

Abstract
Our objective is to identify molecular factors which contribute to the increased risk of smokers for oral squamous cell carcinoma (OSCC). In the present study, we investigated the effects of cigarette smoke condensate (CSC) on gene expression profiles in different human oral cell phenotypes: normal epidermal keratinocytes (NHEK), oral dysplasia cell lines (Leuk1 and Leuk2), and a primary oral carcinoma cell line (101A). We determined differential gene expression patterns in CSC-exposed versus non-exposed cells using high-density microarray RNA expression profiling and validation by quantitative real-time RT-PCR. A set of 35 genes was specifically up- or down-regulated following CSC treatment (25 μg/ml for 24 h) by at least 2-fold in any one cell type. Notably, five genes of the cytochrome P450 (CYP1A1, CYP1B1) and aldo-keto reductase (AKR1C1, AKR1C3, AKR1B10) families were highly increased in expression, some of them 15- to 30-fold. The timing and extent of induction for these genes differed among the four cell phenotypes. A potential biological interaction network for the CSC response in oral cells was derived from these data, proposing novel putative response pathways. These CSC-responsive genes presumably participate in the prevention or repair of carcinogen-induced DNA damage in tobacco-related oral carcinogenesis, and may potentially be exploited for determining the severity of exposure and for correcting mutagenic damage in exposed tissues of the oral cavity.
Keywords: Oral cancer, Aldo-keto reductases, Cytochrome P450, Cigarette smoke condensate, Gene expression, Microarray
1. Introduction
Oral squamous cell carcinoma (OSCC) is the most common malignancy of the head and neck, with a worldwide incidence of 300,000 new cases annually. The major inducer of OSCC is exposure to tobacco, considered to be responsible for 50–90% of cases worldwide, and the incidence of OSCC in cigarette smokers is 7–10 times higher than for never smokers (Sudbo and Reith, 2005; Warnakulasuriya et al., 2005). Many of the chemical carcinogens contained in cigarette smoke are polycyclic aromatic hydrocarbons (PAH), a family of ubiquitous environmental carcinogens that are known to have mutagenic and carcinogenic effects. A variety of PAHs have been shown to cause cellular transformations only after metabolic activation by drug-metabolizing enzymes, such as cytochromes P450 (CYP) and aldo-keto reductases (AKRs) that produce highly reactive carcinogenic electrophiles (Pelkonen and Nebert, 1982; Rubin, 2001; Palackal et al., 2002; Mahadevan et al., 2005).
PAHs are activated to genotoxic intermediates through at least three primary pathways, all of which can lead to the production of G to T transversions (Palackal et al., 2002). In most cases, oxidation of PAHs by CYP enzymes is an initial step in the activation process. Among the various forms of CYPs determined so far, CYP1A1 and CYP1B1 have been shown to be the most important human CYP enzymes in the metabolic activation of PAHs and PAH dihydrodiols (Kim et al., 1998; Shimada et al., 1999; Nebert et al., 2004). It has been hypothesized that genetic variances in CYP expression, inducibility, or activity are responsible for individual susceptibility to cancer (Nebert, 1991; Alexandrie et al., 1994).
The human enzyme CYP1A1 is the most active among the CYPs in metabolizing procarcinogens, like PAHs and aromatic amines, into active species forming DNA adducts (Roberts-Thompson et al., 1993). CYP1A1 variants and cancer risk have been investigated in several studies (Bartsch et al., 2000). CYP1B1 also contributes to aromatic hydrocarbon hydroxylase activity, and interindividual variation in the expression of CYP1B1 has been observed (Shehin et al., 2000; Murray et al., 2001). Human CYP1B1 catalyzes the oxidation of polycyclic aromatic hydrocarbons to yield electrophilic intermediates capable of binding covalently to DNA (Shimada et al., 1996; Crofts et al., 1997), a step believed to be important in the initiation of carcinogenesis. Both CYP1A1 and CYP1B1 may play important roles in the bioactivation of chemically diverse tobacco smoke procarcinogens to reactive metabolites (Kim et al., 2004). Thus, the constitutive and inducible expression of CYP1A1 and CYP1B1 are considered to be important determinants of carcinogenesis, although the exact relationship between CYP1 expression and chemically induced carcinogenesis remains to be established.
One of the pathways involves dihydrodiol dehydrogenases, members of the aldo-keto reductase (AKR) superfamily that includes AKR1A1, AKR1C1, AKR1C2, AKR1C3 and AKR1C4 (Penning et al., 2000; Palackal et al., 2002; Yu et al., 2002). This pathway has been shown experimentally to produce PAH metabolites that form DNA adducts or reactive oxygen species (ROS) leading to oxidative DNA damage (Palackal et al., 2002). AKR1A1 is widely distributed among all mammalian species and can catalyze the reduction of a variety of aromatic and medium-chain aliphatic aldehydes to their corresponding alcohols (Hyndman and Flynn, 1999). AKR1C3 is particularly important in metabolizing potent trans-dihydrodiols containing more than two aromatic rings (Palackal et al., 2002). AKR1B10 is related to human aldose reductase AKR1B1; yet it remains to be determined whether AKR1B10 is induced by tobacco carcinogens or is involved in their metabolism (Penning, 2005).
Identification of genes whose expression is specifically modified by exposure to cigarette smoke will provide a better understanding of their mechanisms of action, and allow the development of sensitive and specific biomarkers for both exposure and susceptibility. Yet the mechanisms and key participants for this process are very poorly understood. The purpose of the present study was to identify molecular factors that may contribute to the pronounced increased risk of smokers for OSCC. We used high-density oligonucleotide microarray expression profiling to determine the effects of the cigarette smoke condensate (CSC) on gene expression in human keratinocytes and OSCC cells in vitro. We focused our analysis on CYPs (1A1 and 1B1) and AKRs (1A1, 1C3, and 1B10), which account for PAH metabolism, and confirmed by microarray and qRT-PCR that these relevant genes have highly induced expression in CSC-exposed cells. Extraction of potential biological interaction networks among those identified and validated genes also suggests novel mechanistic pathways to rationalize the CSC response in the target cells.
2. Materials and methods
2.1. Materials
Cell culture reagents were obtained form the following suppliers: Dulbecco modified Eagle’s medium (DMEM), trypsin (0.25%)–EDTA, penicillin–streptomycin (10 mg/ml), amphotericin B (0.25 mg/ml), HEPES buffer IM, L-glutamine (200 mM) from Cellgro (Herndorn, VA); KGM-2 medium from Cambrex (East Rutherford, NJ); fetal bovine serum (Premium) from Atlanta Biologicals (Atlanta, GA). Dimethyl sulfoxide was from Fisher Biotech (Fair Lawn, NJ), and DEPC-treated water from Ambion (Austin, TX). MTT (1-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliuymbromide) was from Sigma (St. Louis, MO).
