primary antibodies followed by a horseradish peroxidase secondary antibody and developed with enhanced chemiluminescence reagent (Roche) using a UVP gel documentation system.
Immunofluorescence analysis
To determine the effect of ORM on β-catenin localization, 30,000 cells were seeded in 4 wells chamber slide. Next day, ORM (10μM) treatment was given for 24 hrs. Localization of β-catenin was monitor by performing immunofluorescence analysis using confocal microscopy as described (16).
TCF luciferase assay
To investigate the effect of ORM on TCF promoter activity, we performed TCF luciferase assay. The reporter constructs were a generous gift from Dr. R. Moon (University of Washington, Seattle, WA). In this experiment, DU145 cells (1.5×105 cells/well) were plated in triplicate in 12-well plates. Cells were transiently co-transfected with TCF-firefly luciferase reporter constructs (pTOP-FLASH) (1 μg) and Renilla luciferase (200 ng) or (pFOP-FLASH) (1 μg) and Renilla luciferase (200 ng). After 24 hrs, cells were treated with ORM (10 and 20 μM) and lysates prepared at 6 hrs post-treatment. Firefly and Renilla luciferase activity was analyzed by using Dual luciferase kit (Promega). The β-catenin/TCF transcription activity was determined by normalizing the firefly luciferase activity to that of Renilla luciferase activity and calculating the ratio of TOP-FLASH signal to FOP-FLASH signal.
Pulse chase assay
Pulse chase experiment was performed to determine the effect of ORM on β-catenin degradation using translational inhibitor cyclohexamide (CHX). Briefly, 70% confluent DU145 cells were treated with CHX (50 μg/ml) alone or in combination with ORM (15 μM) for 1–24 hrs. Cell lysates was prepared and protein levels of β-catenin was analyzed by Western blot analysis.
Molecular docking
Molecular docking experiments were conducted to know whether and where ORM binds in β-catenin (PDB ID: 4DJS) (17) and GSK3 β (PDB ID: 4ACH) (18) proteins. The 2D and 3D structure of ORM were taken from (https://pubchem.ncbi.nlm.nih.gov/compound/154413#section=Top) Pub Chem. These experiments were performed using autodock 4.2 suit by employing Lamarckian genetic algorithm (19). The grid map illustrating the active site pocket for ligands were calculated by autogrid and the dimension of the grid for 4DJS and 4ACH were 56×50×90 and 60×62×70 grid points respectively with a spacing of 0.375 Aº between the grid points and centered on the ligand. Docking was accomplished by each cycle with an initial population of 150 individuals and the remaining parameter set as default. Ten conformational docking poses were created and the best docked confirmation was selected based on the autodock binding energy (20). The confirmations with the most favorable free binding energy were selected for analyzing the interactions between the target receptor and ligands by visualization with Discovery Studio Software (version 3.5).
Chemoinvasion assay
Cell invasion assay was performed using a cell invasion kit (BD Biocoat™ Matrigel Invasion Chambers; BD Biosciences, San Jose, CA). All procedures were followed as per the manufacturer’s instructions. In brief, PC3 cells (50,000 cells/well) were seeded in an upper chamber containing serum free medium and further treated with ORM (5–15 μM) or 0.1% DMSO as vehicle for 24 hrs. The lower chamber was filled with 500 μl of media containing 20% FBS. Forty eight hrs post-treatment, cells were completely removed from inside the upper chamber by cotton swab. Cells were fixed with methanol and stained with Crystal Violet. Invaded cells were observed by using a light microscope at 100× magnification. Cells that had invaded the matrix membrane were counted in three random fields of view and the experiments were performed in triplicate.
Cell migration assay
Cell migration assay was performed using Boyden’s Chambers (BD Biosciences), as per manufacturer’s protocol. After 48 h incubation, the migrating cells were fixed with methanol and stained with crystal violet and photographed under light microscope. Migratory cells in ORM treated group were compared with control.
Cell proliferation, invasion and migration by real time xCELLigence system
To further confirm the functional impact of ORM on migration, invasion, and proliferation of PrCa cells, real-time proliferation, invasion and migration assays were performed using the xCELLigence system as described (13). Briefly, 5×103 PrCa cells were seeded per chamber of cell proliferation or invasion and 7×104 cells/well for migration. After 24 hrs, ORM (5–15 μM) or the vehicle control (0.1% DMSO) was added and the experiment was allowed to run for 48 hrs. Average baseline cell index of ORM-treated cells was calculated and compared to vehicle-treated control cells.
Agarose bead assay
Effect of ORM on cellular motility was determined by an agarose bead-based cell motility assay as described (21).
Cell cycle analysis
In this experiment, approximately 70% confluent PC3 cells were synchronized by overnight starvation of cells in serum free media. Cells were treated with ORM (10–20 μM) for 24 hrs and cell cycle analysis was performed by flow cytometry as described (12)
Apoptosis analysis
Effect of ORM treatment on apoptosis induction in PrCa cells, was analyzed using Annexin V-7AAD apoptosis kit (BD Biosciences, San Diego, CA) as described (15). In brief, PC3 and DU145 cells (200,000 cells per well) were plated in 6-well plates and allowed to attach overnight. Next day, cells were treated with ORM (10–20 μM) concentrations for 24 hrs. Both floating and adherent cells were collected, washed twice with cold PBS and stained with Annexin V-7AAD (5μL) each/100μL of cell suspension for 20 min in dark at room temperature. Number of apoptotic cells were analyzed by setting FL2 (Annexin V) and FL3 (7AAD) channels in BD Accuri™ flow cytometer (BD Biosciences). To analyze the effect of ORM on mitochondrial membrane potential (Δѱm), we utilized Tetramethyl rhodamine ethyl ester (TMRE) as described (12).
Xenograft study
A total of 12 athymic nude male mice were used to investigate the effect of ORM on PC3 cells derived xenograft tumors. The mice were maintained in a pathogen-free environment and all were carried out as per our approved protocol by the UTHSC Institutional Animal Care and Use Committee (IACUC). Briefly, PC3 cells (2 × 106) were dispersed in 100 μl 1:1 ratio of 1× PBS and 100 μl Matrigel (BD Biosciences) and injected subcutaneously into the dorsal flank of each mouse. The mice were periodically monitored for tumor development and the tumor volume was measured using a digital Vernier caliper. The tumor volume was calculated using the ellipsoid volume formula: tumor volume (mm3) = π/6 × L × W × H, wherein L is length, W is width, and H is height. The mice were given intraperitoneal injection of ORM (250 μg/mice) and 0.2% Ethanol in PBS 3 times a week for 6 consecutive weeks starting from week 1st. The tumor volume was regularly monitored and allowed to grow until the tumor burden reached a maximum volume of 1100 mm3. At the time of sacrifice, the mice tumors were removed, fixed in formalin, embedded in paraffin, and sliced into 5 micron sections for further biochemical analysis.
Immunofluorescence and immunohistochemistry analyses
Immunofluorescence analysis was performed to detect changes occur in the expression of β-catenin in excised xenograft tumors tissues of control and ORM treated mice as described (21).
Statistical analysis
Statistical analysis was performed using an unpaired two-tailed Student t-test and employed to assess the statistical significance between the control and ORM- treated groups. P value < 0.05 was considered as significant.
