

This joint linkage and genome-wide association study comprehensively investigates natural variation in maize grain vitamin E levels using the 5000-line U.S. nested association-mapping panel.
Abstract
Tocopherols, tocotrienols, and plastochromanols (collectively termed tocochromanols) are lipid-soluble antioxidants synthesized by all plants. Their dietary intake, primarily from seed oils, provides vitamin E and other health benefits. Tocochromanol biosynthesis has been dissected in the dicot Arabidopsis thaliana, which has green, photosynthetic seeds, but our understanding of tocochromanol accumulation in major crops, whose seeds are nonphotosynthetic, remains limited. To understand the genetic control of tocochromanols in grain, we conducted a joint linkage and genome-wide association study in the 5000-line U.S. maize (Zea mays) nested association mapping panel. Fifty-two quantitative trait loci for individual and total tocochromanols were identified, and of the 14 resolved to individual genes, six encode novel activities affecting tocochromanols in plants. These include two chlorophyll biosynthetic enzymes that explain the majority of tocopherol variation, which was not predicted given that, like most major cereal crops, maize grain is nonphotosynthetic. This comprehensive assessment of natural variation in vitamin E levels in maize establishes the foundation for improving tocochromanol and vitamin E content in seeds of maize and other major cereal crops.
INTRODUCTION
Tocochromanols are synthesized by all plant tissues but are most abundant in seeds, where they limit the oxidation of membrane and storage lipids, making them essential for seed viability (Sattler et al., 2004; Mène-Saffrané et al., 2010) and overall plant fitness (Maeda et al., 2006; Maeda and DellaPenna, 2007; DellaPenna and Mène-Saffrané, 2011; Inoue et al., 2011). In the human diet, tocochromanols serve as both lipid-soluble antioxidants and the essential nutrient vitamin E (Hussain et al., 2013; Ahsan et al., 2015), with α-tocopherol having the highest vitamin E activity, α-tocotrienol and γ-tocopherol 3- and 6-fold lower activity, respectively, and that of other tocochromanols being negligible (Kamal-Eldin and Appelqvist, 1996; DellaPenna and Mène-Saffrané, 2011). While plant seed oils are the major source of dietary vitamin E, seeds of most crops predominantly contain tocochromanols with low vitamin E activity (DellaPenna and Mène-Saffrané, 2011).
Tocochromanols are synthesized in plastids using various prenyl-diphosphates derived from the plastidic isopentenyl pyrophosphate (IPP) pathway and homogentisic acid (HGA), an intermediate in aromatic amino acid catabolism (Figure 1). Condensation of HGA with phytyl-diphosphate (phytyl-DP), geranylgeranyl-diphosphate (GGDP), or solanesyl-diphosphate (solanesyl-DP) yields committed intermediates that are cyclized and methylated to produce the α, β, γ, and δ isoforms of tocopherols and tocotrienols, and plastochromanol-8 (PC-8), respectively. Tocochromanol biosynthesis is fully elucidated in Arabidopsis thaliana and involves 36 enzymatic activities (encoded by 53 genes) for the biosynthesis of HGA, prenyl-diphosphates, and the core tocochromanol pathway itself (vitamin E [VTE] loci 1 through 6; DellaPenna and Mène-Saffrané, 2011; Lipka et al., 2013). Genes encoding these enzymatic reactions are considered a priori candidates in the Arabidopsis genome that may influence natural variation for tocochromanol traits. Because these 36 enzymatic reactions are conserved across the plant kingdom (Cheng et al., 2003; Sattler et al., 2004; Karunanandaa et al., 2005; Gilliland et al., 2006; Tang et al., 2006; DellaPenna and Mène-Saffrané, 2011; Fritsche et al., 2012; Wang et al., 2012) a priori homologs can be readily identified in both monocot and dicot species (e.g., the maize [Zea mays] genome contains 80 such a priori candidates that encode these 36 activities; Supplemental Data Set 1). In addition, like most monocots, maize also encodes homogentisate geranylgeranyl transferase (HGGT), the committed step in tocotrienol biosynthesis (Cahoon et al., 2003).
Figure 1.

Tocochromanol Biosynthetic Pathways in Maize.
Precursor pathways are summarized in gray boxes. The seven quantified compounds are shown in black text with their corresponding structures. Key a priori genes are in bold italicized text at the pathway step(s) executed by their encoded enzyme with the eight a priori genes identified in this study highlighted in red text. Compound abbreviations: SDP, solanesyl diphosphate; Phytyl-DP, phytyl diphosphate; GGDP, geranylgeranyl diphosphate; HGA, homogentisic acid; MSBQ, 2-methyl-6-solanyl-1,4-benzoquinol; MPBQ, 2-methyl-6-phytyl-1,4-benzoquinol; MGGBQ, 2-methyl-6-geranylgeranyl-1,4-benzoquinol; PQ-9, plastoquinone-9; DMPBQ, 2,3-dimethyl-6-phytyl-1,4-benzoquinol; and DMGGBQ, 2,3-dimethyl-5-geranylgeranyl-1,4-benzoquinol. Gene abbreviations: 1-deoxy-d-xylulose-5-phosphate synthase (dxs2 and 3); arogenate/prephenate dehydrogenase family protein (arodeH2); solanesyl diphosphate synthase (sds); phytol kinase (vte5); phytol phosphate kinase (vte6); p-hydroxyphenylpyruvate dioxygenase (hppd1); homogentisate geranylgeranyltransferase (hggt1); MPBQ/MSBQ/MGGBQ methyltransferase (vte3); and γ-tocopherol methyltransferase (vte4).
The cloning of Arabidopsis VTE genes allowed the core tocochromanol pathway to be engineered for improved nutritional content and composition in various plants (Shintani and DellaPenna, 1998; Savidge et al., 2002; Collakova and DellaPenna, 2003; Karunanandaa et al., 2005; Kumar et al., 2005; Raclaru et al., 2006; Hunter and Cahoon, 2007; Li et al., 2010; DellaPenna and Mène-Saffrané, 2011; Lu et al., 2013; Zhang et al., 2013). Altering the expression of the pathway methyltransferase genes, vte3 and vte4, has profound impacts on the qualitative profiles of specific tocopherols and tocotrienols in leaves and seed without affecting total tocochromanol levels (Shintani and DellaPenna, 1998; Cheng et al., 2003; Van Eenennaam et al., 2003; Karunanandaa et al., 2005; DellaPenna and Mène-Saffrané, 2011; Lu et al., 2013). Engineering total tocotrienol content has proven relatively straightforward, with hggt1 overexpression increasing tocotrienols to levels several times that of tocopherols (Cahoon et al., 2003; Kim et al., 2011; Yang et al., 2011; Zhang et al., 2013; Tanaka et al., 2015). In contrast, engineering total tocopherol content is more difficult, and even with coordinate overexpression of multiple pathway steps, the increases achieved were modest (Savidge et al., 2002; Collakova and DellaPenna, 2003; Karunanandaa et al., 2005; Raclaru et al., 2006; Lu et al., 2013). Identification of the tocopherol-deficient Arabidopsis vte5 and vte6 mutants (Valentin et al., 2006; Vom Dorp et al., 2015), encoding kinases that sequentially phosphorylate phytol to generate phytyl-DP, suggested a mechanism underlying the divergent engineering results for tocotrienols and tocopherols: While tocotrienol biosynthesis can directly utilize GGDP, tocopherol biosynthesis requires phytol to be produced from GGDP and then phosphorylated.
Recent genome-wide association studies (GWAS) in maize and rice (Oryza sativa) grain (Li et al., 2012; Lipka et al., 2013; Wang et al., 2015) showed strong associations of γ-tocopherol methyltransferase (vte4) with α-tocopherol concentrations and much weaker associations of tocopherol cyclase (vte1), hggt1, and an arogenate/prephenate dehydratase with tocotrienol traits in maize grain (Lipka et al., 2013). The panel sizes and density of single-nucleotide polymorphisms (SNPs) in these studies limited both the identification of controlling loci and gene-level resolution of causal variants. In this study, we leveraged the superior statistical power and mapping resolution of the maize nested association mapping (NAM) panel of ∼5000 recombinant inbred lines (RILs) (Yu et al., 2008; McMullen et al., 2009) and the ∼29 million sequence variants of maize HapMap v1 and v2 (Gore et al., 2009; Chia et al., 2012) to comprehensively investigate the quantitative trait loci (QTL) and underlying genes responsible for natural variation in tocochromanol and vitamin E levels in maize grain, one of the most abundantly consumed food staples on the planet.
RESULTS
Genetic Dissection of Tocochromanol Accumulation in Maize Grain
We assessed the genetic basis of tocochromanol traits across the 25 RIL families of the U.S. maize NAM population. Physiologically mature grain samples were quantified for seven tocochromanol compounds by HPLC with fluorescence detection, and the data used to calculate best linear unbiased estimators (BLUEs) for the seven compounds, total tocopherols (ΣT), total tocotrienols (ΣT3), and total tocochromanols (ΣTT3) (Table 1; Supplemental Data Set 2). With the exception of PC-8, all traits had high estimates of heritability (0.71 to 0.89; Table 1). Although the seven tocochromanols are synthesized by a shared biosynthetic pathway (Figure 1), only three pairs of compounds had correlations greater than ∼0.4 (Supplemental Figure 1).
Table 1. Sample Sizes, Ranges, and Heritabilities for Tocochromanol Traits.
| Trait | No. Lines | BLUEs |
Heritabilities |
|||
|---|---|---|---|---|---|---|
| Median | sda | Rangeb | Estimate | sec | ||
| α-Tocopherol (αT) | 4786 | 8.68 | 5.22 | −2.97–33.19 | 0.82 | 0.01 |
| δ-Tocopherol (δT) | 4724 | 1.43 | 1.71 | −1.74–10.05 | 0.71 | 0.02 |
| γ-Tocopherol (γT) | 4789 | 40.08 | 19.63 | −2.78–128.49 | 0.75 | 0.02 |
| Total tocopherols (ΣT) | 4790 | 51.97 | 22.30 | 1.50–153.19 | 0.72 | 0.02 |
| α-Tocotrienol (αT3) | 4784 | 10.15 | 4.18 | −7.62–28.61 | 0.74 | 0.02 |
| δ-Tocotrienol (δT3) | 4689 | 0.76 | 1.15 | −0.86–7.86 | 0.89 | 0.01 |
| γ-Tocotrienol (γT3) | 4770 | 18.49 | 11.69 | −12.23–73.86 | 0.85 | 0.01 |
| Total tocotrienols (ΣT3) | 4765 | 28.91 | 13.87 | −10.33–93.83 | 0.82 | 0.02 |
| Total tocochromanols (ΣTT3) | 4779 | 86.14 | 28.88 | 13.96–221.78 | 0.76 | 0.02 |
| Plastochromanol-8 (PC-8) | 4787 | 1.06 | 0.29 | 0.05–2.38 | 0.18 | 0.02 |
Medians and ranges (in µg g−1 grain) for untransformed BLUEs of 10 tocochromanol grain traits evaluated in the U.S. maize NAM population and estimated heritability on a line-mean basis across 2 years.
sd of the BLUEs.
Negative BLUE values are a product of the statistical analysis. Specifically, it is possible for BLUEs to equal any value from −∞ to +∞.
se of the heritability estimate.
We mapped QTL across the 25 NAM families by joint-linkage (JL) analysis using a composite genetic map of ∼14,000 markers (Ogut et al., 2015). This identified 162 QTL, with eight to 21 QTL per trait (Table 2; Supplemental Data Set 3) and phenotypic variance explained (PVE) of 0.6 to 48.2% (Supplemental Data Sets 4 and 5). Given the biosynthetic relationships of tocochromanols (Figure 1), it seemed likely that multiple traits could be affected by individual QTL and indeed, 90% of overlapping QTL support intervals were also significantly pleiotropic (Supplemental Data Set 6 and Supplemental Figure 2). When overlapping QTL were merged, their numbers were reduced from 162 to 52 unique QTL intervals, of which 31 affected multiple traits (Supplemental Data Set 4).