Cigarette smoke condensate was purchased from Murty Pharmaceuticals Inc. (Lexington, KY) and was prepared using a Phipps-Bird 20-channel smoking machine designed for Federal Trade Commission testing. The particulate matter from Kentucky standard cigarettes (1R3F; University of Kentucky, Lexington, KY) was collected on Cambridge glass fiber filters and the amount of CSC obtained was determined by weight increase of the filter. CSC was prepared by dissolving the collected smoke particulates in dimethyl sulfoxide (DMSO) to yield a 4% solution (w/v). The average yield of CSC was 26.1 mg/cigarette. The CSC was diluted into DMSO and aliquots were kept at −80 °C.
2.2. Cell culture
Normal human epidermal keratinocytes (NHEK) cells (Cambrex, Walkersville, MD) were cultured in KGM-2 medium supplemented with growth factor Bullet Kit (Cambrex). These cells were subcultured by detaching with 0.025% trypsin and transferring into new flasks at a density of 3500 cells/cm2. Exponentially growing NHEK cells were treated and harvested after 2 or 3 passages. MSK Leuk1 and MSK Leuk2 were established from dysplasic leukoplakia lesions adjacent to tongue squamous cell carcinomas in a 46-year-old or 72-year-old female, respectively (gifts from Dr. Peter Sacks, New York University, New York, NY). Cells were routinely maintained in serum-free KGM-2 medium plus supplement bullet kit and passaged using 0.125% trypsin solution. 101A cells were derived from the primary oral squamous cell carcinoma (T3N3M0) involving the tonsillar area of a 65-year-old female (gift from Dr. Thomas E. Carey, University of Michigan, Ann Arbor, MI). This cell line was grown as monolayer in DMEM supplemented with 2 mM L-glutamine, 100 μg/ml penicillin G, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B and 10% FCS. All cell lines were grown to 50–75% confluence at 37 °C in a humidified atmosphere containing 5% CO2. Treatment with CSC (0–100 μg/ml in DMSO) was done for time points of 0, 1, 5, or 24 h; control cells were treated with the same volume of DMSO solvent only. Two biological replicates were used for every treatment regimen. All protocols for the use of human cell lines in this work were approved by the Institutional Review Board of The University of Louisville, Louisville, KY.
2.3. MTT viability assays
Viability of cells after CSC exposure was detected using MTT dye reduction assay. Briefly, 2 × 104 cells/well were seeded in a 96-well plate and cultured for up to 60% confluence. Serial dilutions of CSC were added, and cells were treated for 1, 5, or 24 h. After treatment, 50 μl of MTT solution (5 mg/ml in PBS) were added to each well and incubated for 4 h. The reaction was terminated by addition of 100 μl of 20% SDS in 50% dimethylformamide, and the plate was incubated overnight at 37 °C for total solubilization of reduced MTT. Absorbance was measured at 570 nm in a microplate reader (PowerWave™ Microplate Spectrophotometer, Bio-Tek Instruments Inc., Winooski, VT), and viable cell numbers determined based on a standard curve.
2.4. RNA isolation
Cells were grown to approximately 75% confluence in 6-well plates, with or without CSC treatment (25 μg/ml for 5 or 24 h), and total RNA was isolated with TriZol™ reagent (Invitrogen Corporation, Carlsbad, CA). The purity and integrity of each RNA preparation was evaluated by using RNA Nano Chips on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA).
2.5. Microarray procedures
Micoarray expression analysis was performed on a Microarray Analysis Suite instrument system (Affymetrix, Santa Clara, CA). Biotin-labeled cRNA was synthesized from 10 μg of total RNA using the GeneChip® Expression One-Cycle Target Labeling kit and its standard protocol (Affymetrix). The quality of cRNA preparations was confirmed using the Agilent 2100 BioAnalyzer. Each cRNA (15 μg) sample was fragmented by metal-induced hydrolysis in fragmentation buffer (40 mM Tris-acetate (pH 8.1), 100 mM KOAc, 30 mM Mg(OAc)2), passed through the GeneChip© Sample Cleanup Module, and spiked with hybridization controls (Affymetrix). The labeled cRNA was hybridized for 16 h at 45 °C to Affymetrix HG-U133A arrays, representing probes for ~22,000 annotated genes. The arrays were washed, then stained with streptavidin–phycoerythrin, followed by incubation with biotinylated anti-streptavidin antibody (Vector Laboratories, Burlingame, CA) and a second staining with streptavidin–phycoerythrin. Stained arrays were scanned at 488 nm with an Agilent GeneArray Scanner (Agilent), and the resulting image was analyzed using Microarray Suite 5.0 (MAS5) software (Affymetrix). Two biological replicates were used for each treatment and control.
2.6. Microarray data analysis
All sample data were scaled to a uniform target intensity of 500 to allow quantitative comparisons across experiments. Two-way ANOVA, false discovery rate (FDR) and principle component analysis (PCA) were performed with Partek Pro 6.0 software (Partek Inc., St. Charles, MO).
2.7. Quantitative real-time RT-PCR
Relative quantitation of CYP1A1, CYP1B1, AKR1C1, AKR1C3 and AKR1B10 by real-time reverse-transcriptase PCR (RT-PCR) was done using an ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Foster City, CA). The cDNA prepared for microarray analysis was diluted to 1 ng/μl, and 2 μl was used as template for RT-PCR in a 25 μl reaction. Forward and reverse primer mix was added (3 μl, 1:1 mix, 0.3 μM each) in SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA). RT-PCR cycles consisted of initial denaturing for 10 min at 95 °C, followed by 40 cycles of denaturation at 95 °C/15 s and annealing/extension at 60 °C/1 min. Each reaction was performed in triplicate and ‘no-template’ controls were included in each experiment. Dissociation curves were run to eliminate non-specific amplification, including primer-dimers. The cycle threshold (CT) values were normalized to the house keeping gene β-actin and the fold change was calculated using 2T−ΔΔC method (Livak and Schmittgen, 2001).
