

Abstract
Nowadays, nanotechnology-based modulation of the immune system is presented as a cutting-edge strategy, which may lead to significant improvements in the treatment of severe diseases. In particular, efforts have been focused on the development of nanotechnology-based vaccines, which could be used for immunization or generation of tolerance. In this review, we highlight how different immune responses can be elicited by tuning nanosystems properties. In addition, we discuss specific formulation approaches designed for the development of anti-infectious and anti-autoimmune vaccines, as well as those intended to prevent the formation of antibodies against biologicals.
Keywords: nanotechnology, immune system, tolerance, stimulation, autoimmune disease, vaccine
Graphical abstract
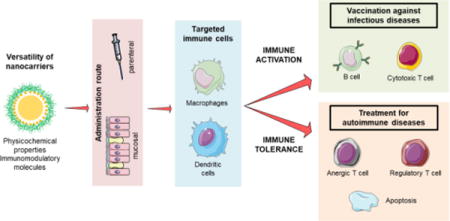
1. Introduction
The modulation of the immune system is the base of new and promising therapies for some of the most prevalent and/or severe diseases of our time, such as cancer, HIV, and type 1 diabetes. The development of treatments based on this modulation is a field in expansion, where the contribution of nanotechnology is growing exponentially [1–3]. Based mainly on the molecular principles that govern the interaction between pathogens and immune cells, the use of nanotechnology represents a new way of communication with the immune system. Both, the composition and the physicochemical characteristics of nanocarriers, can influence their interaction with immune cells. By mimicking the size of microorganisms (bacteria and viruses) and incorporating key molecules involved in immune processes (TLR agonists, cytokines, etc.), nanocarriers can be taken up by the immune cells and modulate their responses. Besides, the use of nanocarriers decorated with targeting moieties can favor their preferential access to specific immune cell populations [2,4–9]. Importantly, the tunable nature of nanotechnology offers the possibility of reinforcing the desired aspect of immunomodulation, which maybe (i) the activation of the immune system in order to generate an immune response against a specific antigen, or (ii) the induction of immunotolerance against antigens and immunoactive drugs. The first option improves the chances of controlling infectious diseases that do not respond well to traditional vaccines, such as HIV or tuberculosis, among others [10–12]. The second, and less explored option, refers to the development of vaccines against autoimmune diseases as well as the targeted administration of immunomodulatory drugs [13–15]. The capacity of nanotechnology to elicit different responses comes from its versatility, gained through the specific combination and meticulous choice of its molecular components, and from the physicochemical properties of the nanosystems.
In this review, we first summarize how nanotechnology may help reaching the desired cell population, and achieving its specific modulation. Then, we offer an overview of the role that nanotechnology has played in the development of new vaccines against infectious diseases, followed by an analysis of its contribution to the treatment of autoimmune diseases. Finally, recent achievements to fight antidrug antibodies are summarized.
2. Access of nanostructures to target cells
For a nanovaccine to be effective, it first needs to access the tissues where the target cells are present. Depending on the administration route, different physiological barriers must be overcome to reach these cells. Thus, nanoparticles (NPs) should be specifically engineered to go preferentially to the target tissue from the site of administration.
2.1. Routes of administration of nanocarriers intended for immunomodulation
Although immune cells are distributed throughout the body, the key cells involved in immunity are concentrated in the lymphoid tissues. Hence, targeting these tissues facilitates the access to immune cells and, consequently, increases the efficacy of administered nanovaccines. Lymphoid tissues are directly accessible through the mucosal surfaces, such as airways, the intestinal tract, or the vagina, although a more straight way to target them is by parenteral injection. The way antigens reach the lymph nodes (LN), following different modalities of administration is illustrated in Fig. 1.
Figure 1. Vaccine administration routes.
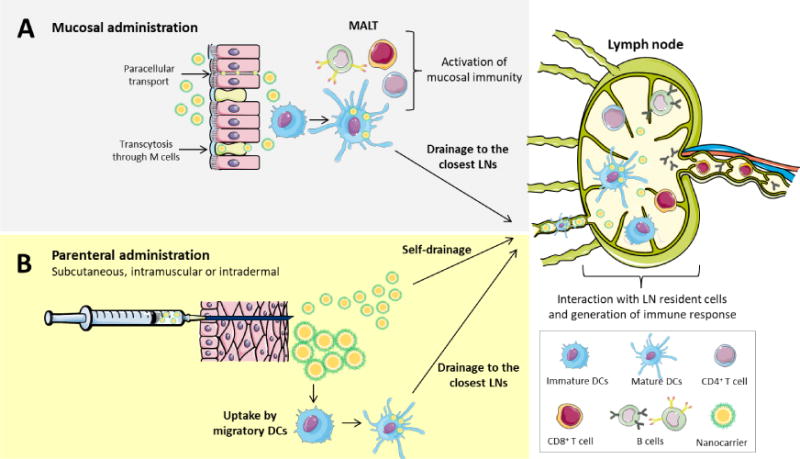
The main administration routes for vaccines are mucosal and parenteral. (A) Mucosal administration refers to the administration mainly through the nasal, oral or vaginal routes. In all these cases, nanocarriers need to reach the mucosal-associated lymphoid tissues (MALT). This can be principally achieved either by a paracellular or transcellular across the microfold (M) cells. At the level of M cells or underneath the epithelium, nanocarriers will encounter the resident dendritic cells and activate them, generating a mucosal immunity while, at the same time, some dendritic cells will drain to the closest lymph node and activate a systemic immune response. (B) Parenteral administration includes subcutaneous, intramuscular or intradermal injection of the nanosystems. The nanocarriers are deposited in the interstitium, where they can have two different fates: self-drain to the closest lymph node or be taken up by migratory dendritic cells, which then will migrate to the closest lymph node.
2.1.1. Mucosal administration
Following mucosal administration, a needle-free and appealing route for vaccination, it is possible to induce both, mucosal and systemic, immune responses [16]. The mucosa-associated lymphoid tissues (MALT) are connected to the mucosal environment through the M cells, which are specialized in the transcytosis of microorganisms and particulate components [3,16]. The activation of mucosal resident T and B cells can be of great importance for an efficient mucosal vaccination [17,18]. This is the reason our group and many others have explored the potential of nanocarriers for the transport of antigens across different mucosae in order to reach a proper stimulation of the immune system. Moreover, the administration of nanocarriers through mucosal routes has also been investigated for tolerance generation [19].
In order to elicit an adequate response, NPs need, first, to overcome the mucus layer that covers the mucosal surfaces. Then, once in contact with the epithelium, nanocarriers are transported either by M cells or by regular epithelial cells [20–22]. NPs can also be internalized by paracellular transport if their composition includes components that can open tight junctions [23]. Moreover, it has also been described that dendritic cells can take up NPs by extending their dendrites into the lumen [24,25].
The specific physiology of the mucosal surfaces is different throughout the body and, hence, the optimal properties for the nanocarriers to cross them may also be different. Initially, bioadhesive nanosystems were thought to be a promising strategy to facilitate the interaction of nanocarriers with the mucus layer and a number of strategies have been described for that purpose [26,27]. For example, Nochi et al. developed adhesive cationic nanogels made of cholesterol-modified pullulan that were able to increase the survival rate of mice after intranasal vaccination against tetanus and the botulin neurotoxin [28]. However, it was also observed that if the systems were retained in the mucus by high adhesive forces, they could be soon eliminated by the clearance mechanisms. This disadvantage led to the engineering of nanocarriers with mucodiffusive properties that would allow them to cross the mucus layer and reach the epithelium. Nowadays, a precise balance between mucoadhesive and mucodiffusive properties is believed to be critical for the effectiveness of nanocarriers delivered through mucosal routes. For good mucodiffusion properties, it has been reported that particle size should be smaller than the mucus mesh size [29]. Although there are studies where microparticles (MPs) showed better results than NPs after oral administration [30,31], in general, the recent trend has been to consider that NPs perform better than MPs [32–37]. In this regard, our group reported that the transport of pegylated polylactic acid (PEG-PLA) NPs across the nasal mucosa was higher than that of MPs. Furthermore, the smaller micrometric sizes (1 and 5 μm) also crossed the epithelium more efficiently than 10 μm particles, with no significant differences between 1 and 5 μm [38]. Interestingly, based on recent in vivo data, very small nanometric sizes (30 nm) may not be as effective as larger ones (200 nm) [39].
Besides the particle size, other nanocarrier’s features may also have important consequences for mucopermeation. For example, in 1998, our group described for the first time that the presence of a PEG coating in NPs made of PEG-PLA had an important role in increasing their transport rate through the nasal [40] and intestinal epithelia [41]. Furthermore, other authors have described that the presence of an adequate PEG coating allows particles with a size in the range 200 – 500 nm to penetrate across the mucus [42,43]. In brief, we may conclude that the size and composition of the nanocarriers, and notably the surface composition, may influence the particle transport across mucosal surfaces.
2.1.2. Parenteral administration
Intramuscular, subcutaneous, and intradermal administrations are the main routes of vaccination. Following these modalities of administration, and depending on their physicochemical properties and composition, NPs can drain directly to the closest lymph node, or stay in the injection site and attract migratory dendritic cells or macrophages. Overall, the main conclusion drawn from several reviews in the literature is that sizes up to 100 nm are able to self-drain to the nearest lymph node, being the drainage usually inversely proportional to the particle size [3,9,34,44–47]. However, very small particles (< 10 nm) can directly drain to blood capillaries [48] and those that reach the lymph nodes have shown limited retention [49]. With regard to the surface charge, some authors have indicated that the drainage of negatively charged NPs to the LN is facilitated by their repulsion with the negatively charged extracellular matrix. This repulsion acts as a driving force moving NPs to the lymphatic system [50–52]. On the other hand, cationic nanosystems tend to form a depot after parenteral administration, being taken up by peripheral and migratory APCs or slowly draining to LNs [53]. Nevertheless, this charge effect may be counterbalanced by the appropriate adjustment of the particle size. For example, Zeng et al. showed that 30 nm cationic micelles were able to self-drain to the closest lymph nodes [54]. Similarly, Kim et al. have reported that both small cationic and anionic poly(γ-glutamic acid)-based nanosystems (30–60 nm) were able to self-drain to the closest lymph node [55]. Finally, the presence of PEG on the surface of the nanocarriers, that usually renders their surface charge close to neutrality, has a positive effect in the drainage to the LN [56–59]. This does not necessarily translate into a higher interaction with immune cells [51,56,60], as the degree of pegylation and the PEG molecular weight may have an impact on the NP opsonization [8,61].
In the case of intravenous (IV) administration, it has been found the possibility to generate a tolerogenic effect by antigen-loaded nanocarriers [62,63]. The hypothesis to explain this result is that NPs delivered by this route are mainly accumulated in the liver and engulfed by Kupffer cells, which are essential for the elimination of apoptotic cells and other debris from the blood, mechanism associated with the maintenance of peripheral tolerance [64]. In this situation, Kupffer cells and liver dendritic cells were shown to have an increased expression of PD-L1 in their surface, which contributes to a higher tolerance [65].
Overall, the conclusion from the reported studies is that the final outcome of the nanocarriers is determined by the simultaneous influence of their properties including particle size, surface charge, shape, hydrophobicity and stiffness, among others (Fig. 2).
Figure 2. Summary of the influence of the physicochemical properties of nanocarriers (particle size and surface charge) in the fate of the nanosystems after administration.
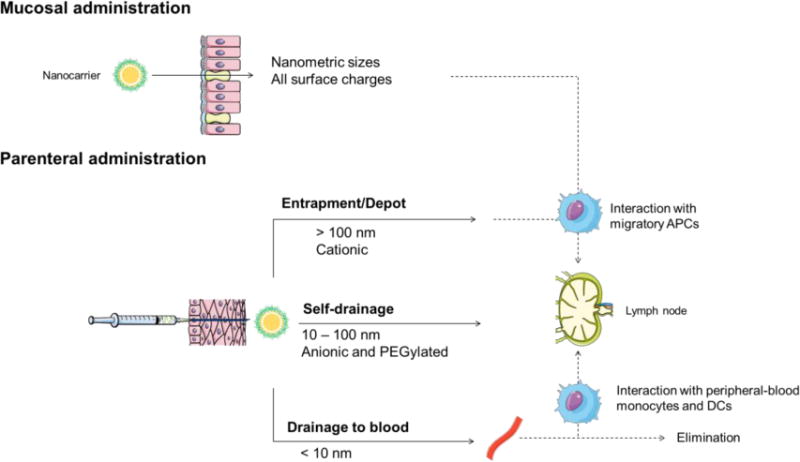
Both particle size and surface charge play an important role in the outcome of nanosystems once administered, either by mucosal or parenteral routes.
2.2. Targeted cell populations in immunomodulation and immunological responses
The immune system is comprised by circulating cells, which are in charge of capturing peripheral antigens (monocytes, macrophages and dendritic cells) and more static cells, such as B and T cells. All these cells are targets of interest for immunomodulation depending on the desired type of response (Fig. 3).
Figure 3. Immune cell network.
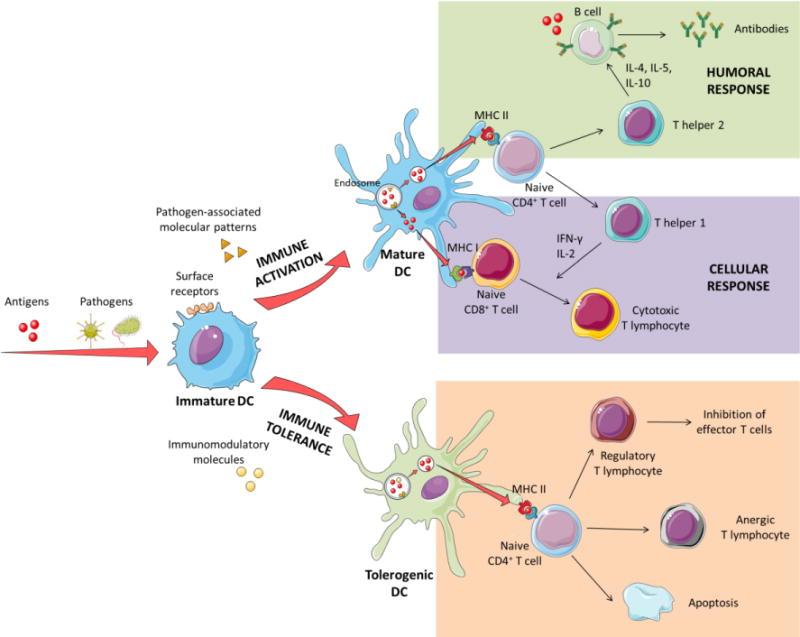
Schematic overview of the generation of different immune responses by dendritic cells. Antigens, pathogens and other molecules are taken up by immature dendritic cells. In the case of pathogens or systems expressing pathogen-associated molecular patterns (PAMPs), their internalization by dendritic cells leads to their presentation by class II major histocompatibility complexes (MHC II) to naïve CD4+ T cells, which activate T helper cells (Th). Th2 cells produce IL-4, IL-5 and IL-10, which stimulate B cells to produce antibodies against the antigen. At the same time, antigens themselves can interact directly with B cells and activate them. Antigens can also be found in the cytosol of dendritic cells, which allows them to be presented by class I major histocompatibility complexes (MHC I), directly activating cytotoxic T lymphocytes. In this case, Th1 cells produce IFN-γ and IL-2, which favor cellular activation and hence, cytotoxic T cell responses.
In the case of antigens presented in the absence of co-stimulatory molecules, or in the presence of immunomodulatory molecules for tolerance, dendritic cells are driven to a state of immune tolerance. In this state, dendritic cells can inhibit T cell activation by different mechanisms. Different stimuli, such as IL-10 or PD-L1 can cause T regulatory cells proliferation that, at the same time, can inhibit effector T cells. Furthermore, the absence of co-stimulatory surface molecules can lead to an unresponsive state in T cells known as anergy. Finally, co-stimulatory Fas-signaling in the immune synapsis can lead to T cell apoptosis and deletion.
In order to generate a biased immune response, two different approaches can be followed, and they involve (i) the design of nanocarriers that can reach preferentially one subset of immune cells, either by passive or active targeting. For this purpose, nanocarriers features, i.e. particle size, surface charge or shape, can be modulated in order to facilitate their passive access to immune cells; however a good discrimination between cells can only be achieved through the use of active targeting ligands. (ii) The use of adjuvants that modify the response given for a specific immune cell subset. These immunomodulatory molecules may mimic pathogen-associated molecular patterns (PAMPs), which are bacterial cell wall components, viral RNA, and CpG DNA. These molecules activate different receptors that will lead to cellular or humoral immune responses. Similarly, different cytokines and other immunomodulatory molecules, such as rapamycin, vitamin D3 or phosphatidylserine (PS), can be loaded into nanosystems to induce tolerogenic responses.
Based on this scheme, the different targeted populations in immunomodulation are monocytes, macrophages and dendritic cells (DCs). Monocytes and macrophages are one of the most common phagocytic cells in the body and represent the first innate defense line. They can be either circulating or resident in tissues, clearing pathogens and apoptotic cells. These cells are able to drain to the injury site, attracted by chemokines, and, hence, they have an important role in presenting antigens and releasing cytokines that modulate the immune response. In addition, they mediate inflammatory processes, which are relevant in a large variety of inflammatory diseases as well as in tumor growth and metastasis.
Several in vitro studies have been conducted in order to determine the characteristics of NPs and MPs that are key for the passive targeting to macrophages. All these studies have shown that both, particle size and shape, may influence the internalization efficiency by macrophages (Table 1A). In this sense, in the past and mainly based on in vitro studies, it was assumed that particles in the micrometric range were well recognized by macrophages [66–72]. Nevertheless, recent studies have questioned this assertion and the current tendency is to believe that NPs can be very efficiently taken up by macrophages [73,74]. On the other hand, regarding the influence of the surface charge on the uptake of NPs by macrophages, several in vitro and in vivo studies have shown different results. Indeed, while in some cases cationic nanosystems were taken up by macrophages at a greater extent than neutral and negative ones [73,75–78], in others, the negative charge was preferable for an efficient uptake [79–84]. For example, Nakanishi et al. reported that positive multilamellar vesicles elicited stronger cellular and humoral immune responses both in vitro and in vivo than neutral or negative systems [78]. On the contrary, Fromen et al. observed that after a pulmonary instillation of anionic and cationic PRINT hydrogels, negative nanosystems were engulfed in a greater manner by pulmonary macrophages [79]. More examples of these somehow contradictory results are summarized in Table 1A.
Table 1A.
Summary of the different strategies considered for a passive targeting to monocytes and/or macrophages
| Studied feature | Composition | Particle size | Surface charge | Key results | Ref. |
|---|---|---|---|---|---|
| Particle size | Non-functionalized polystyrene | 0.9, 1.9, 2.3, 3, 4.3, 5.7, 9 μm | n.d. | ≈ 2 – 3 μm more readily phagocytosed in vitro than smaller (≈ 1 μm) and larger particles (≈ 6 μm) | [68] |
| Polystyrene and PLGA | 1, 2, 3.2, 6.4, 10.1 μm | −87.6 mV/− 9.7 mV | ≈ 6 μm polystyrene particles and ≈ 3 μm PLGA particles were the most efficiently phagocytosed particles in vitro | [70] | |
| PLGA | 1.5, 3.3, 6.1, 10 μm | n.d. | More efficient in vitro delivery of the cargo achieved with ≈ 3 μm particles than with larger (≈ 6 – 10 μm) and smaller ones (≈ 1 μm) | [71] | |
| Carboxylated polystyrene | 0.02, 0.04 and 1 μm | Anionic | 48 h after in vivo administration, more 1 μm particles were co-localized with macrophages in the draining LNs than the smaller particles (0.02 – 0.04 μm) | [72] | |
| Carboxymethyl chitosan grafted and chitosan hydrochloride grafted | 149.2 – 157.3, 300.7 and 456.5 nm | − 38.4 to − 13.2 mV; + 14.8 to + 34.6 mV | Larger NPs were more efficiently taken up in vitro | [73] | |
| Carboxylated polystyrene | 0.5 – 4.5 μm | Anionic | Small-particles group (0.5, 1 and 2 μm) was internalized in vitro at a higher rate than the group of larger particles (3 and 4.5 μm) | [74] | |
| Shape | Non-functionalized polystyrene | Axis from 1 to 12.5 μm | n.d. | Better phagocytosis in vitro in alveolar macrophages for MPs that allow a lower contact angle (ellipsoids and disks) | [66] |
| Particle size and shape | Non-functionalized polystyrene | 0.5 – 3 μm | n.d. | Particles with the longest dimension of 2 – 3 μm showed the maximum attachment to macrophages in vitro | [69] |
| Surface charge | Carboxylated polystyrene covalently coated with BSA or PLL | 1 – 4.5 μm | − 58.3 to − 18.4 mV; + 39.6 to + 49.7 mV | Cationic particles were better taken up than anionic particles by macrophages in vitro | [75] |
| Carboxylated polystyrene covalently coated with BSA, PLL, IgG or PEI | 1 μm | − 21.1 to − 0.8 mV; + 38.2 to + 45.7 mV | Cationic MPs were better taken up by macrophages than negative particles in vitro | [76] | |
| Carboxymethyl chitosan grafted and chitosan hydrochloride grafted | 149.2 – 157.3; 300.7 and 456.5 nm | − 38.4 to +34.6 mV | Positively charged NPs were more efficiently taken up than negatively charged ones in vitro | [73] | |
| DOPC/DODAP and DOPC/DOPS liposomes | 120 nm | Cationic, neutral and anionic | Positively charged liposomes were better taken up than negative and neutral liposomes by rat macrophages in vitro | [77] | |
| PC/Chol/SA, PC/Chol/PA, PC/Chol multilamelar vesicles | n.d. | − 18.6 to + 8.9 mV | Positively charged liposomes showed a higher uptake rate by macrophages in vitro and better immune responses in vivo than neutral and negative liposomes | [78] | |
| PRINT hydrogel, derived from HP4A | 80 nm × 80 nm × 320 nm | Cationic and anionic | After pulmonary instillation, anionic NPs were more efficiently taken up by macrophages than cationic NPs | [79] | |
| Cyanoacrylate NPs coated with dextran or diethylaminoethyl-dextran | 200 nm | + 30 mV/− 20 mV | Anionic NPs showed a higher internalization by macrophages in vitro, and also higher anti-inflammatory properties than the cationic ones | [80] | |
| DSPC/DODAB/Chol or DSPC/DSPG/Chol liposomes | 180 – 190 nm | − 29, 0.1 and + 25 mV | Anionic liposomes were more effective than neutral ones in vivo and in vitro. Cationic liposomes were more potent, but this was associated with a higher cytotoxicity of the forming polymers | [81] | |
| Neutral, carboxylated and aminated polystyrene | 500 nm | − 50, − 0.5 and + 40 mV | Anionic particles decreased the infiltration of inflammatory monocyte-derived macrophages in vivo to a larger extent than cationic or neutral NPs of the same size | [83] |
BSA: bovine serum albumin; Chol: cholesterol; DODAB: dimethyl dioctadecyl ammonium bromide; DODAP: 1,2-dioleoyl-3-dimethylammonium propanediol; DOPC: 1,2-dioleolyl-sn-glycero-3-phosphatidylcholine; DOPS: 1,2-dioleolyl-sn-glycero-3-phosphatidylserine; DSPC: 1,2-distearoyl-sn-glycero- 3-phosphocholine; DSPG: distearoyl-phosphatidylglycerol; HP4A: tetra(ethylene glycol) monoacrylate; IgG: immunoglobulin G; LN: lymph node; MP: microparticle; n.d.: not determined; NP: nanoparticle; PA: L-a-dimyristoyl phosphatidic acid; PEI: polyethylenimine; PC: egg phosphatidylcholine; PLGA: poly(lactic-co-glycolic) acid; PLL: poly-ι-lysine; PRINT: particle replication in non-wetting templates; SA: stearylamine
The studies above-mentioned highlight the lack of a clear conclusion on the best way to target macrophages through the modification of nanocarrier’s particle size and surface charge. Furthermore, their composition is probably an important factor dictating such interaction. To improve this, some authors attempted an active targeting to specific macrophage receptors (Table 1B). For example, iron-oxide NPs coated with IgG, were shown to be taken up by monocytes and macrophages in a much higher extent than the uncoated ones [85]. Other authors have found that targeting the mannose receptor was a way to enhance the interaction of liposomes with tumoral macrophages after IV administration [86].