Table 2. Genetic Association Results for Tocochromanol Traits.
| Trait | No. of JL-QTL | Median Size (sd) of α = 0.01 JL-QTL Support Interval (Mb) | No. of JL-QTL Intervals Containing a Priori Genes | No. of GWAS-Associated Variants in JL-QTL Intervalsa | Maximum RMIP |
|---|---|---|---|---|---|
| α-Tocopherol (αT) | 13 | 4.17 (13.38) | 4 | 57 | 0.98 |
| δ-Tocopherol (δT) | 18 | 3.36 (16.84) | 7 | 65 | 0.92 |
| γ-Tocopherol (γT) | 21 | 7.21 (22.64) | 10 | 72 | 0.95 |
| Total tocopherols (ΣT) | 18 | 8.00 (21.48) | 8 | 58 | 0.90 |
| α-Tocotrienol (αT3) | 17 | 4.65 (14.76) | 4 | 49 | 0.92 |
| δ-Tocotrienol (δT3) | 21 | 5.74 (22.20) | 7 | 68 | 0.91 |
| γ-Tocotrienol (γT3) | 14 | 8.35 (31.01) | 8 | 52 | 0.98 |
| Total tocotrienols (ΣT3) | 12 | 15.47 (24.02) | 8 | 49 | 0.98 |
| Total tocochromanols (ΣTT3) | 20 | 6.05 (21.32) | 6 | 95 | 0.70 |
| Plastochromanol-8 (PC-8) | 8 | 5.05 (21.37) | 4 | 40 | 0.66 |
| JL-QTL Total | 162 | 5.90 (21.43) | 66 | 605 |
Summary of JL-QTL and GWAS variants identified for 10 tocochromanol grain traits evaluated in the U.S. maize NAM population.
GWAS variants residing within JL-QTL support intervals for each trait that exhibited an RMIP of 0.05 or greater.
To more finely resolve these 52 unique QTL, we conducted a GWAS using the ∼29 million variants of maize HapMap v1 and v2 imputed onto the ∼4900 NAM RILs. A total of 1752 marker-trait associations achieved a resample model inclusion probability (RMIP) value ≥0.05 (Valdar et al., 2009; Supplemental Data Set 7). Of these, 34.5% localized to a corresponding trait JL interval (Table 2), with 47 markers having associations with two to four traits, for a total of 605 marker-trait associations. Linkage disequilibrium (LD) decays rapidly in the NAM panel, with the majority of HapMap v1 and v2 polymorphisms (Gore et al., 2009; Chia et al., 2012) showing average LD decay in genic regions to background levels (r2 < 0.2) by 1 kb, but with large variance dependent on allele frequencies (Wallace et al., 2014). As our GWAS-detected markers showed a similar trend of LD decay (Supplemental Figure 3), we limited our candidate gene search space to ±100 kb of GWAS-detected variants, which is appropriate given the high marker density and reported localization of NAM GWAS signals to within a few kilobases of causal variants (Wallace et al., 2014).
To aid in the identification of genes underlying QTL, we employed a triangulation approach (Ritchie et al., 2015) that tested for correlations between (1) genotype of GWAS marker(s), (2) log2-transformed RNA-seq expression levels across six developing kernel stages of the NAM parents for all genes in a search space (Supplemental Figure 4, Supplemental Table 1, and Supplemental Data Set 8), and (3) transformed allelic effect estimates of individual-trait QTL for each family compared with B73, the first maize reference genome and common parent of the U.S. maize NAM population. We initially focused on the 23 unique QTL whose intervals contained one or more of the 81 a priori genes (Supplemental Data Set 1) reasoning that they provide a high-quality set of known targets, which if positively identified in an interval, could guide application of the approach to intervals that lacked a priori candidate genes. Based on the narrow search space defined by LD and GWAS signals in combination with the triangulation data sets, eight a priori genes were determined to underlie a unique QTL (Figure 2; Supplemental Figure 5). These include three genes involved in prenyl group biosynthesis, two in aromatic head group biosynthesis, and three core tocochromanol pathway enzymes (Figures 1 and 3).
Figure 2.

Master Summaries for Selected Identified Genes.
Marker shapes correspond to trait class: circles, tocopherols; triangles, tocotrienols; squares, total tocochromanols, and diamonds, PC-8. Marker colors indicate compound type: yellow, alpha (α); orange, delta (δ); cyan, gamma (γ); purple, PC-8; brown, summed traits (Σ). Gene names are as they appear in Figure 3. Left panels: Directional gene models are depicted as black arrows and the identified gene as a green arrow. Lines with trait names above gene models indicate RMIP of significant GWAS hits ± 100 kb of the peak RMIP variant. Lines below gene models indicate pairwise LD (r2) of each GWAS variant with the peak RMIP variant (dark blue line). The blue ribbon depicts the highest LD, per 200-bp window, to the peak RMIP variant while black ribbons indicate the density of variants tested in GWAS in the 200-bp window (log2 scale). Darker colors correspond to higher values. Right panels: Correlations (r) between JL-QTL allelic effect estimates and expression of the identified gene across six developing kernel time points. Significant correlations are indicated by trait abbreviations above the respective time point. Traits with both JL and GWAS associations appear in black text to the right of the graph and have solid trend lines and symbols, while those with only JL associations are in gray with dashed trend lines and open symbols.
Figure 3.

Percent PVE by a Priori and Novel Causal Genes Underlying JL-QTL.
aeQTL indicates significant correlations between expression values and JL-QTL allelic effect estimates at more than two time points for at least one trait. bBlue shading corresponds to range of PVEs for JL-QTL, with darker blue indicating higher PVEs. cColor coding indicates QTL predominantly affects a trait class (having at least two-thirds of summed PVE).
The Role of a Priori Pathway Genes
The prenyl diphosphates for tocochromanol biosynthesis are made using five-carbon building blocks from the plastidic IPP pathway (eight activities encoded by 15 genes in maize). Only two IPP pathway genes were found to underlie QTL; both encode 1-deoxy-d-xylulose-5-phosphate synthase, the first and committed step of the pathway. dxs2 affected five traits (2.5–5.7% PVE), and dxs3 was specific for PC-8 (2.6% PVE), but unexpectedly, neither was associated with tocopherol traits. Allelic effect estimates and the expression of dxs2, but not dxs3, were strongly correlated from mid-grain development onward, indicating dxs2 is an expression QTL (eQTL; Figures 2 and 3). The maize genome encodes 11 prenyl synthases capable of producing phytyl-DP, GGDP, and solanesyl-DP for the biosynthesis of tocopherols, tocotrienols, and PC-8, respectively, but only one locus, solanesyl-DP synthase (sds), was identified in this study. SDS produces the prenyl tail group for PC-8 and affected PC-8 and two other traits, all with small PVEs. Taken together, these findings indicate that dxs2 and dxs3 function in the primary steps controlling provision of IPP for the biosynthesis of tocotrienol and PC-8 prenyl groups, but surprisingly not for tocopherol prenyl groups.
The aromatic head group for all tocochromanols, HGA, is an intermediate in tyrosine metabolism, and two genes in this pathway were identified as underlying QTL. p-Hydroxyphenylpyruvate dioxgenase1 (hppd1) showed association with eight traits, with PVEs for tocotrienol traits (7.9–10.7% for δT3, γT3, and ΣT3) being much higher than those for the corresponding tocopherol traits. Also identified, with smaller PVEs for αT3 and ΣT3 only, was arogenate dehydrogenase2 (arodeH2); the encoded enzyme carries out the oxidative decarboxylation of l-arogenate to l-tyrosine, which in one additional enzymatic step is converted to HPPD, the substrate for hppd1. Thus, these two genes are the key regulated steps for producing the aromatic head group of individual and total tocochromanols that accumulate in mature maize grain. Two other loci of relevance previously found to be weakly associated with tocochromanol traits in a maize inbred association panel (Lipka et al., 2013), vte1 and one of seven prephenate dehydratases (GRMZM2G437912), were not detected in this study. vte1 was present in multiple JL intervals in the same recombination-suppressed pericentromeric region as hppd1, but signals specific to vte1 could not be resolved due to long-range LD. Prephenate dehydratase association signals were extremely weak in the prior study (Lipka et al., 2013), and this gene was not detected in NAM JL-GWAS.
The three remaining a priori genes identified in this study encode the core tocochromanol pathway enzymes, HGGT1, VTE4, and VTE3. HGGT1 is a prenyl transferase that condenses HGA and GGDP for the biosynthesis of all tocotrienols and had large PVEs for tocotrienols and moderate PVEs for tocopherols (Figure 3), a result consistent with its kinetic preference for GGDP over phytyl-DP (Yang et al., 2011). hggt1 was a strong eQTL, i.e., expression QTL, meaning that the contribution of this QTL to trait variation is significantly associated with variation in expression of the identified gene, through all developing kernel stages analyzed. This gene is strongly expressed in endosperm, the major site of tocotrienol accumulation, and showed little to no expression in other tissues, as also reported for the genes encoding HGGT in rice and barley (Hordeum vulgare) (Cahoon et al., 2003). Consistent with this expression pattern, 83% of the tocochromanols in endosperm of 30 d after pollination (DAP) seed of NAM parents were tocotrienols, while only 2.5% were tocotrienols in embryos (Supplemental Data Set 9). Notably, HGGT was found to have the largest PVEs for three traits, ∼40% for γT3 and ΣT3 and 24% for δT3, suggesting that this locus is indeed the key player for tocotrienol traits, with the exception of αT3. VTE4 catalyzes the final step in αT and αT3 biosynthesis and had the largest PVE for these traits at 48.2% and 32.0%, respectively. This gene was an eQTL with particularly strong correlations with αT allelic effects. These effects spanned a range of 11.58 µg/g, suggesting that vte4 is key for increasing vitamin E levels, a finding concordant with the large PVEs and clear GWAS results obtained for vte4 in this and previous studies (Li et al., 2012; Lipka et al., 2013). The final gene, vte3, encodes a methyltransferase at the branchpoint for δ- and γ-tocochromanols and was the highest-PVE a priori QTL for δT at 8.2%, with smaller PVEs for four other traits.
Novel Large-Effect Loci Control Total Tocopherol and Vitamin E Accumulation
These eight identified a priori genes guided the application of our approach for gene-level resolution in the remaining 44 QTL, which define novel loci affecting tocochromanol traits in maize grain. Like the identified a priori genes, for a gene to be identified in these 44 QTL intervals, it must meet at least two of three criteria: have at least one significant GWAS variant within ±100 kb of the gene, at least two significant FPKM x JL allelic effect estimate correlations, and a compelling biological function for involvement in tocochromanol biosynthesis/accumulation. Applying these criteria to the 44 remaining QTL resulted in the identification of six genes not known to affect tocochromanol traits in any plant system that fall into three categories: metabolism, metabolite transport/storage, and transcriptional regulation.
Both metabolic genes identified for QTL5 and QTL24 encode homologs of protochlorophyllide reductase (POR), a highly regulated step in chlorophyll biosynthesis (Figure 4C). Notably, por2 (QTL24) had the highest PVEs in the panel for γT, δT, ΣT, and ΣTT3 and por1 (QTL5) the second highest PVEs for γT and ΣT (Figure 3); together, they account for the largest allelic effects observed (well beyond those of a priori genes) for tocopherol traits (Figure 3; Supplemental Figure 6). The two por loci were the most robust eQTL in this study and had the largest epistatic interactions (Figure 5), contributing 2.2 to 4.0% additional PVE to the four tocopherol traits.
Figure 4.

Chlorophyll Metabolism in Relation to Phytol Generation and Tocopherol Biosynthesis in Developing Maize Embryos and Endosperm.
(A) Correlation of chlorophyll metabolites and total tocopherol concentrations (pmol g−1).
(B) Compound concentration means (log scale) of NAM parents.
(C) Overview of chlorophyll biosynthesis and degradation and phytol generation in maize embryos and endosperm with the protochlorophyllide reductase expression QTL indicated in red. Compounds measured are in bold black text with the four detectable chlorophyll metabolites in embryos (only chlorophyll a was measurable in endosperm) highlighted in boxes colored as in (A) and (B). Other relevant compounds are in gray and relevant enzymes in black bold italics. Arrow widths represent mean gene expression (FPKM) across embryo development in B73. The black dashed arrows show the proposed route for generating phytol for tocopherol biosynthesis in maize embryos from chlorophyll biosynthetic intermediates, instead of by chlorophyll degradation. Compound abbreviations: ƩT, total tocopherols = αT+δT+γT; Chlide: chlorophyllide; Chl, chlorophyll; Pheo, pheophytin; Protochlide, protochlorophyllide; Phytyl-DP, phytyl diphosphate; GGDP, geranylgeranyl diphosphate.
Figure 5.

Genome-Wide Distribution of Tocochromanol JL-QTL and Their Pairwise Epistatic Interactions.
