

Abstract
The human microbiome has been linked to various host phenotypes, and has been implicated in many complex human diseases. Recent genome-wide association studies (GWAS) have used microbiome variation as a complex trait, and have uncovered human genetic variants that are associated with the microbiome. Here, we summarize results from these studies, and illustrate potential regulatory mechanisms by which host genetic variation can interact with microbiome composition. We argue that, similar to human GWAS, it is important to use functional genomics techniques to gain a mechanistic understanding of causal host-microbiome interactions and its role in human disease. We highlight experimental, functional, and computational genomics methodologies to study the genomic basis of host-microbiome interaction, and describe how these approaches can be utilized to explain how human genetic variation can modulate the effect of the microbiome on the host.
Keywords: microbiome, human genomics, functional genomics, host-microbiome interactions
Microbiome composition associated with human genomics
The microbial communities that live in and on the human body, termed “the human microbiome”, are composed of thousands of species and trillions of microorganismal cells [1]. These communities vary widely across body sites, and their composition is affected by many factors, including host diet, age, sex, weight, population, disease status, medication use, and interaction with other individuals and the environment, as well as host genetics [2–12]. Variation in the microbiome has also been associated with many human diseases and health conditions, such as inflammatory bowel disease (Crohn’s disease and ulcerative colitis), type 2 diabetes, colorectal cancer, in addition to many others [13–17]. Interestingly, many of the diseases that are linked to the microbiome are also controlled by host genetic factors, as has been characterized by more than a decade of genome wide association studies (GWAS). Since both host genetics and the microbiome can affect host traits, understanding the interaction between these two factors is the first step in uncovering their respective roles in disease.
To address this, recent studies have attempted to characterize host genetic determinants of the microbiome. Initial studies to explore this interaction have identified links between the microbiota and host genetic variation in candidate human genes, such as FUT2 (fucosyltransferase 2) and MEFV (mediterranean fever) [18,19]. More recently, researchers began considering the microbiome as a complex human trait [20]. As such, quantitative and statistical genetics approaches can be used to characterize the genetic architecture underlying variation in the microbiome. However, the microbiota is different from traditional quantitative traits, and can be considered a high-dimensional array of complex traits. A microbiome profile is composed of multiple “features”, usually relative abundances of different microbial taxa, pathways, or other functional characteristics of the microbial community. The abundances are usually intercorrelated, and may also have a phylogenetic relationship. Each of these microbial features may be associated with different host genomic loci and through that represent genetic architectures. Moreover, each microbiome feature may be affected by different environmental factors, such as diet [21], and these environmental effects may be stronger than host genetic effects [22]. In addition, microbiome composition may be affected by ecological factors, such as colonization history.
Although it is difficult to control for many of these potential confounders, researchers have recently successfully used genome-wide analysis of host genetic variation to identify loci in the mouse genome that are associated with abundances of microbial taxa in the mouse gut [21,23–25]. Studies in humans have followed, first focusing on examining the heritability of microbiome composition [26]. Using gut microbiome data from hundreds of monozygotic and dizygotic twin pairs, taxa that are significantly heritable were identified. Further, germ-free mice were used to show a potential role for one highly heritable taxon, Christensenellaceae, in host obesity [27]. Moreover, a recent study identified associations between disease-specific risk alleles and gut microbiome composition in inflammatory bowel disease (IBD), and found conserved associations between human genotypes and the microbiome in 49 genetic loci, including the JAK-STAT signaling pathway and host innate immune response [28]. In addition, host genetic variation was identified in 93 individuals using ‘host contamination’ reads in the shotgun metagenomics data generated by the Human Microbiome Project (HMP) [1,29], human SNPs associated with variation in the microbiome were identified [30]. These SNPs are highly enriched in immunity genes and pathways, as well as SNPs that have been associated with microbiome-related complex disease [30]. This study also highlighted an association between human genetic variation in the region of the LCT gene, and the abundance of bifidobacterium in the gut microbiome. LCT encodes the lactase enzyme, which metabolizes lactose, while bifidobacterium uses lactose as a primary carbon source. Studies in the Hutterite population identified loci associated with the abundance of eight bacterial taxa in the gut microbiome during two seasons, summer and winter [31], as well as 37 loci with evidence of association with the abundance of taxa in the airways [32].
More recently, several genome-wide association studies (GWAS) with much larger sample sizes have identified additional loci in the human genome that are associated with microbiome complex traits [33–35]. Although there was little overlap in the loci identified in the three studies, this is not unexpected given the previous difficulty in recovering QTLs in mouse studies [21]. In addition to the unique combinations of environmental factors that affect the different human populations studied, the studies used different sequencing techniques (16S rRNA gene sequencing vs. metagenomics shotgun sequencing), different analysis methods (using diversity vs. abundances of individual bacterial taxa as the complex traits) and populations with somewhat different genetic backgrounds [33–35]. Considering results from all these microbiome GWAS studies, it is evident that (1) the genetic architecture is complex and includes many genes; (2) the effect sizes are small, perhaps as could be expected; (3) sequencing and analysis techniques can have a large influence on the results of these studies; and (4) we do not yet understand how to account for the ecological factors that also contribute to microbiome composition. Clearly, we are in the very early stages of this new field, but it is already apparent that multiple approaches and different types of genetic tools will be necessary to truly understand the genetics of this very complex host-microbiome interaction.
