

Abstract
A strategy to construct different stimuli responsive polymers from post polymerization modifications of a single polymer scaffold via thiol-disulfide exchange has been developed. Here, we report on a random copolymer that enables the design and syntheses of a series of dual or multi-stimuli responsive nanoassemblies using a simple post-polymerization modification step. The reactive functional group involves a side chain monopyridyl disulfide unit, which rapidly and quantitatively reacts with various thiols under mild conditions. Independent and concurrent incorporation of physical, chemical or biologically responsive properties have been demonstrated. We envision that this strategy may open up opportunities to simplify the synthesis of multi-functional polymers with broad implications in a variety of biological applications.
TOC image
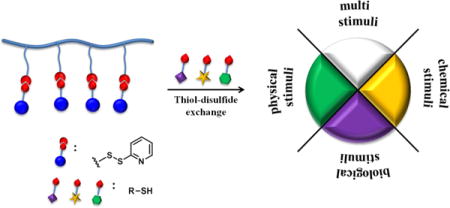
Driven by the increasing demand for multifunctional materials, stimuli responsive molecules or polymers have been intensively developed and used in a wide range of areas such as catalysis, drug delivery and sensing.1–6 Among various stimuli responsive systems, supramolecular assemblies that are capable of responding to two or more environmental changes have attracted particular attentions for two reasons: (i) the multi-responsive feature can be elicited concurrently or sequentially by more than one stimulus, providing better spatiotemporal control;7,8 (ii) since behavioral changes in biological systems are often a result of a combination of environmental changes rather than a single factor, multi-responsive materials offer an ideal artificial platform to mimic biological processes in nature.9–12 Considering the implications, there is a pressing need for simple synthetic methods to obtain multi-stimuli responsive materials with precise control over their architecture and functionality.
Random copolymer based assemblies have recently attracted attention, mainly due to their synthetic simplicity.13 On the other hand, post-polymerization modification (PPM) has emerged as a powerful tool for building functional polymer structures.14–17 PPM circumvents problems associated with direct polymerizations such as the susceptibility or incompatibility of monomers with reaction conditions, since there is no need to optimize the polymerization conditions for all the functional monomers within a polymer. As a result, PPM enables the synthesis of polymeric systems, which are difficult or impossible to produce with direct polymerization.14 PPM has also allowed systematic variations in structures based on a same reactive scaffold, without variables commonly created from the polymerization of individual monomers (e.g., molecular weight and dispersity). This has allowed for a robust development of structure-property correlations.16,17 Thiol-disulfide exchange is of growing interest among several PPM methods,18–26 because: (i) thiol moieties can be conveniently incorporated onto molecules; (ii) thiol-disulfide exchange reactions can occur under mild conditions with quantitative or near quantitative yields; (iii) the resultant disulfide linkage itself provides a moiety that is potentially sensitive to redox environments. Inspired by these advantages, we envisaged building a facile reactive scaffold in which a variety of stimuli responses can be systematically introduced via minimal steps to prepare well-defined dual or multi stimuli-responsive polymeric structures. We demonstrate this possibility by introducing different combinations of physical, chemical or biological stimuli on to a singular polymer scaffold.
The basic scaffold is a methacrylate-based random copolymer backbone that contains modifiable pyridyl disulfide (PDS) and hydrophilic polyethylene glycol (PEG) moieties randomly distributed as side chain functionalities. The polymer, which contains 7:3 ratio of PDS to PEG units, was prepared by reversible addition–fragmentation chain-transfer (RAFT) polymerization. This random copolymer forms an amphiphilic nanoassembly, when distributed in the aqueous phase.27–29 PDS functionality provides a convenient handle for reliably introducing stimuli-responsive groups through thiol-disulfide exchange, because the pyridothione byproduct is not reactive.
To test whether this polymer can be simply used to introduce an orthogonally responsive functional group, we first attempted the reaction of o-nitrobenzyl thiol with the PDS-PEG polymer to synthesize polymer P1 (Figure 1a). o-Nitrobenzyl group has been widely used to construct photo-responsive vehicles for remotely controlled release.30–34 After the addition of o-nitrobenzyl thiol, the solution gradually became yellow as a result of the pyridothione by-product formation. The PDS units were fully converted to o-nitrobenzyl groups, as discerned by the absorption spectrum that measures the pyridothione formation and by 1H NMR (Figure S1). Molecular weights of the polymers, before and after addition of o-nitrobenzyl thiol, were determined by gel permeation chromatography (GPC) and were found to be about 8500 and 9500 g·mol−1 respectively (Figure S5). The Ð of the PDS-PEG polymer and P1 were also found to be similar (1.23 and 1.17, respectively). Most notably, this simple thiol-disulfide exchange method avoids using any additional molecules or harsh reaction conditions to execute the reaction. Also, the product can be purified from the small molecule reactant and the byproduct simply by either precipitation or dialysis, presenting a convenient approach to the functionalization of polymers.
