

Abstract
Conventional cytotoxic chemotherapy is highly effective in certain cancers, but causes dose-limiting damage to normal proliferating cells, especially hematopoietic stem and progenitor cells (HSPCs). Serial exposure to cytotoxics causes a long-term hematopoietic compromise (‘exhaustion’), which limits the use of chemotherapy and success of cancer therapy. Here, we show that the co-administration of G1T28 (trilaciclib), a small-molecule inhibitor of cyclin-dependent kinases 4 and 6 (CDK4/6), contemporaneously with cytotoxic chemotherapy protects murine hematopoietic stem cells (HSCs) from chemotherapy-induced exhaustion in a serial 5-fluorouracil (5FU) treatment model. Consistent with a cell intrinsic effect, we show directly preserved HSC function resulting in a more rapid recovery of peripheral blood counts, enhanced serial transplantation capacity and reduced myeloid skewing. When administered to healthy human volunteers, G1T28 demonstrated excellent in vivo pharmacology and transiently inhibited bone marrow (BM) HSPC proliferation. These findings suggest that the combination of CDK4/6 inhibitors (CDK4/6i) with cytotoxic chemotherapy should provide a means to attenuate therapy-induced BM exhaustion in patients with cancer.
INTRODUCTION
Human BM is sensitive to cell cycle dependent cytotoxic agents, and myelosuppression is the dose-limiting toxicity for most such agents. Myelosuppression causes life-threatening morbidity, augments the cost of care, and compromises therapeutic efficacy, through the induction of treatment delays and reduced therapeutic intensity. It has further been suggested that cytotoxic chemotherapy induces lasting immunosuppression that dampens a beneficial anti-tumor immune response (1). The long-term BM toxicity of cytotoxics is also a major problem for cancer survivors, because it is associated with serious late toxicities of cancer therapy: BM exhaustion, myelodysplastic syndrome (MDS), and acute leukemia. Whereas the depletion of committed hematopoietic progenitor cells (HPCs) is largely responsible for the acute hematopoietic toxicity of chemotherapy, damage and functional attrition of HSCs contribute to therapy-induced late myelotoxicity (2–5).
Existing strategies to manage chemotherapy-induced myelosuppression focus on correcting acute cytopenias through the transfusion of platelets and red cells, and the administration of growth factors: granulocyte-colony stimulating factor (G-CSF) or erythropoietin (EPO). Transfusions decrease quality of life during therapy and are associated with transfusion reactions and risk of infection. Although growth factors ameliorate acute myelosuppression, their use is problematic in that they have substantial acute toxicity of their own (for example, excess mortality and thrombosis for EPO, fever and bone pain for G-CSF) and adverse long-term consequences manifesting as exacerbated HSC exhaustion (6, 7) and an increased risk for MDS and leukemia (8, 9). To date, no therapeutic option is available to prevent or treat chemotherapy-induced functional exhaustion of HSCs.
It has long been suggested that selective inhibition of the proliferation of normal but not cancer cells may provide protection from chemotherapy-induced toxicity (10, 11), but an incomplete understanding of cell cycle regulation has prevented the reduction of this concept to practice. We and others have shown that HSPCs depend on CDK4/6 activity for proliferation (12–14), whereas many human cancers are resistant to CDK4/6 inhibition (for example retinoblastoma (RB)-deficient cancers) (15, 16). This suggests that co-administration of a CDK4/6 inhibitor (CDK4/6i) with cytotoxic chemotherapeutic agents could augment the therapeutic window by protecting normal HSPCs without negating the anti-neoplastic effect against CDK4/6-independent tumors. Using a long-acting oral CDK4/6i, we previously showed that a transient period of “pharmacological quiescence” (PQ) can reduce platinum chemotherapy-induced acute myelosuppression in vivo (17). It is unclear, however, whether long-term BM toxicities of chemotherapy can be attenuated by PQ.
G1T28 (trilaciclib) is a small-molecule CDK4/6i developed for reducing chemotherapy-induced myelosuppression (18). This molecule affords excellent in vivo protection against DNA-damaging agents in rodent models and has favorable potency, selectivity, and pharmacological properties for this purpose (18). In particular, G1T28 is well-suited for chemoprotection applications, which require an agent with a short biological half-life and intravenous (IV) formulation. In this study, we examine the ability of G1T28 to modulate HSPC proliferation and preserve long-term HSC function in humans and mice.
RESULTS
G1T28 induces transient and reversible G1 cell cycle arrest in murine HSPCs
We first determined the pharmacodynamic response of murine HSPCs to G1T28 using 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay (fig. S1–4), focusing on dose and schedule. A single intraperitoneal (IP) dose of G1T28 was sufficient to induce a dose-dependent reduction in EdU incorporation (S-phase cells) in all hematopoietic cell types at 12 hours after treatment (Fig. 1A–C, fig. S2 and S3), without causing other cell cycle effects such as G2/M arrest (fig. S2) or acute cytotoxicity (fig. S4C). This G1T28-induced reduction in S-phase cell frequency was independently confirmed using DNA content analysis (fig. S3B and S4B). HSCs, early HPCs (LSK cells and myeloid progenitors [MPs]), and developing lymphoid cells were the most sensitive to CDK4/6i-induced G1 arrest, showing a significantly lower ED50 (Fig. 1B, p < 0.001) for G1T28 and a more complete G1 cell cycle arrest (Fig. 1C, p < 0.05) compared to maturing erythroid and myeloid cells.
Fig. 1. G1T28 induces transient, reversible G1 cell cycle arrest in both murine and human BM HSPCs.

(A) Rate of murine BM HSC proliferation 12 hours after a single IP injection of the indicated doses of G1T28 (n = 5 mice/dose analyzed in 5 independent experiments, same for B and C). (B) Median effective dose (ED50) of G1T28 to inhibit the in vivo proliferation of the indicated murine hematopoietic cell types with identification scheme shown in fig. S1 (LSK = Lin−Sca-1+c-Kit+ cells, MP = myeloid progenitors, E = erythroid, M = myeloid, B = B lymphocytes, T = T lymphocytes). (C) Relative proliferation rate of each murine hematopoietic cell type at maximum CDK4/6 inhibition, compared to untreated mice. (D) Rate of murine BM HSC proliferation at various time points after a single IP injection of 50 or 100 mg/kg of G1T28 (n = 3-6 mice/dose/time point). (E) Mean plasma concentration curve of G1T28 after single dose IV administration in healthy human volunteers. (F, G) Frequency of total human BM cells (F) and HSC-enriched CD45dimCD34+CD38− BM cells (G) in S/G2/M phase of the cell cycle either before or at 24 and 32 hours after single dose of G1T28 treatment. Data were from Cohort 7 (BED) after single 192 mg/m2 dose of G1T28. Error bars represent SD in A-D and SEM in E-G. The statistical significance of differences between vehicle and G1T28 treated groups in A and D and between indicated groups in B, C, and F was assessed using unpaired, two-tailed Student’s t-tests (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
The cell cycle arrest induced by G1T28 was transient, reversible, and dose-related (Fig. 1A–D, fig. S3, and fig. S4B,D–J). In HSCs, a reduction in EdU incorporation and S/G2/M cell frequency was seen as early as 4 hours after G1T28 treatment, reaching a minimum at 12 hours, and lasting up to 36 hours before recovery (Fig. 1D and S4B). Doses of G1T28 above 50 mg/kg did not cause a further reduction in EdU incorporation (Fig. 1A, fig. S3C–I), but rather extended the duration of cell cycle arrest (Fig. 1D and fig. S4B,D–J), consistent with the measured pharmacokinetics (PK) of G1T28 in mice (fig. S5). Of note, CDK4/6 inhibition not only reduced homeostatic proliferation of HSCs (Fig. 1A, D; S3B and S4B), but also abrogated the potent cell cycle entry of HSCs seen during recovery from cytotoxic myeloablation (fig. S3J), which is essential for maintaining HSC cell cycle arrest during prolonged chemotherapy treatment.
The duration of the G1T28-induced cell cycle arrest differed by hematopoietic cell types, correlating inversely with the sensitivity of each cell type to CDK4/6 inhibition: T cells > HSPCs >= B cells > maturing myeloid and erythroid cells (Fig. 1D and S4D–J). Notably, the proliferation rates of most hematopoietic cell types increased beyond steady state during the early period of recovery (Fig. 1D and fig. S4B,E–J), likely reflecting compensatory proliferation in response to the transient mild reduction of BM cellularity as a result of HSPC cell cycle arrest (fig. S4C).
G1T28 also induces a transient, reversible G1 arrest of human HSPCs
To determine whether G1T28 can induce a similar transient and reversible G1 cell cycle arrest in human HSPCs, as well as to characterize the pharmacological properties and safety of the compound, we completed a first-in-human Phase I clinical trial in healthy volunteers. In total, 45 subjects were enrolled; 36 received G1T28 in seven dosing groups and nine received placebo. On average, demographic characteristics (table S1) were comparable among the seven G1T28 groups and the placebo group with regard to mean age, body mass index (BMI), and body surface area (BSA). G1T28 and placebo (5% dextrose solution) were administered as a 30-minute IV infusion. Of the 33 subjects enrolled in the single ascending dose (SAD) part of the study, 24 received G1T28 at doses of 6, 12, 24, 48, 96, or 192 mg/m2 and 9 subjects received placebo (table S2). Overall, a single IV infusion of G1T28 at all test doses was well tolerated. The most frequently reported treatment emergent adverse events (TEAEs) were headache and nausea (table S3). No TEAEs of severe or life-threatening intensity were reported, and all moderate TEAEs spontaneously resolved within 24 hours. There were no clinically relevant changes in any of the treatment groups with respect to clinical laboratory testing (clinical chemistry, hematology [blood cell counts, including absolute reticulocyte and banded neutrophil counts], and urinalysis), vital signs, ECG, physical examination, or body weight.