Results
ORM treatment inhibits the growth of prostate cancer cells
Metastatic PrCa is the significant cause of mortality and morbidity in PrCa patients (22). Thus, we examined the anti-cancer effects of ORM on human PrCa cell lines (LNCaP, C4-2, PC3 and DU145) by cell counting assay (Supplementary Figure 1). We performed additional cell proliferation assays (MTS assay and real time xCELLigence assay) in two highly metastatic PrCa cell lines (PC3 and DU145). ORM treatment inhibited viability of both PC3 and DU145 cells in a (10-40 μm) dose-dependent manner. The IC50 of ORM in PC3 and DU145 cells was 22 μM (Fig. and 17 μM respectively (Fig. 1Ai–ii), after 24 hrs treatment, while IC50 of ORM was 20 μM in PC3 cells and 15 μM in DU145 cells after 48 hrs treatment respectively (Fig. 1Ai–ii). We next evaluated the effect of ORM treatment on PrCa cell proliferation using xCELLigence assay for the duration of 72 hrs. (Fig.1Bi–ii). This assay monitors cell growth in real time by measuring changes in electric impedance between two golden electrodes embedded in the bottom of the cell culture wells. The impedance, which is converted to a cell index value, is directly proportional to the number of cells and also reflects the cells viability, morphology, and adhesion strength (23). The growth curve, which is presented as a baseline cell index, showed that ORM (10–20 μM) reduced the baseline cell index in PC3 (Fig. 1Bi) and DU145 (Fig. 1Bii) cells in a dose-dependent manner compared to vehicle treated cells. ORM treatment inhibited clonogenic potential of PC3 (Fig. 1Ci–ii), DU145 (Fig. 1Di–ii), and C4-2 (Supplementary Figure 2Ai–ii) cells as determined by independent colony formation assay. Moreover, ORM also inhibited anchorage-dependent growth of C4-2 cells (Supplementary Figure 2 Bi–ii). These results indicate that ORM effectively inhibits growth of PrCa cells including highly aggressive metastatic PrCa cells.
Figure 1. ORM inhibits the growth of hormone refractory PrCa cells.
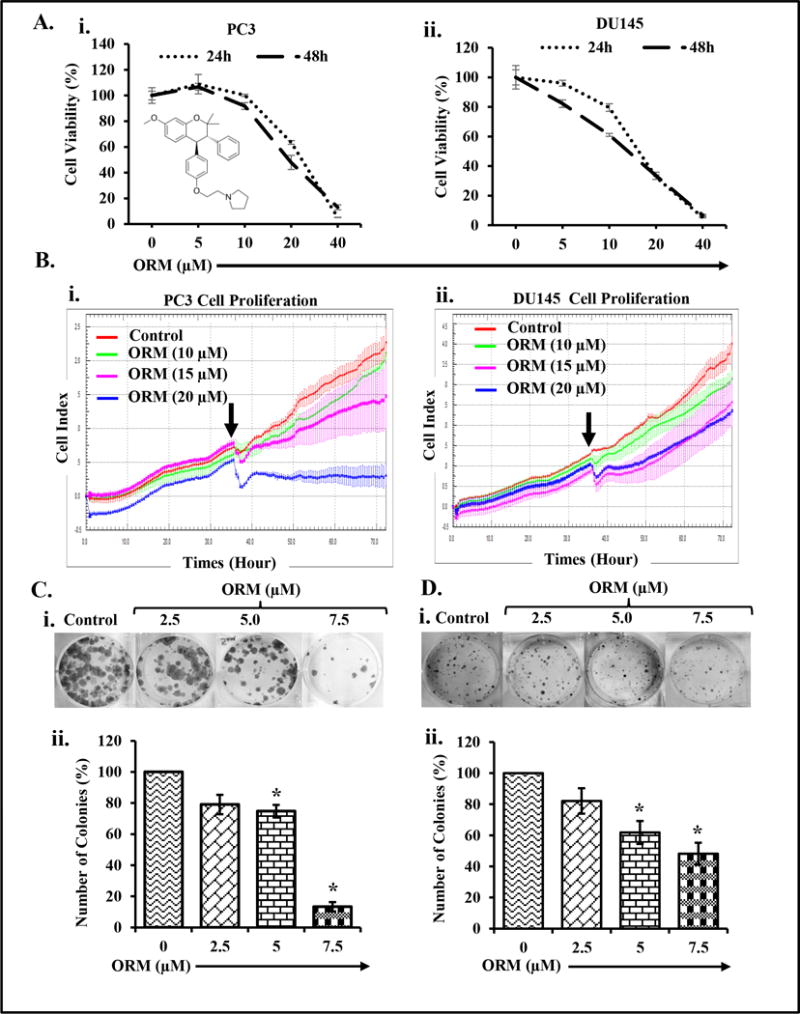
A. Effect of ORM on cell viability of PC3 (i) and DU145 (ii) cells. Briefly, cells (2,500) were seeded in each well of 96-well plate and after overnight incubation, cells were treated with the indicated concentrations of ORM for 24 and 48 hrs. Cell viability was assessed by MTS assay. The line graph represents the percent viable cells compared to the vehicle-treated group cells. Each concentration value is the mean±SE of triplicate wells of each group. B. Effect of ORM on PrCa cells proliferation. Briefly, PrCa cells (5,000 cells/well) were seeded in E-plate (xCELLigence) following the xCELLigence Real Time Cell Analyzer (RTCA) DP instrument manual as provided by the manufacturer. After 38 hrs, ORM or the vehicle control was added and the experiment was allowed to run for 80 hrs. Average baseline cell index for ORM-treated PC3 (Bi) and DU145 (Bii) cells was compared to vehicle treated group. C–D. Effect of ORM on clonogenic potential of PrCa cells. Representative colony images of control and ORM treated PC3 (Ci) and DU145 (Di) cells. Bar graphs indicating quantification of colony formation in PC3 (Cii) and DU145 (Dii) cells. Asterisk (*) denotes the significant value p<0.05.
ORM represses β-catenin signaling in PrCa cells
Since, ORM inhibited proliferation of PrCa cells, thus, we examined the effect of ORM on β-catenin signaling which is a major oncogenic pathway involved in tumorigenesis and metastasis (3,24–27). ORM treatment (10 μM) inhibited nuclear β-catenin in DU145 cells (Fig. 2Ai) through its sequestration in the cytoplasm (Fig. 2Ai) as determined by Western blot analysis. This result was further confirmed by confocal microscopy as ORM showed inhibition of β-catenin translocation into the nucleus of PrCa cells as compared to control (Fig. 2Aii, Supplementary Figure 3Bi–ii). We next evaluated the effect of ORM on lithium chloride (LiCl)-induced β-catenin/TCF promoting activity by transiently co-transfecting the DU145 cells with TCF-firefly luciferase reporter constructs (pTOP-FLASH) and Renilla luciferase or (pFOP-FLASH) and Renilla luciferase. ORM treatment (10 μM) for 6 hrs, significantly (P<0.01) inhibited lithium chloride (LiCl)-induced TCF-4 promoter activity in DU145 cells (Fig. 2iii). We also examined the effect of ORM on activation of GSK3β by Western blot analysis which illustrated a marked increase in phosphorylated GSK3β protein levels in DU145 cells (Fig. 2Bi). Since, we observed activation of GSK3β protein by ORM treatment, thus, we next examined the effect of ORM on β-catenin degradation after using translational inhibitor (cyclohexamide). Results revealed a time-dependent decrease in the protein levels of β-catenin in DU145 cells compared to cyclohexamide treatment alone group (Fig. 2Bii–iii). We next performed molecular docking studies to determine the orientation of ORM bound in the active sites of β-catenin and GSK3β using Discovery Studio software (version 3.5) as described (17). This study revealed that ORM binds with both β-catenin and GSK3β with a considerably high binding energy. ORM binds into the active site of β-catenin (4DJS) and GSK3β (4ACH) with minimum binding energy (∆G), −6.2 kcal/mol and −8.5 kcal/mol respectively (Table inserted as Figure 2C). The docking results also confirmed that ORM strongly binds with amino acid residue of β-catenin at ARG: 469, ARG: 612 (Fig. 2Di–ii), and GSK3β at LYS: 85, ARG 96 (Fig. 2Diii–iv). These amino acid residues actively participate in hydrophobic, hydrophilic and π-π interactions. Overall, these results suggest that ORM is a potent inhibitor of WNT/β-catenin signaling pathway.
Figure 2. Effect of ORM on β-catenin signaling pathway and molecular docking of ORM with β-catenin and GSK3β.