Direction of causality
Despite the knowledge gained from the newly found associations between microbiome composition and host genetics, a remaining open question is the direction of causality. There are several scenarios that can explain these association in the context of human disease (Figure 1A, Key Figure). First, it is entirely possible that for many host phenotypes where there is a correlation between host genetics and the microbiome, it is only host genetics, and not the microbiome, that directly affects the phenotype or disease (Figure 1B). In this scenario, changes in the microbiome can be driven by disease-related physiological and environmental changes, such as inflammation or medication. For example, it has been shown that antidiabetic medication confounds gut microbiome study results [36]. Another potential mechanism that can explain the observed pattern is illustrated in Figure 1C. While it is clear that host genetic variation can control disease phenotypes, only recently it has been shown that this effect can be achieved through changes in the microbiome. An example for this effect is the gene NOD2, in which genetic variants have been strongly associated with inflammatory bowel disease (IBD). Mice lacking NOD2 are prone to colitis, and this effect can be transmitted to wild-type hosts via the microbiota [37].
Figure 1.
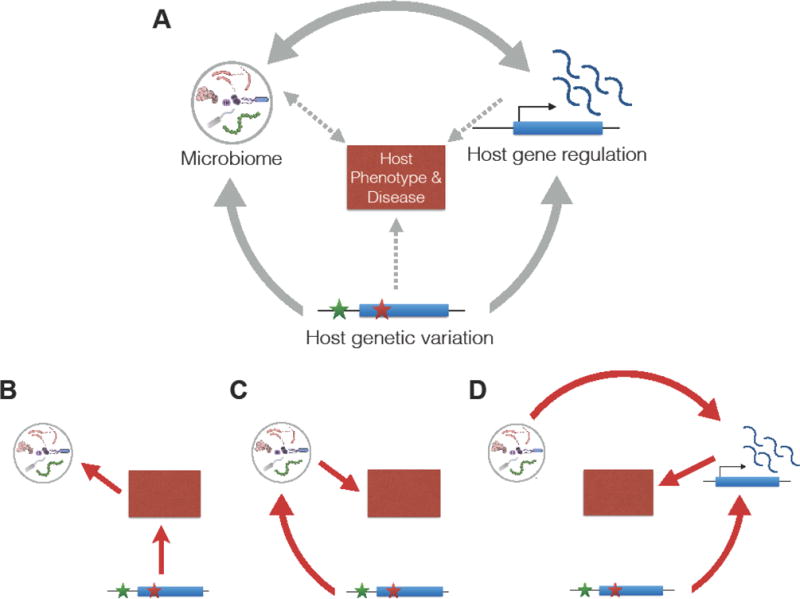
(A) A diagram of possible causal interaction between the microbiome, host genetic variation, and host gene expression to impact host phenotype. (B) Host genetics controls phenotype, which causes an alteration in the microbiome. (C) Host genetics controls the microbiome, which in turn affects host phenotypes. (D) Host genetic variation and the microbiome interact to control host gene regulation, which in turn affects host phenotype.
A third scenario involves an interaction between host genetic variation and the microbiome to affect host gene regulation (Figure 1D). Unlike most other complex traits, microbiome “traits” can also influence host physiology, an effect that can occur at multiple levels, including microbiome-mediated effects on host gene regulation. Recent studies in germ-free and humanized germ-free mice have demonstrated that gene expression in the colon can be modified by microbial exposure in a site-specific manner [38,39]. It has been shown that genome-wide changes in gene expression occur in the colonic epithelium of germ-free mice after colonization with fecal microbiome, including down-regulation of genes involved in transport and metabolism of lipids and other nutrients [39]. The authors found that the host response to microbial colonization varies depending on intestinal region and time post-colonization, and that these changes are mediated by transcription factor (TF) binding without significant changes in the accessible chromatin regions. In addition, a recent study found that microbiome colonization leads to activation or inactivation of hundreds of enhancers in the mouse colon [40]. Similarly, it was found that microbiota treatment of larval threespine sticklebacks leads to change in expression in genes associated with innate immunity [41]. Likewise, an analysis in flies showed that host transcriptional network is determined by the presence of microbiota [42]. Moreover, it was found that among human SNPs associated with microbiome composition, there is an enrichment in SNPs that were identified as expression QTLs (eQTLs; See Glossary) across multiple tissues in the GTEx project [30,43]. These results, along with the fact that microbial composition is tissue-specific [30,44,45] and likely influenced by gene expression in interacting host cells [39], highlights the need for studying host regulatory response to microbial communities. Recent functional genomics studies thus provide an opportunity for delineating the mechanisms by which the microbiome controls host gene regulation.
Host-microbiome interaction through effects on host gene regulation
As the microbiome is associated with both host genetic variation and host gene expression, it is important to consider how host genetic variation can influence gene regulation. In recent years, eQTL mapping studies have identified genetic variants associated with inter-individual differences in gene expression in colon in healthy and disease states [46–54]. Additionally, the GTEx project includes transverse and sigmoid colon samples from 169 and 124 individuals, respectively, which have been used to identify eQTLs [43]. A subset of these eQTLs could potentially also modulate the changes in gene regulation induced by host-microbiome interactions. These changes can be effectively identified through response eQTL studies, which aim to assess how genetic variation affects response to environmental variables. Response eQTL and allele specific expression studies (see Box 1) have identified hundreds of genes whose response to specific environmental perturbations (including pathogens, hormones and pharmacological drugs) is modulated by cis-regulatory polymorphisms. The studies conducted so far have considered only a limited subset of all possible environmental exposures that may be relevant for human health. For example, several human eQTL studies have identified genetic variants that modulate the response to pathogens in immune cells, such as macrophages and dendritic cells [55–58]. These variants may provide a mechanism for immune-related diseases, as they are enriched in risk loci for these traits. The major underlying mechanism for these functional non-coding variants is probably disruption of binding sites for transcription factors activated in response to immunological stimulants. For example, response eQTLs in macrophages treated with live bacterial pathogens are strongly enriched in binding sites for AP1, NFkappaB and IRFs [57]. The success of these studies suggests that similar approaches could potentially shed light on the host regulatory genetics underlying interaction between host genetic variation and commensal microbiomes.