Table 1B.
Summary of the different strategies followed for an active targeting to monocytes and macrophages
| Ligand | Nanosystem | Key results | Ref. |
|---|---|---|---|
| IgG coating | SPIO | Higher in vitro uptake and sustained distribution in lymphoid tissue, in comparison to non-coated SPIO | [85] |
| Mannosylation | Liposomes | Functionalized liposomes accumulated in tumor-associated macrophages better than in other lung areas | [86] |
| Folate | Dendrimer (G5) | High in vitro internalization by macrophages in a receptor-specific manner and great in vivo anti-inflammatory properties | [88] |
| Dextran | Dextran conjugates | After peritoneal administration, larger conjugates selectively associated with macrophages of the adipose tissue | [89] |
G5: generation 5; IgG: immunoglobulin G; SPIO: superparamagnetic iron-oxide nanoparticles
In other studies intended to induce tolerance, authors have taken advantage of the specific expression of folate receptor β in activated macrophages in inflamed joints. For example, folate-functionalized dendrimers showed an increased joint accumulation after IV injection in collagen-induced arthritis (CIA) mice model [87,88]. Similarly, the specific recognition of dextran by scavenger receptors was explored to develop an anti-inflammatory therapy. Namely, dextran NPs containing dexamethasone were used to target pro-inflammatory macrophages from obese patients [89]. On the other hand, hyaluronic acid, has been proposed as a way to specifically interact with the CD44 receptor, found in lymphocytes, among other cells [90,91]. In addition, it has been recently reported that low molecular weight hyaluronic acid exhibits immunostimulant properties [92] and that these properties can be related to the ability of hyaluronan fragments to activate TLR2 and TLR4 [93,94]. Furthermore, recent reports have also claimed the capacity of hyaluronic acid to polarize tumor-associated macrophages from M2 towards a M1 anti-tumoral subtype [95], although further investigation is still needed to determine the impact of these studies.
Dendritic cells (DCs) are the most important antigen-presenting cells (APCs) and have a key role in the modulation of the immune system [96]. As illustrated in Fig. 3, DCs internalize antigens from their surroundings, process them in endosomes/lysosomes and present the resulting peptides through the class II major histocompatibility complex (MHC II), leading to a specific CD4+ T cell activation and proliferation [97]. On the other hand, if the antigens are found in the cytosol of DCs, as in the case of intracellular infections, the peptides will be presented by class I MHC (MHC I) to naïve CD8+ T cells, activating cellular responses. In some cases, external antigens can be translocated from endosomes to the cytosol and, thus, be presented via MHC I, process known as cross-presentation [3,98]. This phenomenon is of great importance in antitumor and infectious disease vaccination where a potent cellular response is required [99]. In both cases, besides antigen presentation, a co-stimulation of T cells through cytokines or co-stimulatory signals is normally needed [45].
Significant attempts have been made to passively (Table 2A) or actively (Table 2B) target DCs using nanocarriers (Fig. 4). DCs have a high phagocytic capacity similar to that of macrophages, however, unlike them, DCs preferentially ingest small virus-size particles [72,100,101]. Therefore, a way to passively target DCs is through the reduction of the nanocarriers’ size. On the other hand, it is also known that providing nanocarriers with a positive surface charge enhances the chances for them to interact with DCs and macrophages [75,79,101–103]. Nevertheless, irrespective of the influence of size and surface charge in the specific uptake of particles by dendritic cells, it seems clear that the most effective approach to precisely target DCs would be providing the nanocarriers with specific targeting ligands (Table 2B) [104]. For example, Cruz et al. systematically studied this possibility by functionalizing pegylated poly(lactic-co-glycolic) acid (PEG-PLGA) NPs with antibodies to target either CD40 (TNF-α family receptor), CD11c (integrin receptor) or DEC-205 (C-type lectin receptor) receptors. All NPs contained an antigen (OVA) and TLR3 and 7 agonists, but only those with a specific ligand showed increased CD8+ T cell activation, both in vitro and in vivo [105]. The targeting of the mannose receptor has also been reported as a strategy to increase the activation of DCs in vitro and in vivo [106,107].
Table 2A.
Summary of the different strategies followed for a passive targeting to dendritic cells
| Studied feature | Composition | Particle size | Surface charge | Key results | Ref. |
|---|---|---|---|---|---|
| Particle size | Carboxylated polystyrene | 0.02, 0.04 and 1 μm | n.d. | 48 h after in vivo administration, smaller sizes (0.02 – 0.04 μm) were found in a higher amount in DCs, in comparison to 1 μm particles | [72] |
| Carboxylated polystyrene plain or coated with PLL, PA, PrS, TT or WGA | 0.1, 0.5, 0.9 and 4.5 μm | − 66.9 to + 41.4 mV | Particles smaller than 0.5 μm had better in vitro uptake by DCs | [101] | |
| Surface charge | PC/PG/Chol, PC/PS/Chol, PC/TAP/Chol liposomes | 180 to 279 nm | − 54.2 to + 44.2 mV | Positively charged liposomes had a greater interaction with DCs in vitro than anionic ones | [102] |
| Carboxylated polystyrene covalently coated with BSA or PLL | 1 – 4.5 μm | − 58.3 to − 18.4 mV; + 39.6 to + 49.7 mV | Cationic particles uptake by DCs in vitro was higher than for anionic particles | [75] | |
| PRINT hydrogel derived from HP4A | 248 to 285 nm | − 38 to + 45 mV | Pulmonary administration of cationic NPs elicited stronger antibody responses than negative NPs administration, which correlates with a higher uptake in vitro of the former by DCs | [103] | |
| PRINT hydrogel derived from HP4A | 80 nm × 80 nm × 320 nm | Cationic and anionic | After pulmonary administration cationic NPs, rather than negative ones, preferentially associated to lung DCs | [79] | |
| Carboxylated polystyrene plain or coated with PLL, PA, PrS, TT or WGA | 0.1, 0.5, 0.9 and 4.5 μm | − 66.9 to + 41.4 mV | Positively charged particles were better internalized by DCS in vitro than negative ones, especially when they were of micrometric sizes | [101] |
Chol: cholesterol; DC: dendritic cell; HP4A: tetra(ethylene glycol) monoacrylate; n.d.: not determined; NP: nanoparticle; PC: dimyristoyl phosphatidylcholine; PG: dimyristoyl phosphatidylglycerol; PA: L-a-dimyristoyl phosphatidic acid; PLL: poly-ι-lysine; PRINT: particle replication in non-wetting templates; PrS: protamine sulphate; TAP: dimyristoyl trimethylammoniumpropane; TT: tetanus toxoid; WGA: wheat germ agglutinin
Table 2B.
Summary of the different strategies adopted for the active targeting to dendritic cells
| Ligand | Nanosystem | Key results | Ref. |
|---|---|---|---|
| Ab for CD40, CD11c, DEC-205 | PEG-PLGA NPs | Only active targeting improved CD8+ T cell activation in vitro and in vivo | [105] |
| Mannosylation | PLGA NPs | More efficient in vitro uptake of NPs by DCs with chemically conjugated mannan than for plain mannan-adsorbed NPs | [106] |
| PLGA NPs | Mannose functionalization stimulated Th1 bias responses, decreasing tumor growth, both in prophylactic and therapeutic treatments | [107] |
Ab: antibody; NP: nanoparticle; PEG-PLGA: pegylated poly(lactic-co-glycolic) acid; PLGA: poly(lactic-co-glycolic) acid; Th1: T helper 1
Figure 4. Nanotechnology-based approaches to modify the immune response to enhance humoral or cellular responses.
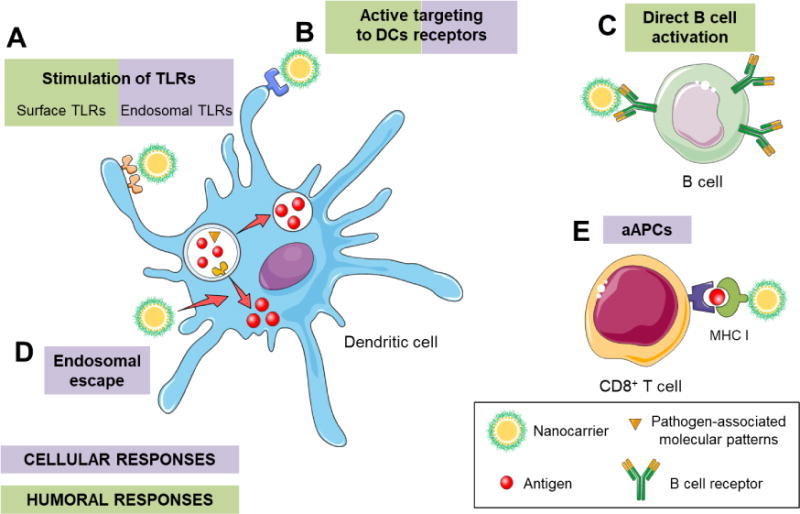
Nanocarriers can drive both humoral and cellular responses, depending on their features and composition. (A) Nanocarriers can deliver toll-like receptor (TLR) agonists that can activate surface or endosomal receptors, driving humoral or cellular responses, respectively. (B) Decorating nanocarriers with antibodies against specific receptors of dendritic cells (DCs) (e.g., CD40, CD11c, DEC-205, mannose, etc.) can activate these cells. (C) The direct targeting to B cells can stimulate them and, thus, favor antibody production and humoral responses. (D) Nanocarriers with properties that promote endosomal escape of the antigens, favor cellular responses. (E) A direct activation of CD8+ T cells through artificial antigen presenting cells (aAPCs) stimulates cytotoxic T lymphocytes.
2.2.1. Cellular responses
In order to fight some infectious diseases (i.e., HIV, malaria) or other diseases, i.e. cancer, the stimulation of a powerful cellular response is necessary. In this context, a correlation between NP size and its ability to favor cross-presentation has been reported [3,9]. In general, studies have shown that smaller sizes enhance cross-presentation and Th1 responses [108–110]. It has been hypothesized that this effect might be related to the capacity of the these NPs to self-drain to the lymph nodes and thus, directly interact with resident CD8+ DCs [111], and also to the specific uptake pathway they follow for internalization. Regarding their uptake, it has been described that particles with sizes similar to virus are endocytosed by DCs through a internalization route that facilitates endosomal escape and drives cellular responses [112–114]. Also, as mentioned above, active targeting to DC205, CD40 or CD11c has shown higher CD8+ T cell activation [105].
As previously discussed, to drive cellular immunity, DCs need to present antigens on MHC I. To achieve this cross-presentation, the antigen has to be present in the cytosol of DCs, thus favoring endosomal escape of the antigen is a requirement for achieving a cellular response (Fig. 4D, Table 2C). This endosomal escape can be promoted by the disruption of the endosome membrane, as discussed by several authors [115,116]. Keller et al. showed how pH-responsive micelles significantly enhance cytotoxic T lymphocyte responses, in comparison to micelles without these properties [117]. This effect was achieved because the forming polymers are protonated at endosomal pH which allows them to interact with the membrane and disrupt the endosome [117,118]. The same tendency was reported in the case of pH-sensitive liposomes, cationic liposomes and bioreducible linkages [119–122]. Other example of cross-presentation and increased cytotoxic T lymphocyte activity has been shown by the ISCOMATRIX adjuvant, both in preclinical and clinical studies, due to a rapid antigen translocation from the endosome [123,124].
Table 2C.
Summary of the different strategies followed in order to facilitate the endosomal escape of antigens in dendritic cells
| Mechanism | Critical feature | Nanosystem | Key results | Ref. |
|---|---|---|---|---|
| Membrane disruption | pH-responsive diblock copolymers | Polyacrylic micelles | pH-responsive micelles caused a higher increase of CD8+ T cell responses in vitro and in vivo than non-pH-responsive controls | [117] |
| Fusion with the membrane | pH-sensitive poly(glycidol) polymers | EPC/DOPE/polymer liposomes | Modified liposomes elicit stronger cellular responses than unmodified systems in vivo | [119] |
| Unknown | Positive lipids (DOTAP or DC-Chol) | DOTAP/Chol/DSPE-mPEG, DC-Chol/DOPE/DSPE-mPEG, EPC/Chol/DSPE-mPEG liposomes | Liposomes with cationic lipids, but not with anionic ones, increased cross-presentation and CD8+ T cell activation in vitro | [120] |
| Membrane disruption (?) | Disulfide crosslinking of the gel | Bioreducible alginate/PEI nanogels | Humoral and cellular responses where enhanced in vitro by the bioreducible nanogel in comparison to the non-reducible one | [121] |
| Unknown | Disulfide bond to nanocarrier | Propylene sulfide NPs | More efficient cross-presentation of the antigen when attached by a reducible link rather than by a non-reducible one | [122] |
| Unknown | ISCOMATRIX adjuvant | ISCOMATRIX + antigen (OVA or E. coli protein) | ISCOMATRIX adjuvant allowed a rapid translocation of the antigen from lysosomes to the cytosol and a greater cross-presentation in vitro, in comparison to immune complexes | [123] |
| Activation of endosomal TLR3 or TLR9 | Poly(I:C), CpG or plasmid DNA | Liposome-Ag-nucleic acid complexes | Complexation of TLR agonists showed an increased CD8+ T cell activation independent of CD4+ T cell help, in comparison to liposomes without TLRs. Also, both prophylactic and therapeutic effects were achieved in two different mice models | [125] |
| Activation of endosomal TLR3 | Poly(I:C) | Cationic adjuvant system (CAF01), composed of DDA and TDB | Immunization with OVA + DDA/TDB/poly(I:C) elicited stronger and longer CD8+ T cell responses in mice than CAF01 alone. In addition, less inflammatory side effects were observed than when administering poly(I:C) alone | [126] |
| Activation of endosomal TLR7/8 | Resiquimod | Temperature-responsive self-assembling particles, based on resiquimod anchored to HPMA or NIPAM scaffolds | Particle formation was key to diminish systemic toxicity and to generate Th1 bias responses, high antibody titers and CD8+ T cell activation in vivo | [127] |
Ag: antigen; CSF21: cationic adjuvant system; Chol: cholesterol; DC-Chol: 3β-[N-(N′,N′-dimethylaminoethane)- carbamoyl] cholesterol; DDA: dimethyldioctadecylammonium; DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DOTAP:, 2-dioleoyl-3-trimethylammonium-propane; DSPE-mPEG: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000]; EPC: egg phosphatylcholine; HPMA: hydrophilic N-(2-hydroxypropyl) methacrylamide; NIPAM: N-isopropylacrylamide; NP: nanoparticle; OVA: ovalbumin; PEI: polyethylenimine; poly(I:C): polyinosinic–polycytidylic acid; TDB: trehalose 6,6′-dibehenate; Th1: T helper 1; TLR: toll-like receptor
With regard to the use of adjuvants, toll-like receptors (TLRs) are extensively used for immunomodulation (Fig. 4A, Table 2C). More specifically, for Th1 biased responses, endosomal TLRs (TLR3, 7, 8 and 9) are an interesting target. These receptors recognize bacterial and viral genetic material, thus their activation will trigger a cellular response, as would a viral or intracellular-bacterial infection. In addition, the combination of nanotechnology and adjuvants has shown a great CD8+ T cell activation with a decrease in the toxicity associated with these molecules [125–127]. Furthermore, since it is known that pathogens normally express several PAMPs at the same time, the combination of several immunomodulatory molecules can further enhance the elicited immune response [7,9,128].
An alternative procedure to generate cellular responses is a direct targeting to CD8+ T cells (Fig. 4E). For this, some authors have employed the so-called artificial antigen presenting cells (aAPCs), which present in their surface major histocompatibility complex molecules and also specific cell markers for T cell recognition and activation [129]. Using this strategy with paramagnetic particles and quantum dots, an increase in CD8+ T cell activation and a decrease in tumor growth were observed [130]. Later on, ellipsoidal PLGA nano-aAPCs were developed, and were shown to be more efficient than the spherical ones in driving CD8+ T cell activation [131].
2.2.2. Humoral responses
Since B cells are in charge of antibody production, a sustained activation of these cells is crucial to guarantee humoral responses. This is the mechanism of action by which most vaccines on the market led to long-lasting antibody responses. Normally, B cell activation is driven by both, the direct interaction of the antigen with the B cell receptor (BCR) and the co-stimulation by CD4+ T cells [132,133].
Some authors have suggested that the location of the antigen on the NP’s structure may influence the resulting humoral response (Table 3). For example, Temchura et al. observed that calcium-phosphate NPs with the antigen covalently attached to their surface, led to a substantial increase in B cell activation in vitro, in comparison to the soluble antigen [134]. Similarly, Moon et al. showed that the display of the antigen onto the surface of multilamellar vesicles provided an enhanced humoral response as compared to the antigen encapsulated [135]. In agreement with these data, several reports showed that the covalent conjugation of the antigen to liposomes could generate stronger antibody responses as compared to those obtained for other types of antigen association (Table 3) [136–146]. Nevertheless, the number of studies on the importance of the linking process of the antigen to the nanocarrier is very limited and it requires further exploration.
Table 3.
