

Abstract
Tumor hypoxia interferes with the efficacy of chemotherapy, radiotherapy, and tumor necrosis factor-α. TRAIL (tumor necrosis factor-related apoptosis inducing ligand) is a potent apoptosis inducer that limits tumor growth without damaging normal cells and tissues in vivo. We present evidence for a central role of lysosomal cathepsins in hypoxia and/or TRAIL-induced cell death in oral squamous cell carcinoma (OSCC) cells. Hypoxia or TRAIL-induced activation of cathepsins (B, D and L), caspases (−3 and −9), Bid cleavage, release of Bax and cytochrome c, and DNA fragmentation were blocked independently by zVAD-fmk, CA074Me or pepstatin A, consistent with the involvement of lysosomal cathepsin B and D in cell death. Lysosome stability and mitochondrial membrane potential were reduced in hypoxia and TRAIL-induced apoptosis. However, TRAIL treatment under hypoxic condition resulted in diminished apoptosis rates compared to treatment under normoxia. This inhibitory effect of hypoxia on TRAIL-induced apoptosis may be based on preventing Bax activation and thus protecting mitochondria stability. Our data show that TRAIL or hypoxia independently triggered activation of cathepsin B and D leading to apoptosis through Bid and Bax, and suggest that hypoxic tissue regions provide a selective environment for highly apoptosis-resistant clonal cells. Molecular therapy approaches based on cathepsin inhibitors need to address this novel tumor-preventing function of cathepsins in OSCC.
Keywords: Hypoxia, TRAIL, Cathepsin, Lysosomes, Apoptosis, Oral cancer
Introduction
Hypoxia, a reduction in the level of tissue oxygen tension, occurs during acute and chronic vascular diseases, pulmonary diseases and cancer, and can lead to apoptotic or necrotic cell death [1]. Hypoxia-induced gene expression can directly promote malignant progression, but may also interfere with cytotoxic tumor therapies (chemotherapy, radiation, immunotherapy, or suicide gene therapy) that are primarily mediated by triggering apoptosis selectively in cancer cells [1, 2].
Apoptosis in response to cancer therapy proceeds through activation of the core apoptotic machinery including the extrinsic cell death receptor and the intrinsic mitochondrial signaling pathway [3]. Both pathways are connected through Bid, a pro-apoptotic Bcl-2 family member which is cleaved during apoptosis by lysosomal proteases [4]. In certain cell types, caspase-8 has been shown to convert Bid from a latent to a pro-apoptotic form capable of inducing cytochrome c release through the action of Bax or Bak and subsequent activation of caspase-9 [5]. This release of cytochrome c into the cytosol triggers caspase-3 activation through formation of the cytochrome c/Apaf-1/caspase-9 apoptosome complex, whereas Smac/Diablo and Omi/HtrA2 promote caspase activation through neutralizing the inhibitory effects of IAPs [6]. TRAIL/Apo2L is a cytotoxic ligand belonging to the TNF superfamily that includes TNF-α and FasL. TRAIL engages the extrinsic apoptotic pathway by binding to its membrane-bound death receptors (DR4 and DR5), which transmit an apoptotic signal via their intracellular death domains. Studies using TRAIL knockout mice have demonstrated that TRAIL plays a critical role in suppressing tumor initiation and metastasis [7]. TRAIL is of special interest for cancer therapy since it has been shown to rapidly and selectively kill cancer cells while sparing normal cells [8].
Although caspases are well established as the main players in apoptosis, other proteases such as calpains, cathepsins, and serine proteases account for alternative types of programmed cell death [9]. Cathepsins are proteases which, under physiologic conditions, are localized intralysosomally. In response to certain signals they are released from the lysosomes into the cytoplasm where they trigger apoptotic cell death via various pathways, including the activation of caspases or the release of pro-apoptotic factors from mitochondria [10]. Many tumors express increased amounts of cathepsins, and the action of TNF is dependent on the ‘lysosomal pathway’ of apoptosis [11]. Active participation of lysosomal proteases has been observed in cell death induced by several stimuli, including oxidative stress, TNF-α and chemotherapeutic drugs [12]. Recently, cathepsin B and D have been shown to play a dominant role in executing the apoptotic program in several tumor cell lines [13–16]. These studies demonstrated the presence of a cathepsin-mediated proteolytic event in the apoptotic pathway triggered by TRAIL. Recent studies suggest that cathepsin leads to Bid cleavage and that Bax translocation is a general mechanism [17, 18]. In culture, cathepsin B and D can trigger cytochrome c release from the mitochondria into the cytosol [4, 15, 19]. The fact that tumors frequently contain high levels of cathepsins may prove useful in selectively targeting tumor cells for apoptosis induction by ligands.
Hypoxia strongly decreases the efficacy of many anticancer drugs; however, little is known about the effects of hypoxia on TRAIL-induced tumor cell apoptosis. Thus, we evaluated whether hypoxia influences the efficacy of TRAIL and the role of lysosomal cathepsins in oral cancer cell apoptosis. Here we report novel evidence that hypoxia treatment of OSCC cells significantly inhibits TRAIL-induced apoptosis by blocking lysosomal release of cathepsins B and D to the cytosol. Our data show that lysosomal cathepsins modulate the apoptotic activity of TRAIL, and that this pathway is less efficient in a hypoxic environment.
Materials and methods
Cell lines and reagents
The oral squamous cell carcinoma cell lines MDA1386Tu (1386Tu) and MDA1386Ln (1386Ln) were obtained from the primary tumor and a lymph node metastasis (tumor stage T4N3B) of a 71 year old male patient with primary hypopharynx tumor (generous gift from Peter Sacks, New York University, New York) [20]. The cells were maintained in DMEM/F12 1:1 (v/v) mix containing 10% fetal bovine serum and 0.4 µg/ml hydrocortisone at 37°C with 5% CO2. Rabbit polyclonal antibodies for human cathepsin B and D were obtained from Athens Research and Technology (Athens, GA), and HRP-conjugated goat anti-rabbit or anti-mouse antiserum was from BD Biosciences (San Diego, CA). Protease inhibitor CA-074, cell permeable CA074Me, and z-Arg-Arg-NHMec were from Peptides International (Louisville, KY); G418 sulphate was from Mediatech Inc. (Herndon, VA). Human recombinant TRAIL was purchased from Biosource International (Camarillo, CA). Cell-permeable inhibitor z-VAD-fmk, rabbit polyclonal IgGs for caspase-3 (H-277), Bax (N-20) and Bid (FL-195) were all from Santa Cruz Biotechnology (Santa Cruz, CA). DEVD-AFC, IETD-AMC, DEVD-CHO and IETD-CHO were from Alexis (San Diego, CA), LEHD-CHO and LEHD-AFC from Biomol (Plymouth Meeting, PA), anti-PARP antibody from Cell Signaling Technology (Beverly, MA). Mouse monoclonal β-actin antibody, phenylmethylsulfonyl fluoride, aprotinin, leupeptin, pepstatin A, z-Phe-Arg-NHMec, acridine orange and MTT reduction assay kit (TOX-1 kit) were from Sigma (St. Louis, MO). Mouse monoclonal anti-cytochrome c IgG and anti-cathepsin L antibodies were from BD Biosciences-Pharmingen (San Diego, CA). Bradford protein assay kit was purchased from Bio-Rad (Hercules, CA), M-PER Mammalian Protein Extraction Reagent from Pierce (Rockford, IL). Protease inhibitor cocktail and Apoptotic DNA Ladder Kit were from Roche (Indianapolis, IN), and enhanced chemiluminescence ECL substrate from Amersham Biosciences (Piscataway, NJ). The mitochondrial potential sensor 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) and Lyso-Tracker Green were from Molecular Probes (Eugene, OR), propidium iodide from Invitrogen (Carlsbad, CA); cathepsin L inhibitors z-FF-FMK and z-FY-CHO and cathepsin B detection kit were purchased from Calbiochem (San Diego, CA). L-Leucyl-L-leucine methyl ester was purchased from Bachem (Torrance, CA). All protocols for the use of human cell lines in this work were approved by the Institutional Review Boards of The University of Louisville and the University of Texas at Houston.
Hypoxia exposure and treatments
Hypoxic conditions were produced by placing logarithmic phase subconfluent monolayer cultures in a modular incubator chamber and equilibrating for 30 min with humidified gas containing 1% oxygen, 5% CO2 and 94% nitrogen. The cultures were maintained under hypoxic conditions for 24 h; control cells were grown in normal oxygen for the same duration. After incubation, media collection and cell harvesting were done immediately within 2–3 min to avoid adaptation of cells to re-oxygenation. Cells were pre-treated in 6-well plates wells in triplicate at 37°C for 24 h with or without recombinant human TRAIL protein (100 ng/ml). All inhibitor pre-incubation were for 24 h in the absence or presence of CA-074Me (25 µM), pepstatin A (5 µM), zFF-fmk (10 µM), or zVAD-fmk (10 µM); control wells had the same volume of DMSO solvent. For lysosomal instability studies, L-Leucyl-L-leucine methyl ester (LeuLeuOMe) was added to a final concentration of 400 µM for 6 h.