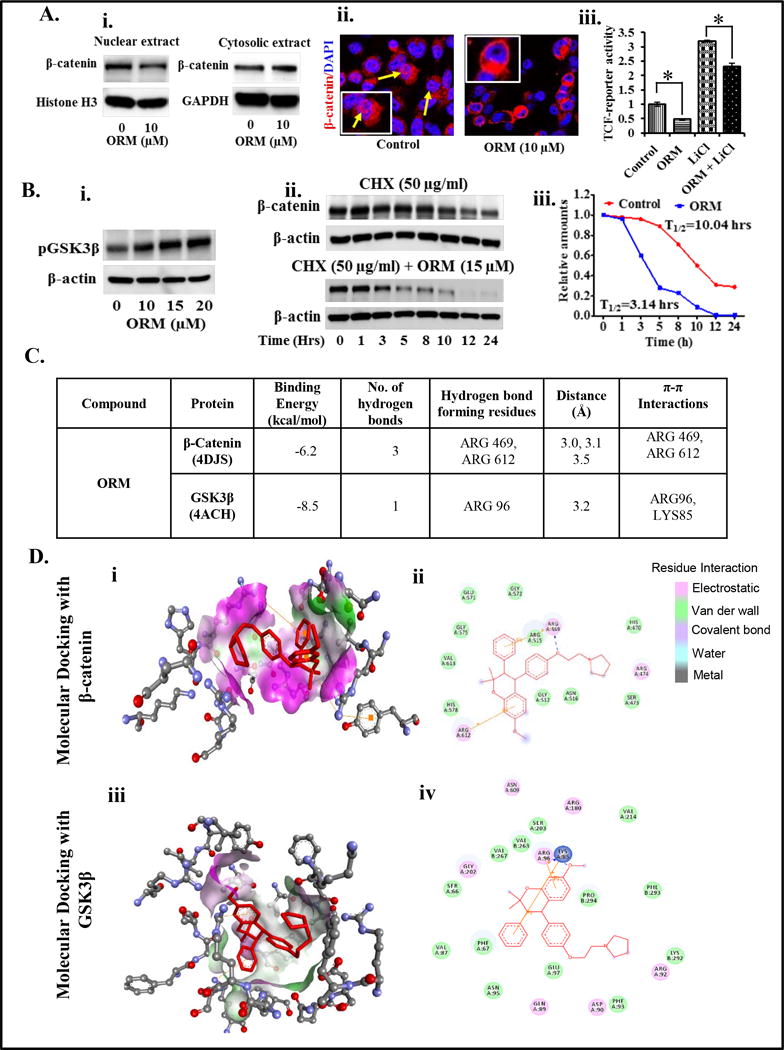
A. Effect of ORM on β-catenin distribution in cytoplasm and nucleus of DU145 cells. Briefly, cells were treated with indicated concentrations of ORM for 24 hrs, nuclear extracts were prepared and subjected for Western blot analysis to detect the protein levels of β-catenin. Results demonstrating decreased expression of nuclear β-catenin and increased expression of β-catenin in the cytoplasm (Ai) of DU145 cells. Blots were re-probed with Histone H3 and GAPDH antibodies as an internal control. Effect of ORM on β-catenin localization in PC3 cells as determined by confocal microscopy (Aii). Yellow arrows indicate localization of β-catenin in control and ORM treated cells after 24 hrs treatment (Original magnification 40×) (Aii). Effect of ORM on TCF-4 promoter activity (Aiii). Cells were transiently co-transfected with TCF-firefly luciferase reporter constructs (pTOP-FLASH) (1 μg) and Renilla luciferase (200 ng) or (pFOP-FLASH) (1 μg) and Renilla luciferase (200 ng). After 24 hrs, cells were treated with LiCl (50 μM) alone or in combination with ORM (10 μM). Cell lysates were prepared 6 hrs post-treatment and firefly and Renilla luciferase activity was analyzed by using Dual luciferase kit (Promega). The β-catenin/TCF transcription activity was determined by normalizing the firefly luciferase activity to that of Renilla luciferase activity and calculating the ratio of TOP-FLASH signal to FOP-FLASH signal. Values in bar graph indicates mean±SE of three wells reading in each group. Asterisk (*) denotes the significant value p<0.01. B. Effects of ORM on protein levels of phospho GSK3β in DU145 as determined by Western blot analysis (Bi). Effect of ORM on β-catenin degradation as analyzed by pulse chase experiment. Briefly, DU145 cells were treated with CHX (50 μg) alone or in combination with ORM (15 μM) at indicated time points. Protein lysates were prepared and subjected for Western blot analysis to analyze the protein levels of β-catenin. Results indicates protein levels of β-catenin in alone CHX-treated (upper blot) and in CHX and ORM-treated (Lower blot) (Bii). Line graph showing quantification of Western blots of Figure Bii. T1/2 denotes time point for 50% β-catenin degradation. C–D. Molecular docking studies of ORM with β-catenin and GSK3β. C. Table showing docking score of ORM with β-catenin and GSK3β. D. Stereo view of ORM binding with β-catenin (Di) and GSK3β (Diii) showing hydrogen bond donor and acceptor residues around component. Schematic diagram of ORM docking with β-catenin (Dii) and GSK3β (Div) showing residues involved in hydrogen-bonding, Pi interactions, charge or polar interactions, Van der Waals interactions which are represented by respective colors.
ORM treatment effectively attenuates metastatic potential of PrCa cells
Epithelial to mesenchymal transition (EMT) is the basic characteristic of cancer cell in which epithelial cells undergo morphological and molecular changes that transform these cells to mesenchymal, highly metastatic (invasive and motile) and drug resistant phenotype (28). Thus, targeting EMT will reduce the invasive phenotypes of a cancer cell and have significant advantage to overcome drug resistance. It has been reported that β-catenin is involved in invasion and metastasis via inducing EMT in various tumor cells including PrCa (29,30). Since, ORM effectively inhibits β-catenin signaling, therefore, we evaluated the effect of ORM treatment on various EMT markers in PrCa cells. ORM treatment revealed marked inhibition of N-cadherin, and Snail expressions in PC3 and DU145 cells (Fig. 3Ai–ii), while it induced the expression of E-cadherin in PC3 PrCa cells (Fig. 3Ai). Activation of MMPs are involved in matrix degradation that facilitate invasion of cancer cells (31), thus we sought to investigate the effect of ORM treatment on MMPs and found that ORM inhibited the expression of MMP2 and MMP3 (Fig. 3Ai–ii). We next performed functional assays (invasion and migration) by Boyden’s chamber to determine whether ORM inhibits the invasive and migratory potential of PrCa cells. Our results revealed that ORM treatment (5–15 μM) effectively inhibited both invasion (Fig. 3Bi) and migration (Fig. 3Ci) of PC3 cells and C4-2 cells (Supplementary Figure 2 Ci–ii). We further investigated the impact of ORM on real time invasion and migration of PC3 cells using xCELLigence system. ORM treatment also effectively decreased invasion (Fig. 3Bii) and migration (Fig. 3Cii) of PC3 cells. We further confirmed effect of ORM on PC3 cells migration (Fig. 3D) by beads assay. Our results revealed a marked decrease in number of migratory PC3 cells after 48 hrs ORM treatment compared to control group (Fig. 3D).
Figure 3.