The pharmacokinetic properties of G1T28 were assessed using standard approaches. After a single IV infusion of G1T28 at doses shown in table S2, the median time (tmax) to reach peak serum concentration (Cmax) ranged between 0.25 and 0.47 h after start of infusion (Fig. 1E and table S4). The geometric mean of Cmax and the estimated area under the curve (AUC0-t and AUC0-inf) for G1T28 increased in a dose-proportional manner over the dose ranges tested (table S4). The elimination of G1T28 appeared to occur in 3 phases after the end of infusion (EOI): a fast α-phase until approximately 4 h, followed by a β-phase until approximately 36 h, and a more gradual terminal γ-phase, suggesting a 3‐compartment model (Fig. 1E). Because the concentrations of the lowest dose groups fell below the quantifiable limit before the last elimination phase, the terminal half-life (t1/2) of the lowest dose groups was determined by the first elimination phase (t1/2 of 5.32 to 9.28 h). The terminal t1/2 of the 48, 96, and 192 mg/m2 dose groups was determined by the second elimination phase, with a t1/2 of 12.9 to 14.7 h (table S4). However, because the contribution of the terminal phase to the effective half‐life of the drug is approximately 15-20%, the longer t1/2 at the higher dose levels is likely to be less relevant. Compartmental analysis at the biologically effective dose (BED) of 192 mg/m2 demonstrated geometric mean values for Cmax and AUC0-inf of 1705 ng/mL and 2991 h*ng/mL, respectively (table S4). The coefficients of variability (%) for these values were all ≤ 15%, demonstrating low inter‐subject variability. Total systemic exposure was independent of gender, body surface area, age, and body weight. These results demonstrate that G1T28 has acceptable in vivo pharmacology for use in human patients.
To determine the effects of G1T28 on human HSPC proliferation, we analyzed the cell cycle status of BM HSPCs in 12 subjects (cohort 7) either before or after G1T28 treatment (BED, 192 mg/m2). A single BM aspirate was obtained from each subject (before dose, n = 5; 24 h, n = 3; 32 h, n = 4), and cell cycle status of total BM, uncommitted BM HSPCs, and lineage-committed BM cells were assessed by cellular DNA content analysis (table S5, fig. S6 and S7). Total BM cells showed a 45% reduction in S/G2/M cell frequency at 24 hours with a partial recovery at 32 hours (Fig. 1F). This extent of G1T28-induced total BM cell cycle arrest is in line with murine data (fig. S4D). The effects of G1T28 on the proliferation of different BM cell lineages were also consistent with the murine findings. The percentage of cells in S/G2/M phase of the cell cycle decreased at 24 hours after G1T28 for all cell populations except the granulocyte lineage (Fig. 1G, fig. S7, and table S5). As in the mouse, the strongest effects were seen on immature HSPCs, with more modest effects on differentiated cells (fig. S7 and table S5). Given that a single dose of G1T28 did not significantly alter peripheral blood (PB) cell counts for up to 14 days after infusion (fig. S8), these data together suggest G1T28-induced HSPC cell cycle arrest is transient and reversible, and well-tolerated in humans.
Transient G1 arrest accelerates hematologic recovery after single dose 5FU treatment
We next examined the effects of a transient HSPC G1 arrest on chemotherapy-induced BM exhaustion using 5FU, a pyrimidine analogue toxic to proliferating cells (19). Although a single dose of 5FU causes transient myelosuppression without directly damaging quiescent HSCs (20), repeated doses of 5FU provide a well-characterized model of HSC exhaustion (21, 22). First, to determine the effects of PQ on 5FU-induced acute myelosuppression, adult C57BL/6 mice were treated with either vehicle or G1T28 30 minutes before a single injection of 5FU. 5FU treatment caused a rapid and transient depletion of all blood cell lineages (Fig. 2A). In accord with published studies (17), G1T28 pre-treatment did not prevent the initial drop in blood cell counts, but significantly accelerated the PB count recovery (Fig. 2A, p < 0.05). Because the acute hematologic toxicity of 5FU is caused by the depletion of rapidly dividing HPCs (18, 20), we determined the effect of G1T28 treatment on the number of BM progenitors at various time points after 5FU administration using the methylcellulose colony formation assay. Although G1T28 treatment did not prevent the decline of total BM cell count at 24 hours after 5FU administration (Fig. 2B), it increased the number of colony-forming cells that survived 1 and 2 days after 5FU treatment (Fig. 2C), resulting in a faster recovery of the number of colony-forming progenitors (Fig. 2C) and an accelerated recovery of BM cell counts (Fig. 2B). Together, these data indicate that a G1T28-induced transient G1 cell cycle arrest attenuates the acute hematopoietic toxicity of cytotoxic agents by protecting rapidly proliferating BM progenitors.
Fig. 2. Transient G1 arrest by G1T28 pretreatment accelerates hematologic recovery after single dose 5FU treatment.
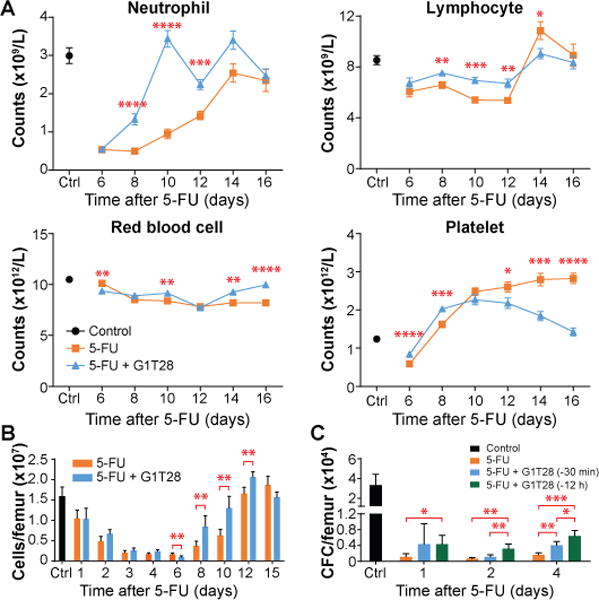
(A, B) Complete blood cell counts (A) and BM cellularity (B) at various time points after a single dose of 5FU with or without G1T28 pretreatment (n = 5-18 mice/treatment/time point). (C) Number of colony-forming cells (CFC) per femur 1 to 4 days after a single treatment with 5FU ± G1T28 (n = 4-11 mice/treatment/time point). Data represent mean ± SEM in A and mean ± SD in B and C. The statistical significance of differences between indicated groups was assessed using unpaired, two-tailed Student’s t-tests (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Transient G1 cell cycle arrest protects mice from repeated 5FU-induced lethal myelosuppression
Single-dose 5FU treatment causes transient myelosuppression, but repeated administration of 5FU at relatively short intervals (7–10 days) causes irreversible morbidity requiring euthanasia, resulting from incomplete recovery of BM hematopoiesis between treatments (23–25). To determine whether PQ at the time of each serial dose of 5FU could afford survival protection, we subjected adult mice to repeated doses of 5FU at intervals of 7 or 10 days. Twelve hours before (−12 h) each 5FU injection, animals received either vehicle, or 50 or 100 mg/kg of G1T28. In all cases, G1T28-treated animals exhibited a marked and dose-dependent enhancement in survival compared to vehicle-treated controls (Fig. 3A, B). At the 100 mg/kg dose, G1T28-treated mice showed a 55% increase in median survival over controls (28 days vs. 18 days) when 5FU was given at 7-day intervals (Fig. 3A), and a 125% increase in median survival (63 days vs. 28 days) when 5FU was given at 10-day intervals (Fig. 3B). Marked protection was also noted in the 10-day assay when G1T28 was given 30 minutes (−30 min) before each 5FU treatment, but not when it was given 24 hours (−24 h) before 5FU treatment (Fig. 3C). G1T28 given twice, at −24 h and −30 min, offered no additional protection over treatment at −30 min alone (Fig. 3C). After intraperitoneal injection, 5FU is rapidly eliminated from the plasma (t1/2 = 10-20 minutes) (26) and then induces DNA damage for several hours post-infusion. Given that the maximum G1 arrest in HSPCs occurs between 12 and 24 hours after G1T28 treatment (Fig. 1D and fig. S4B, D–J), the finding of enhanced protection when G1T28 was administered at −30 minutes or −12 hours, but not −24 hours, suggests that potent protection of HSPCs is best achieved when the period of G1 arrest overlaps with the in vivo biologic effect of 5FU treatment. Other structurally distinct CDK4/6 inhibitors (palbociclib and ribociclib), as well as G-CSF (pegfilgrastim), also demonstrated a survival benefit in this assay (Fig. 3D, table S6). These results suggest that a CDK4/6i-induced G1 cell cycle arrest provides a substantial reduction in lethal myelosuppression caused by serial administration of a cytotoxic agent.
Fig. 3. G1T28 pretreatment protects mice from repeated doses of 5FU challenge.