Box 1. Response eQTL mapping identifies genetic variants that modulate the transcriptional response to environmental perturbations.
Recent studies have shown that genetic variants in regulatory regions not only are associated with gene expression levels across individuals, but also modulate the gene expression response to environmental perturbations. Loci harbouring these variants are defined response or interaction eQTLs. Response eQTLs have been successfully identified in cells treated with microbial pathogens, or with different chemicals, including hormones and drugs [55–58,92–95]. Response eQTLs have also been described as cases of genotype-by-environment interactions for molecular phenotypes. Specifically, they represent cases of variants where the effect of the genotype on the gene expression phenotype varies depending on the cellular context. An example of a response eQTL for pathogen infection would be a locus associated with the expression of a gene only in infected cells. In recent years, the increasing accessibility to and sophistication of next-generation sequencing (NGS) techniques has allowed for the development of allele-specific analyses to identify regulatory variants [96–98]. Allele-specific approaches compare allelic effects within individuals, thereby controlling for same genetic background and cellular environment. Analysis of ASE across different environments or environmental proxies is emerging as a powerful approach to identify genes with response eQTLs in a large number of environmental contexts [99,100].
Considering these studies, it is compelling to consider the microbiome as an environmental exposure for the host. In this case, the microbiome can elicit a host regulatory response in a manner similar to response eQTL studies, which is illustrated in Figure 2. For many host genes, the exposure to the microbiome will not affect gene regulation (Figure 2A). Conversely, some genes will change their expression in response to interaction with the microbiome, in a manner that does not depend on regulatory genetic variation (Figure 2B). However, for a subset of genes, the response can be modulated by genetic variants that are located in the regulatory regions controlling these genes (Figure 2C), or in other words, a microbiome response eQTL. Similar to response eQTL of other environmental stimuli, identifying microbiome response eQTLs would require a large number of samples. However, as described above, the microbiome is a complex community that is highly variable in its taxonomic and genic composition across individuals and health conditions [1]. It is likely that taxonomic and functional variation in the microbiome will affect the function of an eQTL in host cells. For example, the effect size of a given response eQTL can depend on the specific configuration of the microbiota, as illustrated in Figure 3. In this simplified example, the abundance of a given microbe within the microbiome determines the effect, whereby at low abundance there is no change in gene expression and thus no response eQTL. However, increasing abundance of this microbe increases gene expression, but in a manner that is dependent on the host genotype at the eQTL locus. This type of genotype X microbiome effect adds a layer of complexity to functional genomics studies of host-microbiome interactions, because in addition to variation in host genetics, variation in the microbiome also has to be considered and incorporated in the study design. This highlights the need for high-throughput experimental models that can incorporate these two sources of variation.
Figure 2.
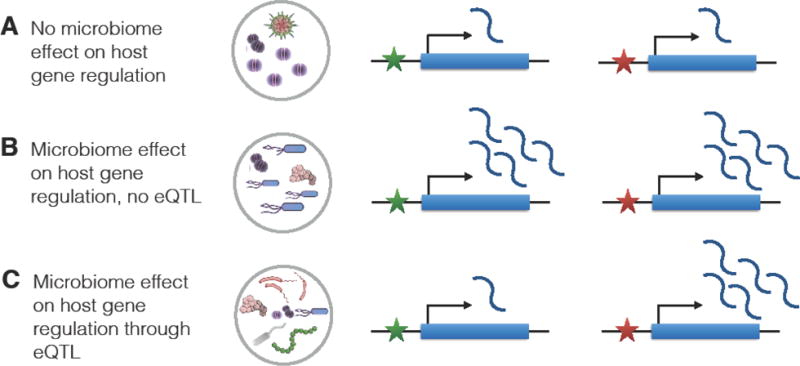
An illustration of microbiome impact on host gene expression for a hypothetical gene, and the effect of an eQTL, which is designated with a star with two variants (red and green). (A) microbiome does not alter gene expression; (B) Microbiome regulates the gene and causes an increase in gene expression in a consistent manner, regardless of genetic variation; (C) Microbiome regulates the gene and causes an increase in gene expression only if the eQTL has the red variant.
Figure 3.
