

Abstract
Despite the urgency for prevention and treatment of lung adenocarcinoma (LUAD), we still do not know drivers in pathogenesis of the disease. Earlier work revealed that mice with knockout of the G-protein coupled receptor Gprc5a develop late onset lung tumors including LUADs. Here, we sought to further probe the impact of Gprc5a expression on LUAD pathogenesis. We first surveyed GPRC5A expression in human tissues and found that GPRC5A was markedly elevated in human normal lung relative to other normal tissues and was consistently down-regulated in LUADs. In sharp contrast to wild type littermates, Gprc5a-/- mice treated chronically with the nicotine-specific carcinogen NNK developed LUADs by six months following NNK exposure. Immunofluorescence analysis revealed that the LUADs exhibited abundant expression of surfactant protein C and lacked the clara cell marker Ccsp, suggesting that these LUADs originated from alveolar type II cells. Next, we sought to survey genome-wide alterations in the pathogenesis of Gprc5a-/- LUADs. Using whole exome sequencing, we found that carcinogen-induced LUADs exhibited markedly higher somatic mutation burdens relative to spontaneous tumors. All LUADs were found to harbor somatic mutations in the Kras oncogene (p. G12D or p. Q61R). In contrast to spontaneous lesions, carcinogen-induced Gprc5a-/- LUADs exhibited mutations (variants and copy number gain) in additional drivers (Atm, Kmt2d, Nf1, Trp53, Met, Ezh2). Our study underscores genomic alterations that represent early events in the development of Kras mutant LUAD following Gprc5a loss and tobacco carcinogen exposure and that may constitute targets for prevention and early treatment of this disease.
Keywords: Lung adenocarcinoma, carcinogenesis, Gprc5a, Kras, whole-exome sequencing
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer-deaths in the United States and worldwide 1, 2. The majority (∼85%) of diagnosed NSCLC cases are attributed to cigarette smoking (tobacco carcinogen exposure) 3, 4. Lung adenocarcinoma (LUAD) represents the most commonly diagnosed histological subtype of NSCLC in former or current smokers 3, 4. While smoking cessation has been shown to reduce the risk of developing LUAD, this risk does not return to a baseline level for former smokers 5, 6. Various mutational profiling studies have revealed disparate molecular subtypes of LUAD 7-10 that are closely associated with tobacco consumption history. While somatic activating mutations in the epidermal growth factor receptor (EGFR) oncogene are prevalent in LUADs that develop in non-smokers, mutations in the kirsten rat sarcoma oncogene (KRAS) are prevalent in smoker LUADs 3, 7-11. Particular molecular subtypes of LUAD (e.g. KRAS mutant LUADs) display dismal clinical outcomes and are resistant to various forms of targeted therapies 12, 13 thus warranting the need for new strategies for early treatment of this malignancy.
Earlier studies have underscored restricted patterns of gene expression in lung epithelial and malignant cells 4. For instance, the embryonic stem cell transcription factor, sry-box 2 (SOX2) is preferentially found in the conducting airway epithelia 14, the presumed cell-of-origin of squamous NSCLCs 15. SOX2 promotes growth of squamous NSCLC cells and has been shown to be specifically up-regulated in squamous NSCLC 14, 16, 17. The transcription factor NK2 homeobox 1 (NKX2-1 also known as TITF-1) is prevalent in peripheral lung cells (alveolar cells) 18, the presumed anatomical origin of LUADs 15. NKX2-1 was demonstrated to be preferentially gained in a significant fraction of LUADs but not in squamous NSCLCs 7, 19, and to function as a lineage-specific oncogene in LUADs 20. The retinoid-inducible G-protein coupled receptor, Gprc5a, was shown to be preferentially expressed in fetal and adult normal lung 21. Mice with knockout of both alleles of Gprc5a (Gprc5a-/-) in the presence or absence of mild exposure (single dose) to the nicotine-specific nitrosamine ketone (NNK) tobacco carcinogen, and in sharp contrast to similarly treated wild type (WT) littermates, develop latent LUADs 22, 23. While these earlier reports suggest that Gprc5a may function as a lung lineage-specific tumor suppressor, the molecular pathogenesis of LUADs, including those linked to smoking, following Gprc5a knockout remains elusive.