Summary of the different antigen attachments used for activation of humoral responses
| Nanosystem | Covalent attachment | Key results | Ref. |
|---|---|---|---|
| CaP NPs loading HEL or BSA | Maleimide bond | NPs were absorbed to B cells in an antigen-specific manner in vitro, inducing their activation | [134] |
| ICMVs with malaria antigen | Maleimide bond | ICMVs with antigen conjugated and encapsulated elicit stronger in vivo humoral responses than MVs with encapsulated antigen alone | [135] |
| DLPC/Chol, DOPC/Chol, PC/Chol, DMPC/Chol, DPPC/Chol, DSPC/Chol liposomes | Diazotisation | No difference in immune responses were found when comparing encapsulation to surface-conjugation of the antigen | [136] |
| DMPC/Chol/DPPE liposomes | Pyridyldithio propionic acid | Covalent linkage of antigen increased both IgG and IgM responses, while encapsulation only elicit IgG responses | [137] |
| PC/SA/Chol liposomes | Diazotisation | Conjugation of antigen to the surface elicit longer and stronger antibody responses than encapsulated or free antigen | [138] |
| DMPC/Chol/DPPE liposomes | Diazotisation | More rapid and prolonged responses obtained with antigen surface linkage than encapsulation | [139] |
| PC/PS/Chol liposomes | Palmitylation of the peptide | Incorporation of antigen conjugated to palmitic acid showed stronger humoral responses in vivo than liposomes with the free antigen | [140] |
| PC/PG/PE/Chol liposomes | Maleimide bond | Conjugation of the antigen to SUV or LUV showed greater responses in vivo than encapsulation, with the best responses for SUV observed with the antigen coupled and MPLA encapsulated | [141] |
| PC/Chol liposomes | n.d. | Surface conjugation of antigen increased the antibody levels faster, while entrapment of the antigen showed stronger secondary responses | [142] |
| DMPC/Chol/DPPE liposomes | Diazotisation | Surface conjugation elicited longer responses in vivo than encapsulation, also presented a different Ig profile | [143] |
| DMPC/Chol/DPPE liposomes | Diazotisation | Both conjugation and encapsulation elicited strong humoral responses in vivo, but conjugation generated a greater blastogenic response | [144] |
| DMPC/DMPG/Chol/LA liposomes | Diazotisation | A high surface display of the antigen generated better humoral responses in vivo | [145] |
| DSPC/Chol/DMPG/MPLA liposomes | Peptidic bond | Physical association was needed for T cell activation, and only surface conjugation induced strong antibody responses | [146] |
BSA: bovine serum albumin; CaP: calcium phosphate; Chol: cholesterol; DLPC: dilinoleoyl phosphatylcholine; DMPC: dimyristoyl phosphatidylcholine; DMPG: dimyristoyl phosphatidylglycerol; DOPC: dioleyl phosphatylcholine; DPPC: phosphatylcholine; DPPE: dipalmitoyl phosphatidylethanolamine; DSPC: distearoyl phosphatylcholine; HEL: hen egg lysozyme; IgG: immunoglobulin G; IgM: immunoglobulin M; MPLA: monophosphoryl lipid A; MVs: multilamellar vesicles; n.d.: not determined; LA: lipid A; LUV: large unillamelar vesicles; NP: nanoparticle; PC: egg phosphatylcholine; PE: phosphatidylethanolamine; PG: phosphatidylglycerol; SA: stearylamide; SUV: small unillamelar vesicles
With regard to the influence of the size on the humoral responses of antigens associated to NPs, it has been reported that for some specific compositions micrometric sizes have a tendency to preferentially generate Th2 responses, in comparison to smaller sizes [112–114]. The mechanism behind this behavior could be related to the uptake pathway. It has been described that for sizes bigger than 500 nm the internalization and processing route of the antigen lead to a more efficient presentation by MHC II, generating stronger humoral responses [113,114].
Another possibility to favor humoral responses could be the administration of TLR2 agonists, since these are able to generate Th2-biased responses [147,148]. Similarly, the activation of surface TLRs (TLR2 and TLR4) showed that they can efficiently inhibit CD8+ T cell activation [149].
2.2.3. Tolerogenic responses
In autoimmune diseases, the generation of tolerance is needed to control the immune response developed against self-antigens. During the last years, different nanotechnology-based approaches have been explored with regard to their capacity to generate tolerogenic profiles (Fig. 5).
Figure 5. Nanotechnology-based antigen-specific approaches for tolerance generation.
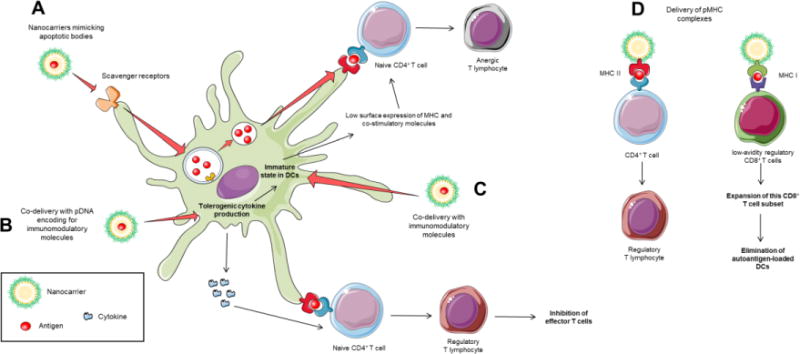
Strategies for antigen-specific tolerance mediated by dendritic cells (DCs) modulation through nanotechnology. (A) Nanocarriers mimicking apoptotic bodies may follow the debris elimination process, where self-antigens presentation induces regulatory T cells. (B) The co-delivery with pDNA encoding for tolerogenic cytokines, i.e. IL-10, may enhance its expression and induce regulatory T cells and anergy. (C) Using immunomodulatory molecules may promote the maintenance of immature state of DCs, while presentation of antigens with low surface density of major histocompatibility complexes (MHC) and costimulatory molecules may promote T cell anergy. (D) The delivery of peptide-MHC (pMHC) complexes to T cells may expand memory T cells with regulatory capacity.
The debris produced during apoptosis, a process of programmed cell death, are eliminated by APCs. The APCs present the processed antigens within a tolerogenic environment, without activating immune responses [150]. Mimicking this environment, nanocarriers can follow debris elimination routes and take advantage of this process to generate tolerance. For the uptake of apoptotic debris, scavenger receptors play the main role in apoptotic signal recognition and debris endocytosis [151]. The incorporation of these apoptosis signal molecules, such as phosphatidylserine (PS), in the nanocarrier composition may enhance its uptake in APCs and allow for a tolerogenic antigen presentation. For example, in one experimental approach, 50 % of mice treated with antigen-loaded PS liposomes could be prevented from acquiring type 1 diabetes (T1D) [152]. Also, experiments show that MARCO-targeted polystyrene MPs follow the debris elimination route, and help to present the antigens loaded in a non-inflammatory way [62]. Interestingly, not only the presence of PS, but also its geometrical surface disposition was found to play a role in tolerance induction. For example, Roberts et al. observed that PLGA NPs displaying a nanorod-presentation were more efficient at inducing tolerogenic responses than the spherical ones [153].
Furthermore, the loading of immunomodulatory molecules in nanocarriers has been shown to help APCs to achieve a tolerogenic state. Molecules such as rapamycin, dexamethasone or vitamin D3 may be co-encapsulated with antigens inside nanocarriers, and promote its presentation in a tolerant environment in APCs [154–156]. Moreover, the delivery of nucleic acids coding for modulatory cytokines has been explored with the goal of inducing tolerogenic profiles in immune cells [157,158].
Finally, the association of antigen-MHC complexes (pMHC) on the surface of iron oxide NPs has been shown to expand autoregulatory T cell memory in different animal models. Indeed, Tsai et al. showed that pMHC class I-coated NPs triggered massive expansions of autoregulatory CD8+ T cells, and these cells were able to suppress polyclonal autoimmune responses by selectively targeting autoantigen-loaded APCs in the target tissue and draining lymph nodes [159]. On the other hand, another report showed that the use of pMHC class II-coated NPs expanded disease-specific regulatory CD4+ T cells.[160].
3. The potential of nanotechnology for vaccination
During the last decades, great efforts have been made to develop systems capable of generating protective immune responses against a variety of antigens. In this section, we present an overview of the work done for specific antigens, such as HIV, malaria or hepatitis B. In this context, it is important to mention that most vaccines currently on the market are based on the generation of humoral protection, which has turned out to be inefficient for some infectious diseases and for cancer, where a strong cellular response is needed. In these particular cases, nanotechnology might be a promising solution. Another article of this special issue is focused on the application of nanotechnology for cancer treatment, which is out of the scope of this review.
The first evidence of the potential of nanotechnology for vaccination was reported 30 years ago by Birrenbanch and Speiser. These authors showed that polyacrylamide NPs could work as adyuvants as they were able to increase the immune response against human IgG and tetanus toxoid after subcutaneous administration to guinea pigs [161]. Years later, Preis and Langer proposed the idea of “single-dose vaccines” based on the possibility to control the release of proteins from polymeric beads [162]. These results were the foundation for the development of controlled antigen delivery systems and nanovaccines.
The development of nanotechnology-based vaccines with a more translational perspective started in the early 90s when the World Health Organization (WHO) proposed the initiative of developing a single-dose vaccine for tetanus toxoid. From this point on, many studies with PLGA-based microsystems were conducted [163]. Unfortunately, despite their good antigen release profiles, a certain protein denaturation was observed due to the pH acidification caused by the degradation of the polymer. To solve this problem different approaches were considered, among them, the use of a protective oil-core surrounded by a PLGA shell or the inclusion of poloxamer 188 to prevent interaction between polymer and antigen [164,165]. At the same time, the potential of nanometric size systems started to gain importance. Almeida et al. developed 500 and 800 nm PLA microspheres for nasal administration of tetanus toxoid with promising results [166]. Later on, our group found that the pegylation of PLA was essential in order to enhance the stability and penetration of the NP across mucosal surfaces [167]. Indeed, the results from experiments using PEG-PLA NPs, did show an increase in the access of the associated antigen to the blood circulation and LNs [40]. Moreover, high and long-lasting anti-tetanus Ig titers were reported with these nanosystems, due to their ability to cross the nasal epithelium [37,168]. Subsequently, more hydrophilic polymers were explored with regard to their ability to transport antigens across mucosal surfaces. In particular, our group pioneered the development of chitosan NPs as alternative candidates for the development of nanovaccines, especially for those administered through mucosal routes [169]. Our studies concluded that the intranasal administration of chitosan NPs loaded with tetanus toxoid resulted in an increase in the humoral and mucosal responses, in comparison to the results obtained with the administration of the free antigen or even with those obtained when the antigen administered was associated to alum [167,170].
As previously mentioned, many studies have tried to develop nanotechnology-based vaccines against a large number of diseases. These diseases include hepatitis B, malaria or HIV, among others, as reported in the following lines.
Our group has also been involved in the development of nanoformulations of the recombinant hepatitis B surface antigen (rHBsAg). In particular, rHBsAg was associated to chitosan NPs and administered by the intramuscular route. The results showed an IgG immunogenic response that was higher than the one observed for the control alum formulation [171]. The same antigen was also adsorbed on chitosan-based nanocapsules [172], a system that was also pioneered by our group [173]. These nanosystems are composed of an oily core surrounded by a chitosan shell, where the protein is adsorbed. After intramuscular administration of rHBsAg attached to chitosan-based nanocapsules, an important antibody responses as well as a more balanced Th1/Th2 profile were obtained [172].
The tendency in the last years has been to design nanosystems that combined the intrinsic targeting properties of nanocarriers with the encapsulation of adjuvants. In this regard, we combined the mucoadhesive properties of chitosan with the adjuvants squalene and imiquimod (TLR7/8 agonist). The results of the intranasal administration of this system showed that the co-encapsulation of antigen and adjuvants was key to generate enhanced and long-lasting IgG levels [174]. More recently, a layer-by-layer approach was evaluated to encapsulate the rHBsAg. This approach consisted on coating the rHBSAg viral particles with a cationic polymer (protamine or polyarginine), followed by an anionic layer of poly(I:C). These nanostructures were able to elicit a more balanced Th1/Th2 ratio after intranasal and intramuscular administration [175].
The development of an effective vaccine against malaria has also attracted a lot of attention in the last decades. In 2015, GSK licensed a vaccine under the name of Mosquirix™, that contains the circumsporozoite protein of Plasmodium falciparum and the liposome-based adjuvant AS01, composed by monophosphoryl lipid A (MPLA) and the saponin QS-21 [176]. This new vaccine has shown good safety profiles and an efficacy rate of 50 % [177], leaving the door open for new improved systems. In this regard, some critical advances have been made thanks to the use of nanotechnology. For example, Moon et al. developed two different formulations of the VMP001-malaria antigen. One of them consisted of PLGA NPs with a phospholipidic coating [178], and the other one of multillamellar vesicles[135], both of them carrying the malaria antigen on the surface. The subcutaneous administration of both formulations in the presence of adjuvant MPLA led to strong humoral and cellular responses, as well as a more balanced Th1/Th2 profile [135,178].
The design of an HIV vaccine is another global challenge, since this disease kills over 1 million people per year according to the World Health Organization. Currently, the most promising vaccine undergoing clinical trials is based on the combination of a viral vector expressing the group antigens (Gag) and the protease (Pro), together with the HIV gp120 envelope recombinant glycoprotein adsorbed onto alum, which has demonstrated a 31 % efficacy [179]. These results highlight the importance of continuing the search for new HIV nanovaccines. The major obstacles for an HIV vaccine are the choice of an effective immunogen and the development of a nanosystem able to generate a potent immune response. The above-mentioned multilamellar vesicles developed by Moon et al. were also evaluated as a potential carrier for the antigen consisting of the envelope glycoprotein (Env) gp140 trimers. This new composition resulted in Th1/Th2 balanced profiles and increased titers against the antigens [180]. A similar strategy based on displaying HIV trimers on the liposomes surface in order to target B cells has been adopted by other authors, showing positive results in terms of neutralizing antibodies responses [181–183]. On the other hand, Hanson et al. co-administered two liposomal formulations, one of them displaying an Env-derived peptide and encapsulating a T-helper peptide, and another one loaded with cyclic di-GMP. Their results showed enhanced CD4+ and CD8+ T cell responses and high-titer and durable humoral responses in mice. However, the immune sera did not neutralize HIV [184]. More recently, Kasturi et al. reported enhanced protection of non-human primates against up to 12 low-dose intravaginal challenges with SIVsmE660. Interestingly, these results were achieved using PLGA-based NPs loading TLR 4/7/8 ligands as adjuvants, in a physical mixture either with the soluble immunogens Env and Gag or displayed in virus-like particles [185].
Important efforts have also been devoted to develop a nanovaccine against Chlamydia trachomatis, an intracellular bacterium that infects over 100 million people annually. For example, Stary et al. reported positive results for NPs made of a triblock copolymer (PLGA-polyhistidine-PEG) and functionalized with the TLR7/8 agonist resiquimod. These nanoparticles exhibited a pH-dependent surface charge, that switched from slightly negative (at pH: 7.4) to positive (at pH below 6.5). This positive charge allowed the adsorption of the NPs to the antigen (inactivated Chlamydia trachomatis bacteria). The formulation was then administered subcutaneously, nasally or intravaginally to mice and, in all cases, strong systemic memory T cell responses were generated. However, only mucosal vaccination effectively protected against a challenge with Chlamydia trachomatis [186].
As a consequence of all these efforts, some NP-based adjuvants have already reached the market. This is the case of MF59, AS03, or the previous mentioned AS01. MF59 is a 160 nm nanoemulsion of squalene, Tween®80 and Span®85 that is part of an Influenza vaccine, commercialized mostly in Europe since 1997 by Novartis under the name of Fluad® [187]. AS03 is also a nanoemulsion-based adjuvant, composed of squalene, tocopherol and Tween®80, property of GSK. Currently, this adjuvant can be found in the pandemic influenza vaccine Prepandrix™, approved in 2008 [188]. Also, Epaxal® and Inflexal® V are two virosome-based vaccines for hepatitis A and influenza, respectively, that are commercialized in some European countries [189].
In addition to these adjuvants and vaccines, a great number of nanoformulations for vaccine delivery are currently in clinical trials and they are illustrated in Table 4.
Table 4.
Summary of some relevant nanovaccine-delivery systems that are being evaluated in clinical trials
| Name/Company | Nanocarrier | Disease | Vaccination route | Clinical Phase | Ref. |
|---|---|---|---|---|---|
| SELA-070/Selecta Biosciences | Synthetic Vaccine Particles (SVP™) | Smoking cessation and relapse prevention | Parenteral (SC) | Phase I | [190, 191] |
| MAS-1/Nova Immunotherapeutics Limited | Nanoparticular emulsion-based adjuvant | Seasonal Influenza | Parenteral | Phase I | [192, 193] |
| FluGem®/Mucosis BV | Bacterium-like particles | Influenza | Mucosal (IN) | Phase I | [194, 195] |
| SynGem®/Mucosis BV | RSV | Mucosal (IN) | Phase I | [194, 196] | |
| VCL-HB01/Vical Inc | Vaxfectin® adjuvant: cationic lipid-based liposomes | HSV-2 | Parenteral (IM) | Phase II | [197, 198] |
| ASP0113/Vical Inc | Poloxamer CRL1005+ DNA | CMV in hematopoietic cell transplant patients | Parenteral (IM) | Phase III | [199, 200] |
| CMV | Phase II | [199, 201] | |||
| HBV003/Vaxine Pty Ltd | Advax: D-inulin MPs | Hepatitis B | Parenteral (IM) | Phase I/II | [202, 203] |
| FLU003/Vaxine Pty Ltd | H5N1 Avian Influenza | Parenteral (IM) | Phase I | [202, 204] | |
| R21 + Matrix M1/University of Oxford & Novavax | Antigen + Matrix M™: saponin-based particles (saponins, synthetic Chol and phospholipids) | Malaria | Parenteral (IM) | Phase I/II | [205, 206] |
| RSV F Vaccine/Novavax | RSV F Vaccine: recombinant F-proteins from RSV that self-assemble to form NPs | RSV | Parenteral (IM) | Phase III | [207, 208] |
| RSV F Vaccine + Matrix M/Novavax | RSV F Vaccine + Matrix M™ | RSV | Parenteral (IM) | Phase II and III | [205, 208, 209] |
| LV305/Immune Design | LVR305: Antigen-specific ZVex® vector (hybrid, reengineered virus designed to carry genetic information of a tumor antigen) | Non-small cell lung cancer, Melanoma and Sarcoma | Parenteral (ID) | Phase I | [210 – 212] |
| CMB305/Immune Design | LV305 + G305(GLA adjuvant system) | Sarcoma, Melanoma, Non-small cell lung cancer and Ovarian cancer | Parenteral (ID and IM) | Phase I | [210, 211, 213] |
| JVRS-100/Juvaris Biotherapeutics Inc | JVRS-100: cationic lipids/non-coding DNA plasmid complexes | Leukaemia | Parenteral (IV) | Phase I | [214, 215] |
| 1790GAHB/GlaxoSmithKline | GMMA: outer membrane particles from bacteria | Dysentery | Parenteral (IM) | Phase I | [216, 217] |
| CTH522-CAF01/Statens Serum Institut | CAF01: cationic adjuvant system composed of DDA and TDB | Chlamydia trachomatis | Parenteral (IM) | Phase I | [126, 218] |
Chol: cholesterol; CMV: cytomegalovirus; DDA: dimethyldioctadecylammonium; GLA: glucopyranosyl lipid A; GMMA: generalized modules of membrane antigen; HSV-2: Herpes Simplex Virus - 2; ID: intradermal; IM: intramuscular; IN: intranasal; MP: microparticle; NP: nanoparticle; RSV: Respiratory Syncital Virus; SC: subcutaneous; TDB: trehalose 6,6′-dibehenate
Taking into consideration the huge efforts made in this field at the research level, it is expected that, in the near future, new nanovaccines will land in the market, and provide hope for defeating devastating illnesses of our generation.
4. The potential of nanotechnology for immunomodulation of autoimmune diseases
As previously mentioned, immunomodulation is a desirable strategy to avoid exacerbated immune responses against ubiquitous molecules, such as self-proteins and, hence, it is of particular interest for the treatment of autoimmune diseases. In autoimmune diseases, autologous proteins are recognized as non-self-antigens by the immune system, leading to the generation of autoreactive T and B cell clones. Currently, the treatment of this kind of diseases is symptomatic and relies on the use of classical anti-inflammatory drugs as well as immunosuppressive therapies. Unfortunately, these therapies are unspecific and lead to significant side effects (Table 5). Due to the complex regulatory network of the immune processes, moving from these therapies to targeted and specific treatments has been found to be an important challenge in biomedical research. In that sense, nanotechnology offers the possibility of the specific delivery of the drug/antigen to the desired cell population, as well as the co-delivery of the targeted drugs with adequate immunomodulatory molecules. Furthermore, nanotechnology offers the possibility to protect the drug from degradation, increasing its half-time life.
Table 5.
Current and most used treatments for selected autoimmune diseases
| Disease | Treatment | Administration route | Mechanism of Action |
|---|---|---|---|
| Multiple sclerosis | IFNβ | SC/IM | Balances the expression of pro- and anti-inflammatory agents in the brain Reduces the number of inflammatory cells that cross the blood brain barrier |
| Glatiramer acetate | SC | Strong promiscuous binding to MHC molecules and consequent competition with myelin antigens for their presentation to T cells | |
| Natalizumab | IV | Blockade of α4 integrin and consequent inhibition of immune cells extravasation | |
| Immunosuppresive agents | Oral/IV | Blockade of immune response at different levels | |
| Type 1 diabetes | Insulin injections | SC | Decrease of glucose levels |
| Rheumatoid arthritis | NSAIDs | Oral | Inhibition of the synthesis of prostaglandins and thromboxanes |
| Corticosteroids | Oral/intra-articular | Regulation of genes related with inflammation and suppression of immune response | |
| TNFα antagonists | SC/IV | Blockade of either TNFα or its receptor | |
| Disease-modifying anti-rheumatic drugs (DMARDs) | Oral/SC/IV | Slow down disease progression by different mechanisms | |
| Inflammatory bowel disease | Aminosalicylates | Oral | Modulation of gene expression and consequently inhibition of cyclooxygenase and NF-κβ and its downstream signals |
| Corticosteroids | Oral | Regulation of genes related with inflammation and suppression of immune response | |
| Immunosuppressive agents | SC/IV | Blockade of immune response at different levels | |
| TNFα antagonists | SC/IV | Blockade of either TNFα or its receptor | |
| Antibiotics | Oral | Decreasing concentrations of bacteria in the gut lumen Altering the composition of intestinal microbiota |
|
| Systemic lupus erythematosus | NSAIDs | Oral | Inhibition of the synthesis of prostaglandins and thromboxanes |
| Antimalarial drugs | Oral | Altering lysosome stability Suppressing antigen presentation Inhibiting prostaglandin and cytokine synthesis Influencing both TLR signaling and leukocyte activation |
|
| Corticosteroids | Oral | Regulation of genes related with inflammation and suppression of immune response | |
| Immunosuppressive agents | SC/IV | Blockade of immune response at different levels |
IM: intramuscular; NSAID: nonsteroidal anti-inflammatory drug; MHC: major histocompatibility complexes; SC: subcutaneous; TLR: toll-like receptor
In this section, we discuss recent advances in nanotechnology regarding immunomodulation to fight against autoimmunity. First, we present the role of the nanocarriers used to enhance the response of immunosuppressant drugs. Next, we focus on more specific approaches evaluating the potential of nanotechnology for antigen-specific therapies in autoimmune diseases with known self-antigens. From the delivery point of view, the common feature of these strategies is that the target cells are the immunocompetent cells.