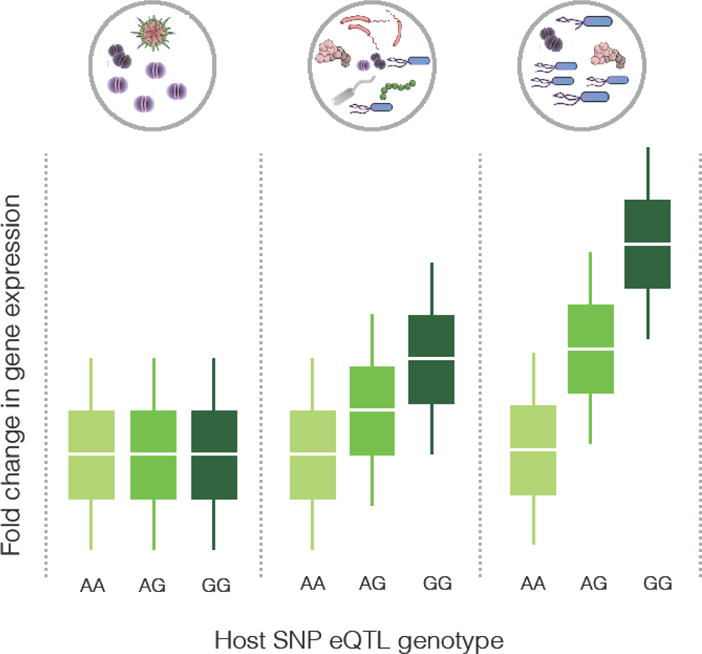
An illustration of interaction between microbiome and host eQTL in regulating a hypothetical gene. The gene’s expression level is shown on the y-axis, and the genotype at the eQTL is on the x-axis. The three panels correspond to three microbiome compositions, with the lowest abundance of the causal microbe on the left, and the highest on the right.
Experimental models for functional genomics of host-microbiome interactions
Animal models offer distinct advantages as experimental systems for the study of host-microbiome interactions. Studies in inbred and outbred mouse strains showed distinct host genotype effects on microbial composition underscoring the importance of host genetic factors on shaping the intestinal microbiome [23,25,59]. However, it is challenging to use animal models to study specific variants identified in human studies of host-microbiome interactions, and animal studies further limited by expense and labor intensity. In humans, few studies have analyzed gene expression changes in colonic biopsies in association with exposure to probiotics [60,61]. While these studies have been able to identify specific host pathways that are modified by the microbial/probiotic treatment, they are limited by the type of treatment that can be administered to healthy subjects, and by the invasiveness of the medical procedure necessary to obtain colonic biopsies.
In vitro model systems allow for controlled experimental conditions, can be derived from specific cell types (e.g., intestinal epithelial cells), and can be co-cultured with other cell types, such as immune cells. Colonic epithelial cell monolayers have been employed to study the responses to various treatments, especially in the context of colorectal cancer [62–66]. Newer models of the gastrointestinal tract, such as Human-Microbiota Interaction (HMI) module [67] and the human gut-on-a-chip [68] have been developed to recapitulate the dynamic nature of the gastrointestinal tract in order to study host responses under more physiological conditions. However, genomic studies of responses to the microbiome have not been done using these models. The application of in vitro models for interrogation of the genomic architecture of host-microbiome interactions is limited by the fact that cell lines are transformed and malignant (e.g., Caco2) and thus might not reflect responses in primary cells.
In vitro primary epithelial cell lines overcome some of these limitations, as was recently demonstrated [69]. Potentially, they are better suited for large-scale studies of host-microbiome interactions, across diverse microbial extracts representative of host physiological and pathological states. This model was tested by developing a novel co-culturing approach that exposes primary colonic epithelial cells to healthy microbiome extracts prepared for microbiome transplants. Using this approach it was demonstrated that the host response to the microbiome in the colon is largely mediated through genes involved in cell junctions, cell adhesion, and immune response. In addition, ASE (Allele Specific Expression) analysis identified 12 genes with genetic variants that modulate the cellular response to the microbiome and may represent mediators of the pathogenic effect of the microbiome on human traits.
Ex vivo systems such as primary tissue culture or intestinal organoids [70] are promising models that could be used to study inter-individual variation in host-microbiome responses, because they have not been transformed and recapitulate in vivo cellular architecture. Primary tissue culture using biopsies obtained from healthy and diseased human colon has been used to assess the effects of probiotics on inflammation elicited by Salmonella infection [71]. Primary colonic tissue culture has been used to study inter-individual and -ethnic differences in transcriptional response to vitamin D [72], and, this ex vivo model has potential for studying the genomic landscape of response phenotypes including host-microbiome interactions. Limitations of primary tissue culture include short-term viability, cell type heterogeneity, and relative invasiveness of the procedure to obtain tissue samples [73]. Maintenance of an aerobic-anaerobic interface using primary gut tissue needs to be evaluated for studying host-microbiome interactions.
Organoids are newly described ex vivo cultures established from tissue-derived [74] or pluripotent [75] stem cells. This technology was enabled by improved understanding of the stem cell niche and has many advantages over animal and in vitro models. Organoids can be derived from many tissue types including the gastrointestinal epithelium (e.g., duodenum, stomach, small and large intestine) as well as other organs (e.g., prostate, liver) from mice and humans. Organoids can be maintained long-term and recapitulate important cellular features such as diversity, function and spatial organization [76]. Furthermore, co-culture of organoids with immune cells [77] and mesenchymal cells [78] allows for study of responses in an environment similar to that in vivo. Human intestinal organoids derived from pluripotent stem cells demonstrated a transcriptional signature most similar to fetal intestine that become more “adult-like” upon in vivo transplantation [79], although whether epigenetic marks are also maintained in organoids has yet to be established. Three-dimensional organoids are grown on an extracellular matrix with the luminal surface on the inside of the spheroid structure. As such, studies of commensal and pathogenic microorganisms have required microinjection into the center of organoids to mimic physiological conditions [80–82]. There has been one study of transcriptional response of murine ileal organoids to short chain fatty-acids and microbial metabolites [83]; however, larger scale studies elucidating the genomic landscape of host-microbiome interactions have not yet been done using organoids. While there are many advantages to organoid culture, disadvantages include limited ability to maintain cultures under anaerobic conditions long-term as well as cost and a learning curve for establishment of organoid cultures.