Our present study was aimed at furthering our understanding of the impact of Gprc5a expression on LUAD pathogenesis. We demonstrate that human GPRC5A is largely preferentially expressed in normal lung epithelia relative to other normal tissues, is down-regulated in smoking-exposed normal-appearing airway cells and is consistently suppressed in LUADs. Following in vivo carcinogenesis analyses, we find that chronic exposure of Gprc5a-/- mice to NNK, in contrast to saline-treated mice with latent tumor development, results in accelerated malignant adenocarcinoma onset within 6 months after exposure. Surveying genome-wide alterations in Gprc5a-/- LUADs by whole-exome sequencing, we report somatic activating mutations in the Kras oncogene in both spontaneous and NNK-associated LUADs. In contrast to spontaneous tumors, carcinogen-induced Gprc5a-/- LUADs exhibited mutations and copy number alterations in additional drivers. Our study highlights genomic aberrations in the early progression of Kras mutant LUAD following Gprc5a knockout and tobacco carcinogen exposure. These aberrations may comprise viable targets for prevention and treatment of this aggressive molecular subtype of LUAD.
Materials And Methods
Chemical and reagents
The tobacco carcinogen NNK with a purity of greater than 99% was purchased from Midwest Research Institute (The National Cancer Institute's Chemical Carcinogen Reference Standard Repository). Hematoxylin and Eosin (H&E) staining reagents were purchased from Sigma-Aldrich.
Animal housing and tobacco carcinogen experiments
We used Gprc5a knockout (Gprc5a−/−) mice, which were generated as described previously23. The mice were maintained according to a protocol approved by the MD Anderson Cancer Center Institutional Animal Care and Use Committee at the institution's specific pathogen-free animal facility. This facility is approved by the American Association for Accreditation of Laboratory Animal Care and is operated in accordance with current regulations and standards of the US Department of Agriculture and the Department of Health and Human Services. Gprc5a-/- (2 months old) were divided into groups (per treatment and sacrifice time point) of 5 or 6 mice and were injected three times per week (for eight weeks) intraperitoneally with 50 mg/kg of body weight NNK (150 mg/kg per week) dissolved in saline or with saline alone as control. Mice were sacrificed at every month for seven months following NNK or saline injection. A subset of mice exposed to control saline were sacrificed at later time points (> 16 months following saline) to capture late onset spontaneous lung adenocarcinomas.
Histopathological analysis of lung lesions
After sacrifice at different time points, lungs were excised and inflated by injection with formalin for lesion histopathological assessment. Lung surface lesions were assessed by macroscopic observation. Formalin-fixed lung specimens were then embedded in paraffin (FFPE). Histological sections (four μm) were prepared and analyzed by H&E staining using standardized criteria24 for characterization and diagnosis of lung lesions (hyperplasias, adenomas and adenocarcinomas) by an experienced pathologist (J.F.). Lesions were enumerated from each slide/section to yield a tumor burden per lung specimen. All H&E slides were scanned by Aperio (Leica biosystems Inc.).
Immunofluorescence (IF) analysis
Lungs were injected with a mixture of 50% optimal cutting temperature (OCT) medium (Tissue-Tek) and 50% PBS. Frozen histologic sections (5 μm thick) from adult mice were fixed in ice-cold 4% buffered paraformaldehyde for 15 min. Embryos (day 16.5) from WT mice were cut and immersed in 10% buffered formalin (two days) for subsequent IF analysis. After washing, sections were first incubated with serum-free protein block (DAKO) for 1 hour. Specimens were then incubated with antibodies raised against clara cell secretory protein (Ccsp/CC10; Santa Cruz Biotechnology; 1:250) and prosurfactant protein C (Sftpc; Chemicon EMD Millipore, 1:200) overnight. After washing, specimens were incubated with FITC-labeled donkey anti-goat secondary antibody (Alexa 488) for 1.5 h in the dark followed by washing and incubation with Texas Red-labeled goat-anti-rabbit secondary antibody (Alexa 594). Slides were then incubated with ProLong® Gold Antifade Reagent with DAPI (Cell Signaling Technology). To assess Gprc5a expression, specimens were incubated after blocking with a mixture of antibodies raised against Gprc5a (Santa Cruz Biotechnology; 1:25), Podoplanin (Pdpn; DSHB; 1:400 dilution) and Sftpc (1:10). The specimens were then washed and incubated with FITC-labeled donkey anti-goat secondary antibody (Alexa 488) and Texas Red-labeled goat anti-rabbit secondary antibody (Alexa 594) or with Texas Red-labeled donkey anti-goat secondary antibody (Alexa 594) and FITC-labeled goat anti-rabbit secondary antibody (Alexa 488) for 1.5 h in the dark. All images were captured using a BX61 immunofluorescent system (Olympus) and merged using the CytoVision workstation (Leica biosystems Inc.) at magnification of 200×.