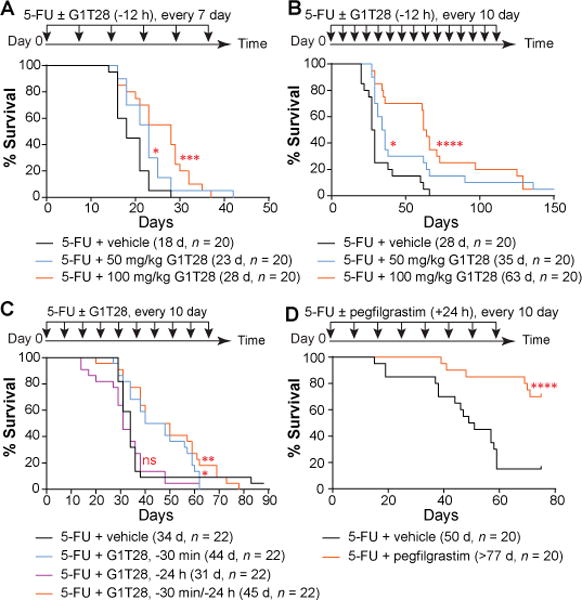
(A–D) Survival curves of mice that were treated with 5FU plus vehicle, G1T28, or pegfilgrastim at 7 (A) or 10 (B-D) day intervals. G1T28 was given 12 hours before 5FU in A and B, and either 30 minutes before, 24 hours before, or both in C. Pegfilgrastim was given 24 hours after 5FU in (D). Numbers in parenthesis indicate median survival (d = day), and animal number (n) in each treatment group. Significance for the pairwise comparison to 5FU + vehicle was calculated using the log-rank test in A, B, and D and Gehan-Breslow-Wilcoxon test in C (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant).
Transient G1 arrest by G1T28 at the time of chemotherapy protects murine HSCs from serial 5FU treatment-induced proliferative exhaustion
Even in the absence of direct cytotoxicity, chemotherapy agents can induce functional attrition of HSCs through a process of proliferative exhaustion. HSCs have an extensive but finite capacity to sustain multilineage hematopoiesis (27), and repeated rounds of proliferation (such as during recovery from 5FU-induced BM ablation) readily exhaust their long-term multilineage reconstitution potential. To determine whether the attenuation of acute chemotherapy-induced myelosuppression by transient G1 arrest (Fig. 2) can reduce the proliferative burden on quiescent HSCs, thereby extending their functional lifespan, we assessed the effect of G1T28 on protecting HSCs against serial 5FU treatment-induced proliferative exhaustion. Young adult B6.SJL (CD45.1+) mice were treated with 4 rounds of 5FU +/− G1T28 at 3-week intervals (Fig. 4A) - a schedule that causes HSC proliferative exhaustion and premature aging (22). As was the case after a single dose of 5FU (Fig. 2A), complete blood count (CBC) analysis after the fourth serial dose of 5FU showed that the G1T28-treated mice exhibited faster count recovery compared to vehicle-treated controls (fig. S9A). After 4 rounds of 5FU followed by count recovery, BM cells were harvested, enumerated, and transplanted as per Fig. 4A. Consistent with a previous report (22), serial 5FU treatment caused a decrease in BM B cell frequency and an increase in BM HSC frequency, similar to those observed during hematopoietic aging (28–31), which were not rescued by G1T28 pre-treatment (fig. S9B, C). These observations suggest that although a transient G1 arrest at the time of 5FU administration affords potent protection of PB counts after each cycle, it does not rescue the aging-like phenotypic expansion of HSCs induced by serial 5FU.
Fig. 4. G1T28-induced PQ protects HSCs from proliferative exhaustion.
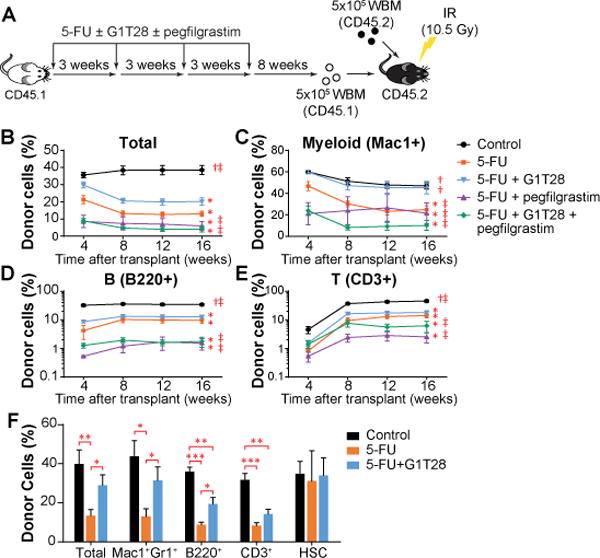
(A) Treatment and BM transplantation schematic. WBM, whole bone marrow cells (B-E) The percentage of donor-derived cells (CD45.1+) in total PB leukocytes (B), or myeloid cell (Mac1+, C), B lymphocyte (B220+, D), and T lymphocyte (CD3+, E) fractions 1 to 4 months after primary BM transplantation. 3-8 donor mice/treatment, 5 recipients/donor. Statistical significance of differences between treatment groups at 16 weeks after transplantation was assessed using one-way ANOVA with correction for multiple comparisons. (*, P < 0.05 compared to control; †, P < 0.05 compared to 5FU + vehicle, ‡, P < 0.05 compared to 5FU + G1T28). (F) The percentages of donor-derived cells in total BM cells, HSCs, and major BM cell lineages 32 weeks after BM transplantation. 5 donor mice/treatment, 5 recipients/donor. Statistical significance was assessed by two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001). Average donor cell reconstitution in B-F was calculated by first averaging the value of the 5 recipients from each donor, followed by calculating the mean of all donors. Error bars represent SEM.
To directly test whether a transient G1 arrest at the time of each dose of serial 5FU protects long-term HSC function, we performed competitive BM transplantation assays 8 weeks after the last dose of 5FU (Fig. 4A). Five hundred thousand BM cells (CD45.1+) harvested from 5FU-only or 5FU+G1T28 treated mice were transplanted against an equal number of untreated CD45.2+ BM cells into lethally irradiated C57BL/6 recipients (CD45.2+). The frequencies of donor-derived cells in recipient PB were monitored monthly for 4 months after transplant. Although serial 5FU treatment significantly reduced the long-term contribution of HSCs to all PB lineages (Fig. 4B–E, p < 0.05), HSCs from G1T28-treated animals demonstrated enhanced reconstitution of all lineages (Fig. 4B–E, p < 0.05). This observation indicates that a transient G1 arrest induced contemporaneously with serial administration of 5-FU provides a sustained, transplantable attenuation of chemotherapy-induced HSC damage.
Given that growth factor treatment is currently used to address chemotherapy-induced myelosuppression, we also directly compared the effects of G-CSF (pegfilgrastim) treatment with G1T28-induced G1 arrest on protecting long-term HSC function from serial 5FU-induced damage. Whereas pegfilgrastim enhanced survival against serial 5FU treatment at short intervals, presumably by stimulating the proliferation of surviving myeloid progenitors (Fig. 3D and table S6), pegfilgrastim treatment 24 and 48 hours after each cycle of 5FU treatment did not benefit long-term HSC function. In fact, G-CSF treatment further reduced the long-term multilineage reconstitution potential of HSCs compared to 5FU treatment alone (Fig. 4B–E). Moreover, the combined use of pegfilgrastim with G1T28 abolished the HSC protective effect of G1T28 (Fig. 4B–E). These data are in line with previous reports that G-CSF treatment exacerbates HSC injury after irradiation (32, 33) and findings that its use is associated with an increased later risk of myelodysplasia or leukemia (8, 9) . Therefore, although G-CSF can accelerate the acute hematologic recovery after BM injury, it appears to also exacerbate HSC exhaustion (7), likely due to disrupting HSC-niche interaction that results in HSC mobilization and proliferation (34). In contrast, the PQ approach by CDK4/6i appears to benefit both acute count recovery and long-term HSC function.
An analysis of the BM of recipient mice at 32 weeks after transplantation showed that G1T28 pre-treatment also increased donor cell reconstitution of all lineages in the BM compared to 5FU treatment alone (Fig. 4F). Analogous to transplantation studies comparing murine HSCs from young and old donors (31, 35, 36), the frequency of donor-derived HSCs in recipient BM was not reduced by 5FU treatment, regardless of G1T28 administration (Fig. 4F). These findings indicate that a period of G1 arrest bracketing the time of each 5FU treatment rescues HSC exhaustion—a decline in HSC function on a per cell basis that is induced by serial exposure to chemotherapy or aging.
To determine the durability of HSC protection afforded by G1T28-induced G1 arrest, we transplanted 107 total BM cells from each primary recipient into lethally irradiated individual secondary recipient mice 32 weeks after the initial transplantation. HSCs from G1T28-treated mice continued to produce significantly greater overall reconstitution in the secondary recipients (Fig. 5A, p < 0.05). Of note, although 5FU-only treated BM cells contributed poorly to PB lymphoid cells in secondary recipients (Fig. 5A), consistent with the myeloid-skewed differentiation pattern of aged or exhausted HSCs (31, 37), G1T28-pretreated cells maintained robust lymphoid reconstitution potential (Fig. 5A). Accordingly, when a separate group of 5FU ± G1T28 treated mice were analyzed 13 months after the fourth dose of 5FU, in the absence of BM transplantation, G1T28-pretreated mice also showed a significantly higher PB lymphoid to myeloid cell ratio compared to 5FU-only treated mice (fig. S9D, p < 0.05). We carefully monitored our colonies for malignancy after primary and secondary transplantation, but did not observe the emergence of secondary hematologic malignancies in any 5FU +/− G1T28 treated mice or BM transplant recipients during a 1-year follow-up period. Together, these results indicate that PQ at the time of serial 5FU treatment protects the long-term reconstitution potential of HSCs and attenuates the myeloid-biased differentiation of damaged or aged HSCs.