Computational analysis of multi-omic host and microbiome data
Joint analysis of host genetics and microbiome data can be used to identify novel biological interactions contributing to host phenotype, microbiome phenotype, or both. These high-dimensional, multi-omic studies are powerful instruments for discovery and hypothesis generation, and can enable discovery of host-microbiome interactions that would be difficult to discover in model systems. These studies can be divided into two general categories of analysis: studies in which certain aspects of host genetics or other features are controlled via experimental design (design-based host-microbiome studies), and studies in which host factors are actually quantitated within experimental blocks for direct association with the microbiota (multi-omic host-microbiome studies). Examples of design-based host-microbiome studies are the use of genetic knockout or knockdown animals [22], or the study of heritability of microbiomes in families or twins [11,27]. These approaches are by far the most common type of host-microbiome study, and do not actually require collection of host genomic data directly. However, it is challenging to generate novel mechanistic hypotheses about host-microbiome interactions through these studies, as the type of host variation under study includes a single gene, thus being limited to well-studied genes. In addition, heritability studies only consider relatedness and do not include information on specific host genes. In contrast, the multi-omic host-microbiome studies represent both a much greater statistical challenge and a higher potential for discovery of novel interactions. Examples are studies that attempt to associate the microbiome directly with host genetic variation (SNP data from sequencing or genotyping arrays), gene expression (RNA-seq or expression microarrays), epigenetics (methylation data), or other types of variation such as immune system activation.
Multi-omic host-microbiome studies can in theory allow direct interrogation of statistical host-microbiome associations by combining high-dimensional host data with high-dimensional microbiome data. These types of analysis are playing an increasingly important role in microbiome studies as the microbiota are influenced heavily by host immunity and metabolism, diet, environmental exposures, host demographics, and clinical history including a wide range of medications with different effects on the microbiota [2]. For example, host genotype has now been linked directly to microbiome variation in several instances, including with replication across cohorts in healthy individuals [30,84] and patients with inflammatory bowel disease [44]. Host gene expression has also been correlated to variation in microbial taxa on skin and in the ileal pouch of patients with ileal pouch-anal anastomosis [85]. However, since environmental, dietary, and ecological factors also describe a large fraction of interindividual variation in gut microbiota [22], and may also be correlated with host genetics, they may confound study results and must be controlled for statistically or through study design [44]. Host genetics can also be used to predict tissue-specific gene expression [86], which may allow interrogation of host-microbiome expression networks in existing data integrating both host genotype with microbiome. As both microbial and host gene expression vary widely across cell types and environmental conditions, deeper exploration into dual-expression datasets will likely reveal more close correlates of host-microbiome interaction.
The importance of host genetics in microbiome-based translational medicine
Although there has been a recent sharp rise in interest in using the microbiome as a potential medical therapeutic, microbiome-based treatments have existed for many years. These treatments mostly included dietary recommendations and supplements of milk-souring bacteria in the form of fermented milk products, tablets, powders, and food additives [87]. These have later evolved into the use of “probiotics”, commonly defined as live microorganisms that benefit health [88]. In addition, some physicians, such as Dr. Benjamin Eiseman, recognized the microbiome as a potential treatment for complications of antibiotics at the beginnings of their widespread usage. As early as 1950s, his team successfully treated a series of patients with pseudomembranous colitis using stool from healthy donors [89]. This procedure has seen a dramatic rise in usage in the recent years as an effective treatment of antibiotic-refractory Clostridium difficile infections. Fecal microbiota preparations have evolved into increasingly standardized products that are becoming acceptable by mainstream medicine [90]. However, C. difficile is but one of a number of multidrug resistant pathogens that constitute a growing threat to modern healthcare. Fecal microbiota transplantation (FMT) is increasingly being considered as an approach to restore colonization resistance in subsets of patients with heavy antibiotic exposure and weakened immune system [91]. Moreover, FMT is currently being examined for use as a therapy for several other microbiome-related conditions, such as inflammatory bowel disease. In this context, it is important to consider the possible interaction of host genetics with this treatment. Indeed, it is likely that a patient’s response to an FMT treatment can be modulated by specific host factors that interact with the microbial community being introduced, including the host genome. We anticipate that future algorithms in precision medicine will be able to incorporate a patient’s genome into selection of optimal microbial treatment regimen to achieve the best clinical outcomes.
Concluding Remarks and Future Perspectives
Human genomic variation has an important role in affecting host-microbiome interaction. Although one of the major mechanisms by which the microbiome can affect the host is by altering host gene regulation, we know little about microbiome-driven alterations of host gene expression, and how human genetic variation can modulate these regulatory interactions. We argue that functional genomics techniques, which have been extensively used in human genomics, can be useful in characterizing these regulatory effects of the microbiome. We outline experimental and computational approaches that can be used, and highlight potential regulatory mechanisms that can be discovered.
Trends Box.
Human genetic variation is associated with variation in microbiome composition across populations and body sites. These microbiome-linked variants are enriched in disease-related genes.
Identification of eQTLs for microbiome traits may provide mechanistic insights into how microbiome can interact with host genetic variation
Novel functional genomics experimental approaches can identify microbiome-controlled eQTLs and describe the combined role of human genetic variation and microbiome composition in controlling complex disease.