Figure 1.
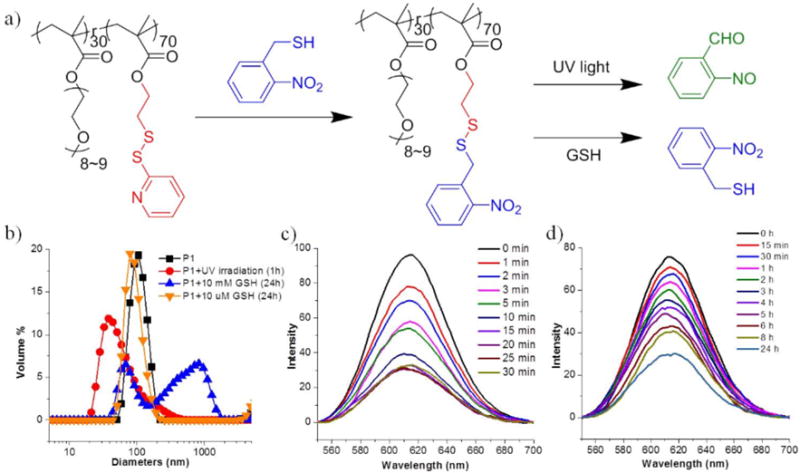
(a) Schematic presentation of P1 preparation by thiol-disulfide exchange and corresponding by-product generation of P1 under different stimuli conditions; (b) DLS profiles of P1 micelle (0.5 mg/mL) in the presence of different stimuli; (c) Variation of emission spectra of Nile Red encapsulated in the P1 micelle (0.5 mg/mL) under UV irradiation with different time; (d) Variation of emission spectra of Nile Red encapsulated in the P1 micelle (0.5 mg/mL) in the presence of 10 mM GSH.
Since P1 has both hydrophilic PEG and hydrophobic o-nitrobenzyl moieties, it is likely to self-assemble in aqueous solution. Once the hydrophobic moieties are cleaved upon the introduction of stimuli, leading to the shift of hydrophilic-hydrophobic balance, the size of the P1 aggregate changes. To investigate the self-assembly features and stimuli-responsive properties, we evaluated the hydrodynamic diameter of P1 in aqueous solution by dynamic light scattering (DLS) (Figure 1b). P1 showed an aggregate size of ~100 nm, which decreased to ~37 nm upon UV irradiation. This change was also verified by absorption spectroscopy, in which the absorbance at 360 nm increased over time due to the cleavage of o-nitrobenzyl group and the concomitant generation of the by-product, o-nitrosobenzaldehyde (Figure S6a). To test the dual-responsive features of P1, this polymer was also treated with glutathione (GSH). A size change was also observed here, which is attributed to the cleavage of the disulfide bond. This change was also found to be concentration-dependent. P1 did not form well-assembled aggregates after the treatment of 10 mM GSH, while only a slight change was observed in the presence of 10 μM GSH. Although not the focus of this work, these concentrations were chosen to demonstrate the ultimate biological relevance of these responsive features; the mM and μM concentrations of GSH were chosen to mimic the cytosolic and extracellular GSH conditions, respectively.35–37
If the observed changes in the size of the assemblies in response to the redox and light stimuli were indicators of the expected reactions, it is likely that the guest encapsulation capabilities of these amphiphilic assemblies would significantly change. This is anticipated, because the nitroaromatic unit is released in both these reactions, which should cause the assembly to lose significant hydrophobicity associated with the assembly. To explore the guest encapsulation and the triggered release possibilities in P1 assembly, Nile red was used as a fluorescence probe. As the o-nitrobenzyl moieties act as the hydrophobic site, P1 provides a hydrophobic pocket to host water-insoluble guest molecules, such as Nile red. If this dye molecule were to be released from the assembly’s hydrophobic pocket, it precipitates in water and therefore the solution should lose its fluorescence. To test whether the light- or redox-induced cleavage of the hydrophobic units would indeed cause a molecular release, we first monitored the temporal evolution of the emission intensity of Nile red upon irradiation at 365 nm. More than 65% of the dye was released within just 30 minutes (Figure 1c), which was also confirmed by monitoring the absorbance of the solution (Figure S6b). Similar phenomenon was observed when the solution of P1 was treated with 10 mM GSH (Figure 1d). A loss of fluorescence intensity was also observed and 60% of release was achieved within 24 hours, while there was no discernible release at a GSH concentration of 10 μM GSH during the same time scale. These observations indicate that P1 is responsive to both light and redox changes in its environment.