4.1. Nanomedicines for the treatment of inflammatory diseases
Inflammation is a common immune process that helps the body to eliminate injury related debris, such as microbes, toxins, and necrotic cells. This mechanism is triggered by extracellular signaling factors that attract plasma proteins, immune cells and phagocytes. This inflammatory response could be either acute or chronic. Chronic inflammation usually lasts longer and leads to complications due to tissue degeneration [219]. The chronic inflammatory diseases include autoimmune diseases and auto-inflammatory diseases. In the case of autoimmune diseases, such as inflammatory bowel disease, rheumatoid arthritis, type 1 diabetes, lupus or multiple sclerosis, T cells are thought to be the main triggers of the disease process. Different cytokines, such as TNFα, play a role in maintaining these autoreactive T cells. On the other hand, auto-inflammatory diseases, such as sepsis, gout or type II diabetes, are mainly mediated by innate immune system effectors, such as macrophages, the complement cascade, and cytokines such as IL-1β [220,221]. In these chronic diseases, the targeted treatment of inflammatory conditions could be considered as an immunomodulatory approach, slowing down disease progression and ameliorating the symptoms by changing the immune response, both directly (using immunosuppressant drugs) or indirectly (using anti-inflammatory drugs). This section focuses on different nanotechnology-based therapies developed for the treatment of inflammation in autoimmune diseases.
Immunosuppressant molecules are frequently used for the treatment of chronic inflammation. The many drugs available on the market for the treatment of inflammatory conditions have shown limited success in controlling disease symptoms due to their non-targeted biodistribution. Moreover, the immunosuppressant therapy is normally associated to off-target organ side effects and systemic toxicity, exacerbated by frequent and long-term dosing. Nanoencapsulation of immunosuppressive agents has been shown to increase the therapeutic success of those drugs based on the principle of passive or active targeting. The targeted delivery of these molecules, mainly to macrophages in the inflammation site, has led to the reduction of their side effects and also to improve their action on the inflammatory signaling routes mediated by immune cells, which can be consider also as immunomodulation. This has been widely reviewed in the literature for pathologies as inflammatory bowel disease, rheumatoid arthritis, or systemic lupus erythematous [15,222,223] (Fig 6).
Figure 6. Main strategies for nanotechnology-based anti-inflammatory treatment of autoimmune diseases.
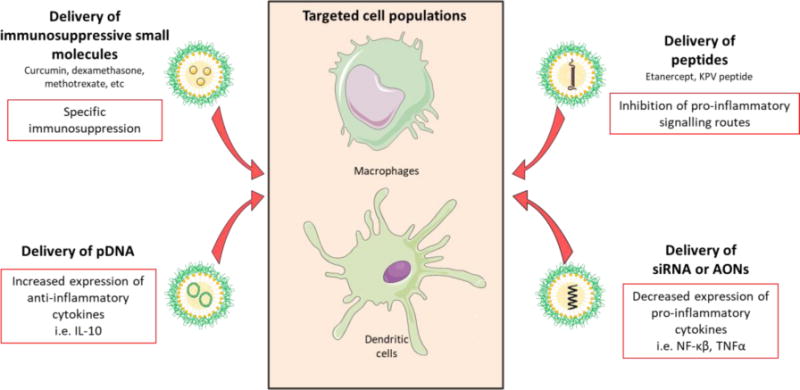
There are four approaches of nanotechnology-based treatments depending on their cargo. First, the delivery of small immunomodulatory molecules has been extensively explored in the suppression of the inflammatory activity of macrophages and dendritic cells (DCs). Second, the delivery of anti-inflammatory peptides has been used in different pathologies. Finally, gene therapy strategies include: pDNA delivery for the expression of anti-inflammatory cytokines, and siRNA or antisense oligonucleotide (AONs) delivery for the downregulation of pro-inflammatory molecules expression.
4.1.1. Inflammatory bowel disease
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the digestive tract, including ulcerative colitis (UC) and Crohn’s disease (CD). UC is confined to the colon, whereas CD can affect any region of the gastrointestinal tract, being the terminal ileum and the colon the most commonly affected areas. Recent research has shown that genetic susceptibility, external environment, intestinal microbial flora and immunological profile are all involved in the pathogenesis of IBD, but the specific causes remain unknown [224]. Current treatments are symptomatic for the induction of remission in acute episodes and avoiding relapsing events. Conventional drugs, including 5-aminosalicylic acid (5-ASA), corticosteroids, immunosuppressant drugs, and anti-TNFα agents are the main treatments today. Depending on localization and activity of the inflammation, these drugs are administered topically, systemically or in combination.
In the case of IBD, colon targeted delivery of immunosuppressive agents is desirable to avoid side effects. For the delivery of small immunosuppressive molecules, polymeric NPs have been widely explored and reviewed in the literature [223,225]. Apart from immunosuppressive drugs, nanotechnology-based siRNA delivery directed to APCs is another approach that has been explored for resolving inflammation in IBD [226,227]. For example, chitosan and its derivatives have been investigated for the siRNA delivery in the colonic region due to its mucoadhesive properties. In one case, chitosan-PLGA NPs were tested orally for the delivery of an antisense oligonucleotide to block NF-κβ factor in an induced-colitis model. The results showed that chitosan-PLGA NPs were selectively accumulated in inflamed tissue and improved the clinical scoring [228]. Similarly, galactosylated trimethylchitosan NPs loaded with a siRNA against mitogen-activated protein kinase (MAPK) showed good in vivo efficacy in induced-colitis mice model after oral administration [229]. Finally, the local delivery of anti-inflammatory peptides or protein antagonists of immune receptors in the inflammation site is a promising approach for the in situ modulation of immune effector cells. For example, the colonic delivery of an alginate-chitosan hydrogel (double oral gavage procedure for in situ gelation) containing KPV peptide-loaded PLGA NPs to an induced-colitis mice model, resulted in a marked amelioration of the inflammatory symptoms. In fact, a considerably lower dose of peptide (12,000-fold) compared to the free peptide, led to a similar therapeutic efficacy. This effect was explained taking into account the better access of the peptide-loaded NPs to the target epithelial and immune cells [230].
4.1.2. Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic autoimmune disorder that primarily affects joints. RA is characterized by synovial inflammation and swelling, autoantibody production as well as cartilage and bone destruction [231]. It has been proposed that the course of the RA development follows a three-step process. Autoimmunity starts to develops in genetic-susceptible individuals, with the presentation of autoantibodies in serum [232]. In a second step, there is an expansion of reactive immune cells that leads the infiltration of inflammatory cells in the joints as a prelude of the chronic inflammatory response. Finally, the patient presents a chronic joint inflammation promoted mainly by macrophages, which constitutes the major hallmark of the third phase of the disease [233]. The systemic delivery of immunosuppressant molecules, both classic small drugs and anti-TNFα antibodies are the main current treatments (Table 5) [231].
The design of nanotechnology-based approaches in RA is focused on increasing the retention time of small immunosuppressive drugs in the joint [222]. For that purpose, a wide variety of nanocarriers have been tested and extensively reviewed in the literature, including polymeric NPs, liposomes, solid-lipid NPs and polymeric micelles [222,234]. Moreover, nanotechnology- based gene therapy has also been explored for the treatment of RA. As in IBD, this therapy is focused in siRNA knockdown of TNFα [226]. Also, the encapsulation of pDNA encoding for IL-10 was widely explored. As an example, Jain et al. showed effective macrophage repolarization from M1 to M2 phenotype in adjuvant-induced arthritis (AIA) mice model after intraperitoneal administration of IL-10-encoding pDNA-loaded alginate NPs [235]. Regarding protein delivery, different anti-inflammatory proteins have been explored. This is the case of self-assembled NPs composed of metracrylate-based copolymers loaded with an IL-1 receptor antagonist (a protein implicated in blocking pro-inflammatory signals). This system was able to maintain the biological activity of IL-1 receptor antagonist in vitro and prolong its retention in rat stifle joint following intra-articular administration in healthy rats [236]. Another example is the nanocomplex of etanercept with succinylated pullulan-g-oligo (L-lactide) polymer. After two months of fortnightly subcutaneous injection of this nanocomplex to a collagen-induced arthritis (CIA) rat model, no cartilage erosion and a depletion of the synovial inflammation were observed [237].
4.1.3. Systemic lupus erythematosus
Systemic lupus erythematous (SLE) is a chronic autoimmune disease characterized by loss of tolerance to self-antigens and production of numerous autoantibodies, due to its heterogenic and non-organ specific origin [238]. The most common treatment strategies are NSAIDs, antimalarial drugs and oral glucocorticoids. Immunosuppressive medications are used to control serious lupus activity that affects major organs (Table 5).
Nanotechnology-based therapies for the treatment of SLE have been reviewed recently [15]. In the following lines, we highlight some of the most significant works in this field. Look et al. developed a liposomal system with a gel-like core containing cyclodextrins surrounded by a lipid bilayer for the delivery of anti-inflammatory agents. Following intraperitoneal administration of this system loaded with mycophenolic acid in a murine lupus model, it was found an increased 2 – 3 months the mean survival time, and this was attributed to the preferential accumulation of the system in DCs [239]. The same group also found that the DCs immunosuppression achieved with this new system was more significant than for the one observed for PLGA NPs loaded with the same drug [240]. In another example, methylprednisolone-loaded liposomes were administered subcutaneously in a murine lupus model and the results showed a reduced the mortality for this group of mice, as compared to that of the group treated with the free drug [241]. Attempts have also been made to treat lupus with gene therapy approaches. For example, following intraperitoneal administration of siRNA anti-MAPK1 (a protein implicated in the pro-inflammatory signaling cascade) loaded into PEG-poly(L-lysine) NPs, in a murine model of lupus nephritis, a significant amelioration of the renal damage was observed [242].
To summarize, different nanotechnology approaches were developed for the treatment of the inflammation in autoimmune diseases (Fig 6). This offers the possibility of controlled and targeted release of immunosupressive drugs, which would avoid the systemic effects of the drugs currently on the market. Furthermore, the change from invasive administration routes (IV) to more patient-friendly ones (mucosal) can also be accomplished by nanotechnology, thus increasing patient compliance.
4.2. Nanovaccines for the treatment of autoimmune diseases
Apart from the symptomatic treatment using anti-inflammatory and immunosuppressive drugs, nanotechnology can contribute with more specific treatments for autoimmune diseases. In this sense, antigen-specific therapies seem a good option to prevent self-antigen recognition that would lead to the activation of auto-reactive T or B cell clones.
The best-known disease-specific self-antigens are: myelin in MS, insulin in T1D, and collagen in RA. Loss of tolerance towards self-antigens is often thought to be the result of both genetic and environmental risk factors, including exposure to infection by particular pathogens, molecular mimicry of endogenous antigens, or bystander activation [243]. However, the molecular mechanisms behind the autoimmune process are not well understood yet. Furthermore, in most of the cases, the self-antigens involved in the physiopathology of the disease remain unknown, limiting these therapies to illnesses with known self-antigens. In a healthy situation, T lymphocytes can distinguish between different antigens with high specificity; however they cannot discriminate between self or non-self-antigens. Central tolerance process occurs in the thymus during the first years of life. During this process, thymic epithelial cells expose in their surface a great variety of self-antigens to T cells. Normally, the T cells that recognize those antigens are eliminated to prevent self-reactivity [244,245]. Besides central tolerance process, peripheral mechanisms regulate these self-reactive T cells if they reach the bloodstream. However, in the case of patients with autoimmune disorders, these peripheral mechanisms fail and the self-reactive T cells stay and cause damage [246].
Different mechanisms for maintenance of peripheral self-tolerance have been proposed. Most of them include DCs and regulatory T cells as the main modulators of self-reactive T cell response [247,248]. The molecular signals in the microenvironment drive DCs homeostasis and function, especially regarding cytokines production and surface expression of co-stimulatory molecules. Differences in the microenvironment can lead to phenotypical changes in DCs, promoting T cell anergy, T cell depletion and regulatory T cell proliferation after immune synapsis formation and antigen recognition [249]. This regulatory T cell expansion promotes the suppression of specific self-reactive T cell clones by different mechanism [248]. Within this context, the “holy grail” of immunotherapy in autoimmune diseases would be the development of antigen-specific treatments targeted to dendritic cells. This approach could maintain the functionality of the immune system whereas specifically blocking the self-reactive T cells which are pathogenic in autoimmune diseases. For this purpose, different protocols where developed during the last decades for the induction of specific tolerance [250].
Trying to simulate the process elicited in allergy treatment, high doses of soluble antigen were injected in order to induce anergy or activation-induced cell death after T cell re-stimulation in autoimmune diseases [251,252]. Unfortunately, although promising result based on this strategy were obtained [253,254], in others, a hyper-sensitivity reaction was observed after the administration of the soluble antigen [255,256]. These contradictory results can be explained by the fact that soluble peptides can induce specific tolerance, but cannot block polyspecific responses in the case of epitope spreading, which is the situation that exists in autoimmune diseases [257].
Based on the high amount of foreign antigens present in food and the general lack of immune reaction against them (except in the case of food allergies), the mucosal administration of soluble antigens has also been explored to induce tolerance. This is thought to happen by different mechanisms dependent on antigen dose. Low-dose of self-antigen is processed by antigen presenting cells in the gastrointestinal tract, promoting the activation of regulatory T cells. On the other hand, high doses of antigens seem to cross the gastrointestinal barrier and promote anergy once in systemic circulation [258]. Studies in animal models led to promising results in terms of blocking disease progression [259–261], however, so far, these results did not translate to a clinical set-up [262].
Nanotechnology is a promising approach to improve vaccination strategies to treat autoimmune diseases. It offers the ability of specific targeting and association of multiple antigens capable of inducing tolerance before epitope spreading happens. Most of the nanotechnology-based strategies are focused on the delivery of self-antigens to DCs, taking advantage of natural peripheral tolerance mechanisms mediated by this cell type (Fig 5). In the next lines, we will summarize and discuss the latest and most relevant nanotechnology-based approaches in antigen-specific therapy against different autoimmune pathologies (Table 6).
Table 6.
Most representative polymeric and lipidic nanocarriers for antigen-specific tolerance generation in autoimmunity
| Nanocarrier type | Loaded molecule | Administration characteristics | Animal model | In vivo results | Ref. |
|---|---|---|---|---|---|
| PLGA NPs | IL-10 or MOG antigen | SC, co-administration of both systems | EAE | Inhibited disease development No vaccination delayed disease onset |
[269] |
| Rapamycin and PLP antigen | SC and IV | R-EAE | Delay in disease onset Complete inhibition of relapse episodes (IV) | [154] | |
| PLP antigen covalently linked | IV | R-EAE | Prevention of disease onset Complete inhibition of relapse episodes | [63, 65] | |
| CII | Oral | CIA | Peyer’s patches accumulation for longer time Reduced plasma levels of CII-antibodies Reduced incidence of arthritis |
[19] | |
| PEG-CII derived peptides | Oral | Healthy | Expansion of IL-4+ and IL-10+ CD4+ T cells | [282] | |
| PLGA MPs | Rapamycin and MOG antigen | Intra-nodal | EAE | Permanently reduction of disease onset and severity | [270] |
| Vitamin D3 and Insulin B, or TGF-β1 and GM-CSF | SC, co-administration of both systems | NOD | Disease onset prevention in 40 % of mice treated Increase in survival time from 19 weeks to 24 weeks | [156] | |
| Liposomes | PS and insulin peptides | IP | NOD | Reduced incidence of T1D Delay in disease onset | [152] |
| PS and MOG peptide | IP | EAE | Reduced clinical score Delay in disease onset | [275] | |
| Methylated BSA and NF-κβ inhibitors | SC | AIA | Reduction in joint swelling severity scores | [283] | |
| Nano-complexes | pDNA encoding for BTLA and MOG antigen | IP injection of pre-treated DCs | EAE | Delay in disease onset Reduction of disease severity | [272] |
| GpG and arginine-modified MOG antigen | SC | EAE | Reduced clinical score In some cases mice remained asymptomatic for the duration of the study (24 days) | [273, 274] |
AIA: adjuvant-induced arthritis; BSA: bovine serum albumin; BTLA: B and T lymphocyte attenuator; CIA: collagen-induced arthritis; CII: type II collagen; DC: dendritic cell; EAE: experimental allergic encephalomyelitis; MOG: myelin oligodendrocyte protein; NOD: non-obese diabetic; PEG: pegylated; PLGA: poly(lactic-co-glycolic) acid; PLP: proteolipid protein; PS: phosphatidylserine; IP: intra-peritoneal; R-EAE: relapsing-EAE; SC: subcutaneous
4.2.1. Multiple sclerosis
Multiple sclerosis (MS) affects around 2.3 million people worldwide, and is the second most common cause of disability in young adults. MS is a central nervous system disorder of autoimmune origin, in which encephalitogenic T cells are involved in damaging the myelin, promoting inflammation, and triggering neuronal and axonal damage [263]. Some self-antigens are known to be related with the pathology, including myelin basic protein (MBP), myelin oligodendrocyte protein (MOG), and proteolipid protein (PLP) [264,265]. The most common treatments for MS are interferon β (IFNβ), glatiramer acetate (GA), and the monoclonal antibody natalizumab, known as disease-modifying therapies (DMT). These treatments are unspecific for MS and often have serious side effects, such as opportunistic infections and tumors [266–268]. As previously indicated, the ideal treatment should be antigen-specific and DCs-targeted, to avoid systemic immunosuppression. In addition, the co-administration of antigen and immunomodulatory molecules using nanotechnology is now emerging as a new therapeutic option for the treatment of MS.
Most of the systems developed for MS treatment are based on PLGA and designed for the co-delivery of the antigen and immunomodulatory molecules such as rapamycin or IL-10. Following subcutaneous or intra-nodal administration in experimental allergic encephalomyelitis (EAE) mice model, it was found that these systems were able to successfully inhibited the progression of the disease [154,269,270]. In another report it was described that a new antigen-coupled PLGA formulation induced liver-dependent tolerance in a relapsing-remitting EAE mice model after IV administration [63]. Similarly, Carambia et al. showed that antigen-coupled poly(maleic anhydride-alt-1-octadecene) polymeric NPs induced also liver-dependent tolerance in an EAE mice model, providing effective control of the disease with a single IV administration due to the efficient induction of regulatory T cells [271].
The ionic complexation of antigenic peptides, or their DNA encoding sequences, and immunomodulatory molecules is nowadays presented as a new nanotechnology approach to induce tolerance. For example, Yuan et al. developed self-assembled NPs using a plasmid encoding for the co-inhibitory receptor B and T lymphocyte attenuator (BTLA) as immunomodulatory signal and MOG antigen modified with the cell penetrating peptide Tat49-57. When DCs pretreated with these NPs were administrated intraperitoneally to an EAE mice model, a decrease in the spinal cord inflammation and inhibition of specific T cell proliferation were observed [272]. A similar approach involved the complexation of arginine-modified MOG antigen and GpG oligonucleotide, an antagonist of TLR9. Studies in EAE mice model showed an improvement in the progression, severity, and incidence of the disease [273,274].
Recently, PS liposomes were also tested as peptide carriers for MS therapy. Liposomes loaded with MOG peptide were administered intraperitoneally (2 boosts) in EAE mice model and the result of this treatment was a decrease in the clinical score and the incidence of the disease [275].
4.2.2. Type 1 Diabetes
Nowadays, 415 million people worldwide have diabetes [276]. Diabetes mellitus is a pandemic group of disorders where insulin metabolism is altered. Within this group, type 1 diabetes (T1D) is considered a chronic autoimmune disease caused by the destruction of β-cells located in the Langerhans islets by the immune system, causing the loss of insulin production in pancreas [277]. Human and murine models have been extensively used to study the pathophysiology of the disease. Results from these studies have shown that the destruction of β cells occurs in a cell-mediated manner, requiring both CD4+ and CD8+ T cells and macrophages [278,279]. Most well-known antigens recognized by T cells in T1D are preproinsulin, glutamic acid decarboxylase and islet-cell antigen-2 [280]. The standard treatment for T1D is the subcutaneous injection of insulin to maintain normoglucemia. As an alternative, antigen-specific treatments aim to avoid the underlying autoimmune response, treating the disease at its origin [250]. The main barriers for the design of antigen-specific approaches are: the complexity of T1D autoantigens map, and the specific targeting to the immune cells involved in disease onset and progression. Nanotechnology offers the possibility of specific targeting and loading multiple antigens at the same time, with or without immunomodulatory molecules. The recently developed nanotechnology-based treatments for T1D are summarized below.