Outstanding Questions Box.
Which human variants, genes, and pathways are associated with variation in microbiome composition across various human populations and body sites? How are these associations affected by non-genetic factors such as diet, medication use, social interaction, environmental contacts, and changes in the microbiome throughout life?
What feature(s) of the microbiome are the most significant “traits” that are affected by host genetic variation? How do ecological factors of the microbiome confound our ability to detect host gene X microbiome interactions?
What are the molecular mechanisms by which host genetic variation affects microbiome composition?
How does inter-individual variation in microbiome composition alter gene expression in host cells, and which host genes and pathways are affected?
How does host genetic variation modulate the effect of microbiome on host gene expression? Can these regulatory effects of the microbiome impact human disease susceptibility?
How can knowledge of the genetic basis of host-microbiome interactions be used to develop microbiome-based therapeutics?
Acknowledgments
R.B. is supported in part by funds from the University of Minnesota College of Biological Sciences, The Randy Shaver Cancer Research and Community Fund, Institutional Research Grant #124166-IRG-58-001-55-IRG53 from the American Cancer Society, and a Research Fellowship from The Alfred P. Sloan Foundation. F.L. is supported in part by funds from the National Institute of Health R01GM109215.
Glossary
- eQTL
Expression quantitative trait locus. A genetic locus associated with variation in the expression of a gene in the population
- Heritability
The fraction of phenotypic variation which can be attributed to a genetic cause
- response-eQTL
A genetic locus associated with the gene expression response to a specific environmental change, for example to a treatment, a disease condition or exposure
- microbiome response eQTL
An eQTL for host gene expression response to a specific microbiome exposure
- Transcription Factor (TF)
A protein that binds the DNA and regulate expression of a gene
- GTEx project
a large study of human gene expression variation across several tissues, aiming to understand the genetic basis of gene expression variation
- Allele-Specific Expression
A departure from the 50:50 ratio in the expression of the two alleles at a heterozygous site in the gene transcript. It is often an indirect way of identifying genes with eQTLs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Consortium, Human Microbiome Project. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhernakova A, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–569. doi: 10.1126/science.aad3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falony G, et al. Population-level analysis of gut microbiome variation. Science. 2016;352:560–564. doi: 10.1126/science.aad3503. [DOI] [PubMed] [Google Scholar]
- 4.Tung J, et al. Social networks predict gut microbiome composition in wild baboons. Elife. 2015;4 doi: 10.7554/eLife.05224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lax S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345:1048–1052. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez A, et al. Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell Rep. 2016;14:2142–2153. doi: 10.1016/j.celrep.2016.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Vangay P, et al. Antibiotics, pediatric dysbiosis, and disease. Cell Host Microbe. 2015;17:553–564. doi: 10.1016/j.chom.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morton ER, et al. Variation in Rural African Gut Microbiota Is Strongly Correlated with Colonization by Entamoeba and Subsistence. PLoS Genet. 2015;11:e1005658. doi: 10.1371/journal.pgen.1005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yassour M, et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med. 2016;8:343ra81. doi: 10.1126/scitranslmed.aad0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodrich JK, et al. Cross-species comparisons of host genetic associations with the microbiome. Science. 2016;352:532–535. doi: 10.1126/science.aad9379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thaiss CA, et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature. 2016 doi: 10.1038/nature20796. [DOI] [PubMed] [Google Scholar]
- 13.Kostic AD, et al. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146:1489–1499. doi: 10.1053/j.gastro.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burns MB, et al. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med. 2015;7:55. doi: 10.1186/s13073-015-0177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baxter NT, et al. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome. 2014;2:20. doi: 10.1186/2049-2618-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qin J, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 17.Gevers D, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tong M, et al. Reprograming of gut microbiome energy metabolism by the FUT2 Crohn’s disease risk polymorphism. ISME J. 2014;8:2193–2206. doi: 10.1038/ismej.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khachatryan ZA, et al. Predominant role of host genetics in controlling the composition of gut microbiota. PLoS One. 2008;3:e3064. doi: 10.1371/journal.pone.0003064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benson AK. The gut microbiome-an emerging complex trait. Nat Genet. 2016;48:1301–1302. doi: 10.1038/ng.3707. [DOI] [PubMed] [Google Scholar]
- 21.Leamy LJ, et al. Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice. Genome Biol. 2014;15:552. doi: 10.1186/s13059-014-0552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmody RN, et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. 2015;17:72–84. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benson AK, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKnite AM, et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS One. 2012;7:e39191. doi: 10.1371/journal.pone.0039191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Org E, et al. Genetic and environmental control of host-gut microbiota interactions. Genome Res. 2015 doi: 10.1101/gr.194118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Opstal EJ, Bordenstein SR. MICROBIOME. Rethinking heritability of the microbiome. Science. 2015;349:1172–1173. doi: 10.1126/science.aab3958. [DOI] [PubMed] [Google Scholar]