DNA primers for RT-PCR were synthesized by Invitrogen Life Technology, after sequences were obtained from the cited references for the following primer pairs: CYP1A1 forward (5′-TGG-ATG-AGA-ACG-CCA-ATG-TC-3′) and reverse (5′-TGG-GTT-GAC-CCA-TAG-CTT-CT-3′) (Huang et al., 1996); CYP1B1 forward (5′-CAC-TGC-CAA-CAC-CTC-TGT-CTT-3′) and reverse (5′-CAA-GGA-GCT-CCA-TGG-ACT-CT-3′) (Huang et al., 1996); AKR1C1 forward (5′-GTA-AAG-CTT-TAG-AGG-CCA-C-3′) and reverse (5′-CAC-CCA-TGC-TTC-TTC-TCG-G-3′) (Penning et al., 2000); AKR1C3 forward (5′-GTA-AAG-CTT-TGG-AGG-TCA-C-3′) and reverse (5′-CAC-CCA-TCG-TTT-GTC-TCG-T′-3′) (Penning et al., 2000); AKR1B10 forward (5′-CCC-AAA-GAT-GAT-AAA-GGT-AAT-GCC-ATC-GGT-3′) and reverse (5′-CGA-TCT-GGA-AGT-GGC-TGA-AAT-TGG-AGA-3′) (Fukumoto et al., 2005). The endogenous control β-actin primers were designed using the PrimerBank software (http://pga.mgh.harvard.edu/primerbank/).
2.8. Identification of molecular pathways
The gene lists (Tables 1 and 2) generated by microarray data analysis was analyzed using PathwayAssist v6.0 software (Stratagene Inc. La Jolla, CA) to assign these genes to specific cellular pathways (Table S1). PathwayAssist software is used for mapping and visualization of biological pathways, gene regulation networks and protein–protein interactions. It identifies biological relationships among genes/proteins of interest based on ResNet, a database of over 500,000 biological interactions built by applying the MedScan text-mining algorithms to all PubMed abstracts, as well as PathArt™, a curated pathway database with over 900 signaling and disease pathways. Microarray expression data were imported into PathwayAssist to graphically display all known relationships and potential interactions between the differentially expressed genes.
Table 1.
CSC-induced genes
| GenBank | Symbol | Gene name | Gene function | Average fold change (increase)
|
|||
|---|---|---|---|---|---|---|---|
| NHEK | Leuk1 | Leuk2 | 101A | ||||
| NM 000499 | CYP1A1 | Cytochrome P450, family 1, subfamily A, polypeptide 1 | Electron transport | 30.68 | 15.8 | 12.57 | 15.46 |
| AU144855 | CYP1B1 | Cytochrome P450, family 1, subfamily B, polypeptide 1 | Electron transport | 10.19 | 14.56 | 14.76 | 4.56 |
| NM 001353 | AKR1C1 | Aldo-keto reductase family 1, member C1 | Xenobiotic metabolism | 6.54 | 5.95 | 7.69 | 9.66 |
| AB018580 | AKR1C3 | Aldo-keto reductase family 1, member C3 | Xenobiotic metabolism | 10.83 | 7.82 | 9.03 | 18.79 |
| NM 020299 | AKR1B10 | Aldo-keto reductase family 1, member B10 | Aldehyde metabolism | 4.07 | 1.09 | 4.0 | 2.31 |
| AA488687 | SLC7A11 | Solute carrier family 7 | Transport | 4.73 | 2.02 | 4.63 | 2.52 |
| NM 000691 | ALDH3A1 | Aldehyde dehydrogenase 3 family, memberA1 | Aldehyde metabolism | 5.90 | 4.11 | 3.17 | 6.09 |
| NM 003662 | PIR | Pirin | Transcription from POL II promoter | 4.31 | 4.18 | 4.23 | 5.10 |
| BC005352 | TNFAIP8 | Tumor necrosis factor, alpha-induced protein 8 | Anti-apoptosis | 4.15 | 1.67 | 2.18 | 1.64 |
| NM 001107 | ACYP1 | Acylphosphatase 1 | Phosphate metabolism | 3.31 | 1.36 | 1.24 | 1.81 |
| AA918224 | KIAA0092 | Translokin | Fibroblast growth factor binding | 3.33 | 1.38 | 1.13 | 1.29 |
| AF119814 | ZNF291 | Zinc finger protein 291 | Unknown | 2.65 | 1.24 | 2.06 | 3.29 |
| N36997 | YT521 | Splicing factor YT521-B | Unknown | 2.65 | 0.99 | 1.32 | 1.00 |
| NM 000081 | CHS1 | Chediak-Higashi syndrome 1 | Intracellular protein transport | 2.62 | 1.34 | 3.64 | 1.19 |
| NM 004915 | ABCG1 | ATP-binding cassette, subfamily G (WHITE), member 1 | Small molecule transport | 2.30 | 2.61 | 8.45 | 2.12 |
| NM 005689 | ABCB6 | ATP-binding cassette, subfamily B (MDR/TAP), member6 | Oncogenesis | 2.24 | 1.29 | 1.81 | 2.55 |
| NM 002395 | ME1 | Malic enzyme 1, NADP(+)-dependent, cytosolic | Malate metabolism | 2.11 | 2.33 | 2.02 | 2.10 |
| AI689429 | PRKCI | Protein kinase C, iota | Intracellular signaling cascade | 2.01 | 1.51 | 3.32 | 1.71 |
| NM 005864 | EFS | Embryonal Fyn-associated substrate | Intracellular signaling cascade | 1.15 | 1.72 | 1.82 | 3.30 |
| NM 022353 | OSGEPL1 | O-sialoglycoprotein endopeptidase-like 1 | Unknown | 1.95 | 1.31 | 2.02 | 2.22 |
| NM 022496 | ACTR6 | Actin-related protein 6 | Unknown | 1.71 | 1.83 | 1.61 | 2.17 |
| BC006471 | AF1Q | ALL1-fused gene from chromosome 1q | Cell growth | 1.83 | 1.56 | 2.18 | 2.08 |
| NM 016951 | CKLF | Chemokine-like factor | Chemotaxis | 1.38 | 2.03 | 2.86 | 1.31 |
| NM 003930 | SCAP2 | src family associated phosphoprotein 2 | Protein complex assembly | 1.65 | 1.56 | 3.04 | 1.44 |
| NM 015416 | HCCR1 | Cervical cancer 1 protooncogene | Unknown | 1.93 | 1.39 | 2.82 | 1.37 |
| AI760629 | CLCN3 | Chloride channel 3 | Chloride transport | 1.65 | 1.36 | 2.81 | 1.75 |
| AU149908 | UBXD2 | UBX domain containing 2 | Regulation of transcription | 1.96 | 1.77 | 2.03 | 1.79 |
The list includes 27 genes overexpressed (≥2-fold; p < 0.005) in CSC-treated (25 μg/ml, 24 h) vs. untreated cells in any one of the cell lines NHEK, Leuk1, Leuk2 and 101A. Assignments of gene functions to biological processes were extracted using PathwayAssist datamining software. Fold changes are derived from the averages of two independent microarray experiments.
Table 2.