Cytotoxicity assay
Cells (1.5 × 104 cells/well in 96-well plates) were treated in triplicate with hypoxia and/or TRAIL protein in the absence or presence of CA-074Me, pepstatin A, zFF-FMK, or zVAD-FMK for 24 h as described above; control assays had the same volume of DMSO. After incubation, cytotoxicity rates were determined by the MTT reduction assay (TOX-1 kit) and absorbance measurement at 570 nm (A570). Cytotoxicity (%) was calculated using the following equation: % cytotoxicity = (C–S)/C × 100, where C and S are the average A570 of untreated control and treated samples, respectively. Each experiment was performed independently at least three times in triplicate, and cytotoxicities are given as means ± SD.
Cathepsin B, D and L activity assay
Cells were grown in 6-well plates to 60–70% confluence, and treated for 24 h with hypoxia and/or TRAIL, with or without pretreatment of CA-074Me, pepstatin A, zFF-FMK, or zVAD-FMK for 24 h as described above. After treatment, the cells were rinsed twice with ice-cold PBS, treated with lysis buffer (400 mM Na-phosphate, 75 mM NaCl, 4 mM EDTA, 0.25% Triton-X 100, pH 6.0) for 1 h on ice, ultrasonicated at 40 W for 1 min (1.0 s on/0.5 s off pulses) (Model 550 Sonic Dismembrator, Fisher Scientific, Pittsburgh, PA), and centrifuged at 25,000 × g (10 min, 4°C) to remove cell debris. Total protein amounts were determined using the Bradford protein assay kit. Cathepsin B and L activity was determined fluorimetrically using the methyl-coumarylamide substrate z-Arg-Arg-NHMec at pH 6.0, and z-Phe-Arg-NHMec at pH 5.5, respectively, as described [21, 22]. Fluorescence was measured with an excitation wavelength of 360 nm and emission wavelength of 460 nm. The cathepsin B substrate was used in conjunction with the cathepsin B inhibitor CA-074 (50 µM) in all control assays; the difference between values without and with CA-074 corresponded to cathepsin B activity. To detect cathepsin L activity, the substrate was used in conjunction with the cathepsin L inhibitor z-FY-CHO (25 µM) as control assays; the difference between values without and with z-FY-CHO corresponded to cathepsin L activity. One unit of enzyme activity was defined as the release of 1 µmol of product/min; specific activity as units/mg protein.
Cathepsin D was determined using acid denatured hemoglobin (16.6 g/l) as substrate in ammonium acetate buffer, pH 3.5 [23]. The reaction was stopped with TCA and the absorbance at 750 nm was determined after addition of Folin-Ciocalteau reagent.
Caspase activity assay
Cells were grown in 6-well plates to 60–70% confluence and treated in triplicate wells as described above for cathepsin assays. Then cells were rinsed once and collected in cold PBS by scraping. After centrifugation, cell pellets were resuspended in caspase lysis buffer (10 mM HEPES, pH 7.4, 2 mM EDTA, 0.1% CHAPS) supplemented with protease inhibitors (5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 10 µg/ml pepstatin A, 10 µg/ml aprotinin, and 20 µg/ml leupeptin). Freeze-thaw cell lysis cycles were performed by alternatively transferring the samples from an ethanol/dry ice bath to a 37°C water bath five times. The supernatant was collected after 20 min of centrifugation at 12,000 rpm at 4°C. Assays were performed in caspase buffer (10 mM PIPES, pH 7.4, 2 mM EDTA, 0.1% CHAPS, 5 mM dithiothreitol), to which 50 µM of substrate and 5 µl of protein extract were added to yield a final volume of 100 µl. Peptide substrates for caspase 3-like (−3 and −7), caspase 8 and caspase 9 assays were Ac-DEVD-AFC, Ac-IETD-AMC and Ac-LEHD-AFC, respectively, each dissolved in dimethyl sulfoxide; the respective specific inhibitors DEVD-CHO, IETD-CHO and LEHD-CHO were used in control reactions. Assays were performed in black-wall, clear bottom plates using a Spectramax Gemini XS Microplate Spectrofluorometer (Molecular Devices, Sunnyvale, CA) reading was at 500 nm after excitation at 405 nm for AFC, and at 380 nm after excitation at 460 nm for AMC. The results were compared against AFC and AMC standard curves generated in parallel. Specific activity was expressed as units, with 1 unit defined as AFC or AMC release of 1 nMol/h/µg protein.
Protein extractions and Western blotting
Treated or control cells were rinsed with ice-cold PBS, scraped into 1 ml of PBS, and centrifuged at 4,000 rpm for 3 min. The pellets were resuspended into RIPA buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, and 1 mM EDTA) containing protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride, 10 µg/ml aprotinin, and 2 µg/ml of both leupeptin and pepstatin). Then, cell extracts were sonicated as described above and cell debris was removed by centrifugation. To obtain cellular proteins for cathepsin B, D and L assays, cells were washed in ice-cold PBS, and extracted using M-PER Mammalian Protein Extraction Reagent with protease inhibitor cocktail. Proteins were quantified using the Bradford protein assay kit and compared with a γ-globulin standard curve. Equal amounts of total proteins were separated on a SDS-polyacrylamide gel and transferred onto nitrocellulose membranes by electroblotting overnight at 20 V. Membranes were blocked in TBS-T (10 mM Tris-HCl, 150 mM NaCl, 0.25% Tween 20, pH 7.5) with 5% fat-free powdered milk at room temperature for 1 h. After rinsing membranes in TBS-T, the following primary antibodies were used: rabbit polyclonal antibodies for human cathepsin B and D, mouse monoclonal antibody for cathepsin L, rabbit polyclonal IgGs for caspase-3, Bid, Bax and PARP, or mouse monoclonal β-actin antibody. After incubation overnight at 4°C or 1 h at room temperature, the membranes were washed four times, 10 min each, in TBS-T. Secondary antibodies used were either horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG, followed by five washes with TBS-T. Bands were detected using ECL substrate. For β-actin detection, previously probed membranes were soaked in stripping buffer (70 mM Tris-HCl, pH 6.8, 2% SDS, 0.1% β-mercaptoethanol) at 60°C for 30 min, and incubated as above.
Cytochrome c release assay
Cells were collected, washed once in ice cold PBS, resuspended in cytosol extraction buffer, and cytosolic extracts were prepared as described previously [24]. Western blotting for cytochrome c was done with mouse monoclonal anti-cytochrome c IgG; the absence of intra-mitochondrial proteins was verified by blotting for mitochondrial cytochrome oxidase with mouse monoclonal anti-cytochrome oxidase IgG.
Mitochondrial membrane potential (Δψm) assessment
Δψm was assessed by using a dye, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) after hypoxia and/or TRAIL exposure, followed by FACS analysis for red and green fluorescence. Cells were also treated in the absence or presence of CA-074Me (25 µM), pepstatin A (5 µM), zFF-FMK (10 µM), or zVAD-FMK (10 µM) for 24 h. The cells were stained with JC-1 for 15 min at 37°C in a 5% CO2 incubator, rinsed with assay buffer, and examined by flow cytometry. The JC-1 monomer and J-aggregates were detected in the flow cytometer FL1 (green fluorescence) and FL2 (red fluorescence) channels, respectively. Red and green fluorescence were expressed as percentages of total gated cells.
Determination of DNA fragmentation
Apoptosis detection by FACS analysis was performed after staining cells with propidium iodide. Cells were trypsinized, washed and resuspended in cold PBS. After centrifugation at 1,000 rpm for 5 min, 5 × 105 cells were fixed with 1 ml 70% ethanol at −20°C and washed with PBS. Each pellet was resuspended in 500 µl propidium iodide buffer (20 µg/ml propidium iodide, 10 mg/ml RNAse in PBS). After incubation for 30 min in the dark at room temperature, cell fluorescence signals were determined using a FACScalibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed with its Cell Quest software. Cells to the left of the first peak at sub-G1 contained hypodiploid DNA and were considered apoptotic, and were expressed as a percentage of total gated cells. Nucleosomal DNA fragments were also detected with the Apoptotic DNA Ladder Kit after electrophoresis on 2% agarose gels for visualizations of apoptosis-indicative DNA ladders.
Lysosomal stability assessment
Cells were assessed for lysosomal stability using acridine orange (AO) uptake and relocation and LysoTracker Green assays. These dye probes accumulate in lysosomes on the basis of proton trapping or low pH. Fluorescence assays were conducted by using AO for uptake and relocation studies according to Antunes et al. [25]. Cells (2 × 105/ml) were stained with AO (5 µg/ml) in RPMI 1640 medium with Hepes buffer (10 mM, pH 7.3) for 15 min at 37°C. Cells were then washed, resuspended in PBS at 1 × 106/ml, and the green (channel 1) and red (channel 2) fluorescence of 104 cells was recorded on a logarithmic scale by flow cytometry. All steps were carried out in the dark. Results are expressed as a percentage of cells in each channel versus total gated cells.
For co-localization studies, cells were treated with Lyso-Tracker Green (50 nM) (Molecular Probes) for 30 min and cathepsin B fluorogenic substrate (red) for 30 min. Hoechst 33342 stain (10 min) was used to label the cell nuclei. Confocal images were collected under a Nicon Inverted Microscope Eclipse TE300 with oil immersion objective lens (× 60), and captured by a cool snap HQ digital B/W CCD camera (Roper Scientific, Trenton, NJ). A LAMDA 10-2 optical filter changer (Sutter Instrument) was used to capture simultaneous images in different fluorescence wavelengths. All images shown are representative of at least 3 individual images collected from at least three independent studies.