Fig. 5. Transient CDK4/6 inhibition protects long-term HSC function against serial 5FU treatment by reducing overall proliferative burden.
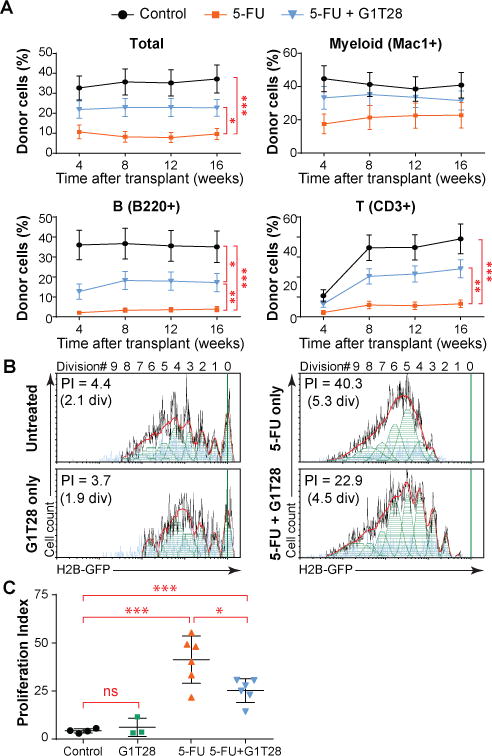
(A) The percentage of donor-derived cells (CD45.1+) in total PB leukocytes or myeloid cell (Mac1+), B lymphocyte (B220+), and T lymphocyte (CD3+) fractions 1 to 4 months after secondary BM transplantation. Data represent the mean ± SEM from 20-22 mice per group. (B, C) Cumulative HSC proliferation after a single dose of 5FU ± G1T28 treatment as determined by H2B-GFP label retention 4 weeks after 5FU administration. Representative H2B-GFP label dilution histograms (and curve fitting) are shown in (B), with quantification in (C). Proliferation index (PI) indicates the average number of daughter cells generated from a single, originally labeled HSC by the time of analysis, such that Log2 (PI) equals the average number of HSC divisions. Data represent mean ± SD from 3-6 mice in 3 experiments. Statistical significance was assessed by unpaired, two-tailed Student’s t-test (*P < 0.05, **P < 0.01, ***P < 0.001). div, division.
G1T28-induced G1 arrest protects long-term HSC function against serial 5FU treatment by reducing HSC proliferative burden
To directly demonstrate that transient G1 arrest attenuates HSC exhaustion through a reduction in proliferative burden, we analyzed the proliferative history of individual HSCs after a single dose of 5FU with or without G1T28 pre-treatment using the histone H2B-green fluorescent protein (H2B-GFP) label dilution assay (38, 39). In this assay, the replicative history of an individual HSC can be inferred by measuring the dilution of GFP-tagged histones, which are conditionally expressed under the control of a doxycycline-inducible promoter. Specifically, adult H2B-GFP;M2-rtTA double transgenic mice were treated with doxycycline for 6 weeks to label all HSCs with H2B-GFP (39). Two weeks after doxycycline removal, mice received vehicle, G1T28, 5FU, or 5FU+G1T28 (with G1T28 given 12 hours before 5FU). H2B-GFP expression in BM HSCs was quantified 4 weeks after 5FU treatment, allowing time for complete hematologic recovery. In mice that received vehicle only, BM HSCs divided 2.1 ± 0.4 times on average, generating 4.3 ± 2.1 daughter HSCs (or a proliferation index [PI] = 4.3 ± 2.1) over the 6 weeks after doxycycline removal (Fig. 5B). G1T28 treatment alone did not affect HSC proliferation over the same period of time (2.4 ± 1.0 divisions, PI = 6.2 ± 2.6) (Fig. 5B, C). In contrast, a single injection of 5FU greatly increased HSC proliferation (5.3 ± 0.5 divisions, PI = 41.3 ± 5.4), which was partially rescued by G1T28 pre-treatment (4.6 ± 0.4 divisions, PI = 25.2 ± 4.7) (Fig. 5B, C). Of note, this benefit was observed after only a single round of 5FU. We were unable to assess the entire proliferation history of individual HSCs after 4 rounds of 5FU treatment because of detection limitations of the H2B-GFP label dilution assay, but we expect that the benefit of PQ would be cumulative with additional cycles. Therefore, PQ at the time of 5FU treatment appears to attenuate HSC exhaustion, at least in part by reducing the number of proliferative events inflicted on an HSC by each round of chemotherapy.
DISCUSSION
In this work, we have shown that G1T28, a potent and selective CDK4/6i, can induce a specific and transient G1 cell cycle inhibition in HSPCs, particularly HSCs. In accord with prior work (17, 18), a transient G1 arrest induced by contemporaneous administration of this CDK4/6i at the time of chemotherapy can markedly ameliorate the acute hematopoietic toxicity of cytotoxic chemotherapy. Of note, we showed that in addition to a beneficial effect on rapidly proliferating hematopoietic progenitors, PQ can also provide lasting protection of the per cell function of quiescent HSCs, thereby ameliorating the long-term toxicity associated with serial exposure to chemotherapy agents (‘exhaustion’).
We also describe first-in-human results of G1T28 in a Phase I trial in healthy volunteers, showing that G1T28 has minimal toxicity and excellent PK and induces a strong proliferative arrest of early human HSPCs. G1T28 is well tolerated after a single IV infusion and has robust pharmacodynamic activity in the BM. At the BED of 192 mg/m2, BM HSPCs exhibited robust G1 cell cycle arrest at 24 and 32 hours after treatment, which should be a sufficient length of time to maintain HSPC quiescence when co-administered with chemotherapy. Standard of care chemotherapy for small cell lung cancer (SCLC) includes carboplatin and etoposide in the first line setting and single agent topotecan in the 2nd/3rd-line. Terminal half-lives for these agents are 6, 7.5, and 3 hours, respectively. Based on these half-lives, a 32-hour G1 arrest would allow approximately 94% (4 half-lives) of the etoposide to be cleared and even more for the other agents. The desired characteristics for a CDK4/6i used for chemoprotection differ from those developed as antineoplastic agents, where typical properties tend to include a longer biological half-life that supports single daily dosing, large volume of distribution, accumulation on repeated dosing, and oral delivery. As expected, considering the dependence of early HSPCs on CDK4/6 for proliferation, continued daily administration of antineoplastic CDK4/6 inhibitors is associated with myelosuppression, particularly neutropenia (40, 41). It is believed that this is not a cytotoxic effect on the HSPCs but a mechanism-based result of continued blockade of HSPC proliferation that decreases the production of new blood cells. In contrast, we have shown that a brief and transient period of CDK4/6 inhibition by G1T28 does not have lingering myelosuppressive effect.
The current study extends earlier work on chemotherapy-induced acute myelosuppression (17) by showing beneficial effects after multiple cycles of chemotherapy treatment that are long-lasting, transplantable, and associated with enhanced HSC function. Unlike lineage-specific strategies used in the clinic to manage acute chemotherapy-induced myelosuppression (such as transfusions and G-CSF/EPO), CDK4/6i-induced PQ protects all blood cell lineages. Moreover, unlike growth factor treatment, which enhances short-term blood cell count recovery at the cost of increasing the risk of leukemia and BM failure (6–9), a CDK4/6i-induced G1 arrest coincident with chemotherapy administration preserves HSC function. Therefore, CDK4/6i-mediated PQ provides the additional benefit of ameliorating the long-term sequelae of cytotoxic agents.
Chemotherapy-associated late BM toxicity results from therapy-induced damage and premature exhaustion of HSCs. Cytotoxics not only can cause direct damage to activated HSCs, but also can induce functional attrition of HSCs indirectly through a process of proliferative exhaustion (Fig. 4, 5 and (22)). This HSC cell intrinsic damage leads to a reduction of long-term multilineage reconstitution potential as well as myeloid-biased differentiation. Reduced long-term HSC function is clinically manifested in cancer survivors as persistent cytopenias, high rates of secondary leukemia and MDS, reduced T cell production and decreased cellular immunity. In the current study, we showed that PQ can attenuate chemotherapy-induced HSC exhaustion and lineage bias by directly modulating the cell cycle of rapidly proliferating activated HSCs at the time of exposure to cytotoxic agents, as well as by indirectly protecting quiescent HSCs by reducing their proliferative burden. Although we were unable to directly assess the effect of CDK4/6 inhibition on the incidence of chemotherapy-induced secondary leukemia, we provide evidence that a G1T28-induced G1 arrest will attenuate most aspects of chemotherapy-induced late BM toxicity.
Beyond reducing late toxicity, the preservation of HSC function would be expected to increase rates of tumor response and long-term cure through a variety of mechanisms. First, dose density and intensity are important predictors of tumor response in several RB-deficient tumors (such as SCLC (42) and triple negative breast cancer (TNBC) (43)), and reduced HSC exhaustion would prevent the dose reductions and treatment delays mandated by the persistent cytopenias that commonly occur in patients after multiple cycles of therapy. Moreover, by reducing HSC myeloid bias and thereby preserving T cell production, this approach could be ‘immune sparing’. An extensive clinical literature suggests that treatment-related lymphopenia is associated with worsened outcome in several types of cancer (44). A preservation of cellular immunity could be especially important in patients who will later receive immune checkpoint inhibitors that induce anti-tumor immunity through the activation of T cells.