From the outermost ring to the center: Black arcs show chromosomes labeled in 20-Mb increments, with small open circles marking centromeres. Gene names are as they appear in Figure 3 and are adjacent to purple and green capsules that indicate a priori and novel gene classes, respectively. Radial, light blue lines show the positions of peak markers for the 162 individual-trait QTL. Lines linking markers show significant epistatic (additive × additive) interactions, with line thickness proportional to phenotypic variance explained by the interaction term (which range from 0.3% to 4.0%). Links are colored by trait class for the interaction: yellow, tocopherols (T); orange, tocotrienols (T3); black, total tocochromanols (ΣTT3).
The strong and specific association of two chlorophyll biosynthetic genes with tocopherols was unexpected for a monocot seed like maize that lacks obvious green coloration during development and is chlorophyll-deficient at physiological maturity (i.e., in dry grain). To assess whether, despite the lack of green coloration in developing grain, chlorophylls might still be present, we dissected embryos and endosperm from the NAM parents at 16, 20, 24, 30, and 36 DAP to quantify the levels of tocochromanols and four major classes of chlorophyll metabolites: chlorophylls a and b, chlorophyllides a and b, pheophytins a and b, and pheophorbides a and b (Supplemental Data Set 9). Embryo tocochromanols are composed of >90% tocopherols, whose absolute levels reflect the extreme diversity of NAM parents, varying by 10- to 100-fold at each developmental stage (Supplemental Figure 7). Surprisingly, developing embryos also contain extremely low, but detectable, levels of chlorophylls a and b, chlorophyllide a, and pheophytin a (Figure 4B), while the other four chlorophyll metabolites assessed were below detection in all samples. Though detectable, total chlorophyll metabolite levels are ∼500 times lower in embryos than in leaves (Ma et al., 2008), and 100- to 1000-fold lower than embryo tocopherols (Figure 4B). The correlation of total tocopherols with chlorophyll a, chlorophyll b, and chlorophyllide a was strong at 16 DAP (r = 0.71–0.76); chlorophyll a correlations with total tocopherols peaked at r = 0.93 at 20 DAP, and with the exception of 30 DAP remained above r = 0.7. Correlations with chlorophyllide a and chlorophyll b with total tocopherols gradually decreased to r = 0.48 and r = 0.36, respectively, at 30 DAP, after which chlorophyll b correlation increased. Pheophytin a is a key intermediate and metabolite marker for the chlorophyll degradation pathway in senescing leaves (Schelbert et al., 2009; Hörtensteiner and Kräutler, 2011; Hörtensteiner, 2013), where it provides phytol for senescence-associated tocopherol biosynthesis (Schelbert et al., 2009; Zhang et al., 2014; Vom Dorp et al., 2015), but pheophytin a only showed weak correlations with total tocopherols at 16 and 20 DAP (r = 0.23 and 0.31, respectively) and was negatively correlated at later developmental stages. Tocopherols accumulated in endosperm to levels <2% that in embryos, and endosperm chlorophyll metabolite levels were similarly reduced, with only chlorophyll a being consistently above the limits of detection (Supplemental Data Set 9; Figure 4B). Nonetheless, correlations of chlorophyll a with total tocopherols in endosperm ranged from r = 0.50 to 0.74 at three stages of development (Figure 4A).
A second group of novel genes has predicted roles in the transport and storage of lipophilic molecules. The identified gene in QTL10, affecting five traits, is one of 12 genes in maize encoding plastid-localized fibrillins, structural proteins that bind hydrophobic molecules and play various roles in their biosynthesis and accumulation in other systems (Deruère et al., 1994; Kim et al., 2015). Fibrillins are prominent components of plastoglobules (Ytterberg et al., 2006; Bréhélin et al., 2007), subcompartments of the chloroplast that also contain tocochromanols, carotenoids, lipids, and various biosynthetic enzymes including tocopherol cyclase. The genetic association of a fibrillin gene family member with tocochromanol content is thus consistent with prior biochemical knowledge that other members of the fibrillin family bind hydrophobic metabolites (e.g., carotenoids). QTL30 also affected multiple traits, and its identified gene encodes a cytosolic glycol(neutral)lipid transfer protein that could participate in the transport of tocochromanols to oil bodies for storage. Finally, QTL6 only affects αT3, and its underlying gene encodes a type of SNARE protein predicted to be plastid-targeted and whose function is consistent with a role in vesicular transport. Of these three genes, only QTL10 was an eQTL. The final gene identified, in QTL39, was an eQTL that affected αT3 and encodes a predicted transcription factor with plant homeodomain zinc finger domains.
DISCUSSION
This study, a comprehensive assessment of natural variation in vitamin E levels in maize grain, provides important insights into the control of tocochromanol content and composition in a global staple crop, with major implications for human nutrition. In total, 52 unique QTL were identified with PVEs as large as 48.2%. We resolved 14 QTL to the gene level using an approach integrating JL-QTL effect estimates, localization of GWAS signals, and RNA-seq data from six stages of developing kernels for the NAM parental genotypes. Only two of the 14 genes identified in this study had been previously associated with natural variation for tocochromanols in maize grain (Li et al., 2012; Lipka et al., 2013). These 14 genes included seven of the nine intervals with largest PVEs (Figure 3; Supplemental Table 2) and in an additive model explained 56 to 93% of phenotypic variation attributed to QTL for the traits analyzed in this study (Supplemental Figure 8). This degree of gene-level resolution of JL-GWAS signals was much greater than in earlier NAM studies (Buckler et al., 2009; Brown et al., 2011; Kump et al., 2011; Poland et al., 2011; Tian et al., 2011; Cook et al., 2012; Peiffer et al., 2014; Wallace et al., 2014; Yan et al., 2015; Zhang et al., 2015) due to three main factors: clear molecular evidence of functional involvement through the incorporation of RNA-seq data, increased marker density provided by the additional 27.4 million HapMap v2 variants, and the tractable genetic architecture of tocochromanol traits (oligogenic and highly heritable). Eight of the 14 identified genes were on a list of 81 a priori maize candidate genes generated based on prior elucidation of precursor and core tocochromanol pathways, primarily in Arabidopsis, while the remaining six encode functions not previously demonstrated to affect tocochromanols in any plant species despite over two decades of molecular genetic studies (Shintani and DellaPenna, 1998; Savidge et al., 2002; Cahoon et al., 2003; Cheng et al., 2003; Sattler et al., 2004; Valentin et al., 2006; DellaPenna and Mène-Saffrané, 2011). With the exception of HGGT, which is only present in the monocot lineage, and the plant homeodomain transcription factor, all other genes identified in this study have clear homologs in major monocot and dicot crop species, providing clear targets to assess in other crops for potential association with desired tocochromanol traits.
In most cases, the eight a priori genes affected tocochromanol traits in ways consistent with the known biochemical activities of their encoded enzymes (Shintani and DellaPenna, 1998; Cahoon et al., 2003; Cheng et al., 2003; Collakova and DellaPenna, 2003; Van Eenennaam et al., 2003; Karunanandaa et al., 2005; Kumar et al., 2005; Tang et al., 2006; Hunter and Cahoon, 2007; DellaPenna and Mène-Saffrané, 2011). For example, the two pathway methyltransferases, vte3 and vte4 (Shintani and DellaPenna, 1998; Cheng et al., 2003; Van Eenennaam et al., 2003), were key for determining the degree of methylation, and hence, the types of tocochromanols accumulated (i.e., α, γ, or δ), but had no impact on total tocochromanol levels. Similarly, the aromatic head group for all tocochromanols, HGA, is produced by hppd1 (Norris et al., 1998; Rippert et al., 2004; DellaPenna and Mène-Saffrané, 2011), which affected nearly every tocochromanol trait, though with larger contributions for tocotrienols. As a group, the eight a priori genes also highlight major differences in the genetic control of tocopherol versus tocotrienol traits in maize grain, particularly in the generation and coupling of their prenyl tail groups to HGA. A single gene for the first and regulated step of the plastidic IPP pathway, dxs2, was strongly and specifically associated with tocotrienol traits, but neither it nor any other IPP pathway gene was associated with tocopherol traits. HGGT, the committed enzyme for tocotrienol biosynthesis, showed extremely strong tocotrienol associations and limited associations with tocopherol traits, a result consistent with its overexpression conferring high levels of tocotrienol production in numerous plant tissues and systems (Cahoon et al., 2003; DellaPenna and Mène-Saffrané, 2011; Zhang et al., 2013; Tanaka et al., 2015) and with the enzyme preferentially condensing HGA with GGDP (Yang et al., 2011). In contrast, the corresponding enzyme that condenses HGA with phytyl-DP for tocopherol biosynthesis, homogentisate phytyltransferase (HPT), lacked association with tocopherol traits. This was unexpected as like hggt overexpression, hpt overexpression increases total tocopherol content in a number of dicot plant systems and tissues (Savidge et al., 2002; Collakova and DellaPenna, 2003; Karunanandaa et al., 2005; Lu et al., 2013). While the genetic control of total tocotrienol content in maize grain is relatively simple, with three, large-effect a priori genes (dxs2, hppd1, and hggt1) collectively accounting for 81% of trait variation, a priori genes account for only 8% of variation in total tocopherol content. Instead, the trait is controlled primarily by novel loci (Supplemental Figure 8), indicating that in maize grain and likely other monocot seed, a fundamentally different process regulates biosynthetic flux to total tocopherols.
Key insight into the regulation of tocopherol biosynthesis in maize grain comes from our finding that two major determinants of tocopherol natural variation in maize grain are homologs encoding POR, which carries out a key reaction in chlorophyll biosynthesis. The two identified por genes accounted for 46% of total tocopherol variation attributed to QTL in an additive model (Supplemental Figure 8), the largest PVEs for δT, γT, and ΣT, and a substantial pairwise epistatic effect that is roughly one-third the dynamic range of their additive effects. The key role of protochlorophyllide reductases in tocopherol biosynthesis in maize grain was especially surprising given that this tissue, like most monocot seed, is nonphotosynthetic and lacks any obvious green coloration.
Our identification of two chlorophyll biosynthetic genes (por homologs) as major determinants of tocopherol content in maize grain and supporting metabolite and expression data in developing embryo are consistent with chlorophyll degradation playing a minor role at best in tocopherol biosynthesis in nonphotosynthetic tissues like maize grain. First, of the four chlorophyll metabolites detectable in developing embryos, pheophytin a, a committed intermediate and metabolic marker for chlorophyll degradation, had the lowest correlation with total tocopherol levels (Figure 4A), opposite of what would be expected if chlorophyll degradation provided the majority of phytol for tocopherol biosynthesis. Instead, chlorophylls a and b and chlorophyllide a, late-stage intermediates in chlorophyll biosynthesis, were strongly and positively correlated with total tocopherol levels throughout embryo development. Additionally, in developing maize embryos (i.e., 30 DAP), the chlorophyll:tocopherol molar ratio is ∼1:800, and as only a single molecule of phytol is released for each chlorophyll degraded, degradation would only provide a trace of the phytol needed for tocopherol biosynthesis, unless massive flux to degradation occurs. While we cannot eliminate this possibility, it seems unlikely, as expression of chlorophyll biosynthetic enzymes prior to POR is extremely low (e.g., 0.1–1% that in leaves; Supplemental Data Set 10) and consistent with the low levels of chlorophyll metabolites in developing embryos (∼0.3% of leaf levels; Ma et al., 2008). In contrast, the two enzymes downstream of POR, chlorophyll synthase (which esterifies GGDP to chlorophyllides a and b) and geranylgeranyl reductase (which reduces the geranylgeranylated intermediates to chlorophylls a and b) are the most highly expressed steps of the pathway in embryos (e.g., 9–18% that in leaves; Supplemental Data Set 10). This suggests that their reactions are strongly favored, which is consistent with chlorophyll a levels being 10- to 100-fold higher than those of other chlorophyll metabolites. Taken together, these findings suggest that aspects of chlorophyll biosynthesis, likely a cycle involving repeated removal of phytol from chlorophyll a followed by efficient reesterification of the resulting chlorophyllides with GGDP and reduction of the geranylgeranylated intermediates to (“phytyl”)-chlorophylls, generate the large amounts of phytol needed for tocopherol biosynthesis in maize embryos (Figure 4C).