Using the same PDS-PEG precursor polymer, we also constructed an amphiphilic polymer that is concurrently responsive to pH and redox variations. In this case, ketal groups were conjugated to the polymer backbone by the reaction of (2,2-dimethyl-1,3-dioxolan-4-yl)methanethiol with PDS-PEG polymer to form polymer P2. Complete conversion of PDS to the ketal-containing side chain was supported by 1H NMR, where the characteristic peaks of pyridine ring disappeared and two diastereotopic methyl peaks appeared at 1.37 ppm and 1.44 ppm (Figure S2). The hydrophobic part of P2 consists of an acid-hydrolyzable ketal functionality, which is sensitive to acidic conditions.38–40 Acid-induced cleavage of ketal groups would transform the hydrophobic ketal units to a more hydrophilic dihydroxyl moiety. Consequently, we envisaged that the hydrophilic-lipophilic balance of the polymer would change, leading to the disruption of micelles (Figure 2a).
Figure 2.
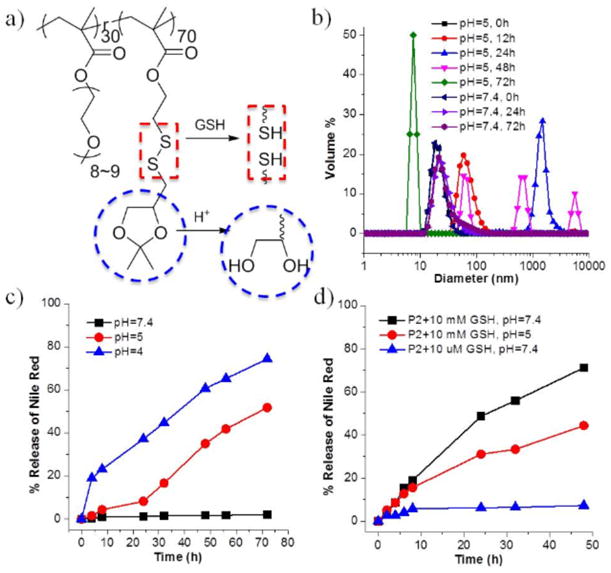
(a) Chemical structure of P2; (b) DLS measurements of P2 micelle solution (0.5 mg/mL) in different pH buffers over time; (c) Nile Red release profiles of P2 (0.5 mg/mL) in different pH (4, 5 and 7.4); (d) Nile Red release profiles of P2 (0.5 mg/mL) in the presence of different concentration of GSH in neutral pH and in the presence of the combination of GSH (10 mM) and low pH.
To examine the acidic sensitivity of P2, we first examined the assembly formed by the amphiphilic polymer in aqueous phase. The polymer formed a 20 nm assembly, the size of which was monitored over time (Figure 2b). At pH 5, the size of these micellar aggregates gradually swelled from 20 nm to over 1000 nm over the initial 24 h. The aggregate size decreased over time after this period, and final size of the aggregates was found to be less than 10 nm with relatively poor correlation after 72 h. In the control experiment, little change in size was observed over the same time period at neutral pH. These results were taken to suggest a slow disassembly of the amphiphilic aggregate under acidic conditions. This assertion is further supported by guest release profiles. Nile red-loaded P2 micelles in buffers of different pH were evaluated by fluorescence spectroscopy over three days. As shown in Figure 2c, the dye release was negligible at neutral pH. At pH 5 however, a significant dye release was observed over 72 h time period. In detail, ~10% of loss in fluorescence intensity was found within the first 24 hours. The release was accelerated afterwards and 35% and 50% of dye was released at 48 hours and 72 hours, respectively. When the pH was even lower (pH = 4), the dye released faster with a higher percentage. The polymer was also observed to be sensitive to redox environment, resulting from the disulfide linkage. As shown in Figure 2d, over 70% of Nile red was released from P2 micelles within 48 h in the presence of 10 mM GSH, with minimal release observed in the presence of 10 μM GSH. Even though the responsiveness of P2 to pH and redox potential has been demonstrated independently, we were interested in investigating the effect of combining the two stimuli upon the molecular release. Accordingly, when P2 was incubated in the presence of 10 mM GSH at pH of 5, less guest molecules was released compared with which in neutral pH. Although surprising at first, this observation is consistent with the fact that GSH is considered to be more active at neutral pH.41
Light is a physical stimulus, while pH is a chemical stimulus. Next, we were interested in exploring the possibility of introducing sensitivity to a biological stimulus in combination with redox sensitivity. Biological imbalances, i.e. variations in specific protein concentration or enzymatic activity, are considered to be primary indicators of pathology, as they are relevant to most biological pathways.42–46 Here, we chose carbonic anhydrase as the model protein, because of its disease relevance.47,48 The specific binding of carbonic anhydrase to sulfonamide moiety is well-established and utilized in binding-induced disassembly.49,50 Therefore, we hypothesized that the introduction of sulfonamide ligand to the polymer would disassemble in the presence of carbonic anhydrase, while maintaining the GSH-responsive features. In this case, we converted part of PDS groups to sulfonamide ligands using the PPM method to obtain polymer P3 (Figure 3a). The ratio of PEG, PDS and sulfonamide in P3 was about 0.3:0.49:0.21. The remaining PDS acts as the hydrophobic part and sulfonamide is the specific ligand to bind with carbonic anhydrase. Specifically, bovine carbonic anhydrase II (bCA-II), with a molecular weight of 30 kD, was used for this study.