A limitation of the PQ approach is that a CDK4/6i-induced G1 arrest at the time of chemotherapy administration could protect tumor cells in addition to normal HSPCs (11, 16). Therefore, it will be important to limit this approach to patients whose cancer proliferates independently of CDK4/6 activity. However, it is worth noting that CDK4/6-independent cancers are not rare: it is estimated in the US alone that there are 300,000 new cases of cancers annually whose proliferation does not depend on CDK4/6 activity (16). Several molecular lesions would be expected to render a cancer CDK4/6-independent, the best described of which is inactivation of the RB protein (15, 17, 45, 46). RB1 is genetically inactivated in ~11% of human cancers (15), including virtually 100% of SCLCs (47) and at high frequency in TNBCs (48) as well as advanced carcinomas of the prostate (49) and bladder (50). Likewise, functional inactivation of RB through expression of the E7 oncoprotein of human papilloma virus is common in squamous malignancies of the head and neck, anus, and cervix (51). Finally, we believe that an even larger fraction of cancers that retain RB expression are functionally deficient in RB through high-level activation of CDK2, which phosphorylates RB, by mechanisms that do not depend on CDK4/6 activity. For example, direct cyclin E amplification has been reported at moderate frequency (>20%) in several human solid tumors (TNBC, ovarian carcinoma, advanced prostate, and bladder cancer)(52). Further evidence for this notion comes from Novartis Cell Line Encyclopedia screens, which have shown that the vast majority of RB-expressing human cell lines are highly resistant to palbociclib (EC50 >2 µM), and therefore likely to be CDK4/6 independent (15). Therefore, even though CDK4/6i-mediated protection of normal HSPCs will not benefit all cancer patients, it offers a potential means to address debilitating short- and long-term side effects of cytotoxic chemotherapy in a large number of patients with CDK4/6 independent cancer.
Another limitation of this work is the schedule-dependent nature of this approach. We have shown that optimal protection is achieved when the period of HSPC G1 arrest overlaps with the in vivo biological effects of chemotherapy, but not for a longer or shorter period. If the period of growth arrest is too short, HSPCs re-enter the cell cycle in the setting of unrepaired DNA damage, causing increased myelosuppression. Likewise, an overly long period of PQ prolongs count nadirs after chemotherapy. Therefore, the safe use of this approach in patients requires the careful timing of HSPC arrest relative to the effective half-life of the chemotherapy agents. These concerns can be largely addressed through the use of an agent like G1T28, with an IV formulation and a relatively short pharmacologic half-life, allowing for precise control of the period of HSPC cell cycle arrest.
In summary, we have shown precise control of human and murine HSPC proliferation using a small moleculeCDK4/6 inhibitor, G1T28. By using this agent to induce a transient cell cycle arrest at the time of exposure to cytotoxics, we have shown long-lasting and cell-intrinsic protection of murine HSCs from chemotherapy-induced exhaustion. We also demonstrated that G1T28 is well tolerated after IV administration in healthy human volunteers and has robust pharmacodynamic activity in inducing transient and reversible cell cycle arrest of BM HSPCs, suggesting that CDK4/6i-induced PQ is feasible in humans. Because G1T28 is currently undergoing testing in this fashion in patients with SCLC (NCT02499770 and NCT02514447) and metastatic triple negative breast cancer (NCT02978716), it will be possible to determine if PQ similarly benefits human HSCs. Such an effect would be expected to increase chemotherapy efficacy as well as reduce late toxicity such as secondary leukemia and MDS in a large fraction of patients receiving cytotoxic chemotherapy.
MATERIALS AND METHODS
Study design
The preclinical portion of the current study aimed to determine whether transient G1 cell cycle arrest induced by pharmacological CDK4/6 inhibition could attenuate the late hematopoietic toxicity of cytotoxic chemotherapy. The well-characterized murine model of 5FU-induced myelosuppression was used to assess the BM protective effect of G1T28. The effect of G1T28 on 5FU-induced HSC exhaustion was assessed by a variety of approaches including serial competitive BM transplantation. The data presented in this study reflect multiple independent experiments performed on separate days using different mice and contain more than 3 biological replicates. No statistical methods were used to predetermine the sample size. No data or outliers were excluded from our analyses. Age and gender-matched mice were randomly treated with vehicle or the test compounds, and lethally irradiated mice were randomly assigned as BM transplantation recipients of variously treated donors. The investigators were not blinded during sample allocation or result analysis. The end point for the survival assay was determined as loss of more than 20% of peak body weight and/or deterioration of body condition. The extent of PB donor chimerism in BM transplantation recipients was monitored for more than 16 weeks to assess long-term multi-lineage reconstitution potential.
The clinical portion of the current study aimed to assess the safety, pharmacokinetics, and pharmacodynamics of IV administered G1T28 in healthy human volunteers. It was designed as a single-center; single-dose first-in-human study (NCT02243150), including a double-blind, randomized, placebo-controlled (3:1 G1T28 vs. placebo administered as a 30 minute IV infusion), SAD part in six 4-8 subject cohorts, and an open-label 12 subject cohort to confirm the BED. The study followed a Continual Reassessment Method (CRM) design, informed by the accrued safety experience throughout the study. Dose escalation between cohorts and the expansion from 4- to 8-subject cohorts were determined by the Safety Monitoring Committee (SMC) based on available safety, PK, and PD data. Up to a 100% dose escalation was allowed between cohorts, if supported by the available safety, PK, and PD data. Dose escalation was planned to continue until the SMC deemed that the BED had been reached or the criteria for halting dose escalation had been met. Dose escalation was to be halted in the following situations: occurrence of drug-related treatment-emergent ≥ Grade 2 non-hematologic adverse events (AEs) or ≥ Grade 3 hematologic AEs in 2 or more subjects in a given cohort, ≥ Grade 3 non-hematologic AEs or ≥ Grade 4 hematologic AEs in a single subject in a given cohort, or a recommendation to halt dose escalation by the Principal Investigator. Based on preclinical PK/PD modeling and available safety, PK, and PD data from cohorts 1-6, 12 subjects were enrolled in Cohort 7 to confirm the BED. All 12 subjects enrolled in cohort 7 had a single BM aspirate obtained at various time points relative to IV dosing of G1T28 to directly measure the effect of G1T28 on BM HSPC proliferation. The protocol and informed consent were reviewed and approved by the Institutional Independent Ethics Committee, and the trial was conducted in accordance with the principles of the Declaration of Helsinki, in compliance with the International Conference on Harmonization E6 Guideline for Good Clinical Practice, and in compliance with the European Union Clinical Trial Directive. All subjects provided written informed consent.
Mice
Mice used in this study were housed in the AAALAC-accredited, specific-pathogen-free animal care facility operated by the Division of Laboratory Animal Medicine at the University of North Carolina in Chapel Hill. Mouse work was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee. C57BL/6NTac (CD45.2) and B6.SJL-Ptprca/BoyAiTac (CD45.1) mice were purchased from Taconic. B6;129S4-Gt(ROSA)26Sortm1(rtTA*M2)JaeCol1a1tm7(tetO-HIST1H2BJ/GFP)Jae/J (H2B-GFP;M2-rtTA) double transgenic mice were purchased from Jackson Laboratory.
Cell isolation and flow cytometry
Murine BM cells were isolated by flushing the femurs, tibias, and pelvic bones using ice-cold staining medium (1× Ca2+- and Mg2+-free Hank’s balanced salt solution (HBSS, Gibco) supplemented with 10 mM EDTA (Corning) and 2% heat-inactivated bovine serum (Gibco)) and filtered through 40 μm nylon mesh (Sefar). Splenocytes and thymocytes were isolated by crushing spleen or thymus tissue between two microscope slides followed by filtering through nylon mesh. The number of viable cells was determined by manually counting with a hemocytometer. To identify HSCs, LSK cells, and myeloid progenitors (MPs) by flow cytometry, BM cells were stained with antibodies against c-Kit (2B8, APC-Efluor780, eBioscience), Sca-1 (E13-161.7, APC), CD48 (HM48-1, PerCP-Cy5.5), CD150 (TC15-12F12.2, biotin), as well as FITC-conjugated antibodies against the following lineage markers: CD2 (RM2–5), CD3e (145–2c11), CD4 (GK1.5), CD5 (53–7.3), CD8a (53–6.7), B220 (RA3-6B2), Mac-1 (M1/70), Gr-1 (RB6-8C5), and Ter119 (TER-119). CD150 staining was developed using PE-Cy7 conjugated streptavidin. Sometimes c-Kit expressing cells were magnetically enriched using anti-APC magnetic beads on the autoMACS system (Miltenyi). To identify mature myeloid, erythroid, and B lineage cells, BM cells were stained with antibodies against Gr-1 (FITC), B220 (PerCP-Cy5.5), Ter119 (Alexa Fluor 700), and Mac-1 (APC-Efluor 780, eBioscience). All antibody staining of live cells was carried out on ice for 1 hour. All primary and secondary antibodies were purchased from Biolegend unless otherwise indicated. Flow cytometry analysis of stained cells was performed on a customized LSR II 7-laser, 17-color flow cytometer (Becton-Dickinson), and recorded data were analyzed in BD FACSDiva 8.0.1, FlowJo 10.0.8, or FCS Express 5 Pro RUO software.
Murine HSPC cell cycle analysis by EdU incorporation assay
EdU-labeled murine BM cells and thymocytes were isolated and stained with primary antibodies as described above. The stained cells were then labeled with LIVE/DEAD Fixable Red Dead Cell Stain (Life Technologies) for 30 minutes, followed by fixation in 5% formalin for 10 minutes on ice and permeabilization in staining medium plus 0.1% saponin for 10 minutes at room temperature. EdU staining was performed in 0.1 M Tris-HCl (pH 7.5), 1 mM CuSO4, 0.1 M ascorbic acid, and 5 μM Alexa Fluor 555 azide (Life Technologies) for 10 minutes at room temperature (53). Stained cells were washed twice with ice-cold staining medium, followed by incubation in secondary antibody and 2 μg/ml of 4′, 6-diamidino-2-phenylindole (DAPI) for 30 minutes on ice before flow cytometry analysis.