Unlike maize grain, developing Arabidopsis seed are green, photosynthetic, and contain high levels of tocopherol at a 2- to 4-fold molar excess to chlorophylls (Zhang et al., 2014), while in unstressed leaves, tocopherol levels are much lower and chlorophyll is often at 20- to 50-fold molar excess to tocopherols (Collakova and DellaPenna, 2003). As each mole of chlorophyll contains one mole of esterified phytol, bulk chlorophyll degradation has long been proposed as the source of phytol for tocopherol biosynthesis in such green, photosynthetic tissues (Rise et al., 1989; Chrost et al., 1999; Valentin et al., 2006). The chlorophyll degradation pathway has recently been elucidated in Arabidopsis (Schelbert et al., 2009; Hörtensteiner and Kräutler, 2011; Hörtensteiner, 2013) (Figure 4C), and the phytol released from pheophytin a by pheophytinase could be esterified to fatty acids to yield fatty acid phytyl esters or phosphorylated by VTE5 and VTE6 to yield phytyl-DP (Valentin et al., 2006; Tanaka et al., 2010; DellaPenna and Mène-Saffrané, 2011; Lippold et al., 2012; Zhang et al., 2014; Vom Dorp et al., 2015) (Figure 4C). This latter route clearly provides phytyl-DP for the large amounts of tocopherol synthesized by senescing Arabidopsis leaves, as mutation of the pheophytinase gene eliminates both chlorophyll degradation and the senescence-associated increases in tocopherol and fatty acid phytyl ester levels (Schelbert et al., 2009; Zhang et al., 2014; Vom Dorp et al., 2015). If flux through this chlorophyll degradation pathway provided the majority of phytol for tocopherol biosynthesis in other tissues and leaf development stages, one would expect a similarly severe impact on tocopherol levels in these tissues. However, in nonsenescing leaves and mature seed, tocopherol content in the pheophytinase mutant was unchanged (Zhang et al., 2014). Chlorophyllases can also remove phytol from chlorophyll a and have been proposed as an alternate route for generating phytol, but mutations disrupting the two Arabidopsis chlorophyllases, singly or in combination with the pheophytinase mutant, again had no effect on seed tocopherol levels (Zhang et al., 2014). These combined data indicate that though phytol is released by chlorophyll degradation late in Arabidopsis seed maturation, this phytol contributes little to tocopherol biosynthesis in developing seed, and instead phytol for tocopherol biosynthesis in seed and nonsenescing leaves of Arabidopsis is provided from another source. Tocopherol biosynthesis from this alternative source of phytol is still dependent on VTE5 (phytol kinase activity), as in vte5 mutants, tocopherol levels are reduced by 80% and leaf tocopherol contents by 65% (Valentin et al., 2006). We suggest that like maize grain, Arabidopsis operates a chlorophyll-based cycle for generating phytol for tocopherol biosynthesis in most tissues and developmental stages that is separate from the bulk chlorophyll pool.
In addition to the por loci, the other novel genes identified in this study provide important insights into the accumulation of tocochromanols in plants, but especially for tocopherols, which have higher vitamin E activities than tocotrienols (Kamal-Eldin and Appelqvist, 1996; DellaPenna and Mène-Saffrané, 2011). For example, we identified proteins with transport and storage functions that are associated with tocochromanols at the genetic level. Three of the novel loci encode such functions, including two, a fibrillin and a lipid transfer protein, affecting multiple tocopherol and tocotrienol traits. Fibrillins are encoded by moderate-sized gene families, with individual members having diverse functions ranging from storage of xanthophylls in fruit and flower chromoplasts to interaction with enzymes involved in plastoquinone biosynthesis (Deruère et al., 1994; Singh and McNellis, 2011; Kim et al., 2015). Tocopherols were reported as minor constituents of fibrillins isolated from red bell pepper (Capsicum annuum) fruit (Deruère et al., 1994), and several members localize to plastoglobuli along with various lipid-soluble compounds and enzymes, including tocopherol cyclase (Ytterberg et al., 2006; Bréhélin et al., 2007). The association of multiple tocochromanol traits with a single member of the maize fibrillin family (GRMZM2G031028) suggests that the encoded protein specializes in tocochromanol storage in grain. Finally, lipid transfer proteins are encoded by large gene families in plants and have likewise been implicated in the movement of various lipophilic compounds between membranes. Here, we show that GRMZM2G060870 is a lipid transfer protein implicated in the transport of tocochromanols. Overexpression and knockout studies of a priori genes in other systems have yielded important insight into their roles in tocochromanol biosynthesis (DellaPenna and Mène-Saffrané, 2011). Analogous experiments with the six novel genes identified in this study would provide additional insights into their roles and functions.
Allelic variation at the 14 genes identified in this study, responsible for 56 to 93% of phenotypic variation attributed to QTL for tocochromanols in maize grain, establishes a near-complete foundation for the genetic improvement of vitamin E and non-vitamin E tocochromanol levels in this major food crop and likely in seed of the other major cereals, which are also nongreen, nonphotosynthetic tissues that synthesize tocochromanols. That a moderate number of genes exerts large control over headgroup and tail biosynthesis and the core tocochromanol pathway itself holds great promise for both breeding and engineering of tocochromanols in staple crops. Because tocopherols and tocotrienols are largely under independent genetic control by seven major-effect loci, genomics-assisted breeding approaches can target total tocotrienols (hggt1, hppd1, and dxs2), total tocopherols (two por homologs), or vitamin E content (vte3 and vte4), separately or in combinatorial fashion. It remains an open question whether the levels of other vitamins and essential nutrients in major crop species are under similarly tractable control. If true, this would greatly accelerate global efforts to simultaneously enhance and balance the levels of multiple essential micronutrients in staple crops to benefit human health.
METHODS
Field Environments and Plant Materials for Genetic Mapping
The genetic and genomic approaches used to design and construct the maize (Zea mays) NAM population have been previously described (Yu et al., 2008; Buckler et al., 2009; McMullen et al., 2009). In brief, 25 families of 200 RILs per family were generated by crossing maize inbred line B73 in a reference design to 25 other diverse inbred lines. These 25 NAM families, the intermated B73 × Mo17 (IBM) family (Lee et al., 2002), and an association mapping panel of 281 diverse inbred lines (Flint-Garcia et al., 2005) were evaluated at the Purdue University Agronomy Center for Research and Education in West Lafayette, IN, under standard agronomic practices in the summers of 2009 and 2010. The experimental field design has been previously described (Chandler et al., 2013). In brief, a sets design was used in each of the two environments, with each set including all lines of a family or the association panel. Each family set was arranged in a 10 × 20 incomplete block α-lattice design, and each incomplete block was augmented by the addition of both parental lines as checks. The association panel had a 14 × 20 incomplete block α-lattice design, with each incomplete block augmented by the inclusion of maize inbred lines B73 and Mo17 as checks. A single replicate of the entire experiment of 5481 lines from the NAM and IBM families as well as the 281-member association panel plus repeated check lines was grown in each environment. An experimental unit consisted of a single line planted in a one-row plot that was 3.05 m in length, with an average of 10 plants per plot. In both environments, a minimum of four plants within a plot was self-pollinated by hand. Self-pollinated ears were harvested at physiological maturity and dried to a grain moisture content of ∼15%. Afterwards, the ears of each plot were shelled and bulked to generate a representative, composite grain sample for quantifying tocochromanol levels.
Phenotypic Data Analysis
Tocochromanols were extracted from ∼50 ground kernels for each plot and quantified by HPLC and fluorometry as previously described (Lipka et al., 2013). We assessed three types of tocochromanols based on HPLC data passing internal quality control measures that were collected on 10,306 grain samples from 4862 NAM and 198 IBM RILs, as well as the repeated parental check lines. The 10 evaluated tocopherol, tocotrienol, and plastochromanol phenotypes were as follows: α-tocopherol (αT), δ-tocopherol (δT), γ-tocopherol (γT), α-tocotrienol (αT3), δ-tocotrienol (δT3), γ-tocotrienol (γT3), total tocopherols (αT + δT + γT), total tocotrienols (αT3 + δT3 + γT3), total tocochromanols (total tocopherols + total tocotrienols), and PC-8 in μg g−1 seed. When the level of a tocochromanol compound for a grain sample was below the minimum detection limit of HPLC, a μg g−1 value was approximated for the sample by assigning a uniform random variable ranging from 0 to the minimum HPLC detection value for that given compound within a family for each environment. The IBM RILs were not included in the JL analysis and GWAS of the 10 tocochromanol traits, as they were produced through intermating and thus exhibit a differential recombination rate relative to NAM RILs. However, the IBM family was still included in the following mixed linear model analysis along with the 25 NAM families to provide additional information on genotype-by-environment variation and within-environment spatial variation.
To screen the 10 traits for phenotypic outliers, we initially performed mixed linear model selection with custom Java code invoking ASReml-W version 3.0 (Gilmour et al., 2009) for each trait that followed the same steps of the two-stage model fitting process previously described (Peiffer et al., 2014). In brief, in the first stage, mixed linear models separately fit for each of the two environments included a fixed effect for the grand mean and random effects including the genotypic effects of family and RIL within family, a laboratory effect for HPLC autosampler plate, and spatial effects for field, set within field, and block within set within field. Genetic, HPLC plate, and spatial effects were not confounded because the repeated parental check lines were considered to be from the association panel. Thus, the experimental design allowed for the estimation of genetic effects separate from the laboratory and spatial effects. A first-order autoregressive (AR1 × AR1) correlation structure was also fitted to account for spatial variation in the direction of rows and columns among plots within each environment. For each environment, a backward elimination procedure based on the likelihood ratio test (Littell et al., 2006) was conducted to remove nongenetic random effects and AR1 × AR1 error structures from the model that were not significant (α = 0.05).
In the second stage, a single mixed linear model across both environments was fitted that included and nested the significant laboratory and spatial effects from the individual first stage models. Additional random effects entering the multienvironment model included environment, the interaction between family and environment, and the interaction between RIL within family and environment. Additionally, the significant AR1 × AR1 error structures within each environment were included in the model. From the final fitted model for each trait, influential phenotypic outliers were detected using the DFFITS criterion (Neter et al., 1996; Belsley et al., 2005), and observations were deleted if they exceeded a conservative DFFITS threshold previously suggested for this experimental design of  where p′ is model degrees of freedom (df) + 1 and n the sample size (Hung et al., 2012) (Supplemental Data Sets 2 and 11 and Supplemental File 1).
where p′ is model degrees of freedom (df) + 1 and n the sample size (Hung et al., 2012) (Supplemental Data Sets 2 and 11 and Supplemental File 1).
Once influential outliers were removed, the two-stage model fitting process was conducted again with minor modifications to estimate a BLUE for each RIL across environments. In this implementation, the genotypic effects of family and RIL within family were fitted as fixed effects. Additionally, unique error variances were not separately modeled for each environment when fitting the interaction between family and environment and between RIL within family and environment. To obtain variance component estimates, all terms except for the grand mean were then fitted as random effects. These variance components were used to estimate heritability on a line-mean basis 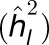 across only the 25 NAM families (Hung et al., 2012), and the standard errors of these estimates were approximated using the delta method (Holland et al., 2003).
across only the 25 NAM families (Hung et al., 2012), and the standard errors of these estimates were approximated using the delta method (Holland et al., 2003).
Prior to conducting JL mapping of QTL in the NAM population, the BLUEs of each trait were screened to detect any remaining statistical outliers using PROC MIXED in SAS version 9.3 (SAS Institute). Specifically, the Studentized deleted residuals (Kutner et al., 2004) were examined, which were obtained from a parsimonious linear model fitted with fixed effects for the grand mean and a single randomly sampled, representative SNP marker (PZA02014.3) from the NAM genetic linkage map of 1106 SNP markers (McMullen et al., 2009). For each trait, a BLUE of each RIL was considered an outlier and removed if it generated a Studentized deleted residual, with n – p – 1 df, that had an absolute value greater than the Bonferroni critical value of t(1 – α/2n; n – p – 1), where t denotes the t-distribution, α the significance level of 0.05, n the sample size of 5460 RILs, and p the number of predictor variables (Supplemental Data Sets 2 and 11, Supplemental Figure 9, and Supplemental File 2). Finally, for the trait δT3, a single RIL was removed that was seen to exert unduly high leverage within the trait JL model, particularly upon the inclusion of interaction terms in epistasis analyses. The observed inflation of allelic effect estimates and PVEs was most severe for a JL peak marker with low alternate allele frequency, as has been previously observed (Rao and Province, 2016).
Next, for each trait, the Box-Cox power transformation (Box and Cox, 1964) was performed on BLUEs with the aforementioned parsimonious model to identify the most appropriate transformation that corrected for unequal variances and non-normality of error terms. This process was conducted using PROC TRANSREG in SAS version 9.3 (SAS Institute) and tested lambda values ranging from −2 to +2 in increments of 0.05 before applying the optimal lambda for each trait. Of the 10 traits, six had a variable number of RILs (range: 3–258) with a BLUE of negative value. Negative values were a product of the shift in location (mean) and scale (sd) of the metabolite trait distributions that takes place in the generation of BLUEs (Burkschat, 2009). A constant of the lowest possible integer needed to make all values positive—a requirement of the Box-Cox power transformation—was added to the BLUEs for a given trait before conducting the transformation procedure. The constants and Box-Cox lambda values applied for each trait are provided in Supplemental Table 3.