Statistical analysis
All data represent at least three independent experiments and are expressed as the means ± SD unless otherwise indicated. ANOVA was used to assess the differences between experimental groups. Significance of TRAIL, hypoxia and TRAIL & hypoxia treated cells were compared with control cells. All inhibitor-treated cells (cathepsin and caspase inhibitors) were compared with TRAIL & hypoxia treated cells unless otherwise indicated.
Results
Expression of cathepsins during TRAIL-induced apoptosis under hypoxic conditions
Several recent studies found that cathepsins B and D are relocalized to the cytoplasm during apoptosis resulting from TRAIL treatment because of lysosomal permeabilization [14, 16, 26]. We recently identified a cathepsin B-activated Bid cleavage pathway as major signaling pathway in TRAIL-induced apoptosis and suggested that cellular factors involved in activation or inhibition of caspases and/or cathepsin release may regulate TRAIL-induced apoptosis [16, 27]. Since little is known about hypoxia-induced genes that play a direct role in either apoptosis or anti-apoptosis, we hypothesized that expression of this cathepsin-mediated pathway may be regulated by hypoxia.
The subcellular relocalizations of cathepsin B, D and L during TRAIL-induced apoptosis or under hypoxic conditions have not been reported before. We reasoned that intracellular activation of cathepsins would be likely if these enzymes play a significant role in TRAIL and/or hypoxia-induced cell death. To test this possibility, we performed immunoblotting of cellular extracts from cells treated for 24 h with TRAIL and/or hypoxia (Fig. 1(A)) and analyzed blots by densitometry and normalization to β-actin (Fig. 1(B)). Hypoxia and/or TRAIL treatment significantly increased the expression of cathepsin B, D and L. By contrast, low levels of intracellular cathepsin B, D and L were detected in untreated or normoxic cells compared to TRAIL- and/or hypoxia-treated cells. The predominant forms of cellular cathepsin B, D and L following treatment were the 31, 34 and 35 kDa active enzymes, respectively; the proenzyme forms did not appear in these cell lines. The specificity of cathepsin down-regulation on TRAIL-induced apoptosis under hypoxic condition was verified by enzyme activity assay for cathepsin B, D and L with specific inhibitors in cell culture (Fig. 1(C)). Hypoxic growth during TRAIL treatment decreased the expression and activity of cathepsin B, D and L by ∼ 20% compared to TRAIL alone (Fig. 1(B)). Cathepsin activation was not restricted to these 1386Tu and 1386Ln cells alone, since we also observed the same effects in related 101A and 686Tu OSCC cells (data not shown).
Fig. 1.
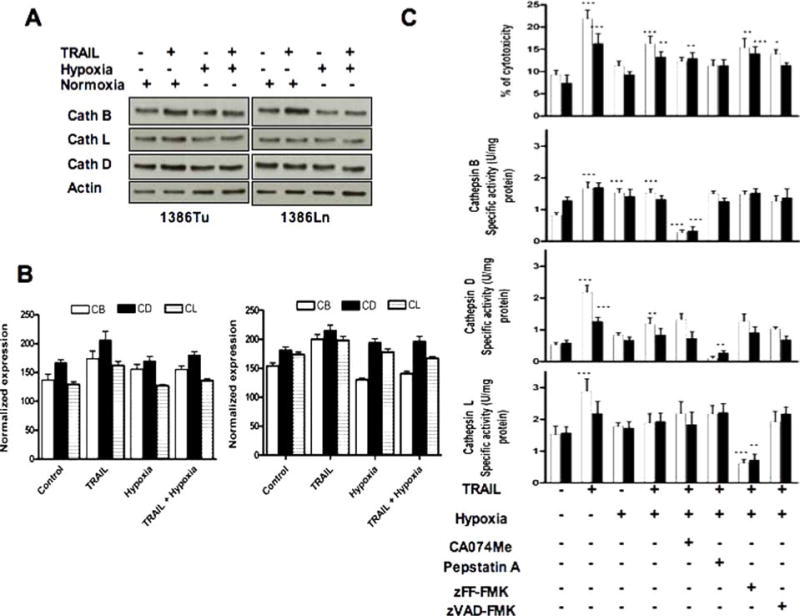
Effects of TRAIL and/or hypoxia on cathepsin expression and activity in OSCC cells. (A) Western blots for 1386Tu (Tu) and 1386Ln (Ln) cells after treatment with TRAIL (100 ng/ml/24 h) and/or hypoxia (1% oxygen, 24 h). Individual blots show active forms of cathepsin B (31 kDa), cathepsin D (34 kDa) and cathepsin L (35 kDa); re-probing for β-actin served as internal loading control. The data are representative of two to three independent experiments with similar results. (B) Densitometic quantitation of cathepsin bands normalized for β-actin, shown as mean ± SD. (C) The 1386Tu (Tu: open bars) and 1386Ln (Ln: closed bars) cells were exposed to TRAIL and/or hypoxia after pretreatment with or without CA074Me (25 µM), pepstatin A (5 µM), zFF-fmk (10 µM) and zVAD-fmk (10 µM) for 24 h, and cytotoxicity and cathepsin activities were measured. The specific activity of cathepsin B, D and L were determined by using Z-Arg-Arg-NHMec, Z-Phe-Arg-NHMec and hemoglobin as a substrate, respectively. The activity results shown are the mean ± SD from three separate experiments; significance of TRAIL, hypoxia and TRAIL & hypoxia treated cells were compared with untreated controls; all other inhibitor-treated cells were compared with TRAIL & hypoxia treatment. ∗P < 0.05,∗∗P < 0.01,∗∗∗P < 0.001; all others are non-significant
We sought to determine whether TRAIL-induced release of cathepsins was dependent on the activation of cellular caspases. Pretreatment of OSCC cells with 10 µM of the pan-caspase inhibitor z-VAD-FMK prior to exposure to TRAIL and/or hypoxia was sufficient to abrogate apoptosis (Fig. 4). However, z-VAD-FMK only partially inhibited the activity of cathepsins (Fig. 1(C)). This agrees with previous data showing that cathepsins, papain and legumain can be inhibited by common caspase-specific inhibitors [28]. We also examined the impact of the cell-permeable cathepsin B inhibitor CA-074Me (25 µM), cathepsin D inhibitor pepstatin A (5 µM), or the cathepsin L inhibitor zFF-fmk (10 µM) during TRAIL and/or hypoxia exposure on cytotoxicity (Fig. 1(C)), caspase activities, and caspase 3 protein expression (Figs. 2 and 3(A)). Cytotoxicity of TRAIL alone or TRAIL and hypoxia combined was significantly higher for Tu cells than for Ln cells (Fig. 1(C)), in agreement with our previous observations [16]. The same differential cytotoxicities for Tu versus Ln cells were observed after treatment with the above cathepsin and caspase inhibitors alone (data not shown). These results indicated that OSCC cells are sensitive to both TRAIL and these protease inhibitors compared to control cells, and that Tu cells are more susceptible than Ln cells.
Fig. 4.
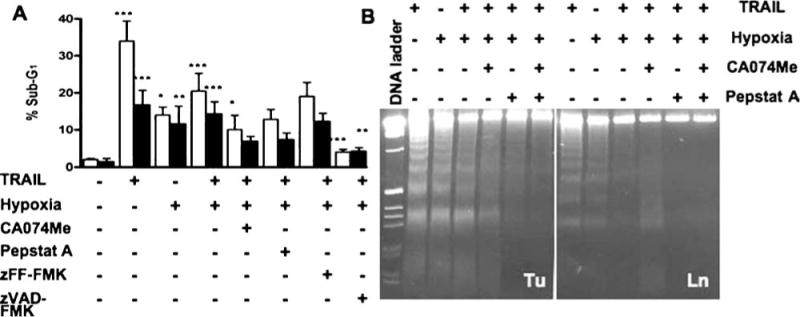
DNA fragmentation in hypoxia and/or TRAIL-induced apoptosis. OSCC cells were exposed to TRAIL (100 ng/ml) and/or hypoxia (1%) after pretreatment with or without CA074Me (25 µM), pepstatin A (5 µM), zFF-fmk (10 µM) and zVAD-fmk (10 µM) for 24 h, and analyzed for nuclear DNA fragmentation. (A) FACS analysis of cells (1386Tu: open bars; 1386Ln: closed bars) labeled with propidium iodide; apoptotic cells with sub-G1 chromosomal DNA content are expressed as a percentage of total gated cells. The FACS data shown are the mean ± SD from three separate experiments; significance of TRAIL, hypoxia and TRAIL & hypoxia treated cells were compared with untreated controls; all other inhibitor-treated cells were compared with TRAIL & hypoxia treatment. ∗P < 0.05,∗∗P < 0.01,∗∗∗P < 0.001; all other are non-significant. (B) 1386Tu (Tu) and 1386Ln (Ln) cells were treated as above, with or without CA074Me and pepstatin inhibitors, and analyzed for nucleosomal DNA fragments by gel electrophoresis. Apoptosis was confirmed by the appearance of a ladder of oligonucleosomal DNA; DNA ladder, DNA 1 kb size marker. The data are representative of two independent experiments with similar results
Fig. 2.