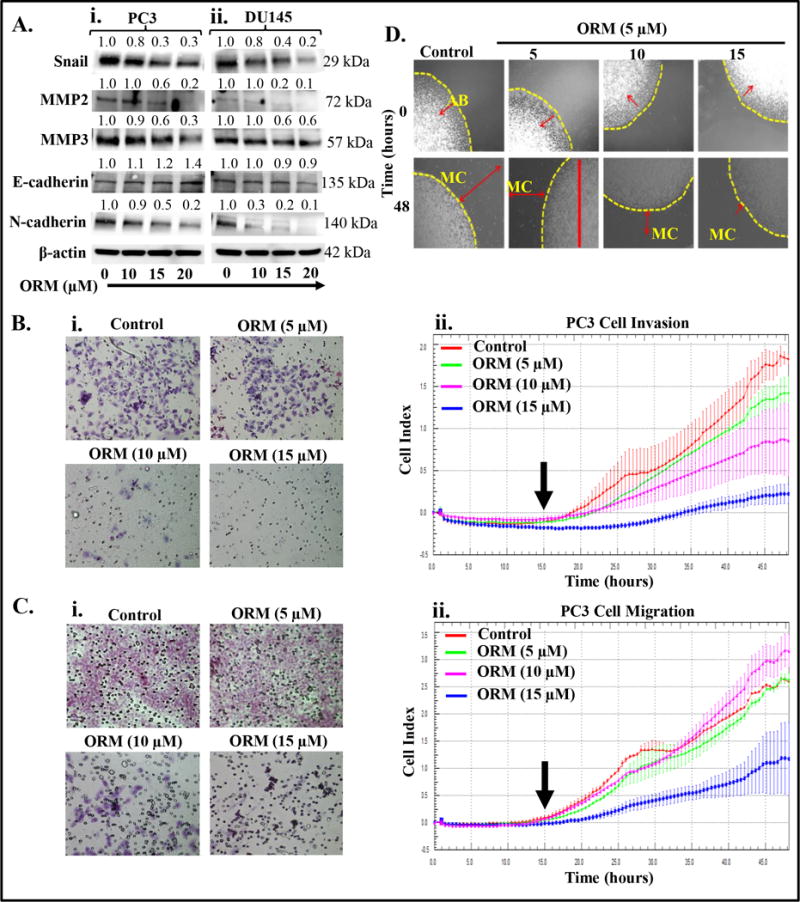
Effect of ORM on cell invasion, migration and EMT markers. Briefly, 70% confluent PrCa cells were treated with ORM (10–20 μM) for 24 hrs. Cell lysates were prepared and subjected for Western blot analysis for EMT markers and MMPs analysis. A. Effect of ORM on EMT markers (E-cadherin, N-Cadherin and Snail) and MMPs (MMP2 and MMP3) in PC3 (i) and DU145 (ii) cells. Values shown above the blots are the densitometry analysis of each protein band normalized with respective β-actin value. B. Effect of ORM on invasion of PC3 cells as determined by Boyden chamber and xCELLigence assays. Representative photographs (20× original magnification) of invaded cells of control and ORM treated PC3 cells as determined by Boyden chamber kit (i). Effect of ORM on real time cell invasion (ii). Briefly, PC3 cells (7×104) were seeded in invasion plate and invasion potential of these cells was determined by xCELLigence instrument as described in material and methods. Results indicate dose-dependent decrease in Cell Index, which correlates inhibition of real time cell invasion by ORM treatment (ii). C. Effect of ORM on cell migration of PC3 cells as determined by Boyden chamber and xCELLigence assays. Representative images (20× original magnification) showing inhibition of PC3 cells migration by Boyden chamber assay (i). Effect of ORM on real time cell migration as determined by xCELLigence assay (ii). Briefly, PC3 cells (7×104) were seeded in migration plate and ORM treatment (5–15 μM) was given after 15 hrs and allowed the plate at 37 °C and 5% CO2 for real time migration assay up to 48 hrs. Results indicate significant decrease in migratory potential of ORM treated PC3 cells compared to control. D. Effect of ORM on motility potential of PC3 cells as determined by agarose bead assay. Representative images of migratory cells (MC) in control and ORM-treated groups at 0 and 48 hrs. AB denotes agarose beads. Images were captured at 20× magnification.
ORM treatment arrests cell cycle via modulation of cell cycle regulatory proteins
Various studies have shown that agents which arrest cell cycle in G0/G1 phase have potential chemotherapeutic effects (32,33). Since, we observed that ORM inhibits the growth of PrCa cells, we sought to determine the effect of ORM on PrCa cell cycle distribution. For this we synchronized PC3 cells and treated with ORM (10–20 μM) for 24 hrs and cell cycle analysis was performed by flow cytometry. ORM treatment arrested PC3 cells cycle in G0–G1 phase in a dose-dependent manner (Fig. 4Ai–ii). ORM treatment resulted in 5%, 20% and 20% increase in cell cycle arrest in G0–G1 phase at 10, 15 and 20 μM dose, respectively, compared to vehicle-control treated cells (insert Table in Fig. 4Aii). Similar result was also observed in DU145 cells (Supplementary Figure 3). We then evaluated the effect of ORM on cell cycle regulatory proteins in PrCa cells. ORM (10–20 μM) inhibited expression of Mcl-1 in both PC3 and DU145 cells (Fig. 4Bi–ii). However, ORM (10–20 μM) treatment showed more effect on cyclin D1 inhibition in DU145 treated cells as compared to PC3 treated cells. ORM treatment induced expressions of cell cycle inhibitory proteins (p21 and p27) in both PC3 and DU145 cells (Fig. 4Bi–ii). These results suggest that ORM arrests cell cycle via modulating key cell cycle regulatory proteins.
Figure 4. Effect of ORM on cell cycle progression of prostate cancer cells.
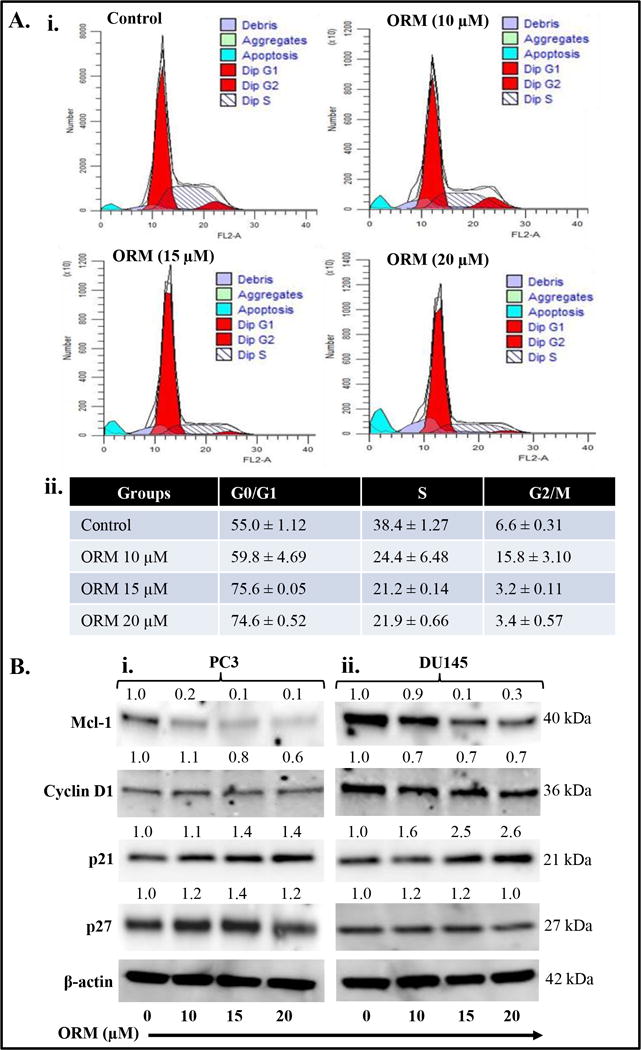
ORM arrests PC3 cell cycle in G0/G1 phase as determined by flow cytometry. A. Histogram (i) and table (ii) represent the cell cycle distribution in PC3 cells. B. Effect of ORM on protein levels of cell cycle regulatory proteins (Cyclin D1, Mcl-1, p21 and p27) in both PC3 (Bi) and DU145 (Bii) cells. Briefly, cells were treated with indicated concentrations of ORM for 24 hrs, total cell lysates were prepared and subjected for Western blot analysis. Equal loading of protein in each lane was determined by probing the blots with β-actin antibody. Values shown above the blots are the densitometry analysis of each protein band normalized with respective β-actin value.
ORM treatment induces apoptosis in PrCa cells
Since, we observed arrest of cell cycle in G0–G1phase, thus we investigated the effect of ORM on apoptotic induction in PrCa cells by flow cytometry analysis. ORM (10–20 μM) dose-dependently increased apoptotic cell populations in both PC3 (Fig. 5Ai) and DU145 (Fig. 5Aii) cells as determined by enhanced Annexin V positive cells. ORM at 20 μM showed 55.6% and 50% apoptotic PC3 and DU145 cells respectively compared to control group (Fig. 5Ai–ii). We next examined the effect of ORM on mitochondrial membrane potential (Δѱm) using TMRE staining (Fig. 5Bi–ii), which is a marker of apoptosis induction through intrinsic pathway. ORM (10–20 μM) dose-dependently decreased TMRE staining in both DU145 and PC3 cells as determined by fluorescence microscopy (Fig. 5Bi) and flow cytometry (Fig. 5Bi–ii), respectively. We also examined the effect of ORM on PARP cleavage which is a marker for apoptosis. ORM (20 μM) induced PARP cleavage as determined by western blot analysis (Fig. 5C). These results suggest apoptosis inducing potential of ORM in PrCa cells.
Figure 5. ORM treatment induces apoptosis in PrCa cells.