JL Analysis
A consensus genetic linkage map comprising 14,772 markers and derived across the 25 NAM families was used for JL analysis. The map was constructed by scoring 4892 available NAM RILs with a genotyping-by-sequencing protocol (Elshire et al., 2011; Glaubitz et al., 2014) and imputing SNP markers at evenly spaced 0.1-cM intervals following a previously described procedure (Ogut et al., 2015). Using this consensus map, a previously described JL analysis procedure (Buckler et al., 2009) was conducted across the 25 families of the NAM population to identify and define positions of QTL controlling phenotypic variability of the 10 tocochromanol traits. In brief, a joint stepwise regression procedure was implemented using modified source code in TASSEL version 3.0 (provided on Github), in which transformed tocochromanol trait BLUEs were the response variable, the family main effect forced into the model first was an explanatory variable, and each of the 14,772 possible marker effects nested within family terms considered for inclusion into the final model were explanatory variables. The model entry or exit selection criterion of marker-by-family effects was based on a permutation procedure, where the transformed BLUEs of each tocochromanol trait were permuted 1000 times and the entry P value thresholds (from a partial F-test) were chosen to control the type I error rate at α = 0.05. These thresholds are listed in Supplemental Table 3. To prevent the simultaneous entry and exit of an effect in the same step, exit thresholds were set equal to twice the value of entry thresholds.
Given that strong linkage between these high-density genetic markers could introduce extensive collinearity among marker genotypes, we developed an additional model fitting approach to correct for multicollinearity and more precisely determine QTL locations and effect estimates thereafter. For the three tocotrienol compounds, their sum, and total tocochromanols, at least one pair of peak JL markers (i.e., the markers in the optimal model determined from stepwise model selection) exhibited an absolute Pearson correlation coefficient (r) greater than 0.8 in their SNP genotype states, calculated using the “pearson” method within the ‘cor’ base function in R. In these cases, the marker with smaller sum of squares within each pair in the corresponding JL model was removed. For each of the remaining peak markers in the JL model for that trait, a rescan procedure was then implemented to test if any closely adjacent markers were more significantly associated with the trait than the peak marker identified in stepwise model selection. Specifically, if a marker on either side was showing, after the multicollinearity correction, a larger sum of squares than the original peak marker, that adjacent marker would replace the original peak marker in the model. This process was repeated until the peak marker under consideration showed the highest sum of squares compared with both of its neighbors, representing a local maximum. All final peak JL markers following rescan, along with the family term, were then refitted to obtain a final JL model for each trait. Allelic effect estimates of these QTL within each family were generated by fitting final trait models using the ‘lm’ function within the lme4 package in R, which also tests the significance of each effect within a family term in two-sided independent t tests. The Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995) was used to control the false discovery rate at 0.05 when identifying potentially significant QTL effects.
For each joint QTL in the final models for all traits, a support interval (using P value threshold of 0.01) was calculated as previously described (Tian et al., 2011). Logarithm of the odds scores were calculated using the ‘logLik’ base function in R. The PVE by each joint QTL was calculated using previous methods (Li et al., 2011), with some modifications. This modified method accounted for within-family variation of allele frequencies by taking a weighted average of an allelic effect of a marker based on its allele frequency within each family and the population size of that family. Solely to assess the true magnitude and direction of QTL allelic effects both within and across NAM families, allelic effect estimates were also generated using untransformed BLUEs, refitting the family term and final JL markers derived from the transformed BLUE model without further model selection or rescan.
GWAS
For each chromosome and each trait, residuals for conducting a GWAS were obtained from the final full JL models with the family term and any joint QTL from that chromosome removed. The genotypic data set used to perform the GWAS in the NAM population consisted of 28.9 million variants (SNPs and short indels of 15 or fewer base pairs in length) contained in HapMap versions 1 and 2, as well as ∼0.8 million copy number variants, as previously described (Wallace et al., 2014). To conduct a GWAS for each trait, these 29.7 million segregating variants were projected onto the NAM RILs based on their genotypic data and the dense 0.1-cM resolution linkage map. Using these projected variants, a forward selection regression procedure was repeated 100 times for each chromosome. This procedure subsampled 80% of the RILs from each family without replacement; this procedure was run separately on chromosome-specific residuals using the NAM-GWAS plug-in in TASSEL version 4.1.32 (Bradbury et al., 2007) as previously described (Wallace et al., 2014). For each trait, the significance threshold for the entry of a marker in the model was empirically determined using a permutation procedure run 1000 times on chromosome-specific residuals. The results of permutations were then averaged across chromosomes (Wallace et al., 2014) to control the genome-wide type I error rate at (α = 0.05). The entry thresholds determined from permutations and used in GWAS are provided in Supplemental Table 3. For each trait and marker, the RMIP value, defined as the proportion of 100 subsamples in which a tested marker was included in the final, forward selection-derived regression model, was calculated. Only markers with an RMIP value of 0.05 or greater were further examined in triangulation analyses.
Growth Environments and Plant Materials for RNA-Seq
A total of three biological replications of the NAM founders were planted on May 10 (rep 1), May 20 (rep 2), and June 1 (rep 3), 2011, at Purdue University’s Agronomy Center for Research and Education in West Lafayette, IN, with ∼15 plants per plot. All plants in each plot were self-pollinated, and pollination dates were recorded. A single ear from a given plot was harvested at (each of) 12, 16, 20, 24, 30, and 36 DAP. Immediately after harvest, whole ears were frozen in liquid nitrogen. The ears were stored at −80°C until kernels could be removed from the still-frozen ears, placed in test tubes, and maintained at −80°C. Kernels from each sample were packed in dry ice and shipped to Michigan State University, from which 30 kernels were randomly sampled and bulked across replicates. For the majority of samples, three biological replicates were available, and 10 seeds were taken from each. In a small number of instances, two or only one replicate was available; in these cases, 15 and 30 seeds were taken from the replicates, respectively.
For root and shoot tissues, seed from the NAM founders were surface-sterilized and germinated on wet filter paper for 4 to 5 d under grow lamps at room temperature. Next, three germinated seedlings were transplanted to 18.93-liter containers with SureMix potting mix (Michigan Grower Products) and fertilized with 1× Hoagland solution. Plants were grown in the greenhouse under long-day conditions for 14 d at 30 to 33°C at which time the plants were removed from pots and rinsed with water to remove the soil. Roots and shoots were harvested separately, immediately frozen in liquid nitrogen, and stored at −80°C until RNA extraction. Equal weights of shoots or roots from the three individual plants were combined into a single sample for RNA isolation.
RNA-Seq and Sample Quality Assessment
Frozen samples were ground to a fine powder in liquid nitrogen. Total RNA from 100 mg of frozen kernel, shoot, and root tissues was isolated using the hot borate protocol (Wan and Wilkins, 1994) except that a Qiagen shredder column was used to filter the lysate prior to the LiCl precipitation step. To assess the quality and concentration of RNA, samples were analyzed using a NanoDrop (Thermo Fisher Scientific) and Bioanalyzer 2100 (Agilent Technologies). mRNA-seq libraries were constructed from total RNA using the Illumina RNASeq kit following the manufacturer’s instructions. Sequencing was performed on the Illumina GAIIx and HiSeq 2000 instruments at the Michigan State University Research Technology Support Facility. Reads (50–55 nucleotides; 11–140 M reads per sample) were generated and their quality evaluated using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). A small number of libraries were sequenced in paired-end mode, but all downstream analyses treated reads as single-end.
Identification of SNPs in RNA-Seq Data
For SNP detection, RNA-seq reads were cleaned for quality using Cutadapt (v 1.4.1) (Martin, 2011). Specifically, Illumina adapter and primer sequences were removed (using the –b option), as well as bases at the 3′ end that had a quality score less than 20 and reads that were fewer than 30 bases in length after cleaning. RNA-seq reads were then aligned to the maize B73 reference genome (AGPv2; http://ftp.maizesequence.org/) using TopHat (v1.4.1) (Langmead et al., 2009) and SAMTools (v0.1.12a) (Li et al., 2009). TopHat was run with a minimum and maximum intron size of five and 60,000 bp, respectively. Indel detection was disabled, and only unique alignments were reported using the -g option; all other options were set to default. BAMTools (v 1.0.2) (Barnett et al., 2011) was used to calculate mapping result statistics. The BAM file for each sample was sorted by leftmost coordinates using the SAMTools sort function (v 0.1.12a) (Li et al., 2009). This file was then indexed using SAMTools index, and a pileup file generated using SAMTools pileup with options –Bcf. An unfiltered matrix file was made and filtered to detect SNPs. SNPs were filtered according to the following requirements: (1) five reads per individual; (2) for an allele to be called within an individual, it had to be in 20% or more of the reads with at least two reads supporting it; (3) be homozygous (monoallelic) in each individual; (4) support by two individuals for an allele to exist; and (5) the position had to be polymorphic (at least two alleles) (Hirsch et al., 2014). The identified SNPs, i.e., those passing all of these filters, were analyzed and clustered by genotype to identify any mislabeled samples. In addition, genetic distances between all samples were calculated as previously described and clustered with seedling transcriptome-derived SNPs identified in the WiDiv 1.0 panel (Hirsch et al., 2014) to further confirm genotype authenticity. All samples passing these quality control steps for an individual genotype were then merged using the SAMTools merge function, and the pipeline repeated from the sort and index step. A total of 175 samples representing 21 root, 21 shoot, and 133 kernel samples (12, 16, 20, 24, 30, and 36 DAP) from 24 of the 26 NAM parents passed quality assessments and were used for SNP detection, and 172 of these were used for triangulation analyses (Supplemental Table 1 and Supplemental Data Set 8).
Gene Expression Analysis
RNA-seq reads were aligned to the maize B73 reference genome (AGPv2; http://ftp.maizesequence.org/) using TopHat (v 1.4.1) (Langmead et al., 2009) and SAMTools (v 0.1.12a) (Li et al., 2009); expression abundances were estimated using Cufflinks (v 1.3.0) (Trapnell et al., 2010) using the RefGen_v2 5b Filtered Gene Set (FGS) (http://ftp.maizesequence.org/release-5b/filtered-set/). When running TopHat, the minimum and maximum intron length was set to 5 and 60,000 bp, respectively, and the same maximum intron length was used for running Cufflinks. The –G and –b options were used when running Cufflinks; all other parameters were left at default. Boundaries of gene models in the AGPv2 annotation were corrected for GRMZM2G012966 (lycopene epsilon cyclase) and GRMZM2G084942 (arogenate dehydrogenase), both which were incorrectly fused with a flanking gene. GRMZM2G012966 (lycopene epsilon cyclase) was split, resulting in a new locus labeled as GRMZM6G010010 (kinase-domain-containing protein). GRMZM2G084942 (arogenate dehydrogenase) was split, resulting in a new locus labeled as GRMZM6G010020 (CBF1 interacting corepressor) and the fragments per kilobase exon model per million fragments mapped (FPKM) values recalculated just for these modified loci. Expression data were reported in FPKM values. A Pearson correlation coefficient (r) was calculated for all pairwise comparisons of all samples using FPKM data. Raw FPKM data were input into R (version 3.1.0) and transformed into a data matrix. Correlations for all observations were calculated using the “pearson” method of the ‘cor’ base function in R. The calculated correlation coefficients were then visualized for all pairwise comparisons using the ‘heatmap.2’ function within the gplots package in R (Supplemental Figure 4).
FPKM Filtering
FPKM reads were annotated by gene not transcripts, mapping to a total of 39,455 loci. The 5b FGS gene set was filtered such that at least one of the kernel developmental samples in at least one sampled founder line had an FPKM greater than 1.0; a total of 27,187 genes remained upon filtering with this criterion. Expression data for genes passing the specified threshold were transformed according to log2(FPKM + 1), where the constant of 1 was added to allow the transformation of “0” values. These log2-transformed values are herein specified as “gene expression.” Within the filtered and transformed transcriptomic data set, early kernel samples correlated more closely in expression abundances with root and shoot samples than with mid- to late-kernel samples (Supplemental Figure 4). The number of aligned 50- or 55-bp reads per sample, both unique and multiple-mapping, had a median of 40 million reads (median 86% of total reads) with sd of 16 million reads (Supplemental Table 1 and Supplemental Data Set 8).