CSC-repressed genes
| GenBank | Symbol | Gene name | Gene function | Average fold change (decrease)
|
|||
|---|---|---|---|---|---|---|---|
| NHEK | Leuk1 | Leuk2 | 101A | ||||
| BC005961 | PTHLH | Parathyroid hormone-like hormone | Cell–cell signaling, cell proliferation | 4.29 | 1.01 | 1.55 | 7.04 |
| U41815 | NUP98 | Nucleoporin 98 kDa | Intracellular protein transport | 3.34 | 1.68 | 1.11 | 3.62 |
| NM 023007 | FLJ12517 | Hypothetical protein FLJ12517 | Unknown | 3.13 | 1.76 | 1.59 | 1.85 |
| NM 024098 | MGC2574 | Hypothetical protein MGC2574 | Regulation of transcription | 2.82 | 1.07 | 1.12 | 2.60 |
| NM 006795 | EHD1 | EH-domain containing 1 | Unknown | 2.15 | 1.01 | 1.60 | 2.39 |
| NM 003017 | SFRS3 | Splicing factor, arginine/serine-rich 3 | Regulation of transcription | 2.03 | 1.21 | 1.58 | 1.86 |
| AF109294 | MTAP | Methylthioadenosine phosphorylase | Regulation of transcription | 1.87 | 1.39 | 2.22 | 1.42 |
| NM 003457 | ZNF207 | Zinc finger protein 207 | Regulation of transcription | 1.69 | 1.02 | 1.49 | 2.83 |
The list includes eight genes down-regulated (≥2-fold; p < 0.005) in CSC-treated (25 μg/ml, 24 h) vs. untreated cells in any one of the cell lines NHEK, Leuk1, Leuk2 and 101A. Assignments of gene functions to biological processes were extracted using PathwayAssist datamining software. Fold changes are derived from the averages of two independent microarray experiments.
3. Results
3.1. Effects of CSC treatment on cell viability
To address whether CSC affected the growth of normal, dysplastic, and tumor cells, the NHEK, Leuk1, Leuk2, and 101A cells were treated with different concentration of CSC (from 0 to 100 μg/ml) for time periods up to 24 h, and viability examined by MTT dye reduction assay (Fig. 1). We observed decreasing cell viability with increasing concentration of CSC, and with increasing exposure time at each dose. Based on these data, we used 25 μg/ml of CSC for microarray and quantitative real-time RT-PCR studies since this dose showed only low toxicity even for the longer time periods.
Fig. 1.

Proliferation of normal human keratinocytes and oral carcinoma cells treated with CSC. A total of 500 cells/well were seeded in 96-well tissue culture plates and treated with 0–100 μg/ml concentrations of CSC for 1 h (■), 5 h (▲) and 24 h (□). After each indicated time/concentration treatment, the growth of cells was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described in Section 2. (A) NHEK; (B) Leuk1; (C) Leuk2; (D) 101A cells. Results are the mean ± S.D. of three independent assays.
3.2. Gene expression analysis
Microarray-based RNA expression profiles were collected for the four cell lines after treatment with 25 mg/ml of CSC and for untreated control cells, each at 0 and 24 h in duplicate samples. A principal component analysis was performed to visualize the similarity in gene expression responses of the different samples and treatments (Fig. 2). All four cell lines as well as the two treatment times separated well into distinguishable groups without mutual overlap, indicating that the resulting expression responses are cell- and treatment-specific. Two-way ANOVA (variables: cell line; treatment) and FDR correction (p < 0.05) identified 232 genes that had changed in all four cell lines in response to CSC treatment. A total of 35 genes were changed significantly (p < 0.005) with at least 2-fold change in any one cell line in response to CSC exposure (Fig. 3). Among them, 27 genes were up-regulated by ≥2-fold (Table 1) and 8 genes were decreased by ≥2-fold (Table 2) in any one cell system. Several of the previously described known CSC-dependent gene products were altered in all cell lines. Interestingly, two genes of CYP and three genes of AKR family were highly increased in expression and were identified as CSC-specific genes (see below). Several novel and previously not recognized CSC-responsive genes were also identified based on their concordant expression profile (Tables 1 and 2).
Fig. 2.

Principal component analysis of microarray data to demonstrate similarity of response of the cell types and treatment times. In the plot, each point represents a cell line (NHEK, Leuk1, Leuk2, or 101A) and treatment time (0 h, black circles; 24 h, gray triangles) of CSC exposure (25 μg/ml). The X, Y and Z-axes are PC#1, PC#2 and PC#3, respectively.
Fig. 3.

CSC-induced gene expression profiles. This heat map shows common genes up-regulated or down-regulated in NHEK, Leuk1, Leuk2 and 101A cell lines after CSC treatment (25 μg/ml), as determined by false discovery rate. Genes are displayed horizontally and samples are displayed as vertical columns for cell lines (each in duplicate) and treatment time period (0 h or 24 h). For each gene, relative expression changes are colored to indicate increased (red) and decreased (blue) levels relative to the average across all samples (yellow). Gene expression profiles for 35 genes were tentatively identified as differentially expressed with at least two-fold change in any one cell line compared.
3.3. Validation of microarray data by quantitative real-time RT-PCR
To confirm the expression changes observed by microarray analysis, quantitative real-time PCR was performed on cDNAs obtained from the NHEK, Leuk1, Leuk2 and 101A cells after 0 and 24 h CSC treatment. Gene-specific primer pairs for CYP1A1, CYP1B1, AKR1C1, AKR1C3 and AKR1B10 were used, and RNA fold changes for 24 h versus 0 h treatment, each normalized to its corresponding β-actin value, were calculated by the 2−ΔΔCT method (Livak and Schmittgen, 2001). The mean fold changes from microarray data were compared to quantitative real-time PCR data (Fig. 4). All gene transcripts showed similar changes in direction and range for the 0 and 24 h treated samples when comparing both techniques.
Fig. 4.

Comparative quantitative real-time RT-PCR analysis. RNA from control (0 h) and treated (24 h, 25 μg/ml CSC) cells were subjected to quantitative real-time RT-PCR analysis using primers specific for CYP1A1, CYP1B1, AKR1C1, AKR1C3, AKR1B10, and the internal standard β-actin. (A) NHEK; (B) Leuk1; (C) Leuk2; (D) 101A cells; open bars, microarray data; solid bars, RT-PCR data. The fold change levels on the Y-axis are relative to the expression in untreated cells. Results are expressed as the mean and range of duplicate determinations for microarray data, and as mean ± S.D. of four independent experiments for qRT-PCR data: *p < 0.05; **p = 0.01; #non-significant.