PLGA NPs were explored for the delivery of self-antigens in combination with immunomodulatory molecules, in the treatment of T1D. For example, Lewis et al. developed a dual-sized PLGA MPs formulation, where non-phagocytosable MPs (30 μm) were loaded with chemoatractive cytokines and phagocytosable MPs (0.5 – 2.5 μm) were loaded with insulin B peptide and vitamin D3. This approach relies on the assumption that the large MPs releasing chemokines stay in the injection site and help to attract immune cells and, hence, to enhance the phagocytosis of small MPs. Once in the APCs, the small MPs deliver the antigen with vitamin D3 for a tolerogenic presentation to the lymphocytes. Using this approach, 40 % of mice were protected from T1D development when the combination of both MPs was injected subcutaneously twice [156]. A different approach involved the use of human denatured insulin-loaded PLGA MPs included in a hydrogel with chemoatractive cytokines. After 3 subcutaneous injections, 40% of non-obese diabetic (NOD) mice were protected [281].
Liposomes containing PS were also developed for the delivery of insulin antigens. As mentioned above, PS was selected as it works as an “eat me” signal in apoptotic cells that can promote the presentation of the antigen with the secretion of tolerogenic cytokines such as PGE2. According to the results, around 50% of NOD mice did not develop diabetes after intraperitoneal liposomes administration [152].
4.2.3. Rheumatoid arthritis
As we stated previously, RA is a long-term autoimmune disorder that primarily affects the joints. Currently, RA treatment is focused on easing the symptoms, or slowing the course of the disease by using immunosuppressant drugs such as corticosteroids or anti-TNF antibodies [231] (Table 5). Although most of the novel approaches for RA treatment are focused on the targeted delivery of immunosuppressant drugs or tissue regeneration, antigen-specific approaches are also being explored as a promising treatment at the onset of the disease through the downregulation of the underlying autoimmune processes, although the number is still limited [19,282,283]. Self-antigens, such as collagen derived peptides, were found to be RA triggers in different animal models [231], and mucosal administration of collagen peptides were found to ameliorate the progression of the disease in patients [284].
Among the different nanotechnology approaches to treat RA, there is the attempt described by Kim et al. based on the oral administration of PLGA NPs loaded with both whole type II collagen (CII) and CII derived peptides. The results showed a reduction of the severity of arthritis after a single oral administration to CIA mice and this positive effect was associated to the accumulation of the CII-loaded PLGA NPs in the Peyer’s patches [19]. Similarly, using CII-derived peptides modified with PEG, Lee et al. developed peptide-loaded PLGA NPs for oral administration. They found that a single administration of the encapsulated PEG-conjugated peptides to healthy DBA/1 mice was able to increase both the rate of IL-4+ CD4+ cells and of IL-10+ CD4+ cells, which could be a promising approach for inducing tolerogenic phenotypes by the oral route [282]. Moreover, liposomes were also explored in antigen-specific therapy for RA by Capini et al.. In a methylated BSA-induced arthritis model, they showed that, after subcutaneous administration of methylated BSA and lipophilic NF-κβ inhibitors (Bay11-7082, curcumin, or quercetin) co-encapsulated in liposomes, all of the combined formulations diminished the score of disease symptoms, compared with untreated mice [283].
In summary, nanotechnology-based antigen-specific approaches are promising for autoimmune diseases therapy and offer the possibility of controlled and targeted release of self-antigens. In addition, the possibility of loading immunomodulatory agents gives nanosystems the ability of enhancing tolerance generation. However, more research is needed for antigen identification and for a better understanding of what causes an autoimmune disease. These studies could help us elucidate the multiple factors that are involved in both epitope spreading and autoimmune response processes. Therefore, multiple antigen approaches could be the best option for efficiently blocking disease progression. To this end, nanotechnology could be a very valuable tool in combining multifactor therapy.
5. The next challenge in immunomodulation: overcoming antidrug antibodies
We are in the era of biologicals, also named as biodrugs or biotherapeutics. The development of the recombinant DNA technology starting in the early 70s and the introduction of recombinant insulin in the market have laid the foundations for the use of biomolecules as therapeutic agents [285,286]. Nowadays, biomolecule-based therapies for a huge variety of diseases are already being used clinically, or are in trials, which shows the great potential of biotherapeutics [287,288]. Linked to this development of biodrugs, one of the major safety concerns that needs to be assessed during preclinical and clinical trials is undesired immunogenicity [289].
One of the first approaches to address unwanted immunogenicity is to measure the formation of antidrug antibodies (ADAs). ADAs recognize different epitopes in a recombinant molecule and bind to them, causing different outcomes in the pharmacological activity of the drug, depending on their neutralizing potential. ADAs formation and their effect on biotherapeutics have been extensively reviewed due to its direct relation with immunogenicity and treatment efficacy [290]. In the case of replacement therapies, ADAs formation could result in cross reactivity with endogenous proteins and, thus, cause severe adverse effects. One highlighted example is erythropoietin, a hormone required for red blood cell development which is used as treatment for anemia in patients with chronic kidney disease [291]. A few clinical subjects were found to develop pure red cell aplasia after erythropoietin infusion, due to ADAs formation and subsequent endogenous erythropoietin recognition [292]. In this case, several factors affected the protein immunogenicity including those depending on product-formulation (leakage of polysorbate 80 from the rubber stoppers of the syringes) and administration (change from IV to subcutaneous administration route) [293–295].
There are multiple factors that influence the immunogenicity of biodrugs and formation of ADAs. Originally, a bacterial or fungi origin of the recombinant proteins could cause immunogenicity, due to the differences in sequence and structure of biomolecules between species. Although nowadays the use of humanized or fully-human biodrugs has greatly contributed to reduce this risk, immunogenicity associated to the aggregation of biodrug molecules and other factors is still a major concern for the optimum exploitation of these modern drugs [296,297]. The association between biodrug molecules upon injection has been thought to be a natural way to enhance antigen processing and presentation in the cells [298,299]. On the other hand, the presence of impurities could also be part of the problem. Finally, the administration route [300,301] and patient-related issues, such as genetic predisposition to ADAs formation or cytokine pattern, could impact immunogenicity [302,303].
Today, there is not a single standard therapy available for ADAs formation. The most frequent therapy is the administration of a prolonged immunosuppressive regimen as in the case of “Pompe disease”, a lisosomal storage disorder [304,305]. Nevertheless, this approach could enhance opportunistic infections and other complications due to systemic immunosuppression. The ideal treatment to avoid ADAs effects would be drug-specific: achieving tolerance to the delivered biotherapeutic molecule, and maintaining its safety and efficacy without systemic immunosuppression. In this field, nanotechnology is emerging as a new approach where a biotherapeutic agent can be specifically delivered together with an immunosuppressive drug, avoiding systemic immunogenic effects and increasing the treatment efficacy. For this purpose, DCs are the usual target, due to their important role in antigens presentation and also because of their relevance in the fate of T cells [247,306]. Indeed, as indicated above, it has been described how the uptake of rapamycin by DCs promotes the differentiation of T cells towards a regulatory phenotype [307]. Furthermore, rapamycin encapsulation, both in PLGA NPs and MPs, enhances the tolerogenic activity of DCs [308,309].
Within this context, it is worthwhile to mention the formulation activity of Selecta Biosciences, in terms of inducing immunotolerance through the use of nanotechnology. They co-administered the coagulation factor VIII together with the immunosuppressive agent rapamycin loaded into PLGA NPs and observed promising in vivo results in terms of maintaining the efficacy of the biologic entity in an hemophilia A mice model [154]. In their search for a more universal approach, they explored the interest of co-administering rapamycin-loaded PLGA NPs together with different proteins (OVA, pegsiticase, adalimumab) and the results showed durable inhibition of ADAs formation [310]. Currently, patients are being recruited for a phase II clinical trial for the evaluation of the co-administration of rapamycin-loaded PLGA NPs with free pegsiticase: a pegylated uricase enzyme implicated in the metabolization of uric acid, that is currently administered for the treatment of hyperuricemia and chronic gout [311].
Despite this original work there is still a limited understanding of the processes that underlie the immunogenicity of biotherapeutics and further research is needed to determine how nanocarriers could modulate immune mechanism to promote tolerance. All this knowledge will help us come up with a rational design of nanotherapeutic agents with better performance for tolerance generation.
Conclusions
In the last decades, nanotechnology has shown an important potential in the immunotherapeutic field. The modulation of a broad variety of immune processes can be achieved with nanotechnology, with promising results not only in vitro but also in vivo. In this review, we have analyzed a significant number of nanotechnology-based formulations for immune activation and tolerance generation. In both cases, the nanocarrier composition and its physicochemical properties have shown to play a crucial role in achieving the desired immune outcome. Despite the knowledge generated, it is quite risky at this point to correlate the physicochemical characteristics of the nanocarriers with their capacity to modulate the immune system. Regarding the composition of the nanocarrier, the use of ligands for specific cell receptors is expected to substantially increase the targeting to a particular subset of immune cells. Besides, the use of immunomodulators could be a useful strategy to effectively polarize the immune response.
As a consequence of all the nanotechnology-based approaches for immune modulation in preclinical studies, a high number of nanoformulations for vaccine delivery and tolerance generation is currently being tested in clinical trials, and a few of them have already reached the market. Indeed, thanks to the significant efforts made in this field at the research level, essential advances have been made to treat diseases like HIV, tuberculosis, type-1 diabetes or multiple sclerosis, among others. A deeper knowledge from the immunological point of view will help to rationally design and engineer new nanosystems that are expected to contribute to find a cure for some of the most threatening illnesses of our time.
Highlights.
- Nanocarriers can be designed to target specific immune cells
- Nanovaccines may help fighting diseases that are elusive to traditional vaccines
- Nanocarriers can bias the immune response from humoral to cellular
- Autoimmune disease treatments can be improved with nanotechnology-based approaches
- The use of nanocarriers may help to avoid ADAs formation against biotherapeutics
Acknowledgments
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health [award number R01AI111805-subaward number 41795-02]; the European Union’s Horizon 2020 research program [grant agreement number 646142]; and the Spanish Ministry of Economy, Industry and Competitiveness [grant number BIO2014-53091-C3-2-R]. Tamara G. Dacoba acknowledges a predoctoral FPU grant from the Spanish Ministry of Education, Culture and Sports (grant number FPU14/05866). Ana Olivera acknowledges a predoctoral FPI grant from the Spanish Ministry of Economy, Industry and Competitiveness (grant number BES-2015-071236). Also, we thank Servier for providing Servier Medical Art, used for the creation of the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–96. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 2.Smith DM, Simon JK, Baker JR. Applications of nanotechnology for immunology. Nat Rev Immunol. 2013;13:592–605. doi: 10.1038/nri3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic nanoparticles for vaccines and immunotherapy. Chem Rev. 2015;115:11109–11146. doi: 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cordeiro AS, Alonso MJ. Recent advances in vaccine delivery. Pharm Pat Anal. 2015;5:49–73. doi: 10.4155/ppa.15.38. [DOI] [PubMed] [Google Scholar]
- 5.Cordeiro AS, Alonso MJ, de la Fuente M. Nanoengineering of vaccines using natural polysaccharides. Biotechnol Adv. 2015;33:1279–1293. doi: 10.1016/j.biotechadv.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Duin D, Medzhitov R, Shaw AC. Triggering TLR signaling in vaccination. Trends Immunol. 2006;27:49–55. doi: 10.1016/j.it.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Gutjahr A, Tiraby G, Perouzel E, Verrier B, Paul S. Triggering intracellular receptors for vaccine adjuvantation. Trends Immunol. 2016;37:573–587. doi: 10.1016/j.it.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Getts DR, Shea LD, Miller SD, King NJC. Harnessing nanoparticles for immune modulation. Trends Immunol. 2015;36:419–427. doi: 10.1016/j.it.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gause KT, Wheatley AK, Cui J, Yan Y, Kent SJ, Caruso F. Immunological principles guiding the rational design of particles for vaccine delivery. ACS Nano. 2017;11:54–68. doi: 10.1021/acsnano.6b07343. [DOI] [PubMed] [Google Scholar]
- 10.Walker BD, Burton DR. Toward an AIDS vaccine. Science (80-) 2008;320:760–764. doi: 10.1126/science.1152622. [DOI] [PubMed] [Google Scholar]
- 11.Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 12.Delany I, Rappuoli R, De Gregorio E. Vaccines for the 21st century. EMBO Mol Med. 2014;6:708–720. doi: 10.1002/emmm.201403876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serra P, Santamaria P. Nanoparticle-based autoimmune disease therapy. Clin Immunol. 2015;160:3–13. doi: 10.1016/j.clim.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Tabansky I, Messina MD, Bangeranye C, Goldstein J, Blitz-Shabbir KM, Machado S, Jeganathan V, Wright P, Najjar S, Cao Y, Sands W, Keskin DB, Stern JNH. Advancing drug delivery systems for the treatment of multiple sclerosis. Immunol Res. 2015;63:58–69. doi: 10.1007/s12026-015-8719-0. [DOI] [PubMed] [Google Scholar]
- 15.Rostamzadeh D, Razavi SR, Esmaeili S, Dolati S, Ahmahi M, Sadreddini S, Jadidi-Niaragh F, Yousefi M, Ahmadi M, Sadreddini S, Jadidi-Niaragh F, Yousefi M, Ahmahi M, Sadreddini S, Jadidi-Niaragh F, Yousefi M. Application of nanoparticle technology in the treatment of systemic lupus erythematous. Biomed Pharmacother. 2016;83:1154–1163. doi: 10.1016/j.biopha.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 16.Csaba N, Garcia-Fuentes M, Alonso MJ. Nanoparticles for nasal vaccination. Adv Drug Deliv Rev. 2009;61:140–157. doi: 10.1016/j.addr.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interface. Annu Rev Immunol. 2011;29:273–293. doi: 10.1146/annurev-immunol-031210-101317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brandtzaeg P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine. 2007;25:5467–5484. doi: 10.1016/j.vaccine.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Kim WU, Lee WK, Ryoo JW, Kim SH, Kim J, Youn J, Min SY, Bae EY, Hwang SY, Park SH, Cho CS, Park JS, Kim HY. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: a novel treatment strategy for induction of oral tolerance. Arthritis Rheum. 2002;46:1109–1120. doi: 10.1002/art.10198. [DOI] [PubMed] [Google Scholar]
- 20.Huang Y, Donovan MD. Microsphere transport pathways in the rabbit nasal mucosa. Int J Pharm Adv. 1996;1:298–309. [Google Scholar]
- 21.Ghirardelli R, Bonasoro F, Porta C, Cremaschi D. Identification of particular epithelial areas and cells that transport polypeptide-coated nanoparticles in the nasal respiratory mucosa of the rabbit. Biochim Biophys Acta - Biomembr. 1999;1416:39–47. doi: 10.1016/S0005-2736(98)00209-0. [DOI] [PubMed] [Google Scholar]
- 22.Amidi M, Romeijn SG, Borchard G, Junginger HE, Hennink WE, Jiskoot W. Preparation and characterization of protein-loaded N-trimethyl chitosan nanoparticles as nasal delivery system. J Control Release. 2006;111:107–116. doi: 10.1016/j.jconrel.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Li M, Zhao M, Fu Y, Li Y, Gong T, Zhang Z, Sun X. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J Control Release. 2016;228:9–19. doi: 10.1016/j.jconrel.2016.02.043. [DOI] [PubMed] [Google Scholar]
- 24.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 25.Niess JH. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science (80-) 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 26.Gupta NK, Tomar P, Sharma V, Dixit VK. Development and characterization of chitosan coated poly-(ε-caprolactone) nanoparticulate system for effective immunization against influenza. Vaccine. 2011;29:9026–9037. doi: 10.1016/j.vaccine.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 27.Pawar D, Mangal S, Goswami R, Jaganathan KS. Development and characterization of surface modified PLGA nanoparticles for nasal vaccine delivery: effect of mucoadhesive coating on antigen uptake and immune adjuvant activity. Eur J Pharm Biopharm. 2013;85:550–559. doi: 10.1016/j.ejpb.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 28.Nochi T, Yuki Y, Takahashi H, Sawada SI, Mejima M, Kohda T, Harada N, Kong IG, Sato A, Kataoka N, Tokuhara D, Kurokawa S, Takahashi Y, Tsukada H, Kozaki S, Akiyoshi K, Kiyono H. Nanogel antigenic protein-delivery system for adjuvant-free intranasal vaccines. Nat Mater. 2010;9:572–578. doi: 10.1038/nmat2784. [DOI] [PubMed] [Google Scholar]
- 29.Olmsted SS, Padgett JL, Yudin AI, Whaley KJ, Moench TR, Cone RA. Diffusion of Macromolecules and Virus-Like Particles in Human Cervical Mucus. Biophys J. 2001;81:1930–1937. doi: 10.1016/S0006-3495(01)75844-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gutierro I, Hernández RM, Igartua M, Gascón AR, Pedraz JL. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine. 2002;21:67–77. doi: 10.1016/S0264-410X(02)00435-8. [DOI] [PubMed] [Google Scholar]
- 31.Mann JFS, Shakir E, Carter KC, Mullen AB, Alexander J, Ferro VA. Lipid vesicle size of an oral influenza vaccine delivery vehicle influences the Th1/Th2 bias in the immune response and protection against infection. Vaccine. 2009;27:3643–3649. doi: 10.1016/j.vaccine.2009.03.040. [DOI] [PubMed] [Google Scholar]
- 32.Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13:1838–1845. doi: 10.1023/A:1016085108889. [DOI] [PubMed] [Google Scholar]
- 33.Shakweh M, Ponchel G, Fattal E. Particle uptake by Peyer’s patches: a pathway for drug and vaccine delivery, Expert Opin. Drug Deliv. 2004;1:141–163. doi: 10.1517/17425247.1.1.141. [DOI] [PubMed] [Google Scholar]
- 34.Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Rev Vaccines. 2010;9:1095–1107. doi: 10.1586/erv.10.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Correia-Pinto JF, Csaba N, Alonso MJ. Vaccine delivery carriers: insights and future perspectives. Int J Pharm. 2013;440:27–38. doi: 10.1016/j.ijpharm.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 36.Bernasconi V, Norling K, Bally M, Höök F, Lycke NY. Mucosal vaccine development based on liposome technology. J Immunol Res. 2016;2016 doi: 10.1155/2016/5482087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vila A, Gill H, McCallion O, Alonso MJ. Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J Control Release. 2004;98:231–244. doi: 10.1016/j.jconrel.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 38.Vila A, Sánchez A, Évora C, Soriano I, McCallion O, Alonso MJ, Évora C, Soriano I, McCallion O, Alonso MJ. PLA-PEG particles as nasal protein carriers: the influence of the particle size. Int J Pharm. 2005;292:43–52. doi: 10.1016/j.ijpharm.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 39.Stano A, Nembrini C, Swartz MA, Hubbell JA, Simeoni E. Nanoparticle size influences the magnitude and quality of immune response after intranasal immunization. Vaccine. 2012;30:7541–7546. doi: 10.1016/j.vaccine.2012.10.050. [DOI] [PubMed] [Google Scholar]