Triangulation Analysis
Genomic regions in which both JL and GWAS associations colocalized were further investigated using the following procedure. First, JL support intervals from two or more individual-trait models that showed physical overlap were merged to form common support intervals. Support intervals detected for a single trait, with no physical overlap within other trait models, were also retained. For each final distinct support interval, Pearson correlations were tested in all pairwise comparisons between (1) QTL effect estimates for that interval in individual-trait JL model(s); (2) genotype state of significant GWAS marker(s) in the interval for the respective trait(s); and (3) log2-transformed expression values of candidate gene(s) directly hit by or within ±100 kb of any of these significant GWAS markers. The use of 100 kb to define the candidate gene search range was established through examination of LD decay and is further elaborated in the results section.
To test for significance of the correlation between JL allelic effect estimates and expression values of each candidate gene proximal to significant GWAS markers, a multiple testing correction to control false discovery rate at 0.05 was imposed on P values of the correlation obtained for each gene. Namely, the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) based on the total number of genes proximal to the GWAS marker (within ± 100 kb) was applied using the GAPIT package (Lipka et al., 2012) in R. For those correlations involving one of the two traits with a negative optimal lambda for the Box-Cox transformation (i.e., an inverse power transformation was applied for δT and δT3), the sign of the correlation was flipped in graphical and tabular representations (Figure 2 and Supplemental Figure 5 for master gene summaries; Supplemental Figure 2 and Supplemental Table 6 for pleiotropy) to represent the true directionality of the relationship between traits.
Epistasis
For each trait, all possible pairwise interactions (additive × additive) between markers comprising the final JL model were individually tested for significance in a model containing all marker main (additive) effects. The P value threshold required for an interaction to enter the model was determined by modeling 1000 null permutations of transformed trait BLUEs with only additive terms in the model to approximate a type I error rate at α = 0.05. Interaction effects passing this threshold were used together with the main effects of markers comprising the final JL model to fit the final epistatic model. Calculations of PVE were performed using effect estimates and allele frequencies within families as described above, except that pairwise genotype scores were collapsed into three classes for interaction terms due to insufficient degrees of freedom to model all possible genotype states in the two-locus case. Specifically, the two vectors of genotype state scores were multiplied to obtain composite scores of −1 (one locus is homozygous for reference allele and the other for minor), 0 (at least one heterozygote, meaning the two alleles are assumed to cancel any interaction), or 1 (both loci are homozygous for reference, or both for minor). Interactions were graphically depicted using the Circos software package (Krzywinski et al., 2009) (Figure 5).
Pleiotropy
Pleiotropy, or shared genetic basis, was assessed between pairs of traits as previously described (Buckler et al., 2009), by applying the JL QTL model for each trait to every other trait. Pearson correlations between allelic effect estimates derived from the final JL model for a trait itself and the model applied from every other trait were evaluated for significance at α = 0.01, which with 23 df means a cutoff of |r| > 0.504. The sharedness, or percentage of shared QTL, between two traits was calculated as the sum of the percentage of significant correlations when the trait 1 model was applied to trait 2 and the percentage when vice versa. Connections among QTL showing sharedness were visualized using the network R package (Butts, 2008, 2015) (Supplemental Figure 2).
Pleiotropy was also examined within common support intervals to validate the merging of individual-trait intervals, a step conducted in previous NAM JL analyses (Tian et al., 2011). In contrast to the above-described pleiotropy analysis, this QTL-level analysis fit the single peak JL marker within the common interval for each trait to every other trait that had a peak JL marker in the interval.
LD Analysis
For each marker showing an RMIP of 0.05 or higher for one or more traits in GWAS, pairwise LD with all other markers within ±1 Mb was estimated through custom Python and R scripts using squared allele-frequency correlations (r2) as previously described (Weir, 1996). A null distribution for LD was generated by performing the same estimation for 50,000 markers selected at random. The same imputed genotypic data set of 29.7 million segregating markers used in JL-GWAS was used to estimate LD.
Standardized Effect Sizes
To more fully compare JL results across traits, effect sizes of JL peak markers were standardized and visualized in relation to the allele frequencies at these markers (Supplemental Figure 6). Given that 12 was the smallest number of QTL detected for a tocochromanol trait aside from PC-8, an outlier in both JL model size (eight QTL) and line-mean heritability (lowest by 4-fold), JL was rerun constraining the number of markers per trait to 12 using transformed BLUEs. Allelic effect estimates were obtained by subsequently refitting these 12-QTL models with untransformed BLUEs and scaled by multiplying by the total heritable variance for each trait (Brown et al., 2011). Total heritable variance was estimated on a by-trait basis, as the line-mean heritability in NAM divided by the sd of untransformed trait BLUEs in the Goodman-Buckler inbred diversity panel (Lipka et al., 2013). Allele frequencies were derived based on founders exhibiting NAM JL allelic effect estimates significantly different from those of B73, using estimates from transformed BLUEs given the involvement of statistical inference.
Preparation and Identification of Standards for Chlorophylls and Their Derivatives
Chlorophylls a and b were isolated from fresh spinach (Spinacia oleracea) leaves as previously described (Canjura and Schwartz, 1991). Chlorophyllides were prepared by grinding fresh spinach leaves in 80% acetone with 20% 40 mM sodium citrate, pH 8, and incubating overnight at room temperature in darkness (Holden, 1961). After centrifuging to pellet debris, the supernatant was extracted twice with diethyl ether. The diethyl ether extracts were pooled, dried over anhydrous sodium sulfate, evaporated, and dissolved in 80:20 methanol:acetone. Pheophytins a and b and pheophorbides a and b were prepared by acidification of their corresponding purified chlorophylls and chlorophyllides, respectively, as described (Schwartz et al., 1981; Canjura and Schwartz, 1991). Each compound was isolated by semipreparative HPLC using a Shimadzu Prominence HPLC and 5 µm Spherisorb ODS-2 column (250 × 4.6 mm) (Orochem Technologies). Pigments were eluted using a linear gradient at 1 mL/min in which Solvent A was 80% methanol in acetone and Solvent B was 80% methanol in 1 M ammonium acetate. The gradient used was 0 to 100% solvent A for 15 min, hold solvent A at 100% for 15 min, and then return to solvent B and reequilibrate for 7 min. Individual compounds were identified and quantified by a combination of their retention and spectral characteristics (Camara, 1985; Lichtenthaler, 1987; Zapata et al., 1987; Milenković et al., 2012).
Extraction and Analysis of Chlorophylls and Derivatives
Embryos and endosperm from each NAM parent were dissected from frozen kernels on a metal plate on dry ice to ensure all tissues remained frozen. Embryo and endosperm tissues were ground in liquid nitrogen and 50 to 60 mg tissue was extracted with 600 μL 10% (v/v) 0.2 M Tris-HCl, pH 8.0, in acetone precooled to −20°C (Schelbert et al., 2009; Christ et al., 2012) that contained 1 mg/mL butylated hydroxytoluene, 1 mg/mL bixin, and 1 mg/mL dl-α-tocopherol acetate as internal recovery controls. Three 3-mm glass beads were added and extraction was done for 5 min by shaking using a commercial paint shaker (HERO). Samples were centrifuged for 5 min at 13,000 RPM in a microfuge, and the supernatant was transferred to a new tube. Three hundred microliters of HPLC-grade water and 500 μL of diethyl ether were added, vortexed, and centrifuged at 13,000 RPM for 2 min to allow for phase separation. The upper (diethyl ether) fraction was transferred to a new microcentrifuge tube, evaporated, and dissolved in 200 μL 100% acetone, which was divided into two aliquots and evaporated. One aliquot was dissolved in 100 μL of 3:1 (v/v) methanol:methyl tert-butyl ether for analyses of tocochromanols as previously described (Lipka et al., 2013). The second aliquot was dissolved in 100 μL of 80:20 (v/v) methanol:acetone and assessed by HPLC for levels of the eight target chlorophyll metabolites as described above. Pheophorbide b and pheophytin b, whose presence indicates artifactual conversions during extraction, were below detection levels in all samples analyzed. For each of the detected chlorophyll metabolites, Pearson correlations with total tocopherol levels were calculated within each time point after removing values (concentration, pmol g−1) that were more than three standard deviations from the mean for the respective compound (the chlorophyll metabolite and/or total tocopherols) within that time point.
Accession Numbers
Genes identified in this study are as listed in Figure 3, using the following accession numbers as available in MaizeGDB: GRMZM2G084942, GRMZM2G493395, GRMZM2G173358, GRMZM2G173641, GRMZM2G112728, GRMZM2G082998, GRMZM2G088396, GRMZM2G035213, GRMZM2G036455, GRMZM2G073351, GRMZM2G039373, GRMZM2G060870, GRMZM2G128176, and GRMZM2G031028. HapMap sequence data, as described by Chia et al. (2012) and Gore et al. (2009), can be found in the NCBI Short Read Archive with accession numbers SRA051245 (HapMap v. 2) and SRP001145 (HapMap v. 1). Both SNP data sets are also available at www.panzea.org. The data reported in this article are tabulated in the supplemental data and archived in the following places: All RNA-seq reads are available at the National Center of Biotechnology Information Sequence Read Archive under BioProject Number PRJNA174231. SNPs and expression abundances (FPKMs) are available from the DRYAD repository (http://dx.doi.org/10.5061/dryad.5t8d9). Scripts used in this study are available on GitHub (https://github.com/GoreLab/Vitamaize_NAM_GWAS_LabVersion.git).
Supplemental Data
Supplemental Figure 1. Pairwise phenotypic correlations between untransformed best linear unbiased estimators of 10 tocochromanol grain traits.
Supplemental Figure 2. Pleiotropy of 162 quantitative trait loci identified in joint-linkage analysis for 10 grain tocochromanol traits in the U.S. maize nested association mapping population.
Supplemental Figure 3. Linkage disequilibrium estimates between GWAS variants in the NAM population.
Supplemental Figure 4. Heat map displaying Pearson’s (r2) correlation coefficient of expression abundances across all samples in this study.
Supplemental Figure 5. Master summaries for the remaining genes identified in this study.
Supplemental Figure 6. Frequency distributions of standardized joint-linkage allelic effect estimates (absolute values) for tocopherol traits and tocotrienol traits, for quantitative trait loci in each of three classes.
Supplemental Figure 7. Concentrations (pmol g−1, log scale) of chlorophyll pathway compounds and total tocopherols in embryos and endosperm of NAM parents analyzed across developing kernel stages.
Supplemental Figure 8. Relative explanation of phenotypic variance for 10 tocochromanol grain traits by each of three classes of joint-linkage quantitative trait loci.
Supplemental Figure 9. Distribution of untransformed best linear unbiased estimators for 10 grain tocochromanol traits across the U.S. maize nested association mapping population.
Supplemental Table 1. Summary of samples used for RNA isolation in this study.
Supplemental Table 2. Percent phenotypic variance explained for unresolved joint-linkage quantitative trait loci.
Supplemental Table 3. Lambda values used in Box-Cox transformation and joint-linkage and genome-wide association study entry thresholds determined from permutations for each trait.
Supplemental Data Set 1. Genomic information for the 81 a priori candidate genes.
Supplemental Data Set 2. Transformed and untransformed best linear unbiased estimators for the U.S. maize nested association mapping population used in the present study.
Supplemental Data Set 3. Final joint-linkage models for the 10 tocochromanol traits evaluated in the U.S. maize nested association mapping population.
Supplemental Data Set 4. Summary of joint linkage-quantitative trait loci and associated GWAS signals for 10 tocochromanol grain traits evaluated in the U.S. maize nested association mapping population.
Supplemental Data Set 5. Summary of joint-linkage quantitative trait locus allelic effect estimates for the 10 tocochromanol traits evaluated in the U.S. maize nested association mapping population.
Supplemental Data Set 6. Summary of pleiotropy analyses.
Supplemental Data Set 7. Complete list of genome wide association study results.
Supplemental Data Set 8. Tissue and RNA-seq information for samples used in this study.
Supplemental Data Set 9. Concentrations (pmol g−1) of tocochromanol and chlorophyll metabolites in developing embryo and endosperm tissue.
Supplemental Data Set 10. Chlorophyll-related gene expression in developing B73 embryos.
Supplemental Data Set 11. Summary of observations and genotypes removed in the “difference in fits” and Studentized deleted residuals outlier detection stages, conducted pre- and postgeneration of best linear unbiased estimators, respectively.
Supplemental File 1. Distribution of raw HPLC phenotypic values for 10 grain tocochromanol traits across and within the U.S. maize nested association mapping population.