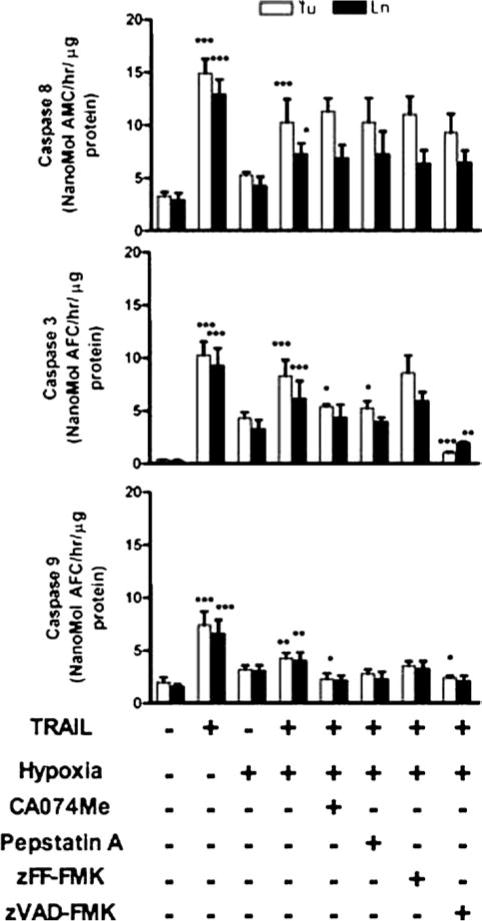
Caspase activities in TRAIL- and/or hypoxia-treated OSCC cells. The 1386Tu (Tu: open bars) and 1386Ln (Ln: closed bars) cells were exposed to TRAIL and/or hypoxia after pretreatment with or without protease inhibitors as described in Fig. 1. Caspase-3/7, -8 and -9 activities were measured by using specific fluorometric substrates, as described in Materials and Methods. The activity results shown are the mean ± SD from three separate experiments; significance of TRAIL, hypoxia and TRAIL & hypoxia treated cells were compared with untreated controls; all other inhibitor-treated cells were compared with TRAIL & hypoxia treatment. ∗P < 0.05,∗∗P < 0.01,∗∗∗P < 0.001; all others are non-significant
Fig. 3.
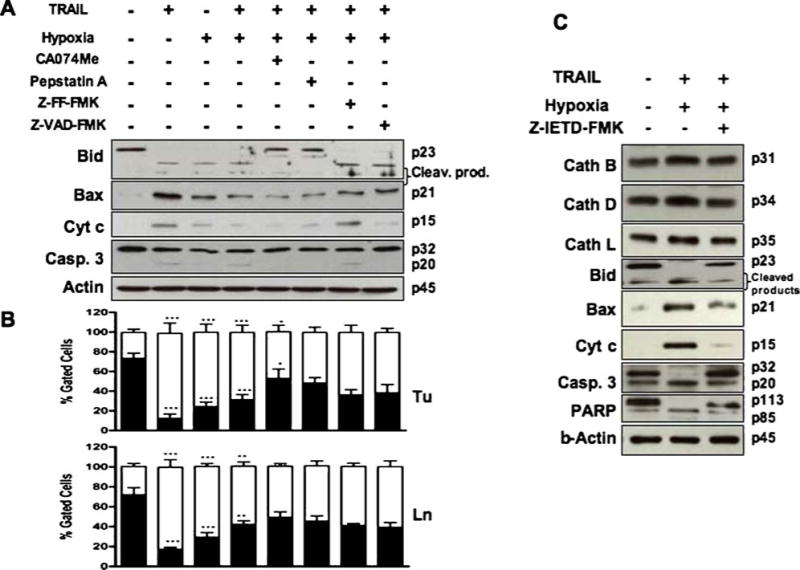
Changes of apoptosis markers in TRAIL and/or hypoxia treated OSCC cells. The 1386Tu cells were exposed to TRAIL and/or hypoxia after pretreatment with or without protease inhibitors as described in Fig. 1. (A) Western blot analysis of 1386Tu cells showing cleavage of Bid (23 KDa) into lower molecular weight products; cleavage of pro-caspase 3 (32 kDa) into the active (20 kDa) form; Bax (21 kDa) activation, and cytochrome c (15 kDa) release into cytosolic fraction; re-probing for β-actin served as internal loading control. The Western blot data are representative of two to three independent experiments with similar results. (B) Mitochondrial membrane potential (Δψm) in 1386Tu (Tu) and 1386Ln (Ln) cells was assessed by using JC-1 dye and FACS analysis for red and green fluorescence, expressed as a percentage of total gated cells. The same treatment regimens as indicated in part A are applied to part B. Closed and open bars represent red and green fluorescence, respectively. The FACS results are the mean ± SD from three separate experiments; significance of TRAIL, hypoxia and TRAIL & hypoxia treated cells were compared with untreated control; all other inhibitor-treated cells were compared with TRAIL & hypoxia treatment. ∗P < 0.05,∗∗P < 0.01,∗∗∗P < 0.001; all others are non-significant. (C) Western blot analysis of 1386Tu cells after TRAIL & hypoxia treatment with or without zIETD-fmk (20 µM) for 24 h. Individual blots show the active form of cathepsin B (31 kDa), cathepsin D (34 kDa), cathepsin L (35 kDa), cleavage of Bid, Bax activation; cytochrome c release, and processing of PARP (113 kDa) into a typical 89 kDa protein; re-probing for β-actin served as internal loading control. Data are representative of two independent experiments with similar results
Alterations of apoptosis markers during TRAIL-induced apoptosis under hypoxic conditions
Experimental inhibition of caspase proteases is known to inhibit TRAIL- or hypoxia-induced cell death in OSCC cells [16, 24]. To determine whether cathepsin B, D and L are important for caspase activation during such apoptosis events, we examined caspase-3/7, -8 and -9 activity, caspase-3 expression, Bid cleavage, Bax and cytochrome c release in Tu and Ln cells in the presence of specific inhibitors for cathepsin B, D and L, and pan-caspase in cell culture (Figs. 2 and 3(A)). Since caspase-3 and -7 have the same substrate specificities in vitro, we hypothesized that caspase-7 could also be activated. TRAIL alone or TRAIL and hypoxia treatment significantly increased caspase-3, -8 and -9 like activities, which were reduced in zVAD-fmk treated cells as expected. Both initiator caspases (caspase-8 and -9) and effector caspases (caspase-3 and -7) seemed to be activated simultaneously. The proform of caspase-3 was cleaved after TRAIL or TRAIL and hypoxia treatment (Figs. 3(A) and (C)) accompanied by an increase in caspase-3 like activity (Fig. 2), indicating that the decrease of pro-caspase-3 resulted from enzyme activation. Interestingly, inhibition of caspase-3 activity with zVAD-fmk by about 70–80% in hypoxia and TRAIL treated cells did not inhibit caspase-8 activity which proves that caspase-8 acts upstream from caspase-3 (Fig. 2). Down-regulation of cathepsin B and D in both Tu and Ln cells by specific inhibitors significantly decreased in TRAIL-induced apoptosis during hypoxic conditions by decreasing caspase-3/7 and -9 activities (Figs. 2, 3(A) and (C)). There was no effect on cathepsin L (Fig. 2). The initiator caspase-8 activated the mitochondrial pathway in these cell lines during TRAIL and/or hypoxia treatment [16], and inhibition of caspase-8 activity with zIETD-fmk partially inhibited caspase-3 cleavage (Fig. 3(C)). Such activation of caspase-3 during TRAIL-induced apoptosis in OSCC cells occurs downstream of Bid cleavage and cytochrome c release from the mitochondria, and zDEVD-fmk markedly inhibited apoptosis [16, 24].
Up-regulation of cathepsin B and D resulted in substantial Bid cleavage, increased Bax and cytochrome c release following 24 h treatment with TRAIL and/or hypoxia. The increase in cathepsin L did not show any response in these events (Fig. 3(A)). We assessed the functional relevance of the hypoxia and/or TRAIL-mediated up-regulation of proapoptotic proteins Bax and Bid in this response. Translocation of Bid to the mitochondria has been shown to engage the intrinsic pathway after stimulus by a death receptor such as TRAIL but not in the hypoxic environment. Western blot analysis showed that Bid was cleaved and Bax up-regulated in response to TRAIL, and this effect was reduced with hypoxia alone (Fig. 3). During combined TRAIL and hypoxia treatment, Bid and Bax were reduced compared to TRAIL or hypoxia alone. This TRAIL and/or hypoxia-induced Bid processing and Bax release was inhibited by treatment with zVAD-fmk, CA074Me or pepstatin A, with similar effects for all three reagents (Fig. 3(A)). This down-regulation of Bid and Bax was further increased in these cells by cathepsin inhibitors CA074Me and pepstatin A, which proves the involvement of cathepsin B and D upstream of Bid and Bax or mitochondria. Also, addition of zIETD-fmk prevented Bid processing and Bax release, consistent with caspase-8 involvement in these processes (Fig. 3(C)). Inhibition of caspase-3, Bid and PARP cleavage, as well as Bax and cytochrome c release by zIETD-fmk all suggest that caspase-8 effects are active upstream of mitochondria. These inhibitor studies show that cathepsins B and D and caspase-8 directly or indirectly participate in activation of Bid, Bax, cytochrome c and PARP in TRAIL and/or hypoxia induced apoptosis. Our data support the model that both cathepsins (B and D) and caspase-8 mediate a TRAIL-induced apoptotic pathway in OSCC cells by activating Bid and Bax upstream of mitochondria.