- 27.Goodrich JK, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knights D, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6:107. doi: 10.1186/s13073-014-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blekhman R, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015;16:191. doi: 10.1186/s13059-015-0759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davenport ER, et al. Genome-Wide Association Studies of the Human Gut Microbiota. PLoS One. 2015;10:e0140301. doi: 10.1371/journal.pone.0140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Igartua C, et al. Host genetic variation in mucosal immunity pathways influences the upper airway microbiome. Microbiome. 2017;5:16. doi: 10.1186/s40168-016-0227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turpin W, et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet. 2016;48:1413–1417. doi: 10.1038/ng.3693. [DOI] [PubMed] [Google Scholar]
- 34.Bonder MJ, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016 doi: 10.1038/ng.3663. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet. 2016;48:1396–1406. doi: 10.1038/ng.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forslund K, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–266. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Couturier-Maillard A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sommer F, et al. Site-specific programming of the host epithelial transcriptome by the gut microbiota. Genome Biol. 2015;16:62. doi: 10.1186/s13059-015-0614-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camp JG, et al. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res. 2014;24:1504–1516. doi: 10.1101/gr.165845.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davison JM, et al. Microbiota regulate intestinal epithelial gene expression by suppressing the transcription factor Hepatocyte nuclear factor 4 alpha. Genome Res. 2017 doi: 10.1101/gr.220111.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Small CM, et al. Host Genotype and Microbiota Contribute Asymmetrically to Transcriptional Variation in the Threespine Stickleback Gut. Genome Biol Evol. 2017;9:504–520. doi: 10.1093/gbe/evx014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobson AJ, et al. The Drosophila transcriptional network is structured by microbiota. BMC Genomics. 2016;17:975. doi: 10.1186/s12864-016-3307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knights D, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6:107. doi: 10.1186/s13073-014-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Connor A, et al. Responsiveness of cardiometabolic-related microbiota to diet is influenced by host genetics. Mamm Genome. 2014;25:583–599. doi: 10.1007/s00335-014-9540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo C, et al. Population-specific genome-wide mapping of expression quantitative trait loci in the colon of Chinese Han people. J Dig Dis. 2016 doi: 10.1111/1751-2980.12399. [DOI] [PubMed] [Google Scholar]
- 47.Zeng C, et al. Identification of Susceptibility Loci and Genes for Colorectal Cancer Risk. Gastroenterology. 2016;150:1633–1645. doi: 10.1053/j.gastro.2016.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peloquin J, et al. O-002 Genes in IBD-Associated Risk Loci Demonstrate Genotype-, Tissue-, and Inflammation-Specific Patterns of Expression in Terminal Ileum and Colon Mucosal Tissue. Inflamm Bowel Dis. 2016;22(Suppl 1):S1. [Google Scholar]
- 49.Hulur I, et al. Enrichment of inflammatory bowel disease and colorectal cancer risk variants in colon expression quantitative trait loci. BMC Genomics. 2015;16:138. doi: 10.1186/s12864-015-1292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Singh T, et al. Characterization of expression quantitative trait loci in the human colon. Inflamm Bowel Dis. 2015;21:251–256. doi: 10.1097/MIB.0000000000000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ongen H, et al. Putative cis-regulatory drivers in colorectal cancer. Nature. 2014;512:87–90. doi: 10.1038/nature13602. [DOI] [PubMed] [Google Scholar]
- 52.Li Q, et al. Expression QTL-based analyses reveal candidate causal genes and loci across five tumor types. Hum Mol Genet. 2014;23:5294–5302. doi: 10.1093/hmg/ddu228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Closa A, et al. Identification of candidate susceptibility genes for colorectal cancer through eQTL analysis. Carcinogenesis. 2014;35:2039–2046. doi: 10.1093/carcin/bgu092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loo LWM, et al. cis-Expression QTL analysis of established colorectal cancer risk variants in colon tumors and adjacent normal tissue. PLoS One. 2012;7:e30477. doi: 10.1371/journal.pone.0030477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barreiro LB, et al. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proceedings of the National Academy of Sciences. 2012;109:1204–1209. doi: 10.1073/pnas.1115761109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fairfax BP, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343:1246949. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nédélec Y, et al. Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell. 2016;167:657–669.e21. doi: 10.1016/j.cell.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 58.Çalışkan M, et al. Host genetic variation influences gene expression response to rhinovirus infection. PLoS Genet. 2015;11:e1005111. doi: 10.1371/journal.pgen.1005111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campbell JH, et al. Host genetic and environmental effects on mouse intestinal microbiota. ISME J. 2012;6:2033–2044. doi: 10.1038/ismej.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Baarlen P, et al. Human mucosal in vivo transcriptome responses to three lactobacilli indicate how probiotics may modulate human cellular pathways. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4562–4569. doi: 10.1073/pnas.1000079107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bron PA, et al. Emerging molecular insights into the interaction between probiotics and the host intestinal mucosa. Nat Rev Microbiol. 2011;10:66–78. doi: 10.1038/nrmicro2690. [DOI] [PubMed] [Google Scholar]
- 62.Farhana L, et al. Bile acid: a potential inducer of colon cancer stem cells. Stem Cell Res Ther. 2016;7:181. doi: 10.1186/s13287-016-0439-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai CC, et al. Increase in apoptosis by combination of metformin with silibinin in human colorectal cancer cells. World J Gastroenterol. 2015;21:4169–4177. doi: 10.3748/wjg.v21.i14.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farhana L, et al. Role of cancer stem cells in racial disparity in colorectal cancer. Cancer Med. 2016;5:1268–1278. doi: 10.1002/cam4.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maderer A, et al. Moguntinones–new selective inhibitors for the treatment of human colorectal cancer. Mol Cancer Ther. 2014;13:1399–1409. doi: 10.1158/1535-7163.MCT-13-0224. [DOI] [PubMed] [Google Scholar]