In vivo G1T28 murine pharmacokinetic (PK) assay
For PK study after IP administration, four- to five-month-old female C57BL/6NHsd mice were given a single injection of 50 mg/kg or 100 mg/kg G1T28. For oral PK study, eight- to ten-week-old male CD1 mice were gavaged with 10, 50, or 100 mg/kg of G1T28. Blood was collected from treated mice via cardiac puncture at 5, 15, and 30 minutes or 1, 2, 4, 8, 12, 18, 24, 36, and 48 hours after drug administration (3 mice/dose/time point). Plasma G1T28 concentration was determined by LC-MS/MS as follows. Murine plasma samples were spiked with G1T28 and the corresponding D3 stable label internal standard (IS) followed by protein precipitation with acetonitrile. After precipitation, a portion of the organic layer was transferred to a 96-well plate, and the extract was chromatographed by reversed phase HPLC on an Allure PFP Propyl column with a mobile phase containing acetonitrile, water, ammonium formate and formic acid. G1T28 and D3 IS were detected by monitoring the precursor and product ions (m/z 447.2 → 336.2 for G1T28, m/z 450.2 → 339.4 for G1T28 D3 IS) with a Sciex 4000 LC MS/MS system. Analyte-to-IS peak area ratios for the standards were used to create linear calibration curves with weighted (1/x2) least-squares regression analysis. The calibration range of the assay was 10 to 10,000 ng/mL with a sample volume of 20 µL of plasma. PK parameters on plasma G1T28 concentration were calculated in Watson (v7.3.0.01, Thermo Inc.).
In vivo G1T28 dose response and pharmacodynamic assay
To determine the dose response of various murine hematopoietic cell types to G1T28-induced cell cycle arrest, 8- to 10-week-old female C57BL/6 mice were treated with a single dose of 0, 10, 25, 50, 100, or 150 mg/kg G1T28 by IP injection. Twelve hours later, each mouse was given a single IP injection of 10 mg/kg EdU and harvested 2 hours later. EdU staining of BM cells and thymocytes was performed as described above. To determine the in vivo pharmacodynamics of G1T28 on murine hematopoietic cell proliferation, 8- to 10-week-old female C57BL/6 mice were given a single IP injection of 50 or 100 mg/kg G1T28, followed by a single IP injection of 10 mg/kg EdU at 0.5, 1, 2, 4, 6, 12, 18, 24, 36, 48, 72, or 168 hours later. BM cells and thymocytes were isolated 2 hours after EdU injection and stained as described above. To determine the effect of G1T28 on actively cycling HSCs, 8-week-old female C57BL/6 mice were given a single IP injection of 150 mg/kg 5FU. 4 days later, half (n = 4) of the treated mice received a single IP injection of 100 mg/kg G1T28, and the other half (n = 4) received vehicle (50 mM citrate buffer) solution. Twelve hours later, each mouse was labeled with EdU for 2 hours. The frequency of EdU+ HSCs in each treatment group was determined as described.
5FU survival assay
Eight- to ten-week-old female C57BL/6 mice were given a single IP injection of 150 mg/kg 5FU once every 7 or 10 days. G1T28 was given to a subset of 5FU-treated animals at 30 minutes, 12 hours, and/or 24 hours before each 5FU treatment at 50 or 100 mg/kg dose by IP injection. Pegfilgrastim (250 μg/kg) was given to a subset of 5FU-treated animals 24 hours after each 5FU treatment by subcutaneous (SC) injection. Treated mice were monitored daily for body condition and/or weight loss, and the end point was determined as severe deterioration of body condition and/or loss of more than 20% of peak body weight.
Methylcellulose colony formation assay
C57BL/6 mice were orally gavaged with vehicle or 150 mg/kg of G1T28 30 minutes or 12 hours before being treated with 150 mg/kg of 5FU. BM progenitor colony-formation assay was performed by plating 1 × 104, 3 × 104, or 6 × 104 unfractionated BM cells harvested at 1, 2, or 4 days after 5FU into methylcellulose culture medium (Methocult GF M3434, StemCell Technologies). Hematopoietic colonies were counted after 8 – 9 days of culture at 37°C in 5% CO2.
Serial 5FU treatment and competitive BM reconstitution assay
Eight-week-old female B6.SJL-Ptprca/BoyAiTac (CD45.1) mice were treated with 4 rounds of vehicle (1x phosphate buffered saline [PBS]) or 150 mg/kg 5FU at 3-week intervals. A subset of the 5FU-treated mice was given a single dose of 150 mg/kg G1T28 by oral gavage 30 minutes before each 5FU injection or 2 doses of 250 μg/kg pegfilgrastim by SC injection at 24 and 48 hours after each 5FU treatment, and the remaining mice received vehicle (50 mM citrate buffer (pH4.0) for gavaging and 0.1% bovine serum albumin in 1x PBS for SC injection) treatment. CBCs were analyzed 4, 7, 11, 14, 18, and 21 days after the last round of 5FU treatment to assess the rate of hematologic recovery. Eight weeks after the last dose of 5FU treatment, BM cells were harvested from the treated mice, and competitive long-term BM reconstitution assay was performed by transplanting 500,000 total BM cells (CD45.1+) from each donor, together with 500,000 competitor BM cells (CD45.2+, from 8-week-old C57BL/6 mice), into lethally irradiated 8-week-old female C57BL/6 (CD45.2+) recipients. Lethal irradiation was carried out using a Gammacell 40 Exactor Cesium137 γ-ray source (MDS Nordia) by giving two doses of 540 rads total body irradiation 2 hours apart. BM cells from each donor mouse were transplanted into 5 recipient mice, and 5 donors were transplanted from each treatment group. PB was collected from the tail veins of recipient mice at 4-week intervals for at least 16 weeks after transplantation. To assess the frequency of donor cells in PB, red blood cells were subjected to ammonium-chloride potassium lysis (54), and remaining leukocytes were stained with antibodies against CD45.2 (104, FITC), CD45.1 (A20, APC-Efluor 780, eBioscience), B220 (PerCP-Cy5.5), Mac-1 (APC), CD3e (PE), and Gr-1 (PE-Cy7). Thirty two weeks after initial transplantation, BM cells were harvested from each primary recipient mouse and analyzed for donor cell contribution to each hematopoietic lineage. For HSC analysis, BM cells were stained with antibodies against lineage markers (FITC), c-Kit (APC-Efluor 780), Sca-1 (PE-Cy7), CD150 (PerCP-Cy5.5), CD48 (PE), CD45.1 (BV421), and CD45.2 (Biotin, followed by streptavidin-Alexa Fluor 647). For analysis of mature myeloid, B, and T cells, BM cells were stained with antibodies against Mac-1 (APC-Efluor 780), Gr-1 (PE-Cy7), B220 (PerCP-Cy5.5), CD3 (PE), CD45.2 (FITC), and CD45.1 (BV421). Secondary BM transplantation was performed by transplanting 1 × 107 total BM cells from each primary recipient into an individual lethally irradiated secondary recipient (CD45.2+). The frequencies of donor CD45.1+ cells in each blood lineage were monitored by analyzing the PB of secondary recipients at 4-week intervals for at least 16 weeks after transplantation.
H2B-GFP label dilution assay
To analyze the proliferation history of HSCs after 5FU and/or G1T28 treatment, adult homozygous H2B-GFP;M2rtTA mice were treated with 2 mg/ml doxycycline in drinking water for 6 weeks to label all HSCs. Two weeks after removing doxycycline, mice were given a single dose of vehicle or 150 mg/kg G1T28 by oral gavage 12 hours before vehicle or 150 mg/kg 5FU treatment. Four weeks after 5FU administration, BM cells were harvested and analyzed for H2B-GFP expression. Unfractionated BM cells were stained with antibodies against lineage markers (Biotin, followed by streptavidin-PE/Cy7), CD150 (PE), CD48 (PerCP-Cy5.5), c-Kit (APC-Efluor 780), and Sca-1 (APC). Curve fitting for HSC H2B-GFP label dilution data was performed using the proliferation analysis module in FCS Express 5 Pro RUO software.
Human study population and safety assessment
Eligible subjects included males and females 18-60 years of age with a body mass index of 18 to 32 kg/m2 (inclusive) and a weight of at least 50 kg. The subjects had to be judged to be in good health based on medical history and physical examination including vital signs, electrocardiogram, and clinical laboratory results. Women of childbearing potential were determined to be in a nongravid state and had agreed to take appropriate precautions to avoid pregnancy from the study enrollment until 3 months after completion of the study. Male subjects with a female partner of childbearing potential were required to use a highly effective form of birth control during the study and for 3 months after completion of the study. Use of medications or herbal products within 14 days before the start of the study was not allowed. Use of investigational medications within 60 days of the start of the study was not allowed.
Safety and tolerability assessments consisted of monitoring AEs, clinical laboratory results, vital signs, 12‐lead electrocardiograms (ECG), and physical examination. Subjects were admitted to the clinic on Day (-1) and remained in the clinic until all study procedures had been completed on Day 5. Subjects returned to clinic for further evaluation on Days 7, 10, and 14. The reporting period for serious adverse events (SAEs) began from the time that the subject provided informed consent up to and including the Day 14 follow‐up visit. AEs were recorded from the time of first dosing up to and including the Day 14 follow‐up visit, and all AEs had to be followed until they had resolved, returned to baseline, or it was deemed that further recovery was unlikely. Toxicity was assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.0 criteria.