- 40.Tobío M, Gref R, Sánchez A, Langer R, Alonso MJ. Stealth PLA-PEG nanoparticles as protein carriers for nasal administration. Pharm Res. 1998;15:270–275. doi: 10.1023/A:1011922819926. [DOI] [PubMed] [Google Scholar]
- 41.Tobío M, Sánchez A, Vila A, Soriano I, Evora C, Vila-Jato JL, Alonso MJ. The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloids Surfaces B Biointerfaces. 2000;18:315–323. doi: 10.1016/S0927-7765(99)00157-5. [DOI] [PubMed] [Google Scholar]
- 42.Lai SK, O’Hanlon DE, Harrold S, Man ST, Wang YY, Cone R, Hanes J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci. 2007;104:1482–1487. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Deliv Rev. 2009;61:158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462:449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- 45.Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gutjahr A, Phelip C, Coolen AL, Monge C, Boisgard AS, Paul S, Verrier B. Biodegradable polymeric nanoparticles-based vaccine adjuvants for lymph nodes targeting. Vaccines 2016. 2016;4:34.4. doi: 10.3390/VACCINES4040034. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abellán-Pose R, Csaba N, Alonso MJ. Lymphatic targeting of nanosystems for anticancer drug therapy. Curr Pharm Des. 2016;22:1194–1209. doi: 10.2174/1381612822666151216150809. [DOI] [PubMed] [Google Scholar]
- 48.Kaminskas LM, Kota J, McLeod VM, Kelly BD, Karellas P, Porter CJ. PEGylation of polylysine dendrimers improves absorption and lymphatic targeting following sc administration in rats. J Control Release. 2009;140:108–116. doi: 10.1016/j.jconrel.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 49.Kourtis IC, Hirosue S, de Titta A, Kontos S, Stegmann T, Hubbell JA, Swartz MA. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS One. 2013;8:1–11. doi: 10.1371/journal.pone.0061646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reddy ST, Berk DA, Jain RK, Swartz MA. A sensitive in vivo model for quantifying interstitial convective transport of injected macromolecules and nanoparticles. J Appl Physiol. 2006;101:1162–1169. doi: 10.1152/japplphysiol.00389.2006. [DOI] [PubMed] [Google Scholar]
- 51.Mueller SN, Tian S, DeSimone JM. Rapid and persistent delivery of antigen by lymph node targeting PRINT nanoparticle vaccine carrier to promote humoral immunity. Mol Pharm. 2015;12:1356–1365. doi: 10.1021/mp500589c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao DA, Forrest ML, Alani AWG, Kwon GS, Robinson JR. Biodegradable PLGA based nanoparticles for sustained regional lymphatic drug delivery. J Pharm Sci. 2010;99:2018–2031. doi: 10.1002/jps.21970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vicente S, Goins BA, Sanchez A, Alonso MJ, Phillips WT. Biodistribution and lymph node retention of polysaccharide-based immunostimulating nanocapsules. Vaccine. 2014;32:1685–1692. doi: 10.1016/j.vaccine.2014.01.059. [DOI] [PubMed] [Google Scholar]
- 54.Zeng Q, Jiang H, Wang T, Zhang Z, Gong T, Sun X. Cationic micelle delivery of Trp2 peptide for efficient lymphatic draining and enhanced cytotoxic T-lymphocyte responses. J Control Release. 2015;200:1–12. doi: 10.1016/j.jconrel.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 55.Kim SY, Noh YW, Kang TH, Kim JE, Kim S, Um SH, Oh DB, Park YM, Lim YT. Synthetic vaccine nanoparticles target to lymph node triggering enhanced innate and adaptive antitumor immunity. Biomaterials. 2017;130:56–66. doi: 10.1016/j.biomaterials.2017.03.034. [DOI] [PubMed] [Google Scholar]
- 56.Carstens MG, Camps MGM, Henriksen-Lacey M, Franken K, Ottenhoff THM, Perrie Y, Bouwstra JA, Ossendorp F, Jiskoot W. Effect of vesicle size on tissue localization and immunogenicity of liposomal DNA vaccines. Vaccine. 2011;29:4761–4770. doi: 10.1016/j.vaccine.2011.04.081. [DOI] [PubMed] [Google Scholar]
- 57.Kaur R, Bramwell VW, Kirby DJ, Perrie Y. Pegylation of DDA:TDB liposomal adjuvants reduces the vaccine depot effect and alters the Th1/Th2 immune responses. J Control Release. 2012;158:72–77. doi: 10.1016/j.jconrel.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 58.Kaur R, Bramwell VW, Kirby DJ, Perrie Y. Manipulation of the surface pegylation in combination with reduced vesicle size of cationic liposomal adjuvants modifies their clearance kinetics from the injection site, and the rate and type of T cell response. J Control Release. 2012;164:331–337. doi: 10.1016/j.jconrel.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 59.De Koker S, Cui J, Vanparijs N, Albertazzi L, Grooten J, Caruso F, De Geest BG. Engineering polymer hydrogel nanoparticles for lymph node-targeted delivery. Angew Chemie - Int Ed. 2016;55:1334–1339. doi: 10.1002/anie.201508626. [DOI] [PubMed] [Google Scholar]
- 60.Takano S, Aramaki Y, Tsuchiya S. Physicochemical properties of liposomes affecting apoptosis induced by cationic liposomes in macrophages. Pharm Res. 2003;20:962–968. doi: 10.1023/A:1024441702398. [DOI] [PubMed] [Google Scholar]
- 61.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 62.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJC, Shea LD, Miller SD. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol. 2012;30:1217–1224. doi: 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, Miller SD. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS Nano. 2014;8:2148–2160. doi: 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heymann F, Tacke F. Immunology in the liver - From homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110. doi: 10.1038/nrgastro.2015.200. [DOI] [PubMed] [Google Scholar]
- 65.McCarthy DP, Yap JWT, Harp CT, Song WK, Chen J, Pearson RM, Miller SD, Shea LD. An antigen-encapsulating nanoparticle platform for Th1/17 immune tolerance therapy. Nanomedicine Nanotechnology, Biol Med. 2017;13:191–200. doi: 10.1016/j.nano.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930–4. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Champion JA, Katare YK, Mitragotri S. Particle shape: a new design parameter for micro- and nanoscale drug delivery carriers. J Control Release. 2007;121:3–9. doi: 10.1016/j.jconrel.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm Res. 2008;25:1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doshi N, Mitragotri S. Macrophages recognize size and shape of their targets. PLoS One. 2010;5:1–6. doi: 10.1371/journal.pone.0010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hasegawa T, Hirota K, Tomoda K, Ito F, Inagawa H, Kochi C, Soma GI, Makino K, Terada H. Phagocytic activity of alveolar macrophages toward polystyrene latex microspheres and PLGA microspheres loaded with anti-tuberculosis agent. Colloids Surfaces B Biointerfaces. 2007;60:221–228. doi: 10.1016/j.colsurfb.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 71.Hirota K, Hasegawa T, Hinata H, Ito F, Inagawa H, Kochi C, Soma GI, Makino K, Terada H. Optimum conditions for efficient phagocytosis of rifampicin-loaded PLGA microspheres by alveolar macrophages. J Control Release. 2007;119:69–76. doi: 10.1016/j.jconrel.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 72.Fifis T, Gamvrellis A, Crimeen-Irwin B, Pietersz GA, Li J, Mottram PL, McKenzie IFC, Plebanski M. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. J Immunol. 2004;173:3148–3154. doi: 10.4049/jimmunol.173.5.3148. [DOI] [PubMed] [Google Scholar]
- 73.He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 74.Pacheco P, White D, Sulchek T. Effects of microparticle size and Fc density on macrophage phagocytosis. PLoS One. 2013;8:1–9. doi: 10.1371/journal.pone.0060989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thiele L, Rothen-Rutishauser B, Jilek S, Wunderli-Allenspach H, Merkle HP, Walter E. Evaluation of particle uptake in human blood monocyte-derived cells in vitro. Does phagocytosis activity of dendritic cells measure up with macrophages? J Control Release. 2001;76:59–71. doi: 10.1016/S0168-3659(01)00412-6. [DOI] [PubMed] [Google Scholar]
- 76.Thiele L, Merkle HP, Walter E. Phagocytosis and phagosomal fate of surface-modified microparticles in dendritic cells and macrophages. Pharm Res. 2003;20:221–228. doi: 10.1023/A:1022271020390. [DOI] [PubMed] [Google Scholar]
- 77.Miller CR, Bondurant B, McLean SD, McGovern KA, O’Brien DF. Liposome-cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry. 1998;37:12875–12883. doi: 10.1021/bi980096y. [DOI] [PubMed] [Google Scholar]
- 78.Nakanishi T, Kunisawa J, Hayashi A, Tsutsumi Y, Kubo K, Nakagawa S, Nakanishi M, Tanaka K, Mayumi T. Positively charged liposome functions as an efficient immunoadjuvant in inducing cell-mediated immune response to soluble proteins. Biochem Biophys Res Commun. 1997;240:793–797. doi: 10.1006/bbrc.1997.7749. [DOI] [PubMed] [Google Scholar]
- 79.Fromen CA, Rahhal TB, Robbins GR, Kai MP, Shen TW, Luft JC, DeSimone JM. Nanoparticle surface charge impacts distribution, uptake and lymph node trafficking by pulmonary antigen-presenting cells, Nanomedicine Nanotechnology. Biol Med. 2016;12:677–687. doi: 10.1016/j.nano.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tomita Y, Rikimaru-Kaneko A, Hashiguchi K, Shirotake S. Effect of anionic and cationic n-butylcyanoacrylate nanoparticles on NO and cytokine production in Raw264.7 cells. Immunopharmacol Immunotoxicol. 2011;33:730–7. doi: 10.3109/08923973.2011.565345. [DOI] [PubMed] [Google Scholar]
- 81.Epstein-Barash H, Gutman D, Markovsky E, Mishan-Eisenberg G, Koroukhov N, Szebeni J, Golomb G. Physicochemical parameters affecting liposomal bisphosphonates bioactivity for restenosis therapy: internalization, cell inhibition, activation of cytokines and complement, and mechanism of cell death. J Control Release. 2010;146:182–195. doi: 10.1016/j.jconrel.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 82.Gutman D, Epstein-Barash H, Tsuriel M, Golomb G. Diagnostinc Res. Springer; Netherlands: 2012. Alendronate liposomes for antitumor therapy: activation of γδ-T cells and inhibition of tumor growth, in: Nano-Biotechnology Biomed; pp. 165–179. [DOI] [PubMed] [Google Scholar]
- 83.Getts DR, Terry RL, Getts MT, Deffrasnes C, Müller M, Van Vreden C, Ashhurst TM, Chami B, McCarthy DP, Wu H, Ma J, Martin A, Shae LD, Witting P, Kansas GS, Kühn J, Hafezi W, Campbell IL, Reilly D, Say J, Brown L, White MY, Cordwell SJ, Chadban SJ, Thorp EB, Bao S, Miller SD, King NJC. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. 2014;6:1–14. doi: 10.1126/scitranslmed.3007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci. 2011;108:1827–1832. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beduneau A, Ma Z, Grotepas CB, Kabanov A, Rabinow BE, Gong N, Mosley RL, Dou H, Boska MD, Gendelman HE. Facilitated monocyte-macrophage uptake and tissue distribution of superparmagnetic iron-oxide nanoparticles. PLoS One. 2009;4:1–12. doi: 10.1371/journal.pone.0004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Locke LW, Mayo MW, Yoo AD, Williams MB, Berr SS. PET imaging of tumor associated macrophages using mannose coated 64Cu liposomes. Biomaterials. 2012;33:7785–7793. doi: 10.1016/j.biomaterials.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakashima-Matsushita N, Homma T, Yu S, Matsuda T, Sunahara N, Nakamura T, Tsukano M, Ratnam M, Matsuyama T. Selective expression of folate receptor β and its possible role in methotrexate transport in synovial macrophages from patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:1609–1616. doi: 10.1002/1529-0131(199908)42:8<1609::AID-ANR7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 88.Thomas TP, Goonewardena SN, Majoros IJ, Kotlyar A, Cao Z, Leroueil PR, Baker JR. Folate-targeted nanoparticles show efficacy in the treatment of inflammatory arthritis. Arthritis Rheum. 2011;63:2671–2680. doi: 10.1002/art.30459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ma L, Liu TW, Wallig MA, Dobrucki IT, Dobrucki LW, Nelson ER, Swanson KS, Smith AM. Efficient targeting of adipose tissue macrophages in obesity with polysaccharide nanocarriers. ACS Nano. 2016;10:6952–6962. doi: 10.1021/acsnano.6b02878. [DOI] [PubMed] [Google Scholar]
- 90.Lesley J, Hyman R, Kincade PW. CD44 and its interaction with extracellular matrix. Adv Immunol. 1993;54:271–335. doi: 10.1016/S0065-2776(08)60537-4. [DOI] [PubMed] [Google Scholar]
- 91.Clark RA, Alon R, Springer TA. CD44 and hyaluronan-dependent rolling interactions of lymphocytes on tonsillar stroma. J Cell Biol. 1996;134:1075–1087. doi: 10.1083/jcb.134.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ke C, Wang D, Sun Y, Qiao D, Ye H, Zeng X. Immunostimulatory and antiangiogenic activities of low molecular weight hyaluronic acid. Food Chem Toxicol. 2013;58:401–407. doi: 10.1016/j.fct.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 93.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 94.Bourguignon LYW, Wong G, Earle CA, Xia W. Interaction of low molecular weight hyaluronan with CD44 and toll-like receptors promotes the actin filament-associated protein 110-actin binding and MyD88-NFκB signaling leading to proinflammatory cytokine/chemokine production and breast tumor invasion. Cytoskeleton. 2011;68:671–693. doi: 10.1002/cm.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Song M, Liu T, Shi C, Zhang X, Chen X. Bioconjugated manganese dioxide nanoparticles enhance chemotherapy response by priming tumor-associated macrophages toward M1-like phenotype and attenuating tumor hypoxia. ACS Nano. 2016;10:633–647. doi: 10.1021/acsnano.5b06779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guermonprez P, Valladeau J, Zitvogel L, Théry C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 97.Roche PA, Furuta K. The ins and outs of MHC class II ‑ mediated antigen processing and presentation. Nat Rev Immunol. 2015;15:203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 99.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- 100.Gamvrellis A, Leong D, Hanley JC, Xiang SD, Mottram P, Plebanski M. Vaccines that facilitate antigen entry into dendritic cells. Immunol Cell Biol. 2004;82:506–516. doi: 10.1111/j.0818-9641.2004.01271.x. [DOI] [PubMed] [Google Scholar]
- 101.Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298:315–322. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 102.Foged C, Arigita C, Sundblad A, Jiskoot W, Storm G, Frokjaer S. Interaction of dendritic cells with antigen-containing liposomes: Effect of bilayer composition. Vaccine. 2004;22:1903–1913. doi: 10.1016/j.vaccine.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 103.Fromen CA, Robbins GR, Shen TW, Kai MP, Ting JPY, DeSimone JM. Controlled analysis of nanoparticle charge on mucosal and systemic antibody responses following pulmonary immunization. Proc Natl Acad Sci. 2015;112:488–493. doi: 10.1073/pnas.1422923112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Macri C, Dumont C, Johnston AP, Mintern JD. Targeting dendritic cells: a promising strategy to improve vaccine effectiveness. Clin Transl Immunol. 2016;5:e66. doi: 10.1038/cti.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Löwik CWGM, Ossendorp F. Targeting nanoparticles to CD40, DEC-205 or CD11c molecules on dendritic cells for efficient CD8+ T cell response: a comparative study. J Control Release. 2014;192:209–218. doi: 10.1016/j.jconrel.2014.07.040. [DOI] [PubMed] [Google Scholar]
- 106.Ghotbi Z, Haddadi A, Hamdy S, Hung RW, Samuel J, Lavasanifar A. Active targeting of dendritic cells with mannan-decorated PLGA nanoparticles. J Drug Target. 2011;19:281–292. doi: 10.3109/1061186X.2010.499463. [DOI] [PubMed] [Google Scholar]
- 107.Silva JM, Zupancic E, Vandermeulen G, Oliveira VG, Salgado A, Videira M, Gaspar M, Graca L, Préat V, Florindo HF. In vivo delivery of peptides and Toll-like receptor ligands by mannose-functionalized polymeric nanoparticles induces prophylactic and therapeutic anti-tumor immune responses in a melanoma model. J Control Release. 2015;198:91–103. doi: 10.1016/j.jconrel.2014.11.033. [DOI] [PubMed] [Google Scholar]
- 108.Hirai T, Yoshioka Y, Takahashi H, Ichihashi K, Yoshida T, Tochigi S, Nagano K, Abe Y, Kamada H, Tsunoda S, Nabeshi H, Yoshikawa T, Tsutsumi Y. Amorphous silica nanoparticles enhance cross-presentation in murine dendritic cells. Biochem Biophys Res Commun. 2012;427:553–556. doi: 10.1016/j.bbrc.2012.09.095. [DOI] [PubMed] [Google Scholar]
- 109.Mant A, Chinnery F, Elliott T, Williams AP. The pathway of cross-presentation is influenced by the particle size of phagocytosed antigen. Immunology. 2012;136:163–175. doi: 10.1111/j.1365-2567.2012.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ahn S, Lee IH, Kang S, Kim D, Choi M, Saw PE, Shin EC, Jon S. Gold nanoparticles displaying tumor-associated self-antigens as a potential vaccine for cancer immunotherapy. Adv Healthc Mater. 2014;3:1194–1199. doi: 10.1002/adhm.201300597. [DOI] [PubMed] [Google Scholar]
- 111.Schmidt ST, Khadke S, Korsholm KS, Perrie Y, Rades T, Andersen P, Foged C, Christensen D. The administration route is decisive for the ability of the vaccine adjuvant CAF09 to induce antigen-specific CD8+ T-cell responses: the immunological consequences of the biodistribution profile. J Control Release. 2016;239:107–117. doi: 10.1016/j.jconrel.2016.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S. Shape and size-dependent immune response to antigen-carrying nanoparticles. J Control Release. 2015;220:141–148. doi: 10.1016/j.jconrel.2015.09.069. [DOI] [PubMed] [Google Scholar]
- 113.Lebre F, Hearnden CH, Lavelle EC. Modulation of immune responses by particulate materials. Adv Mater. 2016;28:5525–5541. doi: 10.1002/adma.201505395. [DOI] [PubMed] [Google Scholar]
- 114.Benne N, van Duijn J, Kuiper J, Jiskoot W, Slütter B. Orchestrating immune responses: how size, shape and rigidity affect the immunogenicity of particulate vaccines. J Control Release. 2016;234:124–134. doi: 10.1016/j.jconrel.2016.05.033. [DOI] [PubMed] [Google Scholar]
- 115.Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 116.Selby LI, Cortez-Jugo CM, Such GK, Johnston APR. Nanoescapology: progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdiscip Rev Nanomedicine Nanobiotechnology. 2017:e1452. doi: 10.1002/wnan.1452. [DOI] [PubMed] [Google Scholar]
- 117.Keller S, Wilson JT, Patilea GI, Kern HB, Convertine AJ, Stayton PS. Neutral polymer micelle carriers with pH-responsive, endosome-releasing activity modulate antigen trafficking to enhance CD8+ T cell responses. J Control Release. 2014;191:24–33. doi: 10.1016/j.jconrel.2014.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Convertine AJ, Diab C, Prieve M, Paschal A, Hoffman AS, Johnson PH, Stayton PS. pH-responsive polymeric micelle carriers for siRNA drugs. Biomacromolecules. 2010;11:2904–2911. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yuba E, Harada A, Sakanishi Y, Watarai S, Kono K. A liposome-based antigen delivery system using pH-sensitive fusogenic polymers for cancer immunotherapy. Biomaterials. 2013;34:3042–3052. doi: 10.1016/j.biomaterials.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 120.Gao J, Ochyl LJ, Yang E, Moon JJ. Cationic liposomes promote antigen cross-presentation in dendritic cells by alkalizing the lysosomal pH and limiting the degradation of antigens. Int J Nanomedicine. 2017;12:1251–1264. doi: 10.2147/IJN.S125866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li P, Luo Z, Liu P, Gao N, Zhang Y, Pan H, Liu L, Wang C, Cai L, Ma Y. Bioreducible alginate-poly(ethylenimine) nanogels as an antigen-delivery system robustly enhance vaccine-elicited humoral and cellular immune responses. J Control Release. 2013;168:271–279. doi: 10.1016/j.jconrel.2013.03.025. [DOI] [PubMed] [Google Scholar]
- 122.Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA, Swartz MA. Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: cross-presentation and T cell activation. Vaccine. 2010;28:7897–7906. doi: 10.1016/j.vaccine.2010.09.077. [DOI] [PubMed] [Google Scholar]
- 123.Schnurr M, Orban M, Robson NC, Shin A, Braley H, Airey D, Cebon J, Maraskovsky E, Endres S. ISCOMATRIX adjuvant induces efficient cross-presentation of tumor antigen by dendritic cells via rapid cytosolic antigen delivery and processing via tripeptidyl peptidase II. J Immunol. 2009;182:1253–1259. doi: 10.4049/jimmunol.182.3.1253. [DOI] [PubMed] [Google Scholar]
- 124.Nicholaou T, Chen W, Davis ID, Jackson HM, Dimopoulos N, Barrow C, Browning J, MacGregor D, Williams D, Hopkins W, Maraskovsky E, Venhaus R, Pan L, Hoffman EW, Old LJ, Cebon J. Immunoediting and persistence of antigen-specific immunity in patients who have previously been vaccinated with NY-ESO-1 protein formulated in ISCOMATRIX. Cancer Immunol Immunother. 2011;60:1625–1637. doi: 10.1007/s00262-011-1041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zaks K, Jordan M, Guth A, Sellins K, Kedl R, Izzo A, Bosio C, Dow S. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J Immunol. 2006;176:7335–7345. doi: 10.4049/jimmunol.176.12.7335. [DOI] [PubMed] [Google Scholar]