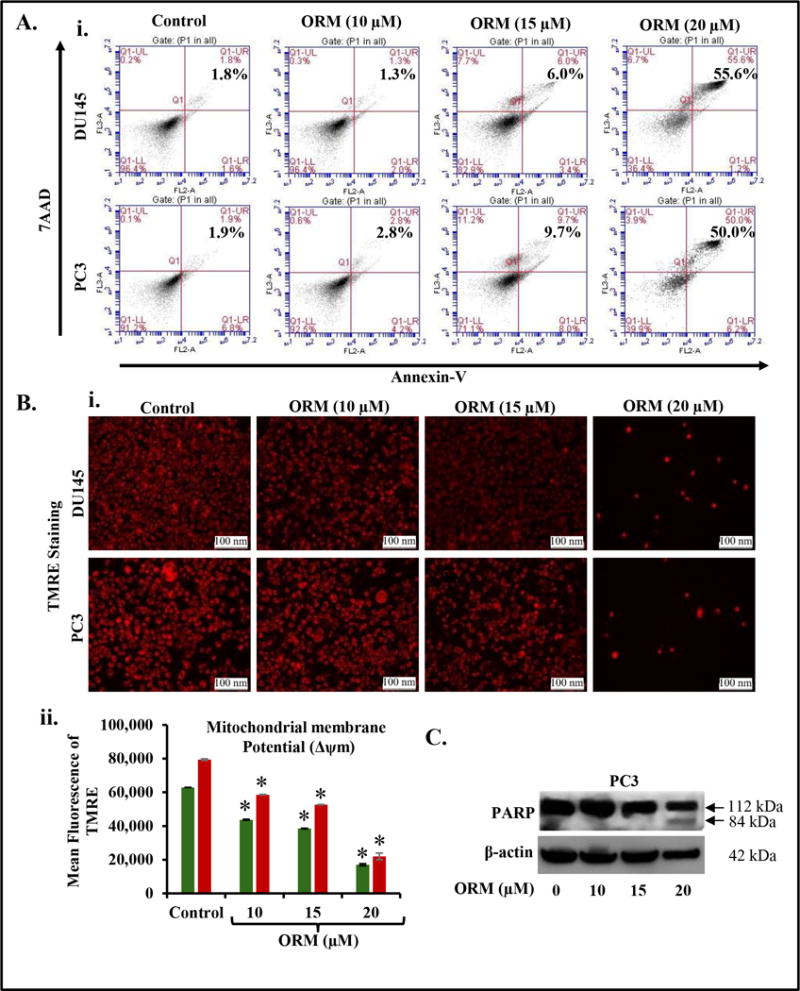
PC3 and DU145 cells were treated with indicated concentration of ORM for 24 hrs and processed for apoptosis analysis using Annexin V-7AAD apoptosis kit. A. Representative FL3-A and FL2-A plots showing dose-dependent increase of apoptosis in DU145 (i) and PC3 cells (ii). B. Effect of ORM on mitochondrial membrane potential (Δѱm) as determined by TMRE staining. Representative Fluorescence images showing dose-dependent effect of ORM on TMRE staining in PC3 and DU145 cells (i). Bar graph indicating dose-dependent inhibition of mitochondrial membrane potential (Δѱm) in ORM-treated PC3 and DU145 cells as determined by flow cytometry. Asterisk (*) denotes the significant value p<0.01. C. Effect of ORM on PARP cleavage. Briefly, 70% confluent PrCa cells were treated with ORM (10–20 μM) for 24 hrs. Whole cell lysates was prepared and subjected for Western blot analysis for full and cleaved PARP.
ORM suppresses tumor growth in xenograft mouse model
To evaluate clinical relevance of our in vitro findings, we subcutaneously implanted PC3 cells in a preclinical xenograft mouse model. Intraperitoneal administration of ORM (250 μg/mice/thrice weekly) significantly (p<0.01) reduced PrCa tumor growth (Fig. 6Ai–iii). To confirm if this tumor growth inhibition is mediated through the suppression of β-catenin, we performed immunofluorescence analysis of excised xenograft tumors for β-catenin. Both cytoplasmic and nuclear β-catenin expression was decreased in ORM treated xenograft tumor cells compared to control tumor cells (Fig. 6B). This result was validated by immunohistochemistry analysis (Fig. 6C). We further determined the expression of EMT markers in excised tumor tissues of control and ORM-treated mice. ORM treatment also inhibited the expression of N-cadherin, Slug, Snail, and Vimentin and induced the expression of E-Cadherin (Fig. 6C). Metastasis-associated Protein 1 (MTA1) is known to be upregulated in many cancer types and is an important metastatic marker for tumor aggressiveness and metastasis (34,35). Interestingly, we observed a decreased expression of MTA1 in ORM treated xenograft tissues as compared to control. Next, we examined the expression of cell proliferative markers, proliferative cell nuclear antigen (PCNA), in control and ORM-treated tumor tissues by immunohistochemistry analysis. We found a marked decrease in nuclear PCNA staining in ORM-treated tumors compared to the tumors in vehicle-treated mice (Fig. 6C). These results reaffirm that ORM has potent therapeutic efficacy against PrCa and could be used for the treatment of metastatic PrCa. The possible molecular mechanisms of ORM to inhibit prostate tumor growth and metastasis have been summarized in a schematic diagram (Supplementary Figure 4) which shows ORM inhibits wnt/β-catenin and EMT signaling and it induces activation of GSK3β, E-cadherin, p21 and p27 signaling pathways, thus decreasing prostate tumor growth and metastasis.
Figure 6. ORM inhibits prostate tumor growth in xenograft mouse model.
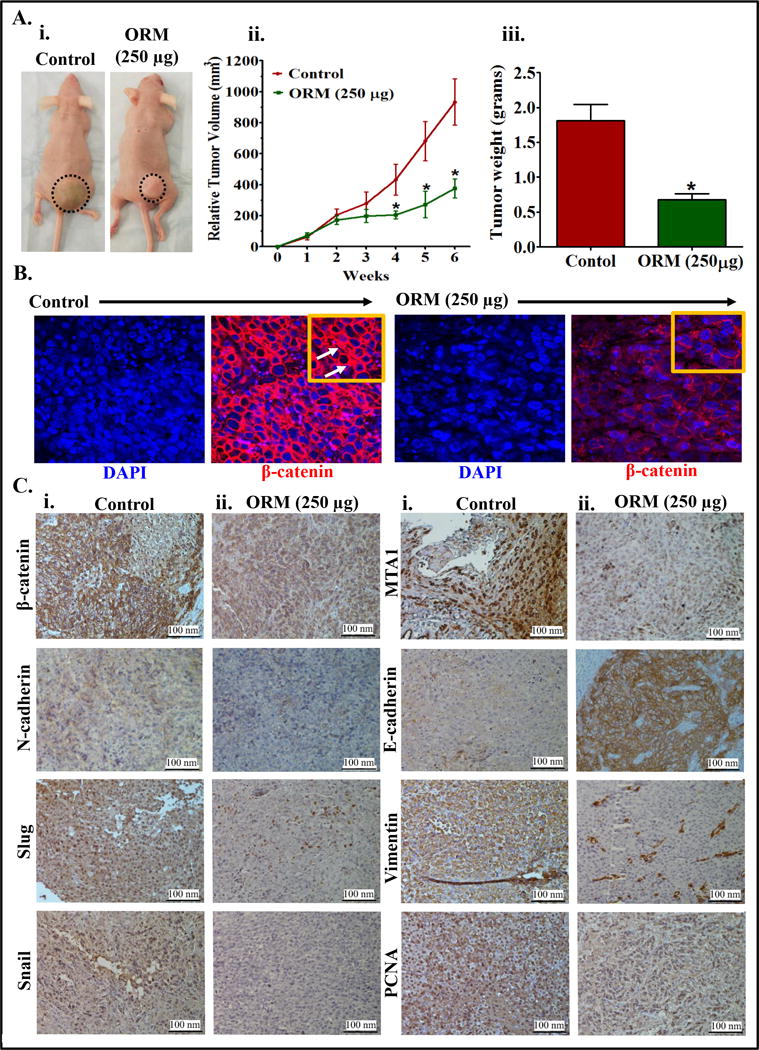
A. Effect of ORM on PC3 cells derived xenograft tumors in athymic nude mice. In brief, a total of 12 mice were used in this experiment and were divided into two groups. 2×106 PC3 cells were injected subcutaneously on dorsal flank of each mouse. ORM (250 μg) was administered (intra-peritoneal; 250 μg/mouse) thrice/week till 6 weeks and control group mice received 0.2% Ethanol in PBS as vehicle control. Mice of both the groups were sacrificed when control mice reached a targeted tumor volume of 1000 mm3. Representative mouse picture of control and ORM treated tumor bearing mouse (i). Average tumor volume of each group mice at different weeks (ii). Bar graph representing tumor weight of each group mice (iii). Value in graph represents mean±SE of 6 mice in each group. Asterisk (*) denotes the significant value p<0.01. B. Effect of ORM on β-catenin expression in xenograft tumors of control and ORM treated mice as determined by immunofluorescence (IF) analysis. White arrows indicate β-catenin accumulation in nucleus of the xenograft tissues. C. Effect of ORM on the expressions of β-catenin, N-cadherin, MTA1, Slug, Snail, Vimentin, and E-cadherin and PCNA in excised tumors of control (i) and ORM (ii) treated mice as determined by immunohistochemistry (IHC) analysis. All images of IF and IHC analyses was captured at 20× magnification.