3.4. Induction of PAH-metabolizing genes
Several of the genes known to be involved in the metabolism of PAHs (CYP1A1, CYP1B1, AKR1C1, AKR1C3 and AKR1B10) were highly up-regulated after exposure to CSC. CYP1A1 and CYP1B1 are known to be key enzymes involved in the metabolism of carcinogens present in the CSC used in this study (Port et al., 2004; Kim et al., 2004; Yang et al., 2005). AKR1C1, AKR1C3 and AKR1B10 have also been shown to be involved in the conversion of PAH trans-dihydrodiols from diol-epoxides to o-quinones that may present an alternative pathway of PAH activation (Penning, 2004, 2005; Fukumoto et al., 2005). In all four CSC-treated cells, CYP1A1 was elevated 12- to 30-fold, and CYP1B1 showed 4.5- to 14.5-fold overexpression compared to untreated samples (Table 1; Fig. 4). We also determined that human AKR1C subfamily members (AKR1C1 and AKR1C3) and AKR1B10 were responsive to CSC exposure, with increases ranging from 2.3-fold to nearly 19-fold (Table 1; Fig. 4). Although there was significant inter-cell line variation in mRNA induction levels of these genes, the CSC-dependent overexpression was clearly consistent for all cell lines. Thus, we demonstrated by two independent techniques a uniform pattern of cell-specific transcriptional activation in terms of time and dose response for CYPs (1A1 and 1B1) and AKRs (1A1, 1C1, and 1B10) in normal (NHEK), pre-malignant (Leuk1, Leuk2) and tumor (101A) cells (Figs. 3 and 4). This suggests that the early induction of these genes in cells after only short-term exposure to CSC is a general phenomenon of the OSCC response to cigarette smoke toxicants.
When comparing the basal and induction levels for the different phenotypes, some important quantitative differences were observed (Fig. 5). The basal transcript levels for the two CYP genes and for AKR1C1 were highest in the normal and pre-malignant cells compared to overall much lower levels in the tumor cell 101A. The AKR1C3 and AKR1B10 transcripts were generally low in all cell lines relative to the other genes (Fig. 5A). Interestingly, both the rate and magnitude of induction by CSC were highest in the non-malignant normal NHEK cells, with CYP1A1 and CYP 1B1 highly elevated prior to induction of the AKR genes. The response of the two dysplasia cell lines was similar yet not as pronounced as for NHEK. On the other hand, the malignant 101A tumor cell line showed only a slow and modest induction beyond its already low basal level (Fig. 5B). These data show that tobacco carcinogens are more potent inducers of xenobiotic metabolizing enzymes CYP1A1 and CYP1B1 in normal keratinocyte cells compared to pre-malignant or tumor cells.
Fig. 5.

CSC-induced expression increases for selected target genes. (A) Basal expression levels in untreated cell lines NHEK, Leuk1, Leuk2, and 101A for the indicated target genes, as measured by microarray-derived target intensities. (B) Time course of induction. For each cell line, target intensities after CSC treatment for 0, 5, and 24 h are plotted for the following genes: CYP1A1 (□); CYP1B1 (▲); AKR1C1 (▼); AKR1C3 (■); AKR1B10 (○).
3.5. CSC-repressed genes
Only a few genes were found to be consistently repressed as a consequence of CSC exposure. These included several transcription regulators as well as proteins involved in intercellular signaling and intracellular protein transport (Table 2); however, none of them had any known involvement in carcinogen metabolism.
3.6. Identification of signaling pathways associated to CSC sensitivity
To identify potential signaling pathways that are associated with CSC response, we analyzed our microarray expression data using PathwayAssist software (Fig. 6). By overlaying microarray expression data onto a biological association network of known protein interactions, co-regulated genes that define a specific signaling pathway can be identified. After importing the 35 genes with induced or decreased expression based on our filtered microarray data (Tables 1 and 2) into PathwayAssist, we identified potential signaling pathways associated with the CSC response in oral keratinocytes. The genes were searched for linkage to cellular processes involving membrane, nucleus and mitochondria. The genes selected in this way to form the proposed molecular networks are listed as supplemental material (Table S1). From this complex interaction gene list, a simplified version of the network for genes associated with drug metabolism/resistance was obtained (Fig. 6). Genes which are either up- or down-regulated are highlighted, and extracellular, cytosolic, nuclear, and mitochondrial factors are displayed according to their cellular distributions. A considerable number of genes associated with the observed CSC response are linked, including CYP (1A1 and 1B1) and AKR (1A1, 1C1, and 1B10) genes which are differentially up-regulated in CSC-treated cells. In this molecular network of genes which are co-regulated, the extracellular proteins TNF, TGFB1, IGF1, FGF2, CCL2, CCL7, PTLH and PGF were identified; affected nuclear genes are SFRS3, HDAC4, NUP98, PIR, CEBPG, EHD1, AHR, NXF1, SFRS12, CHD1, MYOG, HOXA9, POU2F1, AR, CDKN2A, RANBP2, SAFB, PPARG, CCNG1, BCL3 and YT521; responsive mitochondrial genes are ODC1, BCL2 and ALDH1A1. Thus, we have identified a series of interacting genes that constitute potentially important signaling pathways involved in the cellular response to cigarette smoke exposure (Fig. 6, Table S1).
Fig. 6.
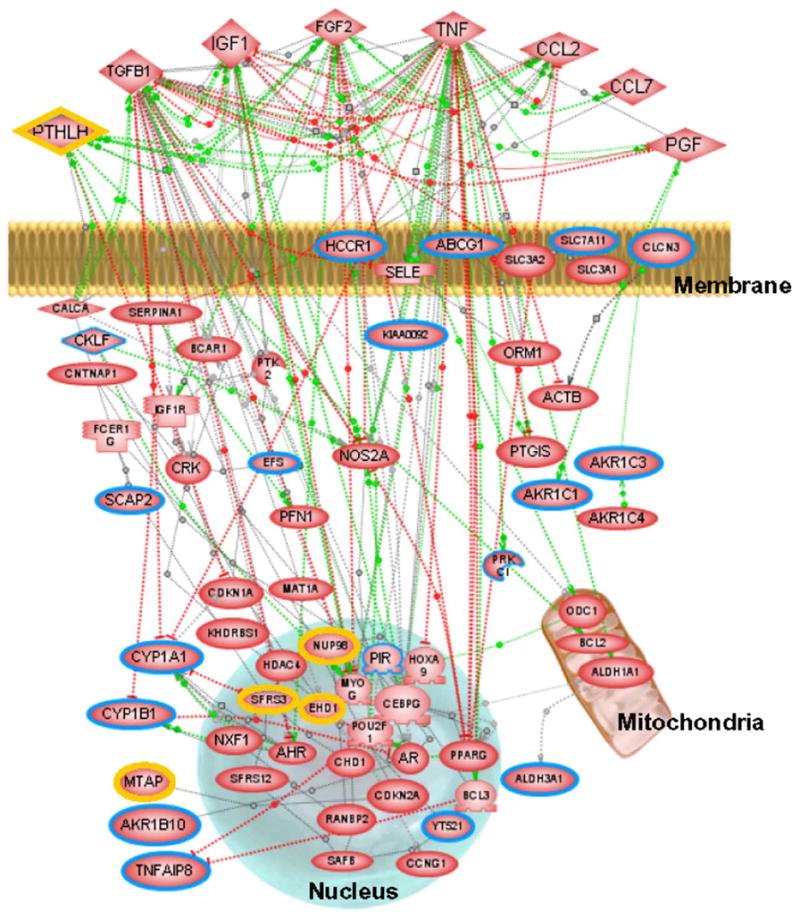
Potential molecular interaction network for the CSC response. A simplified network for genes associated with the CSC response was developed using PathwayAssist. Green lines indicate positive effects, red lines indicate negative effects and gray lines interactions with unknown effect. Colored borders in nodes highlight genes that are up-regulated (blue) or down-regulated (yellow) with at least two-fold change in any one cell type. A complete table of the network genes associated with CSC sensitivity is accessible as Table S1, Supplementary Data online.