Figure 3.
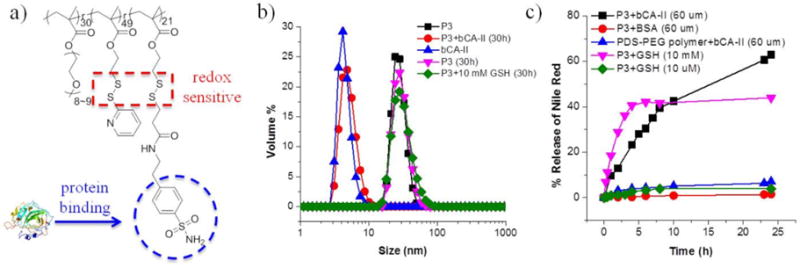
(a) Chemical structure of P3; (b) DLS results of P3 micelle solution (0.5 mg/mL), bCA-II solution (60 μM) and size change of P3 micelle (0.5 mg/mL) in the presence of 60 μM bCA-II or 10 mM GSH after 30 h; (c) Nile Red release profiles of P3 and control polymer PDS-PEG under different circumstances.
To investigate the specific interaction of between the sulfonamide group in P3 and complementary protein, 0.5 mg of P3 was dissolved in water and treated with 60 μM of bCA-II. In aqueous solution, P3 formed an assembly of about 25 nm. After incubating with bCA-II, the size reduced to ~5 nm, which is close to the size of bCA-II by itself (Figure 3b). In the control experiment, no size change was observed in the P3 assembly in the absence of the protein. To further study the protein binding-induced disassembly, a Nile red-encapsulated P3 assembly was treated with bCA-II. A gradual decrease of fluorescence intensity was observed (~60% release over 24 h), while no discernible emission differences were observed in the absence of protein (Figure 3c). To further validate that the guest molecule was released due to specific protein binding, two more control experiments were carried out. First, the P3 assembly was treated with 60 μM bovine serum albumin (BSA), which has no specific interaction with sulfonamide ligands. Second, 60 μM of bCA-II was incubated with the PDS-PEG polymer which has no sulfonamide ligands on the backbone. No significant release of guest molecules was observed in both control experiments (Figure 3c). These data demonstrate that the observed guest release is indeed due to the specific ligand-protein interactions. Similar to P1 and P2, P3 was also found to be sensitive to 10 mM GSH, suggesting that P3 has stimuli responses to both specific protein and redox environment. Note that the GSH-induced release saturates at about 40%, which is lower than the observed guest release in P1 and P2. This is attributed to the possibility that since the hydrophobic groups in this case are the remaining PDS units, the assembly is first crosslinked due to the initially added GSH.29 This possibility is supported by the fact that the size of the assembly is retained in the presence of GSH (Figure 3b).
Finally, we were interested in using the PDS-PEG polymer to introduce concurrent sensitivity to three different stimuli, viz. physical, chemical, and biological stimuli. To this end, we converted majority of the PDS units to o-nitrobenzyl groups and a smaller percentage of the PDS units to sulfonamide ligands via sequential reactions of the PDS-PEG polymer with the respective thiols. The molar ratio of PEG, o-nitrobenzyl and sulfonamide moieties in this polymer (P4) was found to be 0.3:0.5:0.2 from 1H NMR, which is close to the feed ratio of the two thiols. As shown by DLS, P4 assembled in water to form nanoassemblies with a size of ~30 nm (Figure 4b). Since P4 contains light sensitive o-nitrobenzyl moieties, redox-sensitive disulfide linkages and protein-binding ligands, these stimuli would hopefully affect the hydrophobicity-hydrophilicity ratio of P4 and thus induce the disassembly. Indeed, the sizes of P4 solutions all decreased when light, GSH or bCA was applied as a single stimulus, indicating the dissociation of micelles (Figure 4b). In addition, all three stimuli caused significant release of guest molecules on different time scales, as ascertained by Nile red release profiles (Figure 4c–e). These studies for P4 indicate that physical, chemical and biological stimuli responses can be collectively introduced by this PPM strategy to construct multi stimuli-responsive polymers.