Human PK study
G1T28 concentration in human plasma and urine was determined using a validated LC-MS/MS assay. For assay validation, human plasma samples were spiked with G1T28 and the corresponding D3 stable label internal standard (IS), followed by protein precipitation using acetonitrile. A portion of the organic layer after protein precipitation was transferred to a 96-well plate, and the extract was chromatographed by reversed phase HPLC on a Waters XBridge C18 (part #186003021) with a mobile phase containing acetonitrile, water, ammonium acetate and formic acid. G1T28 and the D3 IS were detected by monitoring the precursor and product ions (m/z 447 → 336 for G1T28, m/z 450 → 339 for G1T28 D3 IS) with a Sciex API 4000 LC-MS/MS system. Analyte-to-IS peak area ratios for the standards were used to create linear calibration curves with weighted (1/x2) least-squares regression analysis. The calibration range of the assay was 0.5 to 1,000 ng/mL, with a sample volume of 50 µL of plasma. The precision, accuracy, selectivity, and LLOQs of the assay were determined for G1T28; and the benchtop, freeze/thaw, and freezer stability of the analyte under the conditions of the assay were determined with appropriate QC samples. All of the assessments met the acceptance criteria established for a validated assay. Using this validated assay, plasma (cohorts 1-7) and urine concentrations (cohort 7) of G1T28 were determined with lower limits of quantitation of 0.5 ng/mL and 5.0 ng/mL in plasma and urine, respectively. Blood samples were collected before the dose and at multiple time points after the dose. Standard PK parameters were calculated using both a noncompartmental and a compartmental method (Phoenix WinNonlin 6.3).
Human BM proliferation assay
A single BM aspirate was obtained from all subjects enrolled in the BED cohort (cohort 7, 192 mg/m2, N = 12) to determine the effect of G1T28 on the percentage of various BM progenitor lineages in the G0/G1 or S/G2/M phases of the cell cycle. Using flow cytometry, lineages were identified using the markers CD45, CD34, CD14, CD11b, CD71, and CD61, and the cell cycle phases were evaluated using DRAQ5 DNA dye. BM progenitor cell proliferation was measured before dosing (n = 5), 24 hours after G1T28 dosing (n = 3), or 32 hours after G1T28 dosing (n = 4). Two BM samples (one before a dose and one at 32 hours after a dose) were heavily contaminated with peripheral blood, considered technically inadequate, and were removed from the cell cycle analysis data sets.
Statistical analysis
Statistical significance of differences between two sample groups was assessed using two-tailed Student’s t-test in Graphpad Prism 6 after the similarities in data variance between groups were determined using F-test. The survival analysis of mice after serial 5FU treatment was performed in Graphpad Prism 6 using log-rank test. Sample sizes for all data are given in each figure legend. Error bars represent SEM or SD as described in the figure legends. Unless otherwise indicated, a P-value less than 0.05 was considered statistically significant.
Demographic and safety data for each human subject were tabulated and summarized using descriptive statistics. Summary statistics (means, min-max, standard deviations, coefficients of variation) were calculated using noncompartmental and compartmental methods with WinNonlin software (V6.3, Pharsight) for the concentration versus time data at each sampling and for derived PK parameters. Comparisons of the proliferation of various human hematopoietic stem and progenitor subsets at baseline or after G1T28 treatment were made using two-tailed t-test with a P-value of 0.05.
Supplementary Material
Fig. S1. Flow cytometry gating strategy for various murine BM hematopoietic lineages.
Fig. S2. Representative flow cytometry plots for EdU staining in indicated murine hematopoietic cell types.
Fig. S3. Dose-dependent G1 cell cycle arrest induced by G1T28 in a wide range of murine hematopoietic cell types.
Fig. S4. Transient and reversible cell cycle arrest induced by G1T28 in murine hematopoietic cells.
Fig. S5. In vivo pharmacokinetics of G1T28 after IP or oral administration in mice.
Fig. S6. Flow cytometry gating strategy for various human BM hematopoietic lineages.
Fig. S7. Cell cycle analysis of human BM hematopoietic progenitors at various time points after a single dose of G1T28.
Fig. S8. Changes in peripheral blood cell counts in human subjects after a single dose of G1T28.
Fig. S9. Accelerated hematologic recovery by G1T28 pre-treatment after serial 5FU treatment.
Table S1. Summary of demographic characteristics of healthy volunteers.
Table S2. Summary of G1T28 administration in human subjects.
Table S3. Summary of related TEAEs reported in ≥ 10% of subjects.
Table S4. Summary of plasma G1T28 PK parameters in human.
Table S5. Absolute frequencies of human BM lineage populations in G0/G1 or S/G2/M phases of the cell cycle after G1T28 administration.
Table S6. Median survival extension by CDK4/6i or pegfilgrastim in mice receiving serial 5FU treatment.
One Sentence Summary.
The concomitant use of CDK4/6 inhibitors with cytotoxic agents may prevent chemotherapy-induced bone marrow exhaustion in cancer patients.
Acknowledgments
The authors would like to thank Dr. Kwok-Kin Wong (Dana-Farber Cancer Institute) for advice on experimental design and manuscript comments. We thank Dr. Nancy Fisher and UNC Flow Cytometry Core Facility for assistance with flow cytometry analysis. Part of the Animal Studies were performed within the Lineberger Comprehensive Cancer Center (LCCC) Animal Studies Core Facility at the University of North Carolina at Chapel Hill.
FUNDING: This work is supported by grants from the National Institutes of Health (AG024379, CA163896, CA174074). The UNC Flow Cytometry Core Facility is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The LCCC Animal Studies Core is supported in part by an NCI Center Core Support Grant (CA16086) to the UNC Lineberger Comprehensive Cancer Center.
Footnotes
AUTHOR CONTRIBUTIONS:
S.H, P.J.R and N.E.S conceived various aspects of the project, designed the experiments and interpreted the results with help from J.A.S, J.E.B and J.C.S. Preclinical experiments were performed by S.H, J.A.S, J.E.B and P.J.R. P.J.R, K.M., J.A.S, H.S.W, R.M, W.A.W, R.G.T, H.T, and E.H were responsible for the design, execution, and interpretation of the Phase I healthy volunteer study. The manuscript was written by S.H, P.J.R, J.A.S, J.C.S and N.E.S.
COMPETING INTERESTS:
- Roberts and Sharpless: Cyclin dependent kinase inhibitors and methods of use www.google.com/patents/US20120100100
- Strum, Bisi and Roberts: Transient protection of normal cells during chemotherapy www.google.com/patents/CA2906156A1?cl=en
- Strum, Bisi, Roberts and Sharpless: Transient Protection of Hematopoietic Stem and Progenitor Cells against ionizing radiation www.google.com/patents/US20140274896
- Strum, Bisi, Roberts and Sharpless: Hematopoietic protection against chemotherapeutic compounds using selective cyclin-dependent kinase 4/6 inhibitors www.google.com/patents/WO2010039997A3?cl=en
- Strum: CDK inhibitors www.google.com/patents/US8598186
REFERENCES AND NOTES
- 1.Harris J, Sengar D, Stewart T, Hyslop D. The effect of immunosuppressive chemotherapy on immune function in patients with malignant disease. Cancer. 1976 Feb;37:1058–1069. doi: 10.1002/1097-0142(197602)37:2+<1058::aid-cncr2820370813>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 2.Gardner RV, Lerner C, Astle CM, Harrison DE. Assessing permanent damage to primitive hematopoietic stem cells after chemotherapy using the competitive repopulation assay. Cancer Chemother Pharmacol. 1993;32:450–454. doi: 10.1007/BF00685889. [DOI] [PubMed] [Google Scholar]
- 3.Mauch P, Constine L, Greenberger J, Knospe W, Sullivan J, Liesveld JL, Deeg HJ. Hematopoietic stem cell compartment: acute and late effects of radiation therapy and chemotherapy. Int J Radiat Oncol Biol Phys. 1995 Mar 30;31:1319–1339. doi: 10.1016/0360-3016(94)00430-S. [DOI] [PubMed] [Google Scholar]
- 4.Gardner RV. Long term hematopoietic damage after chemotherapy and cytokine. Front Biosci. 1999 Jul 15;4:e47–57. doi: 10.2741/A479. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Probin V, Zhou D. Cancer therapy-induced residual bone marrow injury-Mechanisms of induction and implication for therapy. Curr Cancer Ther Rev. 2006 Aug 1;2:271–279. doi: 10.2174/157339406777934717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hornung RL, Longo DL. Hematopoietic stem cell depletion by restorative growth factor regimens during repeated high-dose cyclophosphamide therapy. Blood. 1992 Jul 1;80:77–83. [PubMed] [Google Scholar]
- 7.Moore MA. Does stem cell exhaustion result from combining hematopoietic growth factors with chemotherapy? If so, how do we prevent it? Blood. 1992 Jul 1;80:3–7. [PubMed] [Google Scholar]
- 8.Hershman D, Neugut AI, Jacobson JS, Wang J, Tsai WY, McBride R, Bennett CL, Grann VR. Acute myeloid leukemia or myelodysplastic syndrome following use of granulocyte colony-stimulating factors during breast cancer adjuvant chemotherapy. J Natl Cancer Inst. 2007 Feb 7;99:196–205. doi: 10.1093/jnci/djk028. [DOI] [PubMed] [Google Scholar]
- 9.Lyman GH, Dale DC, Wolff DA, Culakova E, Poniewierski MS, Kuderer NM, Crawford J. Acute myeloid leukemia or myelodysplastic syndrome in randomized controlled clinical trials of cancer chemotherapy with granulocyte colony-stimulating factor: a systematic review. J Clin Oncol. 2010 Jun 10;28:2914–2924. doi: 10.1200/JCO.2009.25.8723. [DOI] [PubMed] [Google Scholar]