Discussion
PrCa continues to remain the most common cancer and the second leading cause of cancer-related deaths, in American men. Metastatic PrCa is the end-stage and accounts for the majority of cancer deaths(22). Moreover, men with metastatic PrCa are at a higher risk of developing bone metastasis which results in clinical skeletal morbidity (36). Although PrCa is frequently curable in its early stage by surgical and/or radiation therapy, many patients present locally advanced or metastatic disease for which there are currently no curative treatment options (37). Docetaxel and Cabazitaxel are FDA approved chemotherapeutic drugs for the treatment of metastatic PrCa, but these drugs have severe toxic side effects (38,39). Accumulating evidence suggest that β-catenin signaling pathway and its related oncogenic events play a major role during the development, progression and metastasis of cancer including PrCa (30,40). Thus, there is an urgent need to identify more effective and non-toxic agents or drugs, which can target Wnt/β-catenin and related oncogenic pathways.
ORM has an excellent therapeutic index and is safe for chronic administration in humans (14). The maximum serum concentration (Cmax) of ORM in humans is dose-dependent (Cmax of 55.53 ± 15.43 ng/ml for 30 mg dose and Cmax of 122.57 ± 6.25 ng/ml for 60 mg dose) and is reached within 4–6 hrs(41). Similar Cmax values for ORM also detected in breast cancer patients treated with either 30 mg, twice a week for 12 weeks (Cmax54.98 ± 14.19 ng/ml) or 60 mg of ORM on alternate days for 1 month (Cmax135 ± 15.5 ng/ml). These studies indicate that ORM is a non-toxic, highly bioavailable and shown potent anti-cancer effects against breast cancer (10), HNSCC (11), ovarian (12) and pancreatic cancer (13). However, molecular mechanisms of its anti-cancer properties are not well understood. In this study, we identified a novel molecular mechanism of ORM’s anti-cancer action as it effectively targets wnt/β-catenin and EMT-related oncogenic signaling pathways in PrCa cells.
It is well documented that constitutive activation of β-catenin signaling pathway plays a major role in cancer progression and metastasis (3,24) and drug resistance (42). Accumulating evidences also suggest that β-catenin cross-talks with other oncogenic signaling components leading to more aggressive phenotype of PrCa cells (43). GSK3β-dependent phosphorylation of β-catenin enhances its proteasomal degradation and inhibits its translocation into the nucleus, thus regulates its various downstream target oncogenes. Our results indicate that ORM activates GSK3β, thereby, degrades β-catenin in the cytoplasm and also inhibits nuclear β-catenin translocation and represses TCF-4 promoter activity. It has been reported that ARG 469 is one of the important amino acids involved in β-catenin interaction with TCF4 (44). Our molecular docking results indicate that ORM potentially binds with ARG 469 amino acids of β-catenin (Fig.2Di). It is possible that ORM inhibits β-catenin induced TCF4 promoter activity via binding to this cavity and inhibiting β-catenin/TCF4 interaction. Both LYS 85 and ARG 96 are important amino acids for GSK3β, which plays an important role in ATP binding and phosphoryl transfer (45,46). It has been documented that mutation of LYS85 with ARG inhibits binding of Axin to GSK3β which resulted in inactivation of GSK3β (47). Mutation in ARG 96 leads to complete loss of GSK3β catalytic activity (46). Consistent with this observation, our molecular docking study reveals that ORM effectively docks with LYS 85 and ARG 96 (Fig. 2Dii). It may be possible that ORM induces activation of GSK3β through increased ATP binding at LYS 85 and blocking mutation in these amino acids. More biological studies are warranted to further confirm the importance of these amino acid residues in ORM-induced activation of GSK3β. β-catenin also regulates EMT-related oncogenic signaling though which cancer cells gain mesenchymal and metastatic characteristics (48). EMT is typically accompanied by loss of epithelial markers such as E-cadherin and gain of mesenchymal markers such as N-cadherin, Snail, and vimentin in PrCa (49,50). Our results indicate that ORM induces the expressions of E-cadherin and inhibits Snail and N-cadherin in PrCa cells. These results suggest that ORM effectively blocks EMT progression. MMPs are secreted proteins which help cancer cell to invade through basal lamina by degrading extracellular matrix (51). MMP2/3 plays an important role in progression and metastasis of PrCa (31). Our results indicate that ORM has the ability to inhibit metastatic potential of PrCa cells, through repression of MMP-2 and MMP3. In this study, we have also shown that ORM can effectively inhibit the cell proliferation and clonogenic potential of highly aggressive PrCa cells. Abnormal regulation of cell cycle progression is one of trademark of cancer cells (52). G0/G1 abrogation of the cell cycle prevents cancer cells from repairing DNA and inhibits them from entering the S phase. Thus, the G0/G1 checkpoint has emerged as an attractive therapeutic target for cancer therapy (32). Interestingly, ORM treatment arrested PrCa cells in in G0/G1 phase of cell cycle and induced apoptosis. These results are consistent with similar previous findings in other cancer cells (12,13). It has been shown that cyclins and cyclin-dependent kinases (CDKs), play a critical role in cell cycle progression; their deregulation leads to cell cycle arrest (53). The observed inhibitory effects of ORM on cyclin D1 in PrCa cells clearly demonstrates interference in cell cycle regulatory proteins. It is known that p21/WAF1 and p27/KIP1 regulates CDK activity and our results illustrate increased expression of both p21 and p27 proteins in PrCa cells. These results suggest that ORM has the ability to arrest the PrCa cells in G0/G1 phase via modulating cell cycle regulatory proteins. Thus, ORM is expected to have high therapeutic efficacy against PrCa. Our in vivo therapeutic study showed that ORM significantly (P<0.01) reduces the prostate tumors burden in the preclinical athymic nude mouse model with no apparent toxicity. We also observed effective inhibition of β-catenin and EMT related markers (vimentin, N-cadherin, snail, and slug) in excised xenograft tumors of ORM-treated mice. It has been reported that MTA1 and β-catenin reciprocally activate each other during cancer/metastasis progression (54,55). Inhibition of MTA1 expression correlates with improved clinical outcome in various cancer types. ORM mediated repression of MTA1 in xenograft tumors further indicates its potential to inhibit PrCa metastasis. Taken together, these results confirm that ORM is a potent inhibitor of β-catenin and EMT related signaling pathways and has a potential to inhibit the metastatic phenotype of PrCa cells. These results also suggest that ORM can be repurposed for the treatment of metastatic PrCa. For that, human trials however are warranted in near future.
Conclusion
In summary, we have shown potential anti-cancer effects of ORM against PrCa using cell lines and preclinical mouse models. ORM efficiently targets β-catenin and EMT-related signaling pathways to repress prostate tumor growth and metastatic phenotypes. We conclude that ORM could be used alone or in combination with current therapeutic regimen for the treatment of human PrCa.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. R. Moon (University of Washington, Seattle, WA) for providing TCF/LIF constructs. Confocal microscopy studies were conducted at the imaging core in the Neuroscience Department at The University of Tennessee Health Science Center. This work was supported by Department of Defense (DOD) ARMY GRANT W81XWH-14-1-0154; NIH U01CA162106, NIH R01CA142736, and College of Pharmacy, University of Tennessee Health Science Center Seed Grant.