Figure 4.
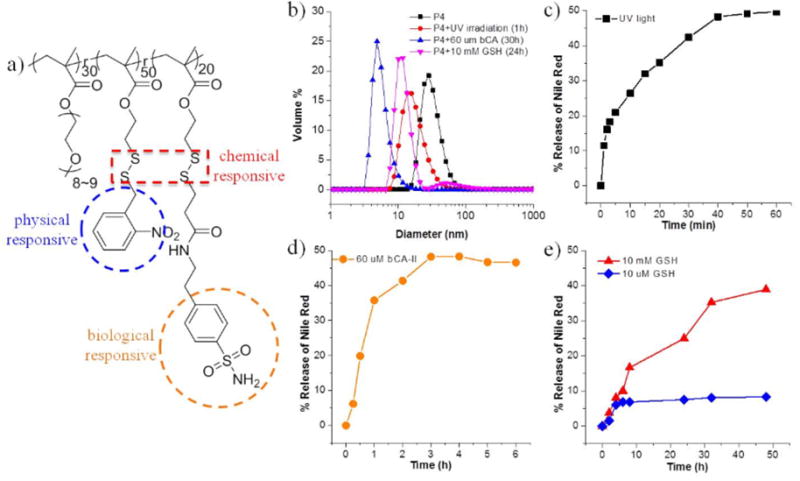
(a) Chemical structure of P4; (b) DLS results of P4 micelle solution (0.5 mg/mL) under different stimuli; Nile Red release profiles of P4 (c) under UV irradiation at 365 nm; (d) in the presence of 60 μM bCA-II and (e) in the presence of 10 mM and 10 μM GSH.
Apart from single stimulus response, we have also evaluated the disassembly, when triggered by multiple stimuli. It is noteworthy that the size of P4 slightly increased when GSH and light were utilized simultaneously, however, the autocorrelation function for reliable DLS measurements could not be obtained, indicating the disruption of most spherical micellar structures (Figure 5a). Effects of the combination of stimuli on guest release are shown in Figure 5b. After treatment of UV light in the presence of 60 μM bCA, the release profile has no obvious difference compared with applying light as a single stimulus. Since light triggered release is much faster than bCA, protein-binding induced disassembly may have little time to be realized before micelle dissociation by light. Interestingly, there is a huge promotion on guest release when light is applied along with 10 mM GSH. This may be due to the continuous reversible exchange/cleavage of disulfide bonds under UV irradiation, which activates disulfide bonds.51,52 In addition, the cleavage of nitrobenzyl group under UV light may loosen the micellar structures, so the water-soluble GSH has more access to disulfide linkages.
Figure 5.
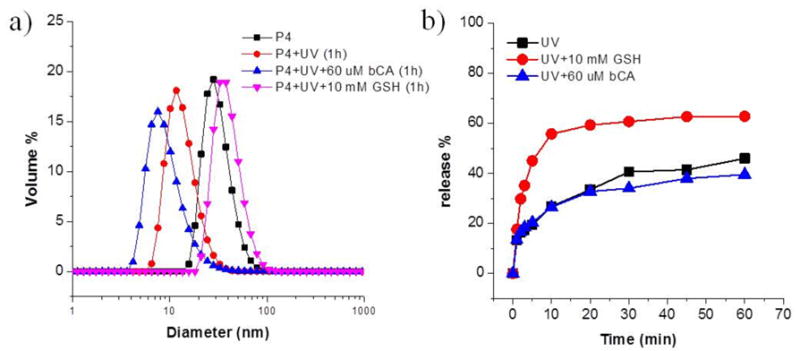
(a) DLS results of P4 micelle solution (0.5 mg/mL) under single stimulus or a combination of different stimuli; (b) Nile Red release profiles of P4 under UV light, the combination of light and bCA or GSH.
In summary, we have designed and synthesized a random copolymer PDS-PEG, the hydrophobic part of which provides opportunities to introduce different functional groups by post-polymerization modification through a simple thiol-disulfide exchange reaction. This method allows the ease of preparation and purification of different “smart” materials. We have engineering this polymer to achieve light/redox, pH/redox and protein/redox dual stimuli-responsive polymers, by simply varying the thiols that react with the PDS units in the precursor polymer. All of these polymers self-assemble in aqueous media to form nanoassemblies, stably encapsulate hydrophobic molecules, and release these guest molecules in response to the specific presence of either of the two stimuli. Additionally, we have utilized this strategy to construct a multi stimuli-responsive polymer, which is sensitive to light/protein/redox environment by sequential replacement of PDS moieties. Since the functionalities of thiols can be altered, we envisage that this approach could have broad applicability or even provide a simple framework for conveniently accessing intelligent materials.