DNA isolation and quality control
Genomic DNA was isolated from lung lesions using 50 μm FFPE sections prepared by the Bio-bank system (Pathology Institute Corp., Toyama, Japan). Genomic DNA was isolated from all samples using the AllPrep DNA/RNA kit from Qiagen according to the manufacturer's instructions. Quality of double-stranded DNA was ascertained using the Picogreen kit according to the manufacturer's instructions.
Whole-exome sequencing (WES)
Sequencing was performed on seven LUADs from different mice (two spontaneous occurring LUADs at 16 months following saline and five LUADs at 5-7 months following NNK exposure; Supplementary Table 1). We also sequenced three tail veins from one wild type and two Gprc5a-/- mice as well as Gprc5a-/- normal lung tissues at one month following saline or NNK for somatic contrasts in the manner described previously 25. DNA libraries, using 200-500 ng of qualified DNA, for massively parallel sequencing were prepared using the SureSelectXT Target Enrichment System for according to the manufacturer's instructions (Agilent Technologies). Library quality and quantity were assessed using a DNA 1000 chip on a 2100 Bioanalyzer. Library fragments were hybridized to the SureSelectXT Mouse All Exon Capture Library (49.6 MB capture) according the manufacturer's protocol. Paired-end cluster generation of denatured templates was performed according to the manufacturer's instructions (Illumina, San Diego, CA) using the HiSeq PE Cluster Kit v4 chemistry and flow cells. WES was performed using the Illumina HiSeq 2500 platform (Illumina, San Diego, CA) utilizing v4 chemistry with paired-end 101 bp reads. Illumina BaseSpace FastQ v1.0.0 was used to de-multiplex and convert the .bcl files to FastQ files for downstream analyses. An average of 196 million unique on-target sequence reads per sample were produced (Table S1).
Identification of somatic variants and copy number variation
Alignment was performed according to Genome Analysis Toolkit (GATK) best practices using burrows-wheeler aligner (BWA) version 0.6.2, GATK-2.6-4, picard-1.114, and samtools-0.1.19 against the mm10 reference with realignments performed using the mouse indel reference mgp.v3.indels.rsIDdbSNPv137. Exomes were filtered against the Agilent M.musculus-SureSelect.Exon.V1.mm10.bed.intervals file corresponding to the capture technology. Somatic variation detection was performed using MuTect-1.1.7 and IndelGenotyper.36.3336 against the same references Annotations were made using variant_tools-2.6.1, using the same references as well as the mouse snp reference mgp.v3.snps.rsIDdbSNPv137. Variants were further filtered for known variants from the database of mouse variation available at ftp-mouse.sanger.ac.uk (release 1303, mgp.v3) 25. Given the scarcity of recurrent variants in the dataset, (thus preventing us from prioritizing genes by adjusting for gene expression and replication timing 26, variants were prioritized for those that occurred in genes described by Vogelstein and colleagues 27 as known to contain bona fide driver mutations in cancer. Mutations in Trp53 as well as in Mtus1 were also prioritized based on the report by Westcott and colleagues 25. Gene homology information between human and mouse genes were obtained from ftp://ftp.informatics.jax.org/pub/reports/index.html#homology. To compute copy number variations (CNVs) from the sequencing data, the control-FREEC algorithm version 7.2 was employed using run settings described previously 28 as well as Sequenza (version 0.0.3) 29. The CNVs were called through tumor-normal comparisons where each LUAD was compared to the normal lung tissues and tail veins.
More details are found in the Supplementary Methods accompanying the manuscript.