In order to determine whether mitochondria are involved below cathepsin in the apoptotic process induced by TRAIL and/or hypoxia, the release of cytochrome c and mitochondrial membrane potential (Δψm) was analyzed by using Western blot (Fig. 3(A)) and JC-1 dye (Fig. 3(B)) in flow cytometry, respectively. Cytochrome c release was inhibited by zVAD-fmk (Fig. 3(A)) or zIETD-fmk (Fig. 3(C)), indicating that caspases are partially responsible. These effects were accompanied by cleavage of caspase-3 and also of PARP, a known substrate for caspase-3. Co-treatment of OSCC cells with TRAIL and/or hypoxia and cathepsin inhibitors (CA074Me or pepstatin A) also resulted in partial inhibition of cytochrome c release and PARP cleavage. Pan-caspase inhibitor (zVAD-fmk) or cathepsin inhibitors (CA074Me and pepstatin A) diminished the changes in Δψm induced by TRAIL and/or hypoxia (Fig. 3(B)), suggesting that both caspase and cathepsin activation are responsible for changes in Δψm.
Taken together, our findings indicate that cathepsin B and D act upstream of mitochondria to promote caspase activation during TRAIL and/or hypoxia-induced apoptosis, and that hypoxia may regulate resistance to TRAIL through lysosomal cathepsins.
Effect of hypoxia on TRAIL-induced nuclear changes and DNA fragmentation
TRAIL or hypoxia induces cell death with morphologic features typical for apoptosis, characterized by cell shrinkage, cleavage of nuclear DNA, condensation of chromatin and fragmentation of nucleus and cytoplasm with subsequent formation of apoptotic bodies [1, 29]. To investigate the effects of hypoxia on TRAIL-induced apoptosis morphology, we exposed human OSCC cells to hypoxic or normoxic conditions for 24 h with or without addition of TRAIL protein. When analyzed by flow cytometry, apoptotic cells with degraded DNA appeared as cells with hypodiploid DNA content, represented in so called “sub-G1” peaks to the left of the main G1 peak on the DNA histograms (Fig. 4(A)). OSCC Tu and Ln cells incubated in hypoxia for 24 h showed apoptotic rates as observed before [24]. In contrast to hypoxia-induced apoptosis, both cell lines were sensitive to TRAIL-induced apoptosis. The Tu cells were the most sensitive to TRAIL, with 30–40% apoptotic Tu cells but only 10–20% apoptotic Ln cells after 24 h of incubation. Clearly, hypoxia decreased the efficacy of TRAIL-induced apoptosis in both cell lines compared to TRAIL alone.
The protective effects of zVAD-fmk, CA074Me, pepstatin A, or zFF-FMK on OSCC cells exposed to TRAIL and/or hypoxia were examined. A large proportion of OSCC cells treated with TRAIL and/or hypoxia exhibited apoptosis-indicative sub-G1 peaks (TRAIL: 34% for Tu and 17% for Ln; hypoxia: 14% for Tu and 12% for Ln, TRAIL and hypoxia: 21% for Tu and 14% for Ln). These apoptosis rates were substantially decreased in the presence of the pan-caspase inhibitor zVAD-fmk (4% for Tu and 4% for Ln), the CB inhibitor CA074Me (11.2% for Tu and 12.6% for Ln), or the cathepsin D inhibitor pepstatin A (13% for Tu and 7% for Ln). Such decreases were not observed with the cathepsin L inhibitor zFF-FMK (19% for Tu and 12% for Ln). CA074Me or pepstatin A reversed the FACS profile of OSCC cells treated with TRAIL and hypoxia and reduced the amount of cells with a sub-G1 DNA-content from 20.5 to ∼11.2% in Tu cells and from 14.2 to ∼7% in Ln cells. Addition of zVAD-fmk to cells reduced the proportion of sub-G1 cells from 20.5% to 3.9% in Tu cells and from 14.2% to 4.2% in Ln cells (Fig. 4(B)). These data indicate that TRAIL and/or hypoxia generate an apoptotic signal in a caspase-independent mode, and confirm the involvement of cathepsin B and D in cell death induced by TRAIL and/or hypoxia.
We further determined the effects of TRAIL and/or hypoxia exposure on nucleosomal DNA fragmentation in OSCC cells as an alternative method for apoptosis detection (Fig. 4(B)). There was a prominent fragmentation of DNA after treatment with TRAIL or hypoxia alone, but the amount of fragmentation was less with combined TRAIL and hypoxia treatment. With CA074Me or pepstatin A, where no residual cathepsin B and D activities were detected (Fig. 1(C)), DNA fragmentation was inhibited (Fig. 4(B)). Importantly, CA074Me or pepstatin A had the same effect as zVAD-fmk and inhibited apoptosis even in the absence of zVAD-fmk. TRAIL protein under hypoxia was not as efficient as under normoxic conditions in killing OSCC cells, indicating that hypoxia affected the pro-apoptotic activity of TRAIL. Therefore, our data suggest that hypoxia may indirectly regulate TRAIL apoptotic signals in OSCC cells through lysosomal cathepsins.
Lysosomal rupture during TRAIL-induced apoptosis under hypoxic conditions
We determined whether the apoptosis-inducing effect was caused by lysosomal leakage, by utilizing the Acridine Orange (AO) uptake and relocation methods [30]. Lysosomal damage was assayed qualitatively by fluorescence microscopy (Fig. 5(A)) and quantitated by flow cytometry assessment (Fig. 5(B)) of the changes in green versus red fluorescence of cells stained with AO before or after exposure to TRAIL and/or hypoxia. AO is a weak base that, due to proton trapping, preferentially distributes within the acidic lysosomal compartment. Due to its metachromatic properties, AO fluoresces red inside intact lysosomes, where it is highly concentrated, and weakly green when released to the cytosol and nucleus, where it is much less concentrated [30].
Fig. 5.
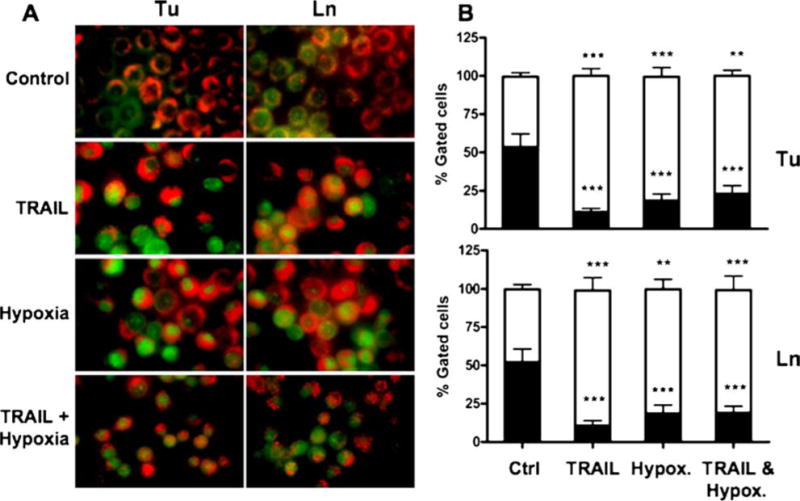
Lysosome stability assessment during TRAIL and/or hypoxia-induced apoptosis. 1386Tu (Tu) and 1386Ln (Ln) cells were exposed to TRAIL (100 ng/ml) and/or hypoxia (1% oxygen) for 24 h, and assessed for lysosomal stability using the AO uptake and relocation method as described in Materials and Methods. (A) Fluorescence microscopy of cells showing merged images from red and green channel. Intact lysosomes appear as red fluorescence (AO uptake), whereas lysosomal rupture will release some AO to the cytosol, shown as green fluorescence (AO relocation); yellow indicate partial AO release in cells. Magnification × 400. (B) Quantitation of fluorescence in AO-stained cells using FACS analysis, expressed as a percentage of total gated cells. Closed and open bars represent red and green fluorescence, respectively. The FACS results shown are the mean ± SD from three separate experiments; significance was calculated compared with untreated control. ∗P < 0.05,∗∗P < 0.01,∗∗∗P < 0.001
Exposure to TRAIL and/or hypoxia for 24 h triggered significant lysosomal disruption, as reflected by the increase in green cytosolic fluorescence (Figs. 5(A) and (B)). Furthermore, the magnitude of observed fluorescence changes correlated well with the degree of ensuing apoptosis observed (Fig. 4). Again, TRAIL was more effective than hypoxia alone, whereas hypoxia reduced the TRAIL-mediated lysosomal release. It is known that apoptosis is accompanied by a slight decrease in cytosolic pH; however, because the relocation of AO to the cytosol (increased green fluorescence) occurred after exposure to TRAIL and/or hypoxia and before any signs of apoptosis, any relation to altered cytosolic pH can be ruled out. Taken together, the results provide a strong correlation between early lysosomal damage and late apoptosis in cells exposed to TRAIL and/or hypoxia.