Supplementary Material
Acknowledgments
The work is supported by the U.S. Army Research Office (W911NF-15-1-0568) to ST. XL gratefully acknowledges the support from the China Scholarship Council.
Footnotes
Supporting Information
Detailed synthetic procedures and characterizations of polymers. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Virezema DM, Comellas Aragones M, Elemans JAAW, Cornelissen JJLM, Rowan AE, Nolte RJM. Self-Assembled Nanoreactors. Chem Rev. 2005;105:1445–1490. doi: 10.1021/cr0300688. [DOI] [PubMed] [Google Scholar]
- 2.Astruc D, Chardac F. Dendritic Catalysts and Dendrimers in Catalysis. Chem Rev. 2001;101:2991–3024. doi: 10.1021/cr010323t. [DOI] [PubMed] [Google Scholar]
- 3.Theato P, Summerlin BS, O’Reilly RK, Epps TH., III Stimuli Responsive Materials. Chem Soc Rev. 2013;42:7055–7056. doi: 10.1039/c3cs90057f. [DOI] [PubMed] [Google Scholar]
- 4.Kataoka K, Harada A, Nagasaki Y. Block Copolymer Micelles for Drug Delivery: Design, Characterization and Biological Significance. Adv Drug Delivery Rev. 2001;47:113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 5.Ge Z, Liu S. Functional Block Copolymer Assemblies Responsive to Tumor and Intracellular Microenvironments for Site-Specific Drug Delivery and Enhanced Imaging Performance. Chem Soc Rev. 2013;42:7289–7325. doi: 10.1039/c3cs60048c. [DOI] [PubMed] [Google Scholar]
- 6.Rapoport N. Physical Stimuli-Responsive Polymeric Micelles for Anti-Cancer Drug Delivery. Prog Polym Sci. 2007;32:962–990. [Google Scholar]
- 7.Cheng R, Meng F, Deng C, Klok HA, Zhong Z. Dual and Multi-Stimuli Responsive Polymeric Nanoparticles for Programmed Site-specific Drug Delivery. Biomaterials. 2013;34:3647–3657. doi: 10.1016/j.biomaterials.2013.01.084. [DOI] [PubMed] [Google Scholar]
- 8.Mura S, Nicolas J, Couvreur P. Stimuli-Responsive Nanocarriers for Drug Delivery. Nat Mater. 2013;12:991–1003. doi: 10.1038/nmat3776. [DOI] [PubMed] [Google Scholar]
- 9.Zhuang J, Gordon MR, Ventura J, Li L, Thayumanavan S. Multi-Stimuli Responsive Macromolecules and Their Assemblies. Chem Soc Rev. 2013;42:7421–7435. doi: 10.1039/c3cs60094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klaikherd A, Nagamani C, Thayumanavan S. Multi-Stimuli Sensitive Amphiphilic Block Copolymer Assemblies. J Am Chem Soc. 2009;131:4830–4838. doi: 10.1021/ja809475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ganta S, Devalapally H, Shahiwala A, Amiji M. A Review of Stimuli-Responsive Nanocarriers for Drug and Gene Delivery. J Controlled Release. 2008;126:187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 12.Mano JF. Stimuli-Responsive Polymeric Systems for Biomedical Applications. Adv Eng Mater. 2008;10:515–527. [Google Scholar]
- 13.Li L, Raghupathi K, Song C, Prasad P, Thayumanavan S. Self-Assembly of Random Copolymers. Chem Commun. 2014;50:13417–13432. doi: 10.1039/c4cc03688c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galvin CJ, Genzer J. Applications of Surface-Grafted Macromolecules Derived from Post-Polymerization Modification Reactions. Prog Polym Sci. 2012;37:871–906. [Google Scholar]
- 15.Gauthier MA, Gibson MI, Klok HA. Synthesis of Functional Polymers by Post-Polymerization Modification. Angew Chem Int Ed. 2009;48:48–58. doi: 10.1002/anie.200801951. [DOI] [PubMed] [Google Scholar]
- 16.Günay KA, Theato P, Klok HA. History of Post-Polymerization Modification. In: Theato, Klok, editors. Fuctional Polymers by Post-polymerization Modification-Concepts, Guidelines and Applications. Verlag: Wiley-VCH; 2012. pp. 1–44. [Google Scholar]
- 17.Günay KA, Theato P, Klok H. Standing on the Shoulders of Hermann Staudinger: Post-Polymerization Modification from Past to Present. J Polym Sci Part A Polym Chem. 2013;51:1–28. [Google Scholar]