Financial Support: This work was supported by Department of Defense (DOD) ARMY GRANT W81XWH-14-1-0154; NIH U01CA162106, NIH R01CA142736, and College of Pharmacy/University of Tennessee Health Science Center Seed Grant.
Footnotes
Conflict of interest: No potential conflicts of interest were disclosed
Author contributions
BBH, AG and MS contributed equally to this work. BBH designed the research, performed mice experiments, and wrote the manuscript; AG performed cell proliferation, invasion, migration assays, IHC, and mice experiments; M.S performed WB analysis experiments. MKT contributed to β-catenin assay & stability studies. ZBH performed docking studies; VKK, participated in xenograft study; NC performed cell cycle and apoptosis analyses; AEM performed MTS assay; FTH synthesized and characterized the ORM; NZ, M.J. and S.C.C. analyzed data, and edited the manuscript.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Silvestris N, Leone B, Numico G, Lorusso V, De Lena M. Present status and perspectives in the treatment of hormone-refractory prostate cancer. Oncology. 2005;69(4):273–82. doi: 10.1159/000089676. [DOI] [PubMed] [Google Scholar]
- 3.Chen G, Shukeir N, Potti A, Sircar K, Aprikian A, Goltzman D, et al. Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer. 2004;101(6):1345–56. doi: 10.1002/cncr.20518. [DOI] [PubMed] [Google Scholar]
- 4.Jaggi M, Nazemi T, Abrahams NA, Baker JJ, Galich A, Smith LM, et al. N-cadherin switching occurs in high Gleason grade prostate cancer. The Prostate. 2006;66(2):193–9. doi: 10.1002/pros.20334. [DOI] [PubMed] [Google Scholar]
- 5.Jaggi M, Johansson SL, Baker JJ, Smith LM, Galich A, Balaji KC. Aberrant expression of E-cadherin and beta-catenin in human prostate cancer. Urologic oncology. 2005;23(6):402–6. doi: 10.1016/j.urolonc.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 6.De P, Carlson JH, Wu H, Marcus A, Leyland-Jones B, Dey N. Wnt-beta-catenin pathway signals metastasis-associated tumor cell phenotypes in triple negative breast cancers. Oncotarget. 2016;7(28):43124–49. doi: 10.18632/oncotarget.8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novak A, Dedhar S. Signaling through beta-catenin and Lef/Tcf. Cellular and molecular life sciences : CMLS. 1999;56(5–6):523–37. doi: 10.1007/s000180050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chesire DR, Ewing CM, Gage WR, Isaacs WB. In vitro evidence for complex modes of nuclear beta-catenin signaling during prostate growth and tumorigenesis. Oncogene. 2002;21(17):2679–94. doi: 10.1038/sj.onc.1205352. [DOI] [PubMed] [Google Scholar]
- 9.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harbor perspectives in biology. 2010;2(2):a002915. doi: 10.1101/cshperspect.a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nigam M, Ranjan V, Srivastava S, Sharma R, Balapure AK. Centchroman induces G0/G1 arrest and caspase-dependent apoptosis involving mitochondrial membrane depolarization in MCF-7 and MDA MB-231 human breast cancer cells. Life sciences. 2008;82(11–12):577–90. doi: 10.1016/j.lfs.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava VK, Gara RK, Bhatt ML, Sahu DP, Mishra DP. Centchroman inhibits proliferation of head and neck cancer cells through the modulation of PI3K/mTOR pathway. Biochemical and biophysical research communications. 2011;404(1):40–5. doi: 10.1016/j.bbrc.2010.11.049. [DOI] [PubMed] [Google Scholar]
- 12.Maher DM, Khan S, Nordquist JL, Ebeling MC, Bauer NA, Kopel L, et al. Ormeloxifene efficiently inhibits ovarian cancer growth. Cancer letters. 2015;356(2 Pt B):606–12. doi: 10.1016/j.canlet.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khan S, Ebeling MC, Chauhan N, Thompson PA, Gara RK, Ganju A, et al. Ormeloxifene suppresses desmoplasia and enhances sensitivity of gemcitabine in pancreatic cancer. Cancer research. 2015;75(11):2292–304. doi: 10.1158/0008-5472.can-14-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh MM. Centchroman, a selective estrogen receptor modulator, as a contraceptive and for the management of hormone-related clinical disorders. Medicinal research reviews. 2001;21(4):302–47. doi: 10.1002/med.1011. [DOI] [PubMed] [Google Scholar]
- 15.Sikander M, Hafeez BB, Malik S, Alsayari A, Halaweish FT, Yallapu MM, et al. Cucurbitacin D exhibits potent anti-cancer activity in cervical cancer. Scientific reports. 2016;6:36594. doi: 10.1038/srep36594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan S, Sikander M, Ebeling MC, Ganju A, Kumari S, Yallapu MM, et al. MUC13 interaction with receptor tyrosine kinase HER2 drives pancreatic ductal adenocarcinoma progression. Oncogene. 2017;36(4):491–500. doi: 10.1038/onc.2016.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grossmann TN, Yeh JT-H, Bowman BR, Chu Q, Moellering RE, Verdine GL. Inhibition of oncogenic Wnt signaling through direct targeting of β-catenin. Proceedings of the National Academy of Sciences. 2012;109(44):17942–7. doi: 10.1073/pnas.1208396109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berg S, Bergh M, Hellberg S, Högdin K, Lo-Alfredsson Y, Söderman P, et al. Discovery of novel potent and highly selective glycogen synthase kinase-3β (GSK3β) inhibitors for Alzheimer’s disease: design, synthesis, and characterization of pyrazines. Journal of medicinal chemistry. 2012;55(21):9107–19. doi: 10.1021/jm201724m. [DOI] [PubMed] [Google Scholar]
- 19.Fuhrmann J, Rurainski A, Lenhof HP, Neumann D. A new Lamarckian genetic algorithm for flexible ligand‐receptor docking. Journal of computational chemistry. 2010;31(9):1911–8. doi: 10.1002/jcc.21478. [DOI] [PubMed] [Google Scholar]
- 20.Ansari MF, Siddiqui SM, Ahmad K, Avecilla F, Dharavath S, Gourinath S, et al. Synthesis, antiamoebic and molecular docking studies of furan-thiazolidinone hybrids. European Journal of Medicinal Chemistry. 2016;124:393–406. doi: 10.1016/j.ejmech.2016.08.053. [DOI] [PubMed] [Google Scholar]
- 21.Chauhan SC, Vannatta K, Ebeling MC, Vinayek N, Watanabe A, Pandey KK, et al. Expression and functions of transmembrane mucin MUC13 in ovarian cancer. Cancer research. 2009;69(3):765–74. doi: 10.1158/0008-5472.CAN-08-0587. [DOI] [PubMed] [Google Scholar]
- 22.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 23.Abassi YA, Xi B, Zhang W, Ye P, Kirstein SL, Gaylord MR, et al. Kinetic cell-based morphological screening: prediction of mechanism of compound action and off-target effects. Chemistry & biology. 2009;16(7):712–23. doi: 10.1016/j.chembiol.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morita N, Uemura H, Tsumatani K, Cho M, Hirao Y, Okajima E, et al. E-cadherin and alpha-, beta- and gamma-catenin expression in prostate cancers: correlation with tumour invasion. British journal of cancer. 1999;79(11–12):1879–83. doi: 10.1038/sj.bjc.6690299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen T, Zhang K, Siegal GP, Wei S. Prognostic Value of E-Cadherin and beta-Catenin in Triple-Negative Breast Cancer. American journal of clinical pathology. 2016 doi: 10.1093/ajcp/aqw183. [DOI] [PubMed] [Google Scholar]