Lysosomal destabilization leading to apoptosis
To confirm lysosomal destabilization after TRAIL and/or hypoxia treatment, OSCC cells were co-loaded with LysoTracker Green, a fluorescent dye that localizes predominantly into lysosomes, and a cell-permeable Cathepsin B red-fluorogenic substrate. There was an increase in lysosomal rupture induced by TRAIL and/or hypoxia, as detected by a redistribution of cathepsin B activity to the cytosol, and partial redistribution of LysoTracker Green fluorescence from a vesicular to a mixed cytosolic/vesicular pattern (Fig. 6(A)). Hypoxia did interfere with the efficacy of TRAIL-induced apoptosis events by decreasing lysosomal rupture. The protein expression (Fig. 3) and DNA analysis (Fig. 4) data for apoptotic events were in agreement with the cathepsin B activity and LysoTracker Green confocal microscopy data (Fig. 6). These data also suggest that TRAIL and/or hypoxia-associated lysosomal permeabilization was nonselective, since two functionally unrelated tracers, LysoTracker Green and Cathepsin B fluoregenic substrate, were both released from lysosomes into the cytosol, implying that multiple enzymes would be released in the cytosol. Thus, our data confirmed for OSCC cells the previously implicated involvement of cathepsins B and D in apoptosis events [10]. These data again demonstrate that hypoxia protects cells from TRAIL-induced lysosome-mediated apoptosis.
Fig. 6.
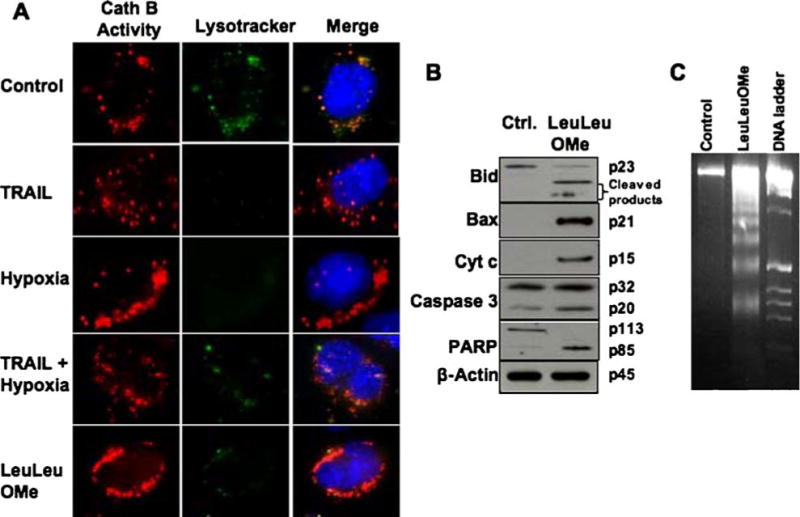
Co-localization of lysosomes and cathepsin B activity in live cells. The 1386Tu cells were exposed to TRAIL and/or hypoxia as described above, and also to Leucyl-Leucine methyl ester (LeuLeuOMe; 400 µM, 6 h) a lysosomotropic agent, as described in Materials and Methods. (A) Upon TRAIL and/or hypoxia stimulation, LysoTracker dye (green) and cathepsin B fluorogenic substrate (red) signals showed redistribution to a diffuse cytosolic pattern. During LeuLeuOMe treatment, cathepsin B was released from disrupted lysosomes. The nucleus was stained with Hoechst 33342 stain (blue). (B) LeuLeuOMe-induced apoptosis in 1386Tu cells. Western blot analysis confirmed Bid cleavage, Bax activation, cytochrome c release, caspase-3 cleavage and PARP processing; re-probing for β-actin served as internal loading control. The data are representative of two independent experiments with similar results. (C) 1386Tu cells were treated as above in B, and analyzed for nucleosomal DNA fragments by gel electrophoresis. Apoptosis was confirmed by the appearance of a ladder of oligonucleosomal DNA; DNA ladder, DNA 1 kb size marker. The data are representative of two independent experiments with similar results
As positive control, the lysosomotropic agent LeuLeuOMe also induced cathepsin B activity release and lysosomal rupture (Fig. 6(A)), as well as activation of apoptosis markers (Figs. 6(B) and (C)). After incubating OSCC cells with LeuLeuOMe, Western blots were done to probe for Bid, caspase 3 and PARP cleavage, and for Bax and cytochrome c release (Fig. 6(B)). Also, treatment of OSCC cells with LeuLeuOMe resulted in DNA fragmentation (Fig. 6(C)). All of these apoptosis makers were activated by lysosomotropic LeuLeuOMe in the absence of TRAIL and/or hypoxia. These results document that TRAIL/hypoxia exposure of OSCC cells leads to apoptosis via lysosomal rupture. Even though the detailed mechanisms are not fully understood, our data suggest that destabilization of lysosomes may be a key mechanism in the cellular response to these conditions.
Discussion
The assumption that hypoxia renders tumor cells resistant to cell death is based on reports of reduced sensitivity of hypoxic tumor cells to radiotherapy and chemotherapy [31, 32]. Hypoxic preconditioning can result in resistance to apoptosis, both short-term via transcriptional and translational adaptive responses, and long-term via selective pressures for mutant cells that have acquired genetic lesions protecting them from hypoxic injury [1, 31]. This model is supported by the failure to target tumor cells with death ligands under less severely hypoxic conditions. When a large panel of tumor cells exposed to 1% oxygen were challenged with TRAIL, their sensitivities were similar to that under normoxia. Notably, hypoxia was not severe enough to induce cell death on its own [33]. Also, pre-conditioning of the human lung carcinoma cell line A549 with 1% oxygen in medium reduced TRAIL-induced cell death, and was accompanied by the up-regulation of Bcl-2 [34, 35]. Although TRAIL has been recognized as a key factor in innate immune surveillance against tumor development and metastasis, as well as a potential cancer therapy target, little is known about the effects of hypoxia on TRAIL-induced apoptosis. In our experimental system, moderate hypoxia per se (1% oxygen for 24 h) induced apoptosis in any of the OSCC tumor cell lines examined. In contrast to the lack of inhibitory effect on TRAIL-induced apoptosis, reduced efficacy of TRAIL-mediated cell death under hypoxic conditions has recently been observed [34–36]. This may be explained in part by methodological differences and cell-specific effects in cell killing.
Lysosomal cathepsins trigger programmed cell death (PCD) via multiple pathways that may overlap with the traditional mediators of apoptosis. Until recently, cathepsins were believed to be primarily involved in nonselective intracellular protein degradation in lysosomes, and their functions outside lysosomes were ignored because of their inactivity at neutral pH [37]. Outside lysosomes, cathepsin activity is regulated by their endogenous inhibitors, the cystatins, which function as threshold inhibitors similar to IAPs, the endogenous inhibitors of caspases [38]. Release of lysosomal cathepsins can overcome the inhibitory potential of cystatins and can lead to apoptosis [27]. Cathepsins B and D are most stable at cytoplasmatic pH and seem to have the most prominent role in apoptotic and necrotic like PCD [12]. We recently demonstrated that hypoxia induces apoptosis in OSCC cells [24] and that cathepsin B is involved in TRAIL-induced apoptosis in these cells [16]. Up-regulation of anti-apoptotic molecules was suggested to associate with hypoxia-induced inhibition of TRAIL-induced apoptosis; however, the functional role of lysosomes in hypoxia-induced anti- or pro-apoptotic molecules in TRAIL-induced apoptosis has not been elucidated. The present study was intended to determine the role of lysosomal cathepsins and lysosomes in TRAIL-induced apoptosis and to explore mechanisms by which hypoxia inhibits TRAIL-induced apoptosis in OSCC cells. We have previously demonstrated that cathepsin B inhibition in these cell lines abrogates the mitochondria-dependent pathway, thereby rendering the cells more resistant to TRAIL-induced apoptosis [16]. Here, we have shown in OSCC cells that hypoxia or TRAIL significantly increased cathepsins B, D and L protein expression and that at the same time hypoxia inhibited TRAIL-induced apoptosis. In other words, decrease in the cathepsin release may also affect the mechanism by which hypoxia inhibits TRAIL-induced apoptosis. Thus, our data suggest the involvement of cathepsins B and D in this process. In addition, although hypoxia induces cathepsin expression, the effect of hypoxia on TRAIL-induced cathepsin expression may be overcome by a simultaneous downregulation of pro-apoptotic proteins of the Bcl protein family.
Both TRAIL and tumor necrosis factor (TNF)-α bind to their respective cell surface receptors, leading to caspase-8 activation. Activated caspase-8 amplifies the apoptotic signal either by directly activating downstream caspases or by cleaving proteins such as Bid [39]. The resulting tBid translocates to the mitochondria from the cytosol to induce mitochondrial damage, cell shrinkage, nuclear condensation and release of cytochrome c [40]. In addition, tBid is able to activate Bax to form Bax multimers in the mitochondria [41], thereby inducing release of cytochrome c. We have shown earlier that caspase-8 triggers Bid cleavage through cathepsin B in TRAIL-induced apoptosis in OSCC cells [16]. Furthermore, after treatment with z-IETD-fmk, CA074Me or pepstatin A, tBid formation was blocked, suggesting that cathepsin B and D work prior to Bid cleavage and that caspase 8 is involved in cathepsin processing. This agrees with other observations in different cell systems [4, 42]. Bax-deficient HCT116 cells were completely resistant to TRAIL regardless of oxygen content, demonstrating a pivotal role of Bax in TRAIL-induced apoptotic signaling [35]. TRAIL-induced cathepsin elevation was decreased by hypoxia to levels lower than those of the untreated conditions (Fig. 1) and the levels of Bid, Bax and cytochrome c release were also affected by hypoxia treatment (Fig. 3). Interestingly, however, increases of cathepsins B and D increased TRAIL-induced apoptosis, suggesting that TRAIL induces cathepsin translocation in a manner independent of Bid cleavage and Bax translocation. This suggests that inhibition of cathepsins, leading to inhibition of Bid cleavage and Bax translocation, may be a general mechanism by which hypoxia inhibits TRAIL-induced apoptosis. Loss of mitochondrial membrane potential contributed to this process as a secondary event. The mechanism by which Bax induces mitochondrial damage and whether the activation of Bax is required for TRAIL-induced apoptosis in hypoxia condition still needs clarification. We suggest that in hypoxic cells, the down-regulation of Bax simultaneously with that of Bid will provide a significant survival advantage [43]. Our data showing that hypoxia and/or TRAIL significantly affect Bax expression levels (Fig. 3), imply that hypoxia may inhibit Bax translocation as reported recently [35]. The key regulator of hypoxia-induced cellular response is believed to be hypoxia inducible factor (HIF-1). HIF-1 can initiate apoptosis by inducing pro-apoptotic proteins such as BNIP3 or NIX, which will inhibit Bcl anti-apoptotic activity. For all OSCC cell lines used here, we observed several-fold increases in HIF-1α expression during hypoxia compared to normoxia [21], confirming a hypoxic response under our experimental conditions. Our data clearly demonstrate that hypoxia protects mitochondrial stability from TRAIL-induced apoptosis by inhibiting Bax activation (Fig. 3(C)).