Supplemental File 2. Distribution of untransformed best linear unbiased estimators for 10 grain tocochromanol traits across and within the U.S. maize nested association mapping population.
Acknowledgments
This research was supported by the National Science Foundation (DBI-0922493 to D.D.P., C.R.B, E.S.B., and T.R. and DBI-0820619 and IOS-1238014 to E.S.B.), by the USDA-ARS (E.S.B.), by Cornell University startup funds (M.A.G.), and by a USDA National Needs Fellowship (C.H.D.). We gratefully acknowledge Evan Klug and Emily McKinney for assistance with sample processing and HPLC, Elodie Gazave for initial Circos scripts, Jason Peiffer for custom Java code calling ASReml, and Pat Brown for pleiotropy plot scripts.
AUTHOR CONTRIBUTIONS
C.H.D., M.A.G., and D.D.P. cowrote the manuscript. C.H.D., C.B.K., and A.E.L. co-led data analysis. M.M.-L. performed HPLC analyses and metabolite quantifications. B.V., E.G.-C., J.H., and J.C. performed transcriptome analysis. J.G.W. generated marker data sets. J.G.W. and D.C.I. coded GWAS, GWAS permutation, and figure scripts. J.C. created website/databases. A.M. made RNA and RNA-seq libraries. P.J.B. modified TASSEL source code and oversaw epistasis calculations. T.R. managed NAM population growth. M.M.-H. managed planting, pollination, harvesting, and processing of the NAM population. B.F.O. and T.T. generated developing kernels for transcriptome analysis. E.S.B. oversaw germplasm, genotyping, and imputation and advised on mapping analysis. C.R.B. oversaw RNA-seq and transcriptome analysis and managed all informatics. M.A.G. oversaw data analysis, project management, design, and coordination. D.D.P. conducted project management and coordination, and oversaw data and metabolite analyses and biological interpretation.
Footnotes
Articles can be viewed without a subscription.
References
- Ahsan H., Ahad A., Siddiqui W.A. (2015). A review of characterization of tocotrienols from plant oils and foods. J. Chem. Biol. 8: 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett D.W., Garrison E.K., Quinlan A.R., Strömberg M.P., Marth G.T. (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27: 1691–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsley D.A., Kuh E., Welsch R.E. (2005). Regression Diagnostics: Identifying Influential Data and Sources of Collinearity. (Hoboken, NJ: John Wiley & Sons; ). [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Met. 57: 289–300. [Google Scholar]
- Box G.E.P., Cox D.R. (1964). An analysis of transformations. J. Roy. Stat. Soc. B Met. 26: 211–252. [Google Scholar]
- Bradbury P.J., Zhang Z., Kroon D.E., Casstevens T.M., Ramdoss Y., Buckler E.S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23: 2633–2635. [DOI] [PubMed] [Google Scholar]
- Bréhélin C., Kessler F., van Wijk K.J. (2007). Plastoglobules: versatile lipoprotein particles in plastids. Trends Plant Sci. 12: 260–266. [DOI] [PubMed] [Google Scholar]
- Brown P.J., Upadyayula N., Mahone G.S., Tian F., Bradbury P.J., Myles S., Holland J.B., Flint-Garcia S., McMullen M.D., Buckler E.S., Rocheford T.R. (2011). Distinct genetic architectures for male and female inflorescence traits of maize. PLoS Genet. 7: e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler E.S., et al. (2009). The genetic architecture of maize flowering time. Science 325: 714–718. [DOI] [PubMed] [Google Scholar]
- Burkschat M. (2009). Linear estimators and predictors based on generalized order statistics from generalized Pareto distributions. Commun. Stat. Theor. M. 39: 311–326. [Google Scholar]
- Butts C. (2008). Network: a package for managing relational data in R. J. Stat. Softw. 24: 1–36.18612375 [Google Scholar]
- Butts C. (2015). Network: Classes for Relational Data. The Statnet Project, http://statnet.org.
- Cahoon E.B., Hall S.E., Ripp K.G., Ganzke T.S., Hitz W.D., Coughlan S.J. (2003). Metabolic redesign of vitamin E biosynthesis in plants for tocotrienol production and increased antioxidant content. Nat. Biotechnol. 21: 1082–1087. [DOI] [PubMed] [Google Scholar]
- Camara B. (1985). Prenylation of chlorophyllide a in Capsicum plastids. Methods Enzymol. 110: 274–281. [Google Scholar]
- Canjura F.L., Schwartz S.J. (1991). Separation of chlorophyll compounds and their polar derivatives by high-performance liquid chromatography. J. Agric. Food Chem. 39: 1102–1105. [Google Scholar]
- Chandler K., Lipka A.E., Owens B.F., Li H., Buckler E.S., Rocheford T., Gore M.A. (2013). Genetic analysis of visually scored orange kernel color in maize. Crop Sci. 53: 189–200. [Google Scholar]
- Cheng Z., Sattler S., Maeda H., Sakuragi Y., Bryant D.A., DellaPenna D. (2003). Highly divergent methyltransferases catalyze a conserved reaction in tocopherol and plastoquinone synthesis in cyanobacteria and photosynthetic eukaryotes. Plant Cell 15: 2343–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia J.M., et al. (2012). Maize HapMap2 identifies extant variation from a genome in flux. Nat. Genet. 44: 803–807. [DOI] [PubMed] [Google Scholar]
- Christ B., Schelbert S., Aubry S., Süssenbacher I., Müller T., Kräutler B., Hörtensteiner S. (2012). MES16, a member of the methylesterase protein family, specifically demethylates fluorescent chlorophyll catabolites during chlorophyll breakdown in Arabidopsis. Plant Physiol. 158: 628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrost B., Falk J., Kernebeck B., Mölleken H., Krupinska K. (1999). Tocopherol biosynthesis in senescing chloroplasts: a mechanism to protect envelope membranes against oxidative stress and a prerequisite for lipid remobilization. In The Chloroplast: From Molecular Biology to Biotechnology, Argyroudi-Akoyunoglou J.H., Senger H., eds (Dordrecht, The Netherlands: Springer Netherlands; ), pp. 171–176. [Google Scholar]
- Collakova E., DellaPenna D. (2003). Homogentisate phytyltransferase activity is limiting for tocopherol biosynthesis in Arabidopsis. Plant Physiol. 131: 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook J.P., McMullen M.D., Holland J.B., Tian F., Bradbury P., Ross-Ibarra J., Buckler E.S., Flint-Garcia S.A. (2012). Genetic architecture of maize kernel composition in the nested association mapping and inbred association panels. Plant Physiol. 158: 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DellaPenna D., Mène-Saffrané L. (2011). Vitamin E. In Advances in Botanical Research, Rebeille F., Douce R., eds (Amsterdam, The Netherlands: Elsevier; ), pp. 179–227. [Google Scholar]
- Deruère J., Römer S., d’Harlingue A., Backhaus R.A., Kuntz M., Camara B. (1994). Fibril assembly and carotenoid overaccumulation in chromoplasts: a model for supramolecular lipoprotein structures. Plant Cell 6: 119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elshire R.J., Glaubitz J.C., Sun Q., Poland J.A., Kawamoto K., Buckler E.S., Mitchell S.E. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6: e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint-Garcia S.A., Thuillet A.C., Yu J., Pressoir G., Romero S.M., Mitchell S.E., Doebley J., Kresovich S., Goodman M.M., Buckler E.S. (2005). Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J. 44: 1054–1064. [DOI] [PubMed] [Google Scholar]
- Fritsche S., Wang X., Li J., Stich B., Kopisch-Obuch F.J., Endrigkeit J., Leckband G., Dreyer F., Friedt W., Meng J., Jung C. (2012). A candidate gene-based association study of tocopherol content and composition in rapeseed (Brassica napus). Front. Plant Sci. 3: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland L.U., Magallanes-Lundback M., Hemming C., Supplee A., Koornneef M., Bentsink L., Dellapenna D. (2006). Genetic basis for natural variation in seed vitamin E levels in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 103: 18834–18841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour A.R., Gogel B.J., Cullis B.R., Thompson R. (2009). ASReml User Guide: Release 3.0. (Hemel Hempstead, UK: VSN International). [Google Scholar]
- Glaubitz J.C., Casstevens T.M., Lu F., Harriman J., Elshire R.J., Sun Q., Buckler E.S. (2014). TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9: e90346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore M.A., Chia J.M., Elshire R.J., Sun Q., Ersoz E.S., Hurwitz B.L., Peiffer J.A., McMullen M.D., Grills G.S., Ross-Ibarra J., Ware D.H., Buckler E.S. (2009). A first-generation haplotype map of maize. Science 326: 1115–1117. [DOI] [PubMed] [Google Scholar]
- Hirsch C.N., Foerster J.M., et al. (2014). Insights into the maize pan-genome and pan-transcriptome. Plant Cell 26: 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden M. (1961). The breakdown of chlorophyll by chlorophyllase. Biochem. J. 78: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland J.B., Nyquist W.E., Cervantes-Martínez C.T. (2003). Estimating and interpreting heritability for plant breeding: an update. In Plant Breeding Reviews, Janick J., ed (Oxford, UK: John Wiley & Sons; ), pp. 9–112. [Google Scholar]
- Hörtensteiner S. (2013). Update on the biochemistry of chlorophyll breakdown. Plant Mol. Biol. 82: 505–517. [DOI] [PubMed] [Google Scholar]
- Hörtensteiner S., Kräutler B. (2011). Chlorophyll breakdown in higher plants. Biochim. Biophys. Acta 1807: 977–988. [DOI] [PubMed] [Google Scholar]
- Hung H.Y., et al. (2012). The relationship between parental genetic or phenotypic divergence and progeny variation in the maize nested association mapping population. Heredity (Edinb.) 108: 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S.C., Cahoon E.B. (2007). Enhancing vitamin E in oilseeds: unraveling tocopherol and tocotrienol biosynthesis. Lipids 42: 97–108. [DOI] [PubMed] [Google Scholar]
- Hussain N., Irshad F., Jabeen Z., Shamsi I.H., Li Z., Jiang L. (2013). Biosynthesis, structural, and functional attributes of tocopherols in planta; past, present, and future perspectives. J. Agric. Food Chem. 61: 6137–6149. [DOI] [PubMed] [Google Scholar]
- Inoue S., Ejima K., Iwai E., Hayashi H., Appel J., Tyystjärvi E., Murata N., Nishiyama Y. (2011). Protection by α-tocopherol of the repair of photosystem II during photoinhibition in Synechocystis sp. PCC 6803. Biochim. Biophys. Acta 1807: 236–241. [DOI] [PubMed] [Google Scholar]
- Kamal-Eldin A., Appelqvist L.-Å. (1996). The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids 31: 671–701. [DOI] [PubMed] [Google Scholar]
- Karunanandaa B., et al. (2005). Metabolically engineered oilseed crops with enhanced seed tocopherol. Metab. Eng. 7: 384–400. [DOI] [PubMed] [Google Scholar]
- Kim E.H., Lee Y., Kim H.U. (2015). Fibrillin 5 is essential for plastoquinone-9 biosynthesis by binding to solanesyl diphosphate synthases in Arabidopsis. Plant Cell 27: 2956–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.H., et al. (2011). Antioxidant activity and inhibition of lipid peroxidation in germinating seeds of transgenic soybean expressing OsHGGT. J. Agric. Food Chem. 59: 584–591. [DOI] [PubMed] [Google Scholar]
- Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R., Raclaru M., Schüsseler T., Gruber J., Sadre R., Lühs W., Zarhloul K.M., Friedt W., Enders D., Frentzen M., Weier D. (2005). Characterisation of plant tocopherol cyclases and their overexpression in transgenic Brassica napus seeds. FEBS Lett. 579: 1357–1364. [DOI] [PubMed] [Google Scholar]
- Kump K.L., Bradbury P.J., Wisser R.J., Buckler E.S., Belcher A.R., Oropeza-Rosas M.A., Zwonitzer J.C., Kresovich S., McMullen M.D., Ware D., Balint-Kurti P.J., Holland J.B. (2011). Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 43: 163–168. [DOI] [PubMed] [Google Scholar]
- Kutner M.H., Nachtsheim C.J., Neter J., Li W. (2004). Applied Linear Statistical Models. (Boston: McGraw Hill Irwin; ). [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M., Sharopova N., Beavis W.D., Grant D., Katt M., Blair D., Hallauer A. (2002). Expanding the genetic map of maize with the intermated B73 x Mo17 (IBM) population. Plant Mol. Biol. 48: 453–461. [DOI] [PubMed] [Google Scholar]