- 126.Nordly P, Rose F, Christensen D, Nielsen HM, Andersen P, Agger EM, Foged C. Immunity by formulation design: induction of high CD8+ T-cell responses by poly(I:C) incorporated into the CAF01 adjuvant via a double emulsion method. J Control Release. 2011;150:307–317. doi: 10.1016/j.jconrel.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 127.Lynn GM, Laga R, Darrah PA, Ishizuka AS, Balaci AJ, Dulcey AE, Pechar M, Pola R, Gerner MY, Yamamoto A, Buechler CR, Quinn KM, Smelkinson MG, Vanek O, Cawood R, Hills T, Vasalatiy O, Kastenmüller K, Francica JR, Stutts L, Tom JK, Ryu KA, Esser-Kahn AP, Etrych T, Fisher KD, Seymour LW, Seder RA. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat Biotechnol. 2015;33:1201–1210. doi: 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, García-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sunshine JC, Green JJ. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine. 2013;8:1173–89. doi: 10.2217/nnm.13.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Perica K, Medero ADL, Durai M, Chiu YL, Bieler JG, Sibener L, Niemöller M, Assenmacher M, Richter A, Edidin M, Oelke M, Schneck J. Nanoscale artificial antigen presenting cells for T cell immunotherapy, Nanomedicine Nanotechnology. Biol Med. 2014;10:119–129. doi: 10.1016/j.nano.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, Green JJ. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small. 2015;11:1519–1525. doi: 10.1002/smll.201402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nat Rev Immunol. 2008;9:15–27. doi: 10.1038/nri2454. [DOI] [PubMed] [Google Scholar]
- 133.Moyer TJ, Zmolek AC, Irvine DJ. Beyond antigens and adjuvants: formulating future vaccines. J Clin Invest. 2016;126:799–808. doi: 10.1172/JCI81083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Temchura VV, Kozlova D, Sokolova V, Überla K, Epple M. Targeting and activation of antigen-specific B-cells by calcium phosphate nanoparticles loaded with protein antigen. Biomaterials. 2014;35:6098–6105. doi: 10.1016/j.biomaterials.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 135.Moon JJ, Suh H, Li V, Ockenhouse CF, Yadava A, Irvine DJ. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci. 2012;109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Davis D, Gregoriadis G. Liposomes as adjuvants with immunopurified tetanus toxoid: influence of liposomal characteristics. Immunol Lett. 1987;61:229–234. [PMC free article] [PubMed] [Google Scholar]
- 137.Shahum E, Thérien HM. Immunopotentiation of the humoral response by liposomes: encapsulation versus covalent linkage. Immunology. 1988;65:315–317. [PMC free article] [PubMed] [Google Scholar]
- 138.Vannier WE, Snyder SL. Antibody responses to liposome-associated antigen. Immunol Lett. 1988;19:59–64. doi: 10.1016/0165-2478(88)90120-4. [DOI] [PubMed] [Google Scholar]
- 139.Thérien HM, Lair D, Shahum E. Liposomal vaccine: influence of antigen association on the kinetics of the humoral response. Vaccine. 1990;8:558–562. doi: 10.1016/0264-410X(90)90008-A. [DOI] [PubMed] [Google Scholar]
- 140.Brynestad K, Babbit B, Huang L, Rouse BT. Influence of peptide acylation, liposome incorporation, and synthetic immunomodulators on the immunogenicity of a 1-23 peptide of glycoprotein D of herpes simplex virus: implications for subunit vaccines. J Virol. 1990;64:680–685. doi: 10.1128/jvi.64.2.680-685.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Frisch B, Muller S, Briand JP, Van Regenmortel MHV, Shuber F. Parameters affecting the immunogenicity of a liposome-associated synthetic hexapeptide antigen. Eur J Immunol. 1991;21:185–193. doi: 10.1002/eji.1830210128. [DOI] [PubMed] [Google Scholar]
- 142.Tan L, Weissig V, Gregoriadis G. Comparison of the immune response against polio peptides covalently-surface-linked to and internally-entrapped in liposomes. Asian Pacific J Allergy Immunol. 1991;9:25–30. [PubMed] [Google Scholar]
- 143.Shahum E, Thérien HM. Correlation between in vitro and in vivo behaviour of liposomal antigens. Vaccine. 1994;12:1125–1131. doi: 10.1016/0264-410X(94)90183-X. [DOI] [PubMed] [Google Scholar]
- 144.Shahum E, Thérien HM. Liposomal adjuvanticity: effect of encapsulation and surface-linkage on antibody production and proliferative response. Int J Immunopharmacol. 1995;17:9–20. doi: 10.1016/0192-0561(94)00082-Y. [DOI] [PubMed] [Google Scholar]
- 145.White WI, Cassatt DR, Madsen J, Burke SJ, Woods RM, Wassef NM, Alving CR, Koenig S. Antibody and cytotoxic T-lymphocyte responses to a single liposome-associated peptide antigen. Vaccine. 1995;13:1111–1122. doi: 10.1016/0264-410X(94)00058-U. [DOI] [PubMed] [Google Scholar]
- 146.Guan HH, Budzynski W, Koganty RR, Krantz MJ, Reddish MA, Rogers JA, Longenecker BM, Samuel J. Liposomal formulations of synthetic MUC1 peptides: effects of encapsulation versus surface display of peptides on immune responses. Bioconjug Chem. 1998;9:451–458. doi: 10.1021/bc970183n. [DOI] [PubMed] [Google Scholar]
- 147.Dillon S, Agrawal A, Van Dyke T, Mccauley L, Koh A, Maliszewski C, Akira S, Pulendran B. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. 2004;172:4733–4743. doi: 10.4049/jimmunol.172.8.4733. [DOI] [PubMed] [Google Scholar]
- 148.Redecke V, Hacker H, Datta SK, Fermin A, Pitha PM, Broide DH, Raz E. Cutting edge: activation of Toll-like receptor 2 induces a Th2 immune response and promotes experimental asthma. J Immunol. 2004;172:2739–2743. doi: 10.4049/jimmunol.172.5.2739. [DOI] [PubMed] [Google Scholar]
- 149.Mandraju R, Murray S, Forman J, Pasare C. Differential ability of surface and endosomal TLRs to induce CD8 T cell responses in vivo. J Immunol. 2014;192:4303–4315. doi: 10.4049/jimmunol.1302244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Poon IKH, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang D, Sun B, Feng M, Feng H, Gong W, Liu Q, Ge S. Role of scavenger receptors in dendritic cell function. Hum Immunol. 2015;76:442–446. doi: 10.1016/j.humimm.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 152.Pujol-Autonell I, Serracant-Prat A, Cano-Sarabia M, Ampudia RM, Rodriguez-Fernandez S, Sanchez A, Izquierdo C, Stratmann T, Puig-Domingo M, Maspoch D, Verdaguer J, Vives-Pi M. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PLoS One. 2015;10 doi: 10.1371/journal.pone.0127057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Roberts RA, Eitas TK, Byrne JD, Johnson BM, Short PJ, McKinnon KP, Reisdorf S, Luft JC, DeSimone JM, Ting JP. Towards programming immune tolerance through geometric manipulation of phosphatidylserine. Biomaterials. 2015;72:1–10. doi: 10.1016/j.biomaterials.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning EA, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A. 2015;112:E156–E165. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng H, Yang Y, Vangundy Z, Papenfuss TL, Lovett-Racke AE, Bachelder EM, Ainslie KM. Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated peptide and dexamethasone in acetalated dextran microparticles. Mol Pharm. 2014;11:828–835. doi: 10.1021/mp4005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin Immunol. 2015;160:90–102. doi: 10.1016/j.clim.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Gallo M, Knox J, Hogeland K, Trucco M, Giannoukakis N. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes. 2008;57:1544–1555. doi: 10.2337/db07-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Basarkar A, Singh J. Poly (lactide-co-glycolide)-polymethacrylate nanoparticles for intramuscular delivery of plasmid encoding interleukin-10 to prevent autoimmune diabetes in mice. Pharm Res. 2009;26:72–81. doi: 10.1007/s11095-008-9710-4. [DOI] [PubMed] [Google Scholar]
- 159.Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity. 2010;32:568–580. doi: 10.1016/j.immuni.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 160.Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, Agrawal S, Keough MB, Yong VW, James E, Moore A, Yang Y, Stratmann T, Serra P, Santamaria P. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016;530:434–440. doi: 10.1038/nature16962. [DOI] [PubMed] [Google Scholar]
- 161.Birrenbach G, Speiser PP. Polymerized micelles and their use as adjuvants in immunology. J Pharm Sci. 1976;65:1763–1766. doi: 10.1002/jps.2600651217. [DOI] [PubMed] [Google Scholar]
- 162.Preis I, Langer RS. A single-step immunization by sustained antigen release. J Immunol Methods. 1979;28:193–197. doi: 10.1016/0022-1759(79)90341-7. [DOI] [PubMed] [Google Scholar]
- 163.Aguado MT, Lambert PH. Controlled-release vaccines - Biodegradable polylactide/polyglycolide (PL/PG) microspheres as antigen vehicles. Immunobiology. 1992;184:113–125. doi: 10.1016/S0171-2985(11)80470-5. [DOI] [PubMed] [Google Scholar]
- 164.Sánchez A, Gupta RK, Alonso MJ, Siber GR, Langer R. Pulsed controlled-released system for potential use in vaccine delivery. J Pharm Sci. 1996;85:547–52. doi: 10.1021/js960069y. [DOI] [PubMed] [Google Scholar]
- 165.Tobío M, Nolley J, Guo Y, McIver J, Alonso MJ. A novel system based on a poloxamer/PLGA blend as a tetanus toxoid delivery vehicle. Pharm Res. 1999;16:682–688. doi: 10.1023/a:1018820507379. [DOI] [PubMed] [Google Scholar]
- 166.Almeida AJ, Alpar HO, Brown MRW. Immune response to nasal delivery of antigenically intact tetanus toxoid associated with poly(L-lactic acid) microspheres in rats, rabbits and guinea-pigs. J Pharm Pharmacol. 1993;45:198–203. doi: 10.1111/j.2042-7158.1993.tb05532.x. [DOI] [PubMed] [Google Scholar]
- 167.Vila A, Sánchez A, Tobío M, Calvo P, Alonso MJ. Design of biodegradable particles for protein delivery. J Control Release. 2002;78:15–24. doi: 10.1016/S0168-3659(01)00486-2. [DOI] [PubMed] [Google Scholar]
- 168.Vila A, Sánchez A, Évora C, Soriano I, Vila Jato JL, Alonso MJ. PEG-PLA nanoparticles as carriers for nasal vaccine delivery. J Aerosol Med. 2004;17:174–185. doi: 10.1089/0894268041457183. [DOI] [PubMed] [Google Scholar]
- 169.Calvo P, Remuñán-López C, Vila-Jato JL, Alonso MJ. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J Appl Polym Sci. 1997;63:125–132. doi: 10.1002/(SICI)1097-4628(19970103)63:1<125::AID-APP13>3.0.CO;2-4. [DOI] [Google Scholar]
- 170.Vila A, Sánchez A, Janes K, Behrens I, Kissel T, Vila-Jato JL, Alonso MJ. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharm Biopharm. 2004;57:123–131. doi: 10.1016/j.ejpb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 171.Prego C, Paolicelli P, Díaz B, Vicente S, Sánchez A, González-Fernández Á, Alonso MJ. Chitosan-based nanoparticles for improving immunization against hepatitis B infection. Vaccine. 2010;28:2607–2614. doi: 10.1016/j.vaccine.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 172.Vicente S, Díaz-Freitas B, Peleteiro M, Sánchez A, Pascual DW, González-Fernández Á, Alonso MJ. A polymer/oil based nanovaccine as a single-dose immunization approach. PLoS One. 2013;8:2–9. doi: 10.1371/journal.pone.0062500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Prego C, Torres D, Alonso MJ. Chitosan nanocapsules: a new carrier for nasal peptide delivery. J Drug Deliv Sci Technol. 2006;16:331–337. doi: 10.1016/S1773-2247(06)50061-9. [DOI] [Google Scholar]
- 174.Vicente S, Peleteiro M, Díaz-Freitas B, Sanchez A, González-Fernández Á, Alonso MJ. Co-delivery of viral proteins and a TLR7 agonist from polysaccharide nanocapsules: a needle-free vaccination strategy. J Control Release. 2013;172:773–781. doi: 10.1016/j.jconrel.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 175.Correia-Pinto JF, Peleteiro M, Csaba N, González-Fernández Á, Alonso MJ. Multi-enveloping of particulated antigens with biopolymers and immunostimulant polynucleotides. J Drug Deliv Sci Technol. 2015;30:424–434. doi: 10.1016/j.jddst.2015.08.010. [DOI] [Google Scholar]
- 176.Didierlaurent AM, Laupèze B, Di Pasquale A, Hergli N, Collignon C, Garçon N. Adjuvant system AS01: helping to overcome the challenges of modern vaccines, Expert Rev. Vaccines. 2017;16:55–63. doi: 10.1080/14760584.2016.1213632. [DOI] [PubMed] [Google Scholar]
- 177.C.T.P. RTS, S Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet. 2015;386:31–45. doi: 10.1016/S0140-6736(15)60721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Moon JJ, Suh H, Polhemus ME, Ockenhouse CF, Yadava A, Irvine DJ. Antigen-displaying lipid-enveloped PLGA nanoparticles as delivery agents for a Plasmodium vivax malaria vaccine. PLoS One. 2012;7 doi: 10.1371/journal.pone.0031472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Guruathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 180.Pejawar-Gaddy S, Kovacs JM, Barouch DH, Chen B, Irvine DJ. Design of lipid nanocapsule delivery vehicles for multivalent display of recombinant env trimers in HIV vaccination. Bioconjug Chem. 2014;25:1470–1478. doi: 10.1021/bc5002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ingale J, Stano A, Guenaga J, Sharma SK, Nemazee D, Zwick MB, Wyatt RT. High-density array of well-ordered HIV-1 spikes on synthetic liposomal nanoparticles efficiently activate B cells. Cell Rep. 2016;15:1986–1999. doi: 10.1016/j.celrep.2016.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Steichen JM, Kulp DW, Tokatlian T, Escolano A, Dosenovic P, Stanfield RL, McCoy LE, Ozorowski G, Hu X, Kalyuzhniy O, Briney B, Schiffner T, Garces F, Freund NT, Gitlin AD, Menis S, Georgeson E, Kubitz M, Adachi Y, Jones M, Mutafyan AA, Yun DS, Mayer CT, Ward AB, Burton DR, Wilson IA, Irvine DJ, Nussenzweig MC, Schief WR. HIV vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity. 2016;45:483–496. doi: 10.1016/j.immuni.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bale S, Goebrecht G, Stano A, Wilson R, Ota T, Tran K, Ingale J, Zwick MB, Wyatt RT. Covalent linkage of HIV-1 trimers to synthetic liposomes elicits improved B cell and antibody responses. J Virol. 2017 doi: 10.1128/JVI.00443-17. JVI.00443-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125:2532–2546. doi: 10.1172/JCI79915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kasturi SP, Kozlowski PA, Nakaya HI, Burger MC, Russo P, Pham M, Kovalenkov Y, Silveira ELV, Havenar-Daughton C, Burton SL, Kilgore KM, Johnson MJ, Nabi R, Legere T, Sher ZJ, Chen X, Amara RR, Hunter E, Bosinger SE, Spearman P, Crotty S, Villinger F, Derdeyn CA, Wrammert J, Pulendran B. Adjuvanting a simian immunodeficiency virus vaccine with Toll-like receptor ligands encapsulated in nanoparticles induces persistent antibody responses and enhanced protection in TRIM5α restrictive macaques. J Virol. 2017;91:e01844–16. doi: 10.1128/JVI.01844-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Stary G, Olive A, Radovic-Moreno AF, Gondek D, Alvarez D, Basto PA, Perro M, Vrbanac VD, Tager AM, Shi J, Yethon JA, Farokhzad OC, Langer R, Starnbach MN, von Andrian UH. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science. 2015;348:aaa8205. doi: 10.1126/science.aaa8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.O’Hagan DT, Ott GS, De Gregorio E, Seubert A. The mechanism of action of MF59 – An innately attractive adjuvant formulation. Vaccine. 2012;30:4341–4348. doi: 10.1016/j.vaccine.2011.09.061. [DOI] [PubMed] [Google Scholar]
- 188.Ledet G, Bostanian LA, Mandal TK. Nanoemulsions as a vaccine adjuvant. In: Tiwari A, Tiwari A, editors. Bioeng Nanomater. CRC press; Boca Raton: 2013. pp. 125–148. [Google Scholar]
- 189.Anselmo AC, Mitragotri S. Nanoparticles in the clinic. Bioeng Transl Med. 2016;1:10–29. doi: 10.1002/btm2.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Safety and pharmacodynamcis of SELA-070 nicotine vaccine in smokers. 2017 https://clinicaltrials.gov/ct2/show/NCT03148925?term=NCT03148925&rank=1 accessed July 4, 2017.
- 191.SVP™ Platform. 2017 http://selectabio.com/platform/svp-platform/ (accessed July 4, 2017)
- 192.Zhang L, Londono P, Grimes S, Blackburn P, Gottlieb P, Eisenbarth GS. MAS-1 adjuvant immunotherapy generates robust Th2 type and regulatory immune responses providing long-term protection from diabetes in late-stage pre-diabetic NOD mice. Autoimmunity. 2014;47:341–350. doi: 10.3109/08916934.2014.910768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.The safety, tolerance, and immunogenicity of MAS-1-adjuvanted seasonal inactivated influenza vaccine (MER4101) 2015 https://clinicaltrials.gov/ct2/show/NCT02500680?term=NCT02500680&rank=1 accessed July 4, 2017.
- 194.Mimopath®. 2017 http://www.mucosis.com/mimopath.php accessed July 4, 2017.
- 195.FluGEM®. 2017 http://www.mucosis.com/flugem.php accessed July 4, 2017.
- 196.Mucosis initiates first-in-human study of SynGEM®, a needle-free nasal spray RSV vaccine. 2016 http://www.mucosis.com/press_releases_07-11-16.php accessed July 4, 2017.
- 197.Vaxfectin® adjuvant. 2017 http://www.vical.com/technology/vaxfectin/default.aspx accessed July 4, 2017.
- 198.Safety and efficacy study of herpes simplex virus type 2 (HSV-2) therapeutic DNA vaccine (HSV-2) 2016 https://clinicaltrials.gov/ct2/show/NCT02837575 accessed July 4, 2017.
- 199.Poloxamer delivery system. 2017 http://www.vical.com/technology/dna-technology/poloxamer/default.aspx accessed July 4, 2017.
- 200.A study to evaluate a therapeutic vaccine, ASP0113, in cytomegalovirus (CMV)-seropositive recipients undergoing allogeneic, hematopoietic cell transplant (HCT) (HELIOS) 2013 https://clinicaltrials.gov/ct2/show/NCT01877655 accessed July 4, 2017.
- 201.A study to evaluate the efficacy and safety of a vaccine, ASP0113, in cytomegalovirus (CMV)-seronegative kidney transplant recipients receiving an organ from a CMV-seropositive donor. 2013 https://clinicaltrials.gov/ct2/show/NCT01974206 accessed July 4, 2017.
- 202.Hayashi M, Aoshi T, Haseda Y, Kobiyama K, Wijaya E, Nakatsu N, Igarashi Y, Standley DM, Yamada H, Honda-Okubo Y, Hara H, Saito T, Takai T, Coban C, Petrovsky N, Ishii KJ. Advax, a delta inulin microparticle, potentiates in-built adjuvant property of co-administered vaccines. EBioMedicine. 2017;15:127–136. doi: 10.1016/j.ebiom.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.A randomised controlled phase 1 study of vaccine therapy for control or cure of chronic hepatitis B virus infection (HBV003) 2017 https://clinicaltrials.gov/ct2/show/NCT03038802 accessed July 4, 2017.
- 204.A phase 1 study to evaluate the immunogenicity and safety of a pandemic avian influenza vaccine in adults (FLU003) 2015 https://clinicaltrials.gov/ct2/show/NCT02335164 accessed July 4, 2017.
- 205.Matrix-M™ technology. (n.d.). http://novavax.com/page/10/matrix-m-adjuvant-technology (accessed July 4, 2017)
- 206.A study to assess the safety and immunogenicity of the malaria vaccine, R21, with Matrix-M1 adjuvant. 2016 https://clinicaltrials.gov/ct2/show/NCT02925403 accessed July 4, 2017.
- 207.Vaccine technology. (n.d.). http://novavax.com/page/8/vaccine-technology (accessed July 4, 2017)
- 208.A study to determine the safety and efficacy of the RSV F vaccine to protect infants via maternal immunization. 2015 https://clinicaltrials.gov/ct2/show/NCT02624947 accessed September 4, 2017.
- 209.Safety and immunogenicity study to evaluate single- or two-dose regimens of RSV F vaccine with and without aluminum phosphate or Matrix-M1™ adjuvants in clinically-stable older adults. 2017 https://clinicaltrials.gov/ct2/show/NCT03026348 accessed July 4, 2017.
- 210.Discovery platforms. 2017 http://www.immunedesign.com/platforms/ accessed July 4, 2017.
- 211.Pipeline. 2017 http://www.immunedesign.com/pipeline/ accessed July 4, 2017.
- 212.Phase 1 study of intradermal LV305 in patients with locally advanced, relapsed or metastatic cancer expressing NY-ESO-1. 2014 doi: 10.1080/2162402X.2020.1847846. https://clinicaltrials.gov/ct2/show/NCT02122861 accessed September 4, 2017. [DOI] [PMC free article] [PubMed]
- 213.A phase 1b safety study of CMB305 (sequentially administered LV305 and G305) in patients with locally advanced, relapsed, or metastatic Cancer expressing NY-ESO-1. 2015 https://clinicaltrials.gov/ct2/show/NCT02387125?term=NCT02387125&rank=1 accessed July 4, 2017.
- 214.Overview of Juvaris’ technology platform. 2014 http://www.juvaris.com/technology/overview.html accessed July 4, 2017.
- 215.JVRS-100 for the treatment of patients with relapsed or refractory leukemia. 2009 https://clinicaltrials.gov/ct2/show/NCT00860522?term=NCT00860522&rank=1 accessed July 4, 2017.
- 216.Gerke C, Colucci AM, Giannelli C, Sanzone S, Vitali CG, Sollai L, Rossi O, Martin LB, Auerbach J, Di Cioccio V, Saul A. Production of a Shigella sonnei vaccine based on generalized modules for membrane antigens (GMMA), 1790GAHB. PLoS One. 2015;10:1–22. doi: 10.1371/journal.pone.0134478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.A study to evaluate safety and immunogenicity of 1 booster dose of 1790GAHB vaccine in healthy adults primed with 3 doses of 1790GAHB vaccine in study H03_01TP compared to 1 vaccination of 1790GAHB in either subjects who received placebo in the same study. 2017 https://clinicaltrials.gov/ct2/show/NCT03089879?term=NCT03089879&rank=1 accessed July 4, 2017.
- 218.Safety of Chlamydia vaccine CTH522 in healthy women aged 18 to 45 years. 2016 https://clinicaltrials.gov/ct2/show/NCT02787109?term=NCT02787109&rank=1 accessed July 4, 2017.