It is possible that other cathepsins may take similar roles as cathepsin B and D in the involvement of lysosomes, due to the ratios of different cathepsins and their cystatin inhibitors in different systems, conditions, tumors, or cell types. Also, other molecular pathways mediated by lysosomal enzymes need to be considered [12]. Our currently proposed chain of events mediated through cathepsin B and D, and particularly lysosomes is as follows (Fig. 7): activated caspase-8 induces cathepsin B and D which are released from lysosomes; these in turn activate cytosolic Bid; tBid is able to activate Bax to form Bax multimers in the mitochondria; Bax induces mitochondrial damage and releases cytochrome c and subsequent apoptosis through caspase-3, -9 and PARP cleavage, with subsequent DNA fragmentation and apoptosis.
Fig. 7.
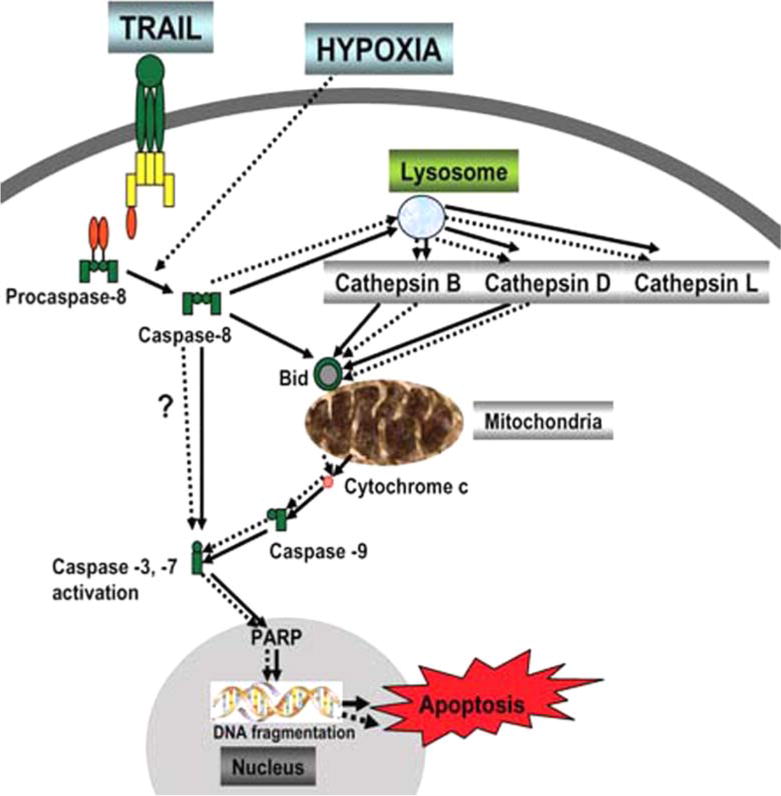
Model for TRAIL/hypoxia induced apoptotic pathway with involvement of lysosomes and lysosomal cathepsins. TRAIL and/or hypoxia activates caspase-8 directly, or indirectly results in disruption of lysosomes and release of cathepsins (B, D and L). The released active cathepsin B and D cleave Bid which translocates from the cytosol to the mitochondria, releases Bax and promotes cytochrome c release. This cascade activates downstream executioner caspases resulting in cell death
Not all target proteins that are cleaved by lysosomal cathepsins have been identified unambiguously since they may vary among the different cathepsins and in different cellular compartments. Cathepsin B can function either as carboxypeptidase at pH 4.5–5.5, like in lysosomes, or as endopeptidase with a pH optimum around 7.4, as present in the cytosol [48]. Since these two catalytic mechanisms have different pH optima, specificity and cleavage mechanism may change in the cytosolic environment [46]. There is evidence that cytosolic Bid is the target for cathepsin B cleavage at the unique Arg65 cleavage site [4, 18]. A similar switch was observed for lysosomal cathepsin H, which usually functions as amino-peptidase but can cleave Bid at Arg71 when cytosolic [18]. Cathepsin D mainly functions as endopeptidase in lysosomes, and there is no clear evidence for Bid cleavage by cathepsin D in lysosomal extracts [46]. However, the proapoptotic Bcl-2 homolog Bax can be the cytosolic target of cathepsin D cleavage, resulting in release of the mitochondrial apoptosis inducing factor (AIF) and subsequent apoptosis [17, 47]. Thus, although differing in their cytosolic target substrates, various cathepsins can contribute to apoptosis initiation by activating different downstream factors in the apoptosis cascade.
Our study provides evidence for the participation of a lysosomal pathway during induced cell death. Bcl-2 family members have been known to control permeabilization of the mitochondrial membrane during apoptosis, but involvement of these proteins in lysosomal membrane permeabilization (LMP) was not considered until recently. TRAIL and/or hypoxia initiated apoptosis through lysosomal disruption [18], and Bid was cleaved and translocated to the mitochondria following lysosomal disruption by lysosomotropic agents. A novel sequence of events was proposed in which cathepsin D triggers Bax activation, Bax induces the selective release of mitochondrial AIF, resulting in an early apoptotic phenotype [17]. It was also suggested that Bax is a mediator of LMP, possibly promoting the release of lysosomal enzymes to the cytosol during apoptosis [26]. After purifying lysosomes from tumor cells and treatment with the lysosomotropic agent LeuLeuOMe, probing for membrane protein expression of Lamp-1 confirmed lysosomal membranes disruption (Nagaraj et al., unpublished data). Lysosomes hold promise as drug targets and mediators of apoptosis signaling which may be less affected by intrinsic or chemotherapy-induced resistance mechanisms [44]. Emerging experimental evidence suggests that alterations in lysosomes may form an “Achilles heel” for cancer cells by sensitizing them to death pathways involving lysosomal membrane permeabilization and release of cathepsins into the cytosol [45].
The data presented clearly showed that hypoxia diminishes TRAIL-induced apoptosis in OSCC tumor cells. Therefore, our findings make TRAIL a less valuable agent for cancer treatment, since inhibition of cathepsin translocation by hypoxia in TRAIL-induced apoptosis may be a mechanism by which tumor cells survive against tumor therapies. Thus, our data suggest that enhancement of cathepsin translocation from the lysosome to the cytosol may be important to increase the efficacy of tumor therapies in hypoxic tissues. Anticancer drugs targeted to survival signaling factors may not be effective if the proapoptotic proteins of the Bcl-2 family are down-regulated to an extent that they cannot couple the apoptotic stimuli to the release of mitochondrial factors that activate the caspase cascade. Lysosome destabilization can be used to trigger tumor cell death even in cells that are resistant to conventional DNA-damaging agents.
Conclusions
We have demonstrated for the first time that hypoxia in OSCC cells inhibits TRAIL-induced apoptosis mediated through lysosomal cathepsins. Despite recently emerging evidence for the significance of cathepsins in apoptosis, the biochemical and signaling mechanisms for cathepsin translocation and activity changes are not fully understood. This study proposes a novel lysosome-mediated pathway for TRAIL and hypoxia-induced apoptosis as an important regulatory event in tumor cells. Our data also are in support of a novel proapoptotic, and thus tumor-preventing function of cathepsins B and D in tumor cells, which opposes the known tumor-promoting role of these proteases. Thus, strategies to target lysosomes appear very promising as successful oral cancer therapy approaches.
Acknowledgments
Supported by NIH grants DE13150 (W.Z.), RO3 DE15723 (N.V), and by Philip Morris USA Inc. and Philip Morris International (W.Z.). Cell lines 1386Tu and 1386Ln were gifts from P. Sacks, New York University. We thank S. Wellhausen and V.R. Jala for help with the FACS and confocal microscope analyses.
Footnotes
No conflict of interest is declared for all listed authors concerning the work presented here.
Contributor Information
Nagathihalli S. Nagaraj, Department of Medicine, James Graham Brown Cancer Center, University of Louisville, Louisville, Kentucky 40202, USA
Nadarajah Vigneswaran, Department of Medicine, James Graham Brown Cancer Center, University of Louisville, Louisville, Kentucky 40202, USA.