Results
Lung lineage-specific tumor suppressor expression patterns for human GPRC5A
The G-protein coupled receptor, Gprc5a, has been previously shown to be preferentially expressed in mouse fetal and adult lung 21. Moreover, mice with knockout of both alleles of this gene (Gprc5a-/-) in the presence or absence of low-dose tobacco carcinogen (single injection of nicotine-specific nitrosamine ketone/NNK), in sharp contrast to wild type littermates, have been demonstrated to develop late onset lung tumors, including LUADs 22, 23, suggesting a tumor suppressor role for this gene in the lung. In the present study, we aimed to better understand the impact of Gprc5a expression on LUAD pathogenesis. We first sought to understand the expression patterns of the human counterpart of the gene (GPRC5A) in normal and malignant human lung tissues. We surveyed a publicly available dataset comprised of human normal tissues from various organs (pan-normal specimens; see Supplementary Methods). This analysis demonstrated that GPRC5A was highly expressed in lung tissues (lung, trachea, bronchus) relative to other organ-specific normal tissues (Figure 1A). Next we probed publicly available datasets comprised of LUADs and uninvolved normal lung tissues (see Supplementary Methods). GPRC5A levels were markedly significantly, and consistently, down-regulated in LUADs compared to normal lung tissues in all datasets tested (all P < 10-3; Figure 1B). We then sought to understand the association between GPRC5A expression and LUAD histopathological subtype. Towards this, we evaluated a tissue microarray of LUADs that we previously studied for GPRC5A immunohistochemical protein expression 30. This analysis revealed no significant differences in GPRC5A protein expression between lepidic, acinar, solid and papillary LUADs (P = 0.29 of the Kruskal-Wallis test; Figure S1). Next we determined to understand expression patterns of GPRC5A in early phases (e.g. “normal-appearing airway field cancerization (ref Kadara)” or “premalignant”) of LUAD pathogenesis. We performed in silico analysis of a dataset we recently reported on the tumor-adjacent cancerization field in the normal-appearing airway 31, and found that GPRC5A was significantly suppressed in both LUADs and adjacent normal-appearing airway cells relative to distant normal lung (P < 10-4; Figure S1). These data suggest plausible airway-lineage expression patterns and properties for GPRC5A in normal and malignant lung epithelial cells.
Figure 1. GPRC5A expression patterns in human normal and malignant lung tissues.
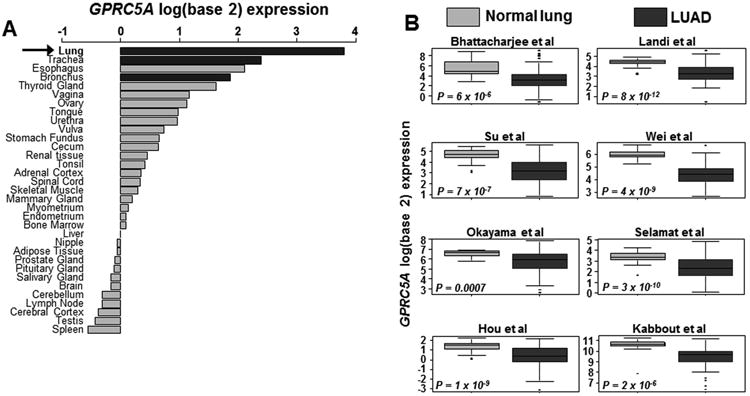
A. Expression levels of GPRC5A mRNA were assessed in an array set comprised of human pan-normal samples (GSE7307 from the gene expression omnibus) as described in Supplementary Methods and in datasets consisting of human LUADs. Normal tissues were ranked (from top to bottom) by decreasing log (base 2) median-centered expression of GPRC5A. B. GPRC5A mRNA expression was also surveyed in datasets 46-52 including a set we previously reported 53 consisting of human LUADs and uninvolved normal tissues. Boxes represent 25% - 75% expression ranges and whiskers constitute maxima and minima. Solid horizontal lines represent median GPRC5A mRNA log (base 2) expression values. P-values were obtained using t-tests.