- Li H., Bradbury P., Ersoz E., Buckler E.S., Wang J. (2011). Joint QTL linkage mapping for multiple-cross mating design sharing one common parent. PLoS One 6: e17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Yang X., Xu S., Cai Y., Zhang D., Han Y., Li L., Zhang Z., Gao S., Li J., Yan J. (2012). Genome-wide association studies identified three independent polymorphisms associated with α-tocopherol content in maize kernels. PLoS One 7: e36807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhou Y., Wang Z., Sun X., Tang K. (2010). Engineering tocopherol biosynthetic pathway in Arabidopsis leaves and its effect on antioxidant metabolism. Plant Sci. 178: 312–320. [Google Scholar]
- Lichtenthaler H.K. (1987). Chlorophylls and carotenoids: pigments of photosynthetic biomembranes. Methods Enzymol. 148: 350–382. [Google Scholar]
- Lipka A.E., Tian F., Wang Q., Peiffer J., Li M., Bradbury P.J., Gore M.A., Buckler E.S., Zhang Z. (2012). GAPIT: genome association and prediction integrated tool. Bioinformatics 28: 2397–2399. [DOI] [PubMed] [Google Scholar]
- Lipka A.E., Gore M.A., Magallanes-Lundback M., Mesberg A., Lin H., Tiede T., Chen C., Buell C.R., Buckler E.S., Rocheford T., DellaPenna D. (2013). Genome-wide association study and pathway-level analysis of tocochromanol levels in maize grain. G3 3: 1287–1299. [DOI] [PMC free article] [PubMed]
- Lippold F., vom Dorp K., Abraham M., Hölzl G., Wewer V., Yilmaz J.L., Lager I., Montandon C., Besagni C., Kessler F., Stymne S., Dörmann P. (2012). Fatty acid phytyl ester synthesis in chloroplasts of Arabidopsis. Plant Cell 24: 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littell R.C., Milliken G.A., Stroup W.W., Wolfinger R.D., Schabenberger O. (2006). SAS® for Mixed Models, 2nd ed. (Cary, NC: SAS Institute; ). [Google Scholar]
- Lu Y., Rijzaani H., Karcher D., Ruf S., Bock R. (2013). Efficient metabolic pathway engineering in transgenic tobacco and tomato plastids with synthetic multigene operons. Proc. Natl. Acad. Sci. USA 110: E623–E632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Baker R.F., Magallanes-Lundback M., DellaPenna D., Braun D.M. (2008). Tie-dyed1 and sucrose export defective1 act independently to promote carbohydrate export from maize leaves. Planta 227: 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H., DellaPenna D. (2007). Tocopherol functions in photosynthetic organisms. Curr. Opin. Plant Biol. 10: 260–265. [DOI] [PubMed] [Google Scholar]
- Maeda H., Song W., Sage T.L., DellaPenna D. (2006). Tocopherols play a crucial role in low-temperature adaptation and Phloem loading in Arabidopsis. Plant Cell 18: 2710–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. [Google Scholar]
- McMullen M.D., et al. (2009). Genetic properties of the maize nested association mapping population. Science 325: 737–740. [DOI] [PubMed] [Google Scholar]
- Mène-Saffrané L., Jones A.D., DellaPenna D. (2010). Plastochromanol-8 and tocopherols are essential lipid-soluble antioxidants during seed desiccation and quiescence in Arabidopsis. Proc. Natl. Acad. Sci. USA 107: 17815–17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenković S.M., Zvezdanović J.B., Anđelković T.D., Marković D.Z. (2012). The identification of chlorophyll and its derivatives in the pigment mixtures: HPLC-chromatography, visible and mass spectroscopy studies. Adv. Technol. 1: 16–24. [Google Scholar]
- Neter J., Kutner M.H., Nachtsheim C.J., Wasserman W. (1996). Applied Linear Statistical Methods. (Chicago: Irwin; ). [Google Scholar]
- Norris S.R., Shen X., DellaPenna D. (1998). Complementation of the Arabidopsis pds1 mutation with the gene encoding p-hydroxyphenylpyruvate dioxygenase. Plant Physiol. 117: 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogut F., Bian Y., Bradbury P.J., Holland J.B. (2015). Joint-multiple family linkage analysis predicts within-family variation better than single-family analysis of the maize nested association mapping population. Heredity (Edinb.) 114: 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer J.A., Romay M.C., Gore M.A., Flint-Garcia S.A., Zhang Z., Millard M.J., Gardner C.A., McMullen M.D., Holland J.B., Bradbury P.J., Buckler E.S. (2014). The genetic architecture of maize height. Genetics 196: 1337–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland J.A., Bradbury P.J., Buckler E.S., Nelson R.J. (2011). Genome-wide nested association mapping of quantitative resistance to northern leaf blight in maize. Proc. Natl. Acad. Sci. USA 108: 6893–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raclaru M., Gruber J., Kumar R., Sadre R., Lühs W., Zarhloul M.K., Friedt W., Frentzen M., Weier D. (2006). Increase of the tocochromanol content in transgenic Brassica napus seeds by overexpression of key enzymes involved in prenylquinone biosynthesis. Mol. Breed. 18: 93–107. [Google Scholar]
- Rao T.J., Province M.A. (2016). A framework for interpreting type I error rates from a product-term model of interaction applied to quantitative traits. Genet. Epidemiol. 40: 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippert P., Scimemi C., Dubald M., Matringe M. (2004). Engineering plant shikimate pathway for production of tocotrienol and improving herbicide resistance. Plant Physiol. 134: 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rise M., Cojocaru M., Gottlieb H.E., Goldschmidt E.E. (1989). Accumulation of α-tocopherol in senescing organs as related to chlorophyll degradation. Plant Physiol. 89: 1028–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M.D., Holzinger E.R., Li R., Pendergrass S.A., Kim D. (2015). Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 16: 85–97. [DOI] [PubMed] [Google Scholar]
- Sattler S.E., Gilliland L.U., Magallanes-Lundback M., Pollard M., DellaPenna D. (2004). Vitamin E is essential for seed longevity and for preventing lipid peroxidation during germination. Plant Cell 16: 1419–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savidge B., Weiss J.D., Wong Y.-H.H., Lassner M.W., Mitsky T.A., Shewmaker C.K., Post-Beittenmiller D., Valentin H.E. (2002). Isolation and characterization of homogentisate phytyltransferase genes from Synechocystis sp. PCC 6803 and Arabidopsis. Plant Physiol. 129: 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelbert S., Aubry S., Burla B., Agne B., Kessler F., Krupinska K., Hörtensteiner S. (2009). Pheophytin pheophorbide hydrolase (pheophytinase) is involved in chlorophyll breakdown during leaf senescence in Arabidopsis. Plant Cell 21: 767–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S.J., Woo S.L., von Elbe J.H. (1981). High-performance liquid chromatography of chlorophylls and their derivatives in fresh and processed spinach. J. Agric. Food Chem. 29: 533–535. [Google Scholar]
- Shintani D., DellaPenna D. (1998). Elevating the vitamin E content of plants through metabolic engineering. Science 282: 2098–2100. [DOI] [PubMed] [Google Scholar]
- Singh D.K., McNellis T.W. (2011). Fibrillin protein function: the tip of the iceberg? Trends Plant Sci. 16: 432–441. [DOI] [PubMed] [Google Scholar]
- Tanaka H., Yabuta Y., Tamoi M., Tanabe N., Shigeoka S. (2015). Generation of transgenic tobacco plants with enhanced tocotrienol levels through the ectopic expression of rice homogentisate geranylgeranyl transferase. Plant Biotechnol. 32: 233–238. [Google Scholar]
- Tanaka R., Rothbart M., Oka S., Takabayashi A., Takahashi K., Shibata M., Myouga F., Motohashi R., Shinozaki K., Grimm B., Tanaka A. (2010). LIL3, a light-harvesting-like protein, plays an essential role in chlorophyll and tocopherol biosynthesis. Proc. Natl. Acad. Sci. USA 107: 16721–16725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S., Hass C.G., Knapp S.J. (2006). Ty3/gypsy-like retrotransposon knockout of a 2-methyl-6-phytyl-1,4-benzoquinone methyltransferase is non-lethal, uncovers a cryptic paralogous mutation, and produces novel tocopherol (vitamin E) profiles in sunflower. Theor. Appl. Genet. 113: 783–799. [DOI] [PubMed] [Google Scholar]
- Tian F., Bradbury P.J., Brown P.J., Hung H., Sun Q., Flint-Garcia S., Rocheford T.R., McMullen M.D., Holland J.B., Buckler E.S. (2011). Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 43: 159–162. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar W., Holmes C.C., Mott R., Flint J. (2009). Mapping in structured populations by resample model averaging. Genetics 182: 1263–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin H.E., Lincoln K., Moshiri F., Jensen P.K., Qi Q., Venkatesh T.V., Karunanandaa B., Baszis S.R., Norris S.R., Savidge B., Gruys K.J., Last R.L. (2006). The Arabidopsis vitamin E pathway gene5-1 mutant reveals a critical role for phytol kinase in seed tocopherol biosynthesis. Plant Cell 18: 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eenennaam A.L., et al. (2003). Engineering vitamin E content: from Arabidopsis mutant to soy oil. Plant Cell 15: 3007–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Dorp K., Hölzl G., Plohmann C., Eisenhut M., Abraham M., Weber A.P.M., Hanson A.D., Dörmann P. (2015). Remobilization of phytol from chlorophyll degradation is essential for tocopherol synthesis and growth of Arabidopsis. Plant Cell 27: 2846–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace J.G., Bradbury P.J., Zhang N., Gibon Y., Stitt M., Buckler E.S. (2014). Association mapping across numerous traits reveals patterns of functional variation in maize. PLoS Genet. 10: e1004845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C.Y., Wilkins T.A. (1994). A modified hot borate method significantly enhances the yield of high-quality RNA from cotton (Gossypium hirsutum L.). Anal. Biochem. 223: 7–12. [DOI] [PubMed] [Google Scholar]
- Wang X., Zhang C., Li L., Fritsche S., Endrigkeit J., Zhang W., Long Y., Jung C., Meng J. (2012). Unraveling the genetic basis of seed tocopherol content and composition in rapeseed (Brassica napus L.). PLoS One 7: e50038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.Q., Yoon M.Y., He Q., Kim T.S., Tong W., Choi B.W., Lee Y.S., Park Y.J. (2015). Natural variations in OsγTMT contribute to diversity of the α-tocopherol content in rice. Mol. Genet. Genomics 290: 2121–2135. [DOI] [PubMed] [Google Scholar]
- Weir B.S. (1996). Genetic Data Analysis II. (Sunderland, MA: Sinauer Associates). [Google Scholar]
- Yan J., Lipka A.E., Schmelz E.A., Buckler E.S., Jander G. (2015). Accumulation of 5-hydroxynorvaline in maize (Zea mays) leaves is induced by insect feeding and abiotic stress. J. Exp. Bot. 66: 593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Cahoon R.E., Hunter S.C., Zhang C., Han J., Borgschulte T., Cahoon E.B. (2011). Vitamin E biosynthesis: functional characterization of the monocot homogentisate geranylgeranyl transferase. Plant J. 65: 206–217. [DOI] [PubMed] [Google Scholar]
- Ytterberg A.J., Peltier J.B., van Wijk K.J. (2006). Protein profiling of plastoglobules in chloroplasts and chromoplasts. A surprising site for differential accumulation of metabolic enzymes. Plant Physiol. 140: 984–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Holland J.B., McMullen M.D., Buckler E.S. (2008). Genetic design and statistical power of nested association mapping in maize. Genetics 178: 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapata M., Ayala A.M., Franco J.M., Garrido J.L. (1987). Separation of chlorophylls and their degradation products in marine phytoplankton by reversed-phase high-performance liquid chromatography. Chromatographia 23: 26–30. [Google Scholar]
- Zhang C., Cahoon R.E., Hunter S.C., Chen M., Han J., Cahoon E.B. (2013). Genetic and biochemical basis for alternative routes of tocotrienol biosynthesis for enhanced vitamin E antioxidant production. Plant J. 73: 628–639. [DOI] [PubMed] [Google Scholar]
- Zhang N., et al. (2015). Genome-wide association of carbon and nitrogen metabolism in the maize nested association mapping population. Plant Physiol. 168: 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Liu T., Ren G., Hörtensteiner S., Zhou Y., Cahoon E.B., Zhang C. (2014). Chlorophyll degradation: the tocopherol biosynthesis-related phytol hydrolase in Arabidopsis seeds is still missing. Plant Physiol. 166: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]