- 219.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 220.Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784–790. doi: 10.1016/j.cell.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dinarello CA. Anti-inflammatory agents: present and future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Gouveia VM, Lima SAC, Nunes C, Reis S. Non-biologic nanodelivery therapies for rheumatoid arthritis. J Biomed Nanotechnol. 2015;11:1701–1721. doi: 10.1166/jbn.2015.2159. [DOI] [PubMed] [Google Scholar]
- 223.Viscido A, Capannolo A, Latella G, Caprilli R, Frieri G. Nanotechnology in the treatment of inflammatory bowel diseases. J Crohn’s Colitis. 2014;8:903–918. doi: 10.1016/j.crohns.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 224.Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. 2014;20:91–99. doi: 10.3748/wjg.v20.i1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Beloqui A, Coco R, Préat V. Targeting inflammatory bowel diseases by nanocarriers loaded with small and biopharmaceutical anti-inflammatory drugs. Curr Pharm Des. 2016;22:6192–6206. doi: 10.2174/1381612822666160211141813. [DOI] [PubMed] [Google Scholar]
- 226.Tran TH, Amiji MM. Targeted delivery systems for biological therapies of inflammatory diseases. Expert Opin Drug Deliv. 2014;12:393–414. doi: 10.1517/17425247.2015.972931. [DOI] [PubMed] [Google Scholar]
- 227.Takedatsu H, Mitsuyama K, Torimura T. Nanomedicine and drug delivery strategies for treatment of inflammatory bowel disease. World J Gastroenterol. 2015;21:11343–11352. doi: 10.3748/wjg.v21.i40.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tahara K, Samura S, Tsuji K, Yamamoto H, Tsukada Y, Bando Y, Tsujimoto H, Morishita R, Kawashima Y. Oral nuclear factor-κB decoy oligonucleotides delivery system with chitosan modified poly(D,L-lactide-co-glycolide) nanospheres for inflammatory bowel disease. Biomaterials. 2011;32:870–878. doi: 10.1016/j.biomaterials.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 229.Zhang J, Tang C, Yin C. Galactosylated trimethyl chitosan-cysteine nanoparticles loaded with Map4k4 siRNA for targeting activated macrophages. Biomaterials. 2013;34:3667–3677. doi: 10.1016/j.biomaterials.2013.01.079. [DOI] [PubMed] [Google Scholar]
- 230.Laroui H, Dalmasso G, Nguyen HTT, Yan Y, Sitaraman SV, Merlin D. Drug-loaded nanoparticles targeted to the colon with polysaccharide hydrogel reduce colitis in a mouse model. Gastroenterology. 2010;138:843–853. doi: 10.1053/j.gastro.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 231.Sardar S, Andersson Å. Old and new therapeutics for rheumatoid arthritis: in vivo models and drug development. Immunopharmacol Immunotoxicol. 2016;38:2–13. doi: 10.3109/08923973.2015.1125917. [DOI] [PubMed] [Google Scholar]
- 232.Rantapää-Dahlqvist S, de Jong BAW, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 233.Holmdahl R, Malmström V, Burkhardt H. Autoimmune priming, tissue attack and chronic inflammation - The three stages of rheumatoid arthritis. Eur J Immunol. 2014;44:1593–1599. doi: 10.1002/eji.201444486. [DOI] [PubMed] [Google Scholar]
- 234.Dolati S, Sadreddini S, Rostamzadeh D, Ahmadi M, Jadidi-Niaragh F, Yousefi M. Utilization of nanoparticle technology in rheumatoid arthritis treatment. Biomed Pharmacother. 2016;80:30–41. doi: 10.1016/j.biopha.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 235.Jain S, Tran TH, Amiji M. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials. 2015;61:162–177. doi: 10.1016/j.biomaterials.2015.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Whitmire RE, Scott Wilson D, Singh A, Levenston ME, Murthy N, García AJ. Self-assembling nanoparticles for intra-articular delivery of anti-inflammatory proteins. Biomaterials. 2012;33:7665–7675. doi: 10.1016/j.biomaterials.2012.06.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jung YS, Park W, Na K. Temperature-modulated noncovalent interaction controllable complex for the long-term delivery of etanercept to treat rheumatoid arthritis. J Control Release. 2013;171:143–151. doi: 10.1016/j.jconrel.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 238.Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–939. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 239.Look M, Stern E, Wang QA, DiPlacido LD, Kashgarian M, Craft J, Fahmy TM. Nanogel-based delivery of mycophenolic acid ameliorates systemic lupus erythematosus in mice. J Clin Invest. 2013;123:1741–1749. doi: 10.1172/JCI65907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Look M, Saltzman WM, Craft J, Fahmy TM. The nanomaterial-dependent modulation of dendritic cells and its potential influence on therapeutic immunosuppression in lupus. Biomaterials. 2014;35:1089–1095. doi: 10.1016/j.biomaterials.2013.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Moallem E, Koren E, Ulmansky R, Pizov G, Barlev M, Barenholz Y, Naparstek Y. A liposomal steroid nano-drug for treating systemic lupus erythematosus. Lupus. 2016;25:1209–1216. doi: 10.1177/0961203316636468. [DOI] [PubMed] [Google Scholar]
- 242.Shimizu H, Hori Y, Kaname S, Yamada K, Nishiyama N, Matsumoto S, Miyata K, Oba M, Yamada A, Kataoka K, Fujita T. siRNA-based therapy ameliorates glomerulonephritis. J Am Soc Nephrol. 2010;21:622–633. doi: 10.1681/ASN.2009030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Atassi MZ, Casali P. Molecular mechanisms of autoimmunity. Autoimmunity. 2008;41:123–132. doi: 10.1080/08916930801929021. [DOI] [PubMed] [Google Scholar]
- 244.Kurd N, Robey EA. T-cell selection in the thymus: a spatial and temporal perspective. Immunol Rev. 2016;271:114–126. doi: 10.1111/imr.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see) Nat Rev Immunol. 2014;14:377–91. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ring GH, Lakkis FG. Breakdown of self-tolerance and the pathogenesis of autoimmunity. Semin Nephrol. 1999;19:25–33. [PubMed] [Google Scholar]
- 247.Lewis KL, Reizis B. Dendritic cells: arbiters of immunity and immunological tolerance. Cold Spring Harb Perspect Biol. 2012;4:a007401. doi: 10.1101/cshperspect.a007401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol. 2010:111–165. doi: 10.1016/B978-0-12-380995-7.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol. 2007;7:665–677. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- 251.Gaur A, Wiers B, Liu A, Rothbard J, Fathman CG. Amelioration of autoimmune encephalomyelitis by myelin basic protein synthetic peptide-induced anergy. Science. 1992;258:1491–1494. doi: 10.1126/science.1279812. [DOI] [PubMed] [Google Scholar]
- 252.Critchfield JM, Racke MK, Zúñiga-Pflücker JC, Cannella B, Raine CS, Goverman J, Lenardo MJ. T cell deletion in high antigen dose therapy of autoimmune encephalomyelitis. Science. 1994;263:1139–1143. doi: 10.1126/science.7509084. [DOI] [PubMed] [Google Scholar]
- 253.Mukherjee R, Chaturvedi P, Qin HY, Singh B. CD4+CD25+ regulatory T cells generated in response to insulin B:9-23 peptide prevent adoptive transfer of diabetes by diabetogenic T cells. J Autoimmun. 2003;21:221–237. doi: 10.1016/S0896-8411(03)00114-8. [DOI] [PubMed] [Google Scholar]
- 254.Coon B, An LL, Whitton JL, von Herrath MG. DNA immunization to prevent autoimmune diabetes. J Clin Invest. 1999;104:189–194. doi: 10.1172/JCI7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Smith CE, Eagar TN, Strominger JL, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2005;102:9595–9600. doi: 10.1073/pnas.0504131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Genain CP, Abel K, Belmar N, Villinger F, Rosenberg DP, Linington C, Raine CS, Hauser SL. Late complications of immune deviation therapy in a nonhuman primate. Science. 1996;274:2054–2057. doi: 10.1126/science.274.5295.2054. [DOI] [PubMed] [Google Scholar]
- 257.Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 258.Friedman A, Weiner HL. Induction of anergy or active suppression following oral tolerance is determined by antigen dosage. Proc Natl Acad Sci U S A. 1994;91:6688–6692. doi: 10.1073/pnas.91.14.6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Meyer AL, Benson JM, Gienapp IE, Cox KL, Whitacre CC. Suppression of murine chronic relapsing experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. J Immunol. 1996;157:4230–4238. [PubMed] [Google Scholar]
- 260.Karpus WJ, Kennedy KJ, Smith WS, Miller SD. Inhibition of relapsing experimental autoimmune encephalomyelitis in SJL mice by feeding the immunodominant PLP139-151 peptide. J Neurosci Res. 1996;45:410–423. doi: 10.1002/(SICI)1097-4547(19960815)45:4<410::AID-JNR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 261.Bai XF, Shi FD, Xiao BG, Li HL, van der Meide PH, Link H. Nasal administration of myelin basic protein prevents relapsing experimental autoimmune encephalomyelitis in DA rats by activating regulatory cells expressing IL-4 and TGF-beta mRNA. J Neuroimmunol. 1997;80:65–75. doi: 10.1016/s0165-5728(97)00133-1. [DOI] [PubMed] [Google Scholar]
- 262.Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, Hafler DA. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science. 1993;259:1321–1324. doi: 10.1126/science.7680493. [DOI] [PubMed] [Google Scholar]
- 263.National Multiple Sclerosis Society. What is MS? (n.d.). http://www.nationalmssociety.org/What-is-MS (accessed April 18, 2017)
- 264.Johnson D, Hafler DA, Fallis RJ, Lees MB, Brady RO, Quarles RH, Weiner HL. Cell-mediated immunity to myelin-associated glycoprotein, proteolipid protein, and myelin basic protein in multiple sclerosis. J Neuroimmunol. 1986;13:99–108. doi: 10.1016/0165-5728(86)90053-6. [DOI] [PubMed] [Google Scholar]
- 265.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 266.Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014;275:350–363. doi: 10.1111/joim.12203. [DOI] [PubMed] [Google Scholar]
- 267.Rudick RA, Sandrock A. Natalizumab: alpha 4-integrin antagonist selective adhesion molecule inhibitors for MS. Expert Rev Neurother. 2004;4:571–580. doi: 10.1586/14737175.4.4.571. [DOI] [PubMed] [Google Scholar]
- 268.Ragonese P, Aridon P, Vazzoler G, Mazzola MA, Lo Re V, Lo Re M, Realmuto S, Alessi S, D’Amelio M, Savettieri G, Salemi G. Association between multiple sclerosis, cancer risk, and immunosuppressant treatment: a cohort study. BMC Neurol. 2017;17:155. doi: 10.1186/s12883-017-0932-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Dianzani C, Boldorini R, Chiocchetti A, Renò F, Dianzani U. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. 2014;32:5681–5689. doi: 10.1016/j.vaccine.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 270.Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM. Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific. Cell Rep. 2016;16:2940–2952. doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, Reimer R, Heine M, Huber S, Waurisch C, Eychmüller A, Wraith DC, Korn T, Nielsen P, Weller H, Schramm C, Lüth S, Lohse AW, Heeren J, Herkel J. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J Hepatol. 2015;62:1349–1356. doi: 10.1016/j.jhep.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 272.Yuan B, Zhao L, Fu F, Liu Y, Lin C, Wu X, Shen H, Yang Z. A novel nanoparticle containing MOG peptide with BTLA induces T cell tolerance and prevents multiple sclerosis. Mol Immunol. 2014;57:93–99. doi: 10.1016/j.molimm.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 273.Tostanoski LH, Chiu YC, Andorko JI, Guo M, Zeng X, Zhang P, Royal W, Jewell CM. Design of polyelectrolyte multilayers to promote immunological tolerance. ACS Nano. 2016;10:9334–9345. doi: 10.1021/acsnano.6b04001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Hess KL, Andorko JI, Tostanoski LH, Jewell CM. Polyplexes assembled from self-peptides and regulatory nucleic acids blunt toll-like receptor signaling to combat autoimmunity. Biomaterials. 2016;118:51–62. doi: 10.1016/j.biomaterials.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Pujol-Autonell I, Mansilla MJ, Rodriguez-Fernandez S, Cano-Sarabia M, Navarro-Barriuso J, Ampudia RM, Rius A, Garcia-Jimeno S, Perna-Barrul D, Martinez-Caceres E, Maspoch D, Vives-Pi M. Liposome-based immunotherapy against autoimmune diseases: therapeutic effect on multiple sclerosis. Nanomedicine (Lond) 2017;12:1231–1242. doi: 10.2217/nnm-2016-0410. [DOI] [PubMed] [Google Scholar]
- 276.International Diabetes Federation. IDF Diabetes Atlas. (7th) 2015 doi: 10.1289/image.ehp.v119.i03. [DOI] [Google Scholar]
- 277.Morran MP, Omenn GS, Pietropaolo M. Immunology and genetics of type 1 diabetes. Mt Sinai J Med A J Transl Pers Med. 2008;75:314–327. doi: 10.1002/msj.20052. [DOI] [PubMed] [Google Scholar]
- 278.Yoon JW, Jun HS, Santamaria P. Cellular and molecular mechanisms for the initiation and progression of beta cell destruction resulting from the collaboration between macrophages and T cells. Autoimmunity. 1998;27:109–122. doi: 10.3109/08916939809008041. [DOI] [PubMed] [Google Scholar]
- 279.Dilts SM, Solvason N, Lafferty KJ. The role of CD4 and CD8 T cells in the development of autoimmune diabetes. J Autoimmun. 1999;13:285–288. doi: 10.1006/jaut.1999.0323. [DOI] [PubMed] [Google Scholar]
- 280.Arvan P, Pietropaolo M, Ostrov D, Rhodes CJ. Islet autoantigens: structure, function, localization, and regulation. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA, Keselowsky BG. A combination hydrogel microparticle-based vaccine prevents type 1 diabetes in non-obese diabetic mice. Sci Rep. 2015;5 doi: 10.1038/srep13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Lee WK, Park JY, Jung S, Yang CW, Kim WU, Kim HY, Park JH, Park JS. Preparation and characterization of biodegradable nanoparticles entrapping immunodominant peptide conjugated with PEG for oral tolerance induction. J Control Release. 2005;105:77–88. doi: 10.1016/j.jconrel.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 283.Capini C, Jaturanpinyo M, Chang HI, Mutalik S, McNally A, Street S, Steptoe R, O’Sullivan B, Davies N, Thomas R. Antigen-specific suppression of inflammatory arthritis using liposomes. J Immunol. 2009;182:3556–3565. doi: 10.4049/jimmunol.0802972. [DOI] [PubMed] [Google Scholar]
- 284.Trentham DE, Dynesius-Trentham RA, Orav EJ, Combitchi D, Lorenzo C, Sewell KL, Hafler DA, Weiner HL. Effects of oral administration of type II collagen on rheumatoid arthritis. Science. 1993;261:1727–1730. doi: 10.1126/science.8378772. [DOI] [PubMed] [Google Scholar]
- 285.Cohen SN, Chang ACY, Boyert HW, Hellingt RB. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci. 1973;70:3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Johnson IS. Human insulin from recombinant DNA technology. Science. 1983;219:632–637. doi: 10.1126/science.6337396. [DOI] [PubMed] [Google Scholar]
- 287.Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28:917–924. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 288.Walsh G. Biopharmaceutical benchmarks 2014. Nat Biotechnol. 2014;32:992–1000. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 289.U.S. Food and Drug Administration. Guidance for industry: immunogenicity assessment for therapeutic protein products. 2014 [Google Scholar]
- 290.Yin L, Chen X, Vicini P, Rup B, Hickling TP. Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products. Cell Immunol. 2015;295:118–126. doi: 10.1016/j.cellimm.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 291.Eschbach JW, Kelly MR, Haley NR, Abels RI, Adamson JW. Treatment of the anemia of progressive renal failure with recombinant human erythropoietin. N Engl J Med. 1989;321:158–163. doi: 10.1056/NEJM198907203210305. [DOI] [PubMed] [Google Scholar]
- 292.Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, Michaud P, Papo T, Ugo V, Teyssandier I, Varet B, Mayeux P. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346:469–475. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]
- 293.Boven K, Knight J, Bader F, Rossert J, Eckardt KU, Casadevall N. Epoetin-associated pure red cell aplasia in patients with chronic kidney disease: solving the mystery. Nephrol Dial Transplant. 2005;20:33–40. doi: 10.1093/ndt/gfh1072. [DOI] [PubMed] [Google Scholar]
- 294.Hermeling S, Schellekens H, Crommelin DJA, Jiskoot W. Micelle-associated protein in epoetin formulations: a risk factor for immunogenicity? Pharm Res. 2003;20:1903–1907. doi: 10.1023/B:PHAM.0000008034.61317.02. [DOI] [PubMed] [Google Scholar]
- 295.Bennett CL, Luminari S, Nissenson AR, Tallman MS, Klinge SA, McWilliams N, McKoy JM, Kim B, Lyons EA, Trifilio SM, Raisch DW, Evens AM, Kuzel TM, Schumock GT, Belknap SM, Locatelli F, Rossert J, Casadevall N. Pure red-cell aplasia and epoetin therapy. N Engl J Med. 2004;351:1403–1408. doi: 10.1056/NEJMoa040528. [DOI] [PubMed] [Google Scholar]
- 296.Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol. 2014;11:99–109. doi: 10.3109/1547691X.2013.821564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, Topp EM. Immunogenicity of therapeutic protein aggregates. J Pharm Sci. 2016;105:417–430. doi: 10.1016/j.xphs.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 298.Ahmadi M, Bryson CJ, Cloake EA, Welch K, Filipe V, Romeijn S, Hawe A, Jiskoot W, Baker MP, Fogg MH. Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm Res. 2015;32:1383–1394. doi: 10.1007/s11095-014-1541-x. [DOI] [PubMed] [Google Scholar]
- 299.Rombach-Riegraf V, Karle AC, Wolf B, Sordé L, Koepke S, Gottlieb S, Krieg J, Djidja MC, Baban A, Spindeldreher S, Koulov AV, Kiessling A. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. PLoS One. 2014;9:e86322. doi: 10.1371/journal.pone.0086322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Singh SK. Impact of product-related factors on immunogenicity of biotherapeutics. J Pharm Sci. 2011;100:354–387. doi: 10.1002/jps.22276. [DOI] [PubMed] [Google Scholar]
- 301.Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016;2016:18. doi: 10.1155/2016/1298473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Hoffmann S, Cepok S, Grummel V, Lehmann-Horn K, Hackermüller J, Stadler PF, Hartung HP, Berthele A, Deisenhammer F, Wassmuth R, Hemmer B. HLA-DRB1*0401 and HLA-DRB1*0408 are strongly associated with the development of antibodies against interferon-beta therapy in multiple sclerosis, Am. J Hum Genet. 2008;83:219–227. doi: 10.1016/j.ajhg.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Bartelds GM, Wijbrandts CA, Nurmohamed MT, Wolbink GJ, de Vries N, Tak PP, Dijkmans BAC, Crusius JBA, vand der Horst-Bruinsma IE. Anti-adalimumab antibodies in rheumatoid arthritis patients are associated with interleukin-10 gene polymorphisms. Arthritis Rheum. 2009;60:2541–2542. doi: 10.1002/art.24709. [DOI] [PubMed] [Google Scholar]
- 304.Lacaná E, Yao LP, Pariser AR, Rosenberg AS. The role of immune tolerance induction in restoration of the efficacy of ERT in Pompe disease. Am J Med Genet Part C Semin Med Genet. 2012;160C:30–39. doi: 10.1002/ajmg.c.31316. [DOI] [PubMed] [Google Scholar]
- 305.Kishnani PS, Dickson PI, Muldowney L, Lee JJ, Rosenberg A, Abichandani R, Bluestone JA, Burton BK, Dewey M, Freitas A, Gavin D, Griebel D, Hogan M, Holland S, Tanpaiboon P, Turka LA, Utz JJ, Wang YM, Whitley CB, Kazi ZB, Pariser AR. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol Genet Metab. 2016;117:66–83. doi: 10.1016/j.ymgme.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 306.Tostanoski LH, Gosselin EA, Jewell CM. Engineering tolerance using biomaterials to target and control antigen presenting cells. Discov Med. 2016;21:403–410. [PubMed] [Google Scholar]
- 307.Fischer R, Turnquist HR, Taner T, Thomson AW. Dendritic Cells. Springer Berlin Heidelberg; Berlin, Heidelberg: 2009. Use of rapamycin in the induction of tolerogenic dendritic cells; pp. 215–232. [DOI] [PubMed] [Google Scholar]
- 308.Haddadi A, Elamanchili P, Lavasanifar A, Das S, Shapiro J, Samuel J. Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells. J Biomed Mater Res - Part A. 2008;84A:885–898. doi: 10.1002/jbm.a.31373. [DOI] [PubMed] [Google Scholar]
- 309.Jhunjhunwala S, Raimondi G, Thomson AWW, Little SRR. Delivery of rapamycin to dendritic cells using degradable microparticles. J Control Release. 2009;133:191–197. doi: 10.1016/j.jconrel.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Kishimoto TK, Ferrari JD, Lamothe RA, Kolte PN, Griset AP, O’Neil C, Chan V, Browning EA, Chalishazar A, Kuhlman W, Fu FN, Viseux N, Altreuter DH, Johnston L, Maldonado RA. Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol. 2016;11:890–899. doi: 10.1038/NNANO.2016.135. [DOI] [PubMed] [Google Scholar]
- 311.Multi-dose safety/pharmacodynamic study of SEL-212/SEL-037 in subjects with symptomatic gout & elevated blood uric acid. 2016 https://clinicaltrials.gov/ct2/show/NCT02959918 (accessed July 4, 2017)