4. Discussion
Tobacco use is a major independent risk factor for the development of oral and pharyngeal cancer and other malignancies of the upper aerodigestive tract (Warnakulasuriya et al., 2005; Almahmeed et al., 2004). We have documented here the effects of CSC, a complex mixture of carcinogens, on gene expression profiles in different human cell lines ranging from normal keratinocytes to pre-malignant oral dysplasia and OSCC tumor cells. We particularly focused on genes known to be involved in the metabolism of PAHs, like CYP1A1, CYP1B1, AKR1C1, AKR1C3 and AKR1B10 (Shimada and Fujii-Kuriyama, 2004; Jiang et al., 2005), since to date the expression of these genes in OSCC is largely unexplored. Our gene expression data, obtained by microarray analysis and validated by real-time PCR, indicated increased expression (mostly >5-fold and up to 30-fold) for the CYP1A1, CYP1B1, AKR1C1, AKR1C3 and AKR1B10 genes in response to CSC exposure. This suggests that CSC specifically induces expression of a unique set of PAH-metabolizing genes in response to this treatment.
Our objective was also to determine response differences among the cell phenotypes in the OSCC progression following exposure to CSC. Basal CYP1 and AKR1 expression was negligible compared to their CSC-induced levels; yet, distinct intercellular differences in CYP1 and AKR1 mRNA levels were observed in untreated cells. Among the five genes analyzed, CYP1B1 and AKR1C1 had the highest basal expression in all cell lines, with CYP1B1 and AKR1C1 showing several fold higher constitutive levels than CYP1A1 and AKR1C3 or AKR1B10. These basal levels appear to decrease with increasing malignancy progression from dysplasia to tumorigenic cells. In all CSC-treated cell lines studied, we observed consistent overexpression of CYP1A1 and CYP1B1. Qualitatively, the induction profiles of these genes were similar among the cell types, although the extents of induction were different and may be determined by the basal expression levels which varied depending on malignancy and tumor stage. The non-malignant cells clearly had the strongest induction, followed by the dysplasia cells, and the tumorigenic cell line had the weakest response.
Our data are in agreement with several previous reports. An RT-PCR-based study showed elevation of CYP1B1 expression in bronchial epithelial cells of smokers versus non-smokers, and showed interindividual variation in basal CYP1A1 and CYP1B1 levels in human lung cells (Willey et al., 1997). The levels of CYP1A1 and CYP1B1 expression in most non-smokers were found to be very low (Willey et al., 1997; Kim et al., 2004), and correlations were found for CYP1A1 variant expression and risk for colorectal carcinoma in smokers (Slattery et al., 2004). Also, there is evidence that tobacco smoke-specific compounds relevant to head and neck carcinogenesis are metabolically activated through CYP1B1 (Thier et al., 2002). In order to minimize the insults caused by such xenobiotics, various tissues are well equipped with diverse Phase I (CYPs) and Phase II (including AKRs) enzymes which are present in abundance either at the basal level or after induction following xenobiotic exposure. CYP1B1 metabolizes numerous PAHs as well as many N-heterocyclic amines, arylamines, amino azo dyes, and several other carcinogens (Guengerich, 2000). Also, many of the inducers are in turn metabolized by CYP1A1. On the other hand, AHR is a ligand-activated transcription factor that controls several genes, including up-regulation of CYP1A1 and CYP1B1 (Nebert et al., 2000). Therefore, the role of CYP1 in detoxification versus activation to cause toxicity is likely to depend on the cell type and the amount of Phase II metabolism.
Pathway analysis of our microarray data showed a significant positive association between CYP1A1 and CYP1B1 expression, in agreement with mechanistic data showing the regulation of these genes by the Ah receptor (Whitlock, 1999). The CYP1A1 enzyme is responsible for activation of many PAHs and related chemicals to DNA-binding species and has, therefore, been considered to be involved in initiation steps of carcinogenesis. The concerted induction of CYP1 and AKR1 would increase the flux of trans-dihydrodiols to redox-active o-quinones and enhance the production of reactive oxygen species, leading to further overexpression of AKR1C isozymes. On one hand, AKR1C isozymes will provide a defense mechanism against the harmful effects of ROS, since they are efficient catalysts for reduction of 4-hydroxy-2-nonenal, a decomposition product of lipid hydroperoxides (Burczynski et al., 2001). On the other hand, induced AKR1C isozymes can convert PAH trans-dihydrodiols produced by CYP1 to the deleterious o-quinones. It was shown that AKR-derived PAH o-quinones will cause change-in-function mutations in the p53 tumor suppressor gene, and that these mutations result from ROS (Yu et al., 2002). If o-quinones and the ROS they generate are not eliminated, they have the potential to cause covalent and oxidative DNA lesions, increasing the mutational load of PAH-exposed oral cells. Together, this sequence of events may contribute to PAH-induced oral carcinogenesis.