- 66.Rabineau M, et al. Contribution of soft substrates to malignancy and tumor suppression during colon cancer cell division. PLoS One. 2013;8:e78468. doi: 10.1371/journal.pone.0078468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marzorati M, et al. The HMI™ module: a new tool to study the Host-Microbiota Interaction in the human gastrointestinal tract in vitro. BMC Microbiol. 2014;14:133. doi: 10.1186/1471-2180-14-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HJ, et al. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci U S A. 2016;113:E7–15. doi: 10.1073/pnas.1522193112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richards AL, et al. Genetic and Transcriptional Analysis of Human Host Response to Healthy Gut Microbiota. mSystems. 2016;1:e00067–16. doi: 10.1128/mSystems.00067-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schweiger PJ, Jensen KB. Modeling human disease using organotypic cultures. Curr Opin Cell Biol. 2016;43:22–29. doi: 10.1016/j.ceb.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 71.Tsilingiri K, et al. Probiotic and postbiotic activity in health and disease: comparison on a novel polarised ex-vivo organ culture model. Gut. 2012;61:1007–1015. doi: 10.1136/gutjnl-2011-300971. [DOI] [PubMed] [Google Scholar]
- 72.Alleyne D, et al. Colonic transcriptional response to 1α,25(OH)2 vitamin D3 in African- and European-Americans. J Steroid Biochem Mol Biol. 2017;168:49–59. doi: 10.1016/j.jsbmb.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mapes B, et al. Ex vivo culture of primary human colonic tissue for studying transcriptional responses to 1α,25(OH)2 and 25(OH) vitamin D. Physiol Genomics. 2014;46:302–308. doi: 10.1152/physiolgenomics.00194.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 75.Spence JR, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dedhia PH, et al. Organoid Models of Human Gastrointestinal Development and Disease. Gastroenterology. 2016;150:1098–1112. doi: 10.1053/j.gastro.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nozaki K, et al. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. J Gastroenterol. 2016;51:206–213. doi: 10.1007/s00535-016-1170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bertaux-Skeirik N, et al. Co-culture of Gastric Organoids and Immortalized Stomach Mesenchymal Cells. Methods Mol Biol. 2016;1422:23–31. doi: 10.1007/978-1-4939-3603-8_3. [DOI] [PubMed] [Google Scholar]
- 79.Finkbeiner SR, et al. Transcriptome-wide Analysis Reveals Hallmarks of Human Intestine Development and Maturation In Vitro and In Vivo. Stem Cell Reports. 2015 doi: 10.1016/j.stemcr.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilson SS, et al. A small intestinal organoid model of non-invasive enteric pathogen-epithelial cell interactions. Mucosal Immunol. 2015;8:352–361. doi: 10.1038/mi.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leslie JL, et al. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect Immun. 2015;83:138–145. doi: 10.1128/IAI.02561-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karve SS, et al. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS One. 2017;12:e0178966. doi: 10.1371/journal.pone.0178966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lukovac S, et al. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. MBio. 2014;5 doi: 10.1128/mBio.01438-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goodrich JK, et al. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe. 2016;19:731–743. doi: 10.1016/j.chom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morgan XC, et al. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol. 2015;16:67. doi: 10.1186/s13059-015-0637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gamazon ER, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vikhanski L. Immunity: How Elie Metchnikoff Changed the Course of Modern Medicine. Chicago Review Press; 2016. [Google Scholar]
- 88.Anukam KC, Reid G. Probiotics: 100 years 1907–2007 after Elie Metchnikoff’s observation. Communicating current research. 2007 at < http://www.basicknowledge101.com/pdf/Probiotics%20100%20years.pdf>.
- 89.Eiseman B, et al. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery. 1958;44:854–859. [PubMed] [Google Scholar]
- 90.Sonnenburg ED, Sonnenburg JL. Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014;20:779–786. doi: 10.1016/j.cmet.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khoruts A, Sadowsky MJ. Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol. 2016;13:508–516. doi: 10.1038/nrgastro.2016.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mangravite LM, et al. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature. 2013;502:377–380. doi: 10.1038/nature12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maranville JC, et al. Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genet. 2011;7:e1002162. doi: 10.1371/journal.pgen.1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maranville JC, et al. Genetic mapping with multiple levels of phenotypic information reveals determinants of lymphocyte glucocorticoid sensitivity. Am J Hum Genet. 2013;93:735–743. doi: 10.1016/j.ajhg.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Siddle KJ, et al. A genomic portrait of the genetic architecture and regulatory impact of microRNA expression in response to infection. Genome Res. 2014;24:850–859. doi: 10.1101/gr.161471.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pastinen T. Genome-wide allele-specific analysis: insights into regulatory variation. Nat Rev Genet. 2010;11:533–538. doi: 10.1038/nrg2815. [DOI] [PubMed] [Google Scholar]
- 97.Kasowski M, et al. Variation in transcription factor binding among humans. Science. 2010;328:232–235. doi: 10.1126/science.1183621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McDaniell R, et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 2010;328:235–239. doi: 10.1126/science.1184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Knowles DA, et al. Allele-specific expression reveals interactions between genetic variation and environment. Nat Methods. 2017 doi: 10.1038/nmeth.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moyerbrailean GA, et al. High-throughput allele-specific expression across 250 environmental conditions. Genome Res. 2016 doi: 10.1101/gr.209759.116. [DOI] [PMC free article] [PubMed] [Google Scholar]