- 18.Markovic I, Stantchev TS, Fields KH, Tiffany LJ, Tomic M, Weiss CD, Broder CC, Strebel K, Clouse KA. Thiol/Disulfide Exchage is a Prerequisite for CXCR4-Tropic HIV-1 Envelope-Mediated T-cell Fusion During Viral Entry. Blood. 2004;103:1586–1594. doi: 10.1182/blood-2003-05-1390. [DOI] [PubMed] [Google Scholar]
- 19.Wedemeyer WJ, Welker E, Narayan M, Scheraga HA. Disulfide bonds and Protein Folding. Biochemistry. 2000;39:4207–4216. doi: 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh S, Basu S, Thayumanavan S. Simultaneous and Reversible Functionalization of Copolymers for Biological Applications. Macromolecules. 2006;39:5595–5597. [Google Scholar]
- 21.Wang L, Kristensen J, Ruffner DE. Delivery of Antisense Oligonucleotides Using HPMA Polymer: Synthesis of a Thiol Polymer and Its Conjugation to Water-Soluble Molecules. Bioconjugate Chem. 1998;9:749–757. doi: 10.1021/bc980034k. [DOI] [PubMed] [Google Scholar]
- 22.Wong L, Sevimli S, Zareie HM, Davis TP, Bulmus V. PEGylated Functional Nanoparticles from a Reactive Homopolymer Scaffold Modified by Thiol Addition Chemistry. Macromolecules. 2010;43:5365–5375. [Google Scholar]
- 23.Wong L, Boyer C, Jia Z, Zareie HM, Davis TP, Bulmus V. Synthesis of Versatile Thiol-Reactive Polymer Scaffolds via RAFT Polymerization. Biomacromolecules. 2008;9:1934–1944. doi: 10.1021/bm800197v. [DOI] [PubMed] [Google Scholar]
- 24.Bulmus V, Woodward M, Lin L, Murthy N, Stayton P, Hoffman A. A New pH-Responsive Glutathione-Reactive, Endosomal Membrane-Disruptive Polymeric Carrier for Intracellular Delivery of Biomolecular Drugs. J Controlled Release. 2003;93:105–120. doi: 10.1016/j.jconrel.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Zugates GT, Amderson DG, Little SR. Synthesis of Poly(β-amino ester)s with Thiol-Reactive Side Chains for DNA Delivery. J Am Chem Soc. 2006;128:12726–12734. doi: 10.1021/ja061570n. [DOI] [PubMed] [Google Scholar]
- 26.Talelli M, Vicent MJ. Reduction Sensitive Poly(L-glutamic acid) (PGA)-Protein Conjugates Designed for Polymer Masked-Unmasked Protein Therapy. Biomacromolecules. 2014;15:4168–4177. doi: 10.1021/bm5011883. [DOI] [PubMed] [Google Scholar]
- 27.Ryu J-H, Jiwpanich S, Chacko R, Bickertion S, Thayumanavan S. Surface-Functionalizable Polymer Nanogels with Facile Hydrophobic Guest Encapsulation Capabilities. J Am Chem Soc. 2010;132:8246–8247. doi: 10.1021/ja102316a. [DOI] [PubMed] [Google Scholar]
- 28.Jiwpanich S, Ryu J-H, Bickerton S, Thayumanavan S. Non-covalent Encapsulation Stabilites in Supramolecular Nanoassemblies. J Am Chem Soc. 2010;132:10683–10685. doi: 10.1021/ja105059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryu J-H, Chacko R, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. Self-Cross-Linked Polymer Nanogels: A Versatile Nanoscopic Drug Delivery Platform. J Am Chem Soc. 2010;132:17227–17235. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]
- 30.Bochet CG. Photolabile Protecting Groups and Linkers. J Chem Soc Perkin Trans. 2002;1:125–142. [Google Scholar]
- 31.Jochum FD, Theato P. Temperature- and Light-Responsive Smart Polymer Materials. Chem Soc Rev. 2013;42:7468–7483. doi: 10.1039/c2cs35191a. [DOI] [PubMed] [Google Scholar]
- 32.Zhao A, Sterner E, Coughlin EB, Theato P. o-Nitrobenzyl Alcohol Derivatives: Opportunities in Polymer and Materials Science. Macromolecules. 2012;45:1723–1736. [Google Scholar]
- 33.Zhao Y. Light-Responsive Block Copolymer Micelles. Macromolecules. 2012;45:3647–3657. [Google Scholar]
- 34.Liu G, Liu W, Dong CM. UV- and NIR-Responsive Polymeric Nanomedicines for On-Demand Drug Delivery. Polym Chem. 2013;4:3431–3443. [Google Scholar]
- 35.Russo A, DeGraft W, Friedman N, Mitchell JB. Selective Modulation of Glutathione Levels in Human Normal Versus Tumor Cells and Subsequent Differential Response to Chemotherapy Drugs. Cancer Res. 1986;46:2845–2848. [PubMed] [Google Scholar]