The data presented here clearly show that CYP1A1 and 1B1 are the major CYP enzymes induced in CSC-mediated oral carcinogenesis. Each of our cell types responded to CSC treatment by overexpressing CYP1A1 and CYP1B1, suggesting that they play a significant role in the activation of procarcinogens found in cigarette smoke. Based on the metabolic capacity of CYP1A1 for PAHs, it can be concluded that it may primarily be responsible for the activation in human oral cells of many procarcinogens and xenobiotics present in cigarette smoke. CYP1A1 induction observed in smokers and non-smokers (Kim et al., 2004), in addition to greater exposure to carcinogens, may account for increased cancer susceptibility among those individuals who are high responders. Thus, CYP1B1 and 1A1 expression levels are sensitive to cigarette smoke exposure, as was also observed in human OSCC cells exposed to the tobacco smoke carcinogen benzo[a]pyrene (Wen and Walle, 2005).
The mechanistic pathways for the CSC response, identified with PathwayAssist, also associate a number of gene products expressed in the nucleus or extracellularly. The secreted gene products include mainly growth factors, proinflammatory cytokines and chemokines. Several altered genes are regulated through the inflammatory cytokine TNF pathway, although the mechanism for TNF activation by CSC and its contribution to carcinogenesis and tumorigenesis are not clear. More detailed functional analyses of these CSC-responsive genes will be necessary to determine their precise mechanistic contributions to the different elements of this pathway.
In summary, our global analysis of gene expression profiles in CSC-treated versus untreated human oral cells identified 35 genes that are significantly affected. Importantly, members of the human CYP1 and AKR1 gene families are highly induced by short-term exposure to CSC components. Induction of some of these genes was previously observed in lung, colon, or rectal tumor tissues of smokers. However, their behavior in differently staged oral tissue cells, which in smokers are directly exposed to cigarette smoke components, has not been characterized before. Our microarray and qRT-PCR results, interpreted in the context of a database of published observations, allowed us to identify members of a pathway that have not previously been associated with OSCC. Thus, we showed that combining gene expression profiles with PathwayAssist analysis provides a powerful tool for the identification of potentially novel signaling pathways. Interactions associated with other biologic processes, such as cell cycle progression, apoptosis, or metastasis, can similarly be identified from our expression profiling data, leading to further insight into the disease etiology and risk factors. The gene response patterns described here shed new light on the mechanisms of carcinogen-induced oral cancer. Furthermore, any of the genes identified within this response pathway may be potential targets for novel therapeutic or preventive agents.
Supplementary Material
Acknowledgments
This work was supported by NIH grant DE13150 and by Philip Morris USA Inc. and Philip Morris International (W.Z.). The Leuk1 and Leuk2 cells were kind gifts from Dr. P. Sacks, New York University, New York, NY; 101A cells were kindly provided by Dr. T. Carey, University of Michigan, Ann Arbor, MI.
Abbreviations
- AKR
aldo-keto reductase
- ANOVA
analysis of variance
- CSC
cigarette smoke condensate
- CYP
cytochrome P450
- DMEM
Dulbecco modified Eagle’s medium
- DMSO
dimethyl sulfoxide
- FDR
false discovery rate
- NHEK
normal human epidermal keratinocytes
- OSCC
oral squamous cell carcinoma
- PAH
polycyclic aromatic hydrocarbon
- PCA
principle component analysis
- PCR
polymerase chain reaction
- RNA
ribonucleic acid
- RT-PCR
reverse transcription-PCR
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.toxlet.2006.03.008.
References
- Alexandrie AK, Sundberg MI, Seidegard J, Tornling G, Rannug A. Genetic susceptibility to lung cancer with special emphasis on CYP1A1 and GSTM1: a study on host factors in relation to age at onset, gender and histological cancer types. Carcinogenesis. 1994;15:1785–1790. doi: 10.1093/carcin/15.9.1785. [DOI] [PubMed] [Google Scholar]
- Almahmeed T, Boyle JO, Cohen EG, Carew JF, Du B, Altorki NK, Kopelovich L, Fang JL, Lazarus P, Subbaramaiah K, Dannenberg AJ. Benzo[a]pyrene phenols are more potent inducers of CYP1A1, CYP1B1 and COX-2 than benzo[a]pyrene glucuronides in cell lines derived from the human aerodigestive tract. Carcinogenesis. 2004;25:793–799. doi: 10.1093/carcin/bgh078. [DOI] [PubMed] [Google Scholar]
- Bartsch H, Nair U, Risch A, Rojas M, Wikman H, Alexandrov K. Genetic polymorphism of CYP genes, alone or in combination, as a risk modifier of tobacco-related cancers. Cancer Epidemiol Biomarkers Prev. 2000;9:3–28. [PubMed] [Google Scholar]
- Burczynski ME, Sridhar GR, Palackal NT, Penning TM. The reactive oxygen species and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the alpha, beta-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. J Biol Chem. 2001;276:2890–2897. doi: 10.1074/jbc.M006655200. [DOI] [PubMed] [Google Scholar]
- Crofts FG, Strickland PT, Hayes CL, Sutter TR. Metabolism of 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP) by human cytochrome P4501B1. Carcinogenesis. 1997;18:1793–1798. doi: 10.1093/carcin/18.9.1793. [DOI] [PubMed] [Google Scholar]
- Fukumoto S, Yamauchi N, Moriguchi H, Hippo Y, Watanabe A, Shibahara J, Taniguchi H, Ishikawa S, Ito H, Yamamoto S, Iwanari H, Hironaka M, Ishikawa Y, Niki T, Sohara Y, Kodama T, Nishimura M, Fukayama M, Dosaka-Akita H, Aburatani H. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin Cancer Res. 2005;11:1776–1785. doi: 10.1158/1078-0432.CCR-04-1238. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Metabolism of chemical carcinogens. Carcinogenesis. 2000;21:345–351. doi: 10.1093/carcin/21.3.345. [DOI] [PubMed] [Google Scholar]
- Huang Z, Fasco MJ, Figge HL, Keyomarsi K, Kaminsky LS. Expression of cytochromes P450 in human breast tissue and tumors. Drug Metab Dispos. 1996;24:899–905. [PubMed] [Google Scholar]
- Hyndman D, Flynn TG. The aldo-keto reductases and their role in cancer. Adv Exp Med Biol. 1999;463:427–434. doi: 10.1007/978-1-4615-4735-8_53. [DOI] [PubMed] [Google Scholar]