- 26.Chen Z, He X, Jia M, Liu Y, Qu D, Wu D, et al. beta-catenin overexpression in the nucleus predicts progress disease and unfavourable survival in colorectal cancer: a meta-analysis. PloS one. 2013;8(5):e63854. doi: 10.1371/journal.pone.0063854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tseng RC, Huang WR, Lin SF, Wu PC, Hsu HS, Wang YC. HBP1 promoter methylation augments the oncogenic beta-catenin to correlate with prognosis in NSCLC. Journal of cellular and molecular medicine. 2014;18(9):1752–61. doi: 10.1111/jcmm.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakazawa M, Kyprianou N. Epithelial-mesenchymal-transition regulators in prostate cancer: Androgens and beyond. The Journal of steroid biochemistry and molecular biology. 2016 doi: 10.1016/j.jsbmb.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 29.Zhao JH, Luo Y, Jiang YG, He DL, Wu CT. Knockdown of beta-Catenin through shRNA cause a reversal of EMT and metastatic phenotypes induced by HIF-1alpha. Cancer investigation. 2011;29(6):377–82. doi: 10.3109/07357907.2010.512595. [DOI] [PubMed] [Google Scholar]
- 30.Jiang YG, Luo Y, He DL, Li X, Zhang LL, Peng T, et al. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. International journal of urology : official journal of the Japanese Urological Association. 2007;14(11):1034–9. doi: 10.1111/j.1442-2042.2007.01866.x. [DOI] [PubMed] [Google Scholar]
- 31.Stearns M, Stearns ME. Evidence for increased activated metalloproteinase 2 (MMP-2a) expression associated with human prostate cancer progression. Oncology research. 1996;8(2):69–75. [PubMed] [Google Scholar]
- 32.Gupta S, Hussain T, Mukhtar H. Molecular pathway for (−)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Archives of biochemistry and biophysics. 2003;410(1):177–85. doi: 10.1016/s0003-9861(02)00668-9. [DOI] [PubMed] [Google Scholar]
- 33.Johnson JJ, Petiwala SM, Syed DN, Rasmussen JT, Adhami VM, Siddiqui IA, et al. alpha-Mangostin, a xanthone from mangosteen fruit, promotes cell cycle arrest in prostate cancer and decreases xenograft tumor growth. Carcinogenesis. 2012;33(2):413–9. doi: 10.1093/carcin/bgr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li DQ, Pakala SB, Reddy SD, Peng S, Balasenthil S, Deng CX, et al. Metastasis-associated protein 1 is an integral component of the circadian molecular machinery. Nature communications. 2013;4:2545. doi: 10.1038/ncomms3545. [DOI] [PubMed] [Google Scholar]
- 35.Li DQ, Pakala SB, Nair SS, Eswaran J, Kumar R. Metastasis-associated protein 1/nucleosome remodeling and histone deacetylase complex in cancer. Cancer research. 2012;72(2):387–94. doi: 10.1158/0008-5472.can-11-2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valdespino V, Tsagozis P, Pisa P. Current perspectives in the treatment of advanced prostate cancer. Medical oncology (Northwood, London, England) 2007;24(3):273–86. doi: 10.1007/s12032-007-0017-9. [DOI] [PubMed] [Google Scholar]
- 37.So A, Gleave M, Hurtado-Col A, Nelson C. Mechanisms of the development of androgen independence in prostate cancer. World journal of urology. 2005;23(1):1–9. doi: 10.1007/s00345-004-0473-1. [DOI] [PubMed] [Google Scholar]
- 38.Schweizer MT, Gulati R, Mostaghel EA, Nelson PS, Montgomery RB, Yu EY, et al. Docetaxel-related toxicity in metastatic hormone-sensitive and metastatic castration-resistant prostate cancer. Medical oncology (Northwood, London, England) 2016;33(7):77. doi: 10.1007/s12032-016-0793-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bahl A, Masson S, Malik Z, Birtle AJ, Sundar S, Jones RJ, et al. Final quality of life and safety data for patients with metastatic castration-resistant prostate cancer treated with cabazitaxel in the UK Early Access Programme (EAP) ( NCT01254279) BJU international. 2015;116(6):880–7. doi: 10.1111/bju.13069. [DOI] [PubMed] [Google Scholar]
- 40.Chesire DR, Dunn TA, Ewing CM, Luo J, Isaacs WB. Identification of aryl hydrocarbon receptor as a putative Wnt/beta-catenin pathway target gene in prostate cancer cells. Cancer research. 2004;64(7):2523–33. doi: 10.1158/0008-5472.can-03-3309. [DOI] [PubMed] [Google Scholar]
- 41.Lal J, Asthana OP, Nityanand S, Gupta RC. Pharmacokinetics of centchroman in healthy female subjects after oral administration. Contraception. 1995;52(5):297–300. doi: 10.1016/0010-7824(95)00213-t. [DOI] [PubMed] [Google Scholar]
- 42.Flores ML, Castilla C, Gasca J, Medina R, Perez-Valderrama B, Romero F, et al. Loss of PKCdelta Induces Prostate Cancer Resistance to Paclitaxel through Activation of Wnt/beta-Catenin Pathway and Mcl-1 Accumulation. Molecular cancer therapeutics. 2016;15(7):1713–25. doi: 10.1158/1535-7163.mct-15-0951. [DOI] [PubMed] [Google Scholar]
- 43.Petre-Draviam CE, Cook SL, Burd CJ, Marshall TW, Wetherill YB, Knudsen KE. Specificity of cyclin D1 for androgen receptor regulation. Cancer research. 2003;63(16):4903–13. [PubMed] [Google Scholar]
- 44.Trosset JY, Dalvit C, Knapp S, Fasolini M, Veronesi M, Mantegani S, et al. Inhibition of protein-protein interactions: the discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins. 2006;64(1):60–7. doi: 10.1002/prot.20955. [DOI] [PubMed] [Google Scholar]
- 45.Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, et al. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(20):11074–9. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang N, Jiang Y, Zou J, Yu Q, Zhao W. Structural basis for the complete loss of GSK3beta catalytic activity due to R96 mutation investigated by molecular dynamics study. Proteins. 2009;75(3):671–81. doi: 10.1002/prot.22279. [DOI] [PubMed] [Google Scholar]
- 47.Ikeda S, Kishida M, Matsuura Y, Usui H, Kikuchi A. GSK-3beta-dependent phosphorylation of adenomatous polyposis coli gene product can be modulated by beta-catenin and protein phosphatase 2A complexed with Axin. Oncogene. 2000;19(4):537–45. doi: 10.1038/sj.onc.1203359. [DOI] [PubMed] [Google Scholar]
- 48.Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer research. 2012;72(19):4883–9. doi: 10.1158/0008-5472.can-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Figiel S, Vasseur C, Bruyere F, Rozet F, Maheo K, Fromont G. Clinical significance of epithelial-mesenchymal transition (EMT) markers in prostate cancer. Human pathology. 2016 doi: 10.1016/j.humpath.2016.10.013. [DOI] [PubMed] [Google Scholar]
- 50.Wang M, Ren D, Guo W, Huang S, Wang Z, Li Q, et al. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. International journal of oncology. 2016;48(2):595–606. doi: 10.3892/ijo.2015.3270. [DOI] [PubMed] [Google Scholar]
- 51.Hadler-Olsen E, Winberg JO, Uhlin-Hansen L. Matrix metalloproteinases in cancer: their value as diagnostic and prognostic markers and therapeutic targets. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2013;34(4):2041–51. doi: 10.1007/s13277-013-0842-8. [DOI] [PubMed] [Google Scholar]
- 52.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 53.McDonald ER, 3rd, El-Deiry WS. Cell cycle control as a basis for cancer drug development (Review) International journal of oncology. 2000;16(5):871–86. [PubMed] [Google Scholar]
- 54.Lu Y, Wei C, Xi Z. Curcumin suppresses proliferation and invasion in non-small cell lung cancer by modulation of MTA1-mediated Wnt/beta-catenin pathway. In vitro cellular & developmental biology Animal. 2014;50(9):840–50. doi: 10.1007/s11626-014-9779-5. [DOI] [PubMed] [Google Scholar]
- 55.Rao Y, Wang H, Fan L, Chen G. Silencing MTA1 by RNAi reverses adhesion, migration and invasiveness of cervical cancer cells (SiHa) via altered expression of p53, and E-cadherin/beta-catenin complex. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban. 2011;31(1):1–9. doi: 10.1007/s11596-011-0141-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.