Wolfgang Zacharias, Department of Medicine, James Graham Brown Cancer Center, University of Louisville, Louisville, Kentucky 40202, USA; Department of Pharmacology and Toxicology, University of Louisville, Louisville, Kentucky 40202, USA.
References
- 1.Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 2.Fulda S, Debatin KM. Apoptosis signaling in tumor therapy. Ann N Y Acad Sci. 2004;1028:150–156. doi: 10.1196/annals.1322.016. [DOI] [PubMed] [Google Scholar]
- 3.Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98:2603–2614. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- 4.Stoka V, Turk B, Schendel SL, et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem. 2001;276:3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 5.Borner C. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol. 2003;39:615–647. doi: 10.1016/s0161-5890(02)00252-3. [DOI] [PubMed] [Google Scholar]
- 6.Van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002;9:1031–1042. doi: 10.1038/sj.cdd.4401088. [DOI] [PubMed] [Google Scholar]
- 7.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol. 2002;168:1356–1361. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- 8.LeBlanc HN, Ashkenazi A. Apo-2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 9.Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol. 2001;2:589–598. doi: 10.1038/35085008. [DOI] [PubMed] [Google Scholar]
- 10.Chwieralski CE, Welte T, Buhling F. Cathepsin-regulated apoptosis. Apoptosis. 2006;11:143–149. doi: 10.1007/s10495-006-3486-y. [DOI] [PubMed] [Google Scholar]
- 11.Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 12.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11:3155–3162. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 13.Foghsgaard L, Wissing D, Mauch D, et al. Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J Cell Biol. 2001;153:999–1010. doi: 10.1083/jcb.153.5.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liaudet-Coopman E, Beaujouin M, Derocq D, et al. Cathepsin D: newly discovered functions of a long-standing aspartic protease in cancer and apoptosis. Cancer Lett. 2006;237:167–179. doi: 10.1016/j.canlet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Beaujouin M, Baghdiguian S, Glondu-Lassis M, Berchem G, Liaudet-Coopman E. Overexpression of both catalytically active and -inactive cathepsin D by cancer cells enhances apoptosis-dependent chemo-sensitivity. Oncogene. 2006;25:1967–1973. doi: 10.1038/sj.onc.1209221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagaraj NS, Vigneswaran N, Zacharias W. Cathepsin B mediates TRAIL-induced apoptosis in oral cancer cells. J Cancer Res Clin Oncol. 2006;132:171–183. doi: 10.1007/s00432-005-0053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bidere N, Lorenzo HK, Carmona S, et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 18.Cirman T, Oresic K, Droga-Mazovec G, et al. Selective disruption of lysosomes in HeLa cells triggers apoptosis mediated by cleavage of Bid by multiple papain-like lysosomal cathepsins. J Biol Chem. 2004;279:3578–3587. doi: 10.1074/jbc.M308347200. [DOI] [PubMed] [Google Scholar]
- 19.Guicciardi ME, Deussing J, Miyoshi H, et al. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest. 2000;106:1127–1137. doi: 10.1172/JCI9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sacks PG. Cell, tissue and organ culture as in vitro models to study the biology of squamous cell carcinomas of the head and neck. Cancer Metast Rev. 1996;15:27–51. doi: 10.1007/BF00049486. [DOI] [PubMed] [Google Scholar]
- 21.Wickramasinghe NS, Banerjee K, Nagaraj NS, Vigneswaran N, Zacharias W. Hypoxia alters cathepsin B/inhibitor profiles in oral carcinoma cell lines. Anticancer Res. 2005;25:2841–2849. [PubMed] [Google Scholar]
- 22.Wickramasinghe NS, Nagaraj NS, Vigneswaran N, Zacharias W. Cathepsin B promotes both motility and invasiveness of oral carcinoma cells. Arch Biochem Biophys. 2005;436:187–195. doi: 10.1016/j.abb.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 23.Turk V, Lah T, Kreger I, Cathepsin D, Cathepsin E. In: Methods of enzymatic analysis. Bergmayer HU, editor. Vol. 5. Verlag Chemie; Weinheim: 1984. pp. 211–222. [Google Scholar]
- 24.Nagaraj NS, Vigneswaran N, Zacharias W. Hypoxiamediated apoptosis in oral carcinoma cells occurs via two independent pathways. Mol Cancer. 2004;3:1–14. doi: 10.1186/1476-4598-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antunes F, Cadenas E, Brunk UT. Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem J. 2001;356:549–555. doi: 10.1042/0264-6021:3560549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kagedal K, Johansson AC, Johansson U, et al. Lysosomal membrane permeabilization during apoptosis–involvement of Bax? Int J Exp Pathol. 2005;86:309–321. doi: 10.1111/j.0959-9673.2005.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vigneswaran N, Wu J, Nagaraj N, Adler-Storthz K, Zacharias W. Differential susceptibility of metastatic and primary oral cancer cells to TRAIL-induced apoptosis. Int J Oncol. 2005;26:103–112. [PubMed] [Google Scholar]
- 28.Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, et al. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: when reaction mechanism is more important than specificity. Cell Death Differ. 2003;10:881–888. doi: 10.1038/sj.cdd.4401247. [DOI] [PubMed] [Google Scholar]
- 29.Nagane M, Huang HJ, Cavenee WK. The potential of TRAIL for cancer chemotherapy. Apoptosis. 2001;6:191–197. doi: 10.1023/a:1011336726649. [DOI] [PubMed] [Google Scholar]
- 30.Brunk UT, Dalen H, Roberg K, Hellquist HB. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic Biol Med. 1997;23:616–626. doi: 10.1016/s0891-5849(97)00007-5. [DOI] [PubMed] [Google Scholar]
- 31.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 32.Wouters BG, Van Den Beucken T, Magagnin MG, Lambin P, Koumenis C. Targeting hypoxia tolerance in cancer. Drug Resist Updat. 2004;7:25–40. doi: 10.1016/j.drup.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Weinmann M, Marini P, Jendrossek V, et al. Influence of hypoxia on TRAIL-induced apoptosis in tumor cells. Int J Radiat Oncol Biol Phys. 2004;58:386–396. doi: 10.1016/j.ijrobp.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 34.Park SY, Billiar TR, Seol DW. Hypoxia inhibition of apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) Biochem Biophys Res Commun. 2002;291:150–153. doi: 10.1006/bbrc.2002.6421. [DOI] [PubMed] [Google Scholar]
- 35.Kim M, Park SY, Pai HS, Kim TH, Billiar TR, Seol DW. Hypoxia inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by blocking Bax translocation. Cancer Res. 2004;64:4078–4081. doi: 10.1158/0008-5472.CAN-04-0284. [DOI] [PubMed] [Google Scholar]
- 36.Mayes PA, Campbell L, Ricci MS, Plastaras JP, Dicker DT, El-Deiry WS. Modulation of TRAIL-induced tumor cell apoptosis in a hypoxic environment. Cancer Biol Ther. 2005;4:1068–1074. doi: 10.4161/cbt.4.10.2255. [DOI] [PubMed] [Google Scholar]
- 37.Castino R, Demoz M, Isidoro C. Destination ‘lysosome’: a target organelle for tumour cell killing? J Mol Recognit. 2003;16:337–348. doi: 10.1002/jmr.643. [DOI] [PubMed] [Google Scholar]
- 38.Tenev T, Zachariou A, Wilson R, Ditzel M, Meier P. IAPs are functionally non-equivalent and regulate effector caspases through distinct mechanisms. Nat Cell Biol. 2005;7:70–77. doi: 10.1038/ncb1204. [DOI] [PubMed] [Google Scholar]
- 39.Kruidering M, Evan GI. Caspase-8 in apoptosis: the beginning of “the end”? IUBMB Life. 2000;50:85–90. doi: 10.1080/713803693. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 41.Roucou X, Montessuit S, Antonsson B, Martinou JC. Bax oligomerization in mitochondrial membranes requires tBid (caspase-8-cleaved Bid) and a mitochondrial protein. Biochem J. 2002;368:915–921. doi: 10.1042/BJ20020972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boya P, Gonzalez-Polo RA, Poncet D, et al. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene. 2003;22:3927–3936. doi: 10.1038/sj.onc.1206622. [DOI] [PubMed] [Google Scholar]
- 43.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–191. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 44.Linder S, Shoshan MC. Lysosomes and endoplasmic reticulum: targets for improved, selective anticancer therapy. Drug Resist Updat. 2005;8:199–204. doi: 10.1016/j.drup.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 45.Fehrenbacher N, Jaattela M. Lysosomes as targets for cancer therapy. Cancer Res. 2005;65:2993–2995. doi: 10.1158/0008-5472.CAN-05-0476. [DOI] [PubMed] [Google Scholar]
- 46.Tardy C, Codogno P, Autefage H, Levade T, Andrieu-Abadie N. Lysosomes and lysosomal proteins in cancer cell death (new players of an old struggle) Biochim Biophys Acta. 2006;1765:101–125. doi: 10.1016/j.bbcan.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Johansson AC, Steen H, Ollinger K, Roberg K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003;10:1253–1259. doi: 10.1038/sj.cdd.4401290. [DOI] [PubMed] [Google Scholar]
- 48.Linebaugh BE, Sameni M, Day NA, Sloane BF, Keppler D. Exocytosis of active cathepsin B enzyme activity at pH 7.0, inhibition and molecular mass. Eur J Biochem. 1999;264:100–109. doi: 10.1046/j.1432-1327.1999.00582.x. [DOI] [PubMed] [Google Scholar]