- Jiang H, Shen YM, Quinn AM, Penning TM. Competing roles of cytochrome P450 1A1/1B1 and aldo-keto reductase 1A1 in the metabolic activation of (+/−)-7,8-dihydroxy-7,8-dihydro-benzo[a]pyrene in human bronchoalveolar cell extracts. Chem Res Toxicol. 2005;18:365–374. doi: 10.1021/tx0497245. [DOI] [PubMed] [Google Scholar]
- Kim JH, Stansbury KH, Walker NJ, Trush MA, Strickland PT, Sutter TR. Metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-diol by human cytochrome P450 1B1. Carcinogenesis. 1998;19:1847–1853. doi: 10.1093/carcin/19.10.1847. [DOI] [PubMed] [Google Scholar]
- Kim JH, Sherman ME, Curriero FC, Guengerich FP, Strickland PT, Sutter TR. Expression of cytochromes P450 1A1 and 1B1 in human lung from smokers, non-smokers, and ex-smokers. Toxicol Appl Pharmacol. 2004;199:210–219. doi: 10.1016/j.taap.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC (T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mahadevan B, Keshava C, Musafia-Jeknic T, Pecaj A, Weston A, Baird WM. Altered gene expression patterns in MCF-7 cells induced by the urban dust particulate complex mixture standard reference material 1649a. Cancer Res. 2005;65:1251–1258. doi: 10.1158/0008-5472.CAN-04-2357. [DOI] [PubMed] [Google Scholar]
- Murray GI, Melvin WT, Greenlee WF, Burke MD. Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu Rev Pharmacol Toxicol. 2001;41:297–316. doi: 10.1146/annurev.pharmtox.41.1.297. [DOI] [PubMed] [Google Scholar]
- Nebert DW. Role of genetics and drug metabolism in human cancer risk. Mutat Res. 1991;247:267–281. doi: 10.1016/0027-5107(91)90022-g. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Dalton TP, Okey AB, Gonzalez FJ. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J Biol Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. Activation of polycyclic aromatic hydrocarbon trans-dihydrodiol proximate carcinogens by human aldo-keto reductase (AKR1C) enzymes and their functional overexpression in human lung carcinoma (A549) cells. J Biol Chem. 2002;277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- Pelkonen O, Nebert DW. Metabolism of polycyclic aromatic hydrocarbons: etiologic role in carcinogenesis. Pharmacol Rev. 1982;34:189–222. [PubMed] [Google Scholar]
- Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TM. Aldo-keto reductases and formation of polycyclic aromatic hydrocarbon o-quinones. Methods Enzymol. 2004;378:31–67. doi: 10.1016/S0076-6879(04)78003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penning TM. AKR1B10: a new diagnostic marker of non-small cell lung carcinoma in smokers. Clin Cancer Res. 2005;11:1687–1690. doi: 10.1158/1078-0432.CCR-05-0071. [DOI] [PubMed] [Google Scholar]
- Port JL, Yamaguchi K, Du B, De Lorenzo M, Chang M, Heerdt PM, Kopelovich L, Marcus CB, Altorki NK, Subbaramaiah K, Dannenberg AJ. Tobacco smoke induces CYP1B1 in the aerodigestive tract. Carcinogenesis. 2004;25:2275–2281. doi: 10.1093/carcin/bgh243. [DOI] [PubMed] [Google Scholar]
- Roberts-Thompson SJ, McManus ME, Tukey RH, Gonzalez FF, Holder GM. The catalytic activity of four expressed human cytochrome P450s towards benzo[a]pyrene and the isomers of its proximate carcinogen. Biochem Biophys Res Commun. 1993;192:1373–1379. doi: 10.1006/bbrc.1993.1568. [DOI] [PubMed] [Google Scholar]
- Rubin H. Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: a bio-historical perspective with updates. Carcinogenesis. 2001;22:1903–1930. doi: 10.1093/carcin/22.12.1903. [DOI] [PubMed] [Google Scholar]
- Shehin SE, Stephenson RO, Greenlee WF. Transcriptional regulation of the human CYP1B1 gene. J Biol Chem. 2000;275:6770–6776. doi: 10.1074/jbc.275.10.6770. [DOI] [PubMed] [Google Scholar]
- Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Guengerich FP, Sutter TR. Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1. Cancer Res. 1996;56:2979–2984. [PubMed] [Google Scholar]
- Shimada T, Tsumura F, Yamazaki H. Prediction of human liver microsomal oxidations of 7-ethoxycoumarin and chlorzox-azone with kinetic parameters of recombinant cytochrome P-450 enzymes. Drug Metab Dispos. 1999;27:1274–1280. [PubMed] [Google Scholar]
- Shimada T, Fujii-Kuriyama Y. Metabolic activation of poly-cyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004;95:1–6. doi: 10.1111/j.1349-7006.2004.tb03162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery ML, Samowtiz W, Ma K, Murtaugh M, Sweeney C, Levin TR, Neuhausen S. CYP1A1, cigarette smoking, and colon and rectal cancer. Am J Epidemiol. 2004;160:842–852. doi: 10.1093/aje/kwh298. [DOI] [PubMed] [Google Scholar]
- Sudbo J, Reith A. The evolution of predictive oncology and molecular-based therapy for oral cancer prevention. Int J Cancer. 2005;115:339–345. doi: 10.1002/ijc.20896. [DOI] [PubMed] [Google Scholar]
- Thier R, Bruning T, Roos PH, Bolt HM. Cytochrome P450 1B1, a new keystone in gene–environment interactions related to human head and neck cancer? Arch Toxicol. 2002;76:249–256. doi: 10.1007/s00204-002-0349-3. [DOI] [PubMed] [Google Scholar]
- Warnakulasuriya S, Sutherland G, Scully C. Tobacco, oral cancer, and treatment of dependence. Oral Oncol. 2005;41:244–260. doi: 10.1016/j.oraloncology.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Wen X, Walle T. Preferential induction of CYP1B1 by benzo[a]pyrene in human oral epithelial cells: impact on DNA adduct formation and prevention by polyphenols. Carcinogenesis. 2005;26:1774–1781. doi: 10.1093/carcin/bgi127. [DOI] [PubMed] [Google Scholar]
- Whitlock JP. Induction of cytochrome P4501A1. Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- Willey JC, Coy EL, Frampton MW, Torres A, Apostolakos MJ, Hoehn G, Schuermann WH, Thilly WG, Olson DE, Hammersley JR, Crespi CL, Utell MJ. Quantitative RT-PCR measurement of cytochromes p450 1A1, 1B1, and 2B7, microsomal epoxide hydrolase, and NADPH oxidoreductase expression in lung cells of smokers and nonsmokers. Am J Respir Cell Mol Biol. 1997;17:114–124. doi: 10.1165/ajrcmb.17.1.2783. [DOI] [PubMed] [Google Scholar]
- Yang CK, Sakai S, Akutagawa T, Matsumoto Y. CYP1A1 inducing potential of airborne particulate extracts collected during a 25-year period (1975–2000) Asian Pac J Cancer Prev. 2005;6:153–158. [PubMed] [Google Scholar]
- Yu D, Berlin JA, Penning TM, Field J. Reactive oxygen species generated by PAH o-quinones cause change-in-function mutations in p53. Chem Res Toxicol. 2002;15:832–842. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.