- 10.Tubiana M, Carde P, Frindel E. Ways of minimising hematopoietic damage induced by radiation and cytostatic drugs–the possible role of inhibitors. Radiother Oncol. 1993 Oct;29:1–17. doi: 10.1016/0167-8140(93)90167-7. [DOI] [PubMed] [Google Scholar]
- 11.Blagosklonny MV, Pardee AB. Exploiting cancer cell cycling for selective protection of normal cells. Cancer Res. 2001 Jun 1;61:4301–4305. [PubMed] [Google Scholar]
- 12.Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004 Aug 20;118:493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 13.Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, Akashi K, Sicinski P. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004 Aug 20;118:477–491. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 14.Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, Ramsey MR, Jin J, Wong KK, Su L, Zhou D, Sharpless NE. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010 Jul;120:2528–2536. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012 Mar 29;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015 Feb;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, Zamboni WC, Wong KK, Perou CM, Sharpless NE. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012 Mar 21;104:476–487. doi: 10.1093/jnci/djs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bisi JE, Sorrentino JA, Roberts PJ, Tavares FX, Strum JC. Preclinical Characterization of G1T28: A Novel CDK4/6 Inhibitor for Reduction of Chemotherapy-Induced Myelosuppression. Mol Cancer Ther. 2016 May;15:783–793. doi: 10.1158/1535-7163.MCT-15-0775. [DOI] [PubMed] [Google Scholar]
- 19.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003 May;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 20.Lerner C, Harrison DE. 5-Fluorouracil spares hemopoietic stem cells responsible for long-term repopulation. Exp Hematol. 1990 Feb;18:114–118. [PubMed] [Google Scholar]
- 21.Harrison DE, Lerner CP. Most primitive hematopoietic stem cells are stimulated to cycle rapidly after treatment with 5-fluorouracil. Blood. 1991 Sep 1;78:1237–1240. [PubMed] [Google Scholar]
- 22.Beerman I, Bock C, Garrison BS, Smith ZD, Gu H, Meissner A, Rossi DJ. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell. 2013 Apr 4;12:413–425. doi: 10.1016/j.stem.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Haan G, Donte B, Engel C, Loeffler M, Nijhof W. Prophylactic pretreatment of mice with hematopoietic growth factors induces expansion of primitive cell compartments and results in protection against 5-fluorouracil-induced toxicity. Blood. 1996 Jun 1;87:4581–4588. [PubMed] [Google Scholar]
- 24.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000 Mar 10;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 25.Ehninger A, Boch T, Medyouf H, Mudder K, Orend G, Trumpp A. Loss of SPARC protects hematopoietic stem cells from chemotherapy toxicity by accelerating their return to quiescence. Blood. 2014 Jun 26;123:4054–4063. doi: 10.1182/blood-2013-10-533711. [DOI] [PubMed] [Google Scholar]
- 26.Peters GJ, Lankelma J, Kok RM, Noordhuis P, van Groeningen CJ, van der Wilt CL, Meyer S, Pinedo HM. Prolonged retention of high concentrations of 5-fluorouracil in human and murine tumors as compared with plasma. Cancer Chemother Pharmacol. 1993;31:269–276. doi: 10.1007/BF00685670. [DOI] [PubMed] [Google Scholar]
- 27.Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008 Feb;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 28.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996 Sep;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 29.de Haan G, Nijhof W, Van Zant G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997 Mar 1;89:1543–1550. [PubMed] [Google Scholar]
- 30.Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. 2004 Feb;5:133–139. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 31.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005 Jun 28;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chua HL, Plett PA, Sampson CH, Katz BP, Carnathan GW, MacVittie TJ, Lenden K, Orschell CM. Survival efficacy of the PEGylated G-CSFs Maxy-G34 and neulasta in a mouse model of lethal H-ARS, and residual bone marrow damage in treated survivors. Health Phys. 2014 Jan;106:21–38. doi: 10.1097/HP.0b013e3182a4df10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li C, Lu L, Zhang J, Huang S, Xing Y, Zhao M, Zhou D, Li D, Meng A. Granulocyte colony-stimulating factor exacerbates hematopoietic stem cell injury after irradiation. Cell Biosci. 2015;5:65. doi: 10.1186/s13578-015-0057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bendall LJ, Bradstock KF. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev. 2014 Aug;25:355–367. doi: 10.1016/j.cytogfr.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 35.Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45:286–290. doi: 10.1016/j.exger.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beerman I, Maloney WJ, Weissmann IL, Rossi DJ. Stem cells and the aging hematopoietic system. Curr Opin Immunol. 2010 Aug;22:500–506. doi: 10.1016/j.coi.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Snoeck HW. Aging of the hematopoietic system. Curr Opin Hematol. 2013 Jul;20:355–361. doi: 10.1097/MOH.0b013e3283623c77. [DOI] [PubMed] [Google Scholar]
- 38.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008 Dec 12;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 39.Foudi A, Hochedlinger K, Van Buren D, Schindler JW, Jaenisch R, Carey V, Hock H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009 Jan;27:84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy CG, Dickler MN. The Role of CDK4/6 Inhibition in Breast Cancer. Oncologist. 2015 May;20:483–490. doi: 10.1634/theoncologist.2014-0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vidula N, Rugo HS. Cyclin-Dependent Kinase 4/6 Inhibitors for the Treatment of Breast Cancer: A Review of Preclinical and Clinical Data. Clin Breast Cancer. 2015 Jul 26; doi: 10.1016/j.clbc.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Sheehan RG, Balaban EP, Frenkel EP. The impact of dose intensity of standard chemotherapy regimens in extensive stage small cell lung cancer. Am J Clin Oncol. 1993 Jun;16:250–255. doi: 10.1097/00000421-199306000-00012. [DOI] [PubMed] [Google Scholar]
- 43.Rampurwala MM, Rocque GB, Burkard ME. Update on adjuvant chemotherapy for early breast cancer. Breast Cancer (Auckl) 2014;8:125–133. doi: 10.4137/BCBCR.S9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ray-Coquard I, Cropet C, Van Glabbeke M, Sebban C, Le Cesne A, Judson I, Tredan O, Verweij J, Biron P, Labidi I, Guastalla JP, Bachelot T, Perol D, Chabaud S, Hogendoorn PC, Cassier P, Dufresne A, Blay JY, R. European Organization for, T. Treatment of Cancer Soft, G. Bone Sarcoma Lymphopenia as a prognostic factor for overall survival in advanced carcinomas, sarcomas, and lymphomas. Cancer Res. 2009 Jul 1;69:5383–5391. doi: 10.1158/0008-5472.CAN-08-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010 Jul 15;29:4018–4032. doi: 10.1038/onc.2010.154. [DOI] [PubMed] [Google Scholar]
- 46.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, Howe E, Fulton LE, Mulvey HE, Bernardo LA, Mohamoud F, Miyoshi N, VanderLaan PA, Costa DB, Janne PA, Borger DR, Ramaswamy S, Shioda T, Iafrate AJ, Getz G, Rudin CM, Mino-Kenudson M, Engelman JA. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.N. Cancer Genome Atlas. Comprehensive molecular portraits of human breast tumours. Nature. 2012 Oct 04;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010 Jul 13;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.N. Cancer Genome Atlas Research. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014 Mar 20;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011 Aug 1;10:2497–2503. doi: 10.4161/cc.10.15.16776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schraml P, Bucher C, Bissig H, Nocito A, Haas P, Wilber K, Seelig S, Kononen J, Mihatsch MJ, Dirnhofer S, Sauter G. Cyclin E overexpression and amplification in human tumours. J Pathol. 2003 Jul;200:375–382. doi: 10.1002/path.1356. [DOI] [PubMed] [Google Scholar]
- 53.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008 Feb 19;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994 Nov;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Flow cytometry gating strategy for various murine BM hematopoietic lineages.
Fig. S2. Representative flow cytometry plots for EdU staining in indicated murine hematopoietic cell types.
Fig. S3. Dose-dependent G1 cell cycle arrest induced by G1T28 in a wide range of murine hematopoietic cell types.
Fig. S4. Transient and reversible cell cycle arrest induced by G1T28 in murine hematopoietic cells.
Fig. S5. In vivo pharmacokinetics of G1T28 after IP or oral administration in mice.
Fig. S6. Flow cytometry gating strategy for various human BM hematopoietic lineages.
Fig. S7. Cell cycle analysis of human BM hematopoietic progenitors at various time points after a single dose of G1T28.
Fig. S8. Changes in peripheral blood cell counts in human subjects after a single dose of G1T28.
Fig. S9. Accelerated hematologic recovery by G1T28 pre-treatment after serial 5FU treatment.
Table S1. Summary of demographic characteristics of healthy volunteers.
Table S2. Summary of G1T28 administration in human subjects.
Table S3. Summary of related TEAEs reported in ≥ 10% of subjects.
Table S4. Summary of plasma G1T28 PK parameters in human.
Table S5. Absolute frequencies of human BM lineage populations in G0/G1 or S/G2/M phases of the cell cycle after G1T28 administration.
Table S6. Median survival extension by CDK4/6i or pegfilgrastim in mice receiving serial 5FU treatment.