- 36.Saito G, Swanson JA, Lee KD. Drug Delivery Strategy Utilizing Conjugation via Reversible Disulfide Linkages: Role and Site of Cellular Reducing Activities. Adv Drug Delivery Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 37.Balendiran GK, Dabur R, Fraser D. The Role of Glutathione in Cancer. Cell Biochem Funct. 2004;22:343–352. doi: 10.1002/cbf.1149. [DOI] [PubMed] [Google Scholar]
- 38.Heffernan MJ, Murthy N. Polyketal Nanoparticles: A New pH-Sensitive Biodegradable Drug Delivery Vehicle. Bioconjugate Chem. 2005;16:1340–1342. doi: 10.1021/bc050176w. [DOI] [PubMed] [Google Scholar]
- 39.Shim MS, Kwon YJ. Controlled Delivery of Plasmid DNA and siRNA to Intracellular Targets Using Ketalized Polyethylenimine. Biomacromolecules. 2008;9:444–455. doi: 10.1021/bm7007313. [DOI] [PubMed] [Google Scholar]
- 40.Lee SJ, Min KH, Lee HJ, Koo AN, Rim HP, Jeon BJ, Jeong SY, Heo JS, Lee SC. Ketal Cross-Linked Poly(ethylene glycol)-Poly(amino acid)s Copolymer Micelles for Efficient Intracellular Delivery of Doxorubicin. Biomacromolecules. 2011;12:1224–1233. doi: 10.1021/bm101517x. [DOI] [PubMed] [Google Scholar]
- 41.Moskaug JO, Sandvig K, Olsnes S. J Biol Chem. 1987;262:10339–10345. [PubMed] [Google Scholar]
- 42.Savariar EN, Ghosh S, González DC, Thayumanavan S. Disassembly of Noncovalent Amphiphilic Polymers with Proteins and Utility in Pattern Sensing. J Am Chem Soc. 2008;130:5416–5417. doi: 10.1021/ja800164z. [DOI] [PubMed] [Google Scholar]
- 43.Takaoka Y, Sakamoto T, Tsukiji S, Narazaki M, Matsuda T, Tochio H, Shirakawa M, Hamachi I. Self-Assembling Nanoprobes that Display Off/On 19F Nuclear Magnetic Resonance Signals for Protein Detection and Imaging. Nat Chem. 2009;1:557–561. doi: 10.1038/nchem.365. [DOI] [PubMed] [Google Scholar]
- 44.Azagarsamy MA, Yesilyurt V, Thayumanavan S. Disassembly of Dendritic Micellar Containers Due to Protein Binding. J Am Chem Soc. 2010;132:4550–4551. doi: 10.1021/ja100746d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yesilyurt V, Ramireddy R, Azhagarsamy MA, Thayumanavan S. Accessing Lipophilic Ligands in Dendrimer-Based Amphiphilic Supramolecular Assemblies for Protein-Induced Disassembly. Chem Eur J. 2012;18:223–229. doi: 10.1002/chem.201102727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo J, Zhuang J, Wang F, Raghupathi KR, Thayumanavan S. Protein AND Enzyme Gated Supramolecular Disassembly. J Am Chem Soc. 2014;136:2220–2223. doi: 10.1021/ja4108676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krishnamurthy VM, Kaufman GK, Urbach AR, Gitlin I, Gudiksen KL, Weibel DB, Whitesides GM. Carbonic Anhydrase as A Model for Biophysical and Physical-Organic Studies of Proteins and Protein-Ligand Binding. Chem Rev. 2008;108:946–1051. doi: 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alterio V, Di Fiore A, D’Ambrosio K, Supuran CT, De Simone G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms. Chem Rev. 2012;112:4421–4468. doi: 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- 49.Molla MR, Prasad P, Thayumanavan S. Protein-Induced Supramolecular Disassembly of Amphiphilic Polypeptide Nanoassemblies. J Am Chem Soc. 2015;137:7286–7289. doi: 10.1021/jacs.5b04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H, Zhuang J, Raghupathi KR, Thayumanavan S. A Supramolecular Dissociation Strategy for Protein Sensing. Chem Commun. 2015;51:17265–17268. doi: 10.1039/c5cc07408h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li L, Song C, Jennings M, Thayumanavan S. Photoinduced Heterodisulfide Metathesis for Reagent-Free Synthesis of Polymer Nanoparticles. Chem Commun. 2015;51:1425–1428. doi: 10.1039/c4cc08000a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michal BT, Spencer EJ, Rowan SJ. Stimuli-Responsive Reversible Two-Level Adhesion from a Structurally Dynamic Shape-Memory Polymer. ACS Appl Mater Interfaces. 2016;8:11041–11049. doi: 10.1021/acsami.6b01251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.