LUAD development following tobacco carcinogen exposure in Gprc5a-/- mice
As mentioned earlier, we found that single dose (104 mg/kg body weight) of NNK lead to development of late onset spontaneous LUADs in Gprc5a-/- mice 22. We determined to perform a relatively more chronic NNK exposure in Gprc5a-/- mice in an attempt to better emulate tobacco carcinogen-associated LUAD development. Gprc5a-/- mice were divided into groups of five to six mice (per treatment and time point) and were injected intraperitoneally three times per week (for eight weeks) with 50 mg/kg of body weight NNK (150 mg/kg per week) dissolved in saline (0.9% NaCl) or with saline alone as control (Figure 2A). Mice were then sacrificed at each month for seven months after saline or NNK treatment (Figure 2A). Histopathological analysis was performed at each time point to probe the development of hyperplasias, adenomas and adenocarcinomas (Figure S3) based on previously reported criteria for classification of proliferative mouse lung lesions24 including presence of nuclear atypia in adenocarcinoma tumors (Figure S3). In sharp contrast to mice treated with saline, NNK-treated Gprc5a-/- mice developed LUADs as early as 3 months following NNK exposure (Figure 2B). Moreover, by six months following NNK, all (100%) Gprc5a-/- mice were found to develop LUADs. Of note, saline treated Gprc5a-/- mice did not exhibit LUADs across all time points but rather displayed lung adenomas or hyperplasias (Figure 2B, Figure S3). Of note, we also found significantly (P < 0.05) increased numbers of LUADs per mouse at seven relative to six months following NNK treatment (Figure 2C). A subset of saline treated mice were followed up for extended periods and were sacrificed at the time of spontaneous/latent LUAD development (see Materials and Methods). None of the mice developed locoregional or distant metastasis, an observation similar to what was previously reported for mice engineered to express mutant Kras 32.
Figure 2. Tobacco carcinogen-mediated LUAD pathogenesis in the Gprc5a-/- mouse.
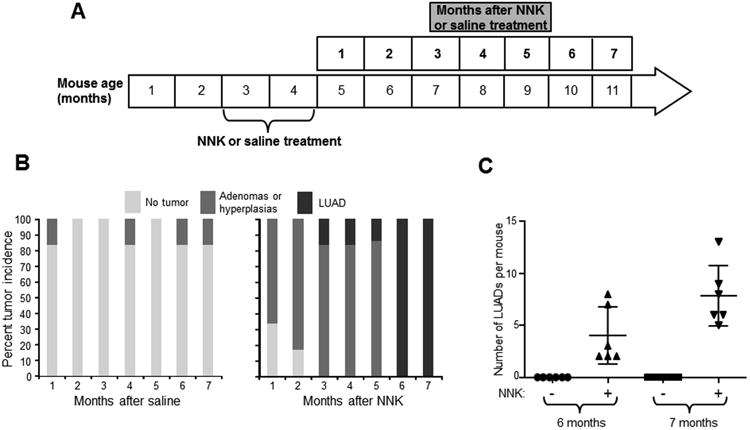
A. Schematic depicting timeline of NNK (three times 50mg/kg body weight per week for eight weeks) or saline treatment of eight weeks old Gprc5a-/- mice (see Methods). Mice were divided into groups of five to six mice (per treatment and time point) and were sacrificed at every month for seven months following saline and NNK treatment. B. At each of the indicated time points, mice lungs were histopathologically evaluated for the development of lesions. The lesions were pathologically categorized (hyperplasias, adenomas and adenocarcinomas) based on previously reported guidelines for murine lung lesions (23) and were then enumerated and compared and contrasted across time points and between treatment groups. C. LUAD burdens, indicated by the number of LUADs per mouse, were statistically compared and contrasted at six and seven months following NNK treatment.
Next we sought to understand the airway lineage cell-of-origin of LUADs in the NNK-treated Gprc5a-/- mice. We performed immunofluorescence (IF) analysis of lesions and adjacent normal bronchi probing for the expression of prototypical markers of airway lineages including surfactant protein C (Sftpc), indicative of alveolar type II (AT2) cells, and clara cell secretory protein (Ccsp/Cc10), indicative of clara/club cells. In contrast to normal bronchial regions, lung lesions were found to lack Ccsp and abundantly express Sftpc suggesting that the cells of origin for these lesions were AT2 cells. Of note, IF analysis of normal wild type (WT) adult (Figure 3B) and embryonic (Figure S4) lung tissue as well as quantitative-real time PCR of CD45-/CD31-/Epcam+ WT normal lung cells sorted based on Podoplanin (Pdpn) expression (Figure S5) revealed that Gprc5a itself was restricted to Pdpn-expressing AT1 cells. Our findings demonstrate that tobacco carcinogen (NNK) exposure accelerates Gprc5a-/- LUAD development, and suggest that these lesions originate from AT2 cells.
Figure 3. Immunofluorescence analysis of airway lineage markers in lung lesions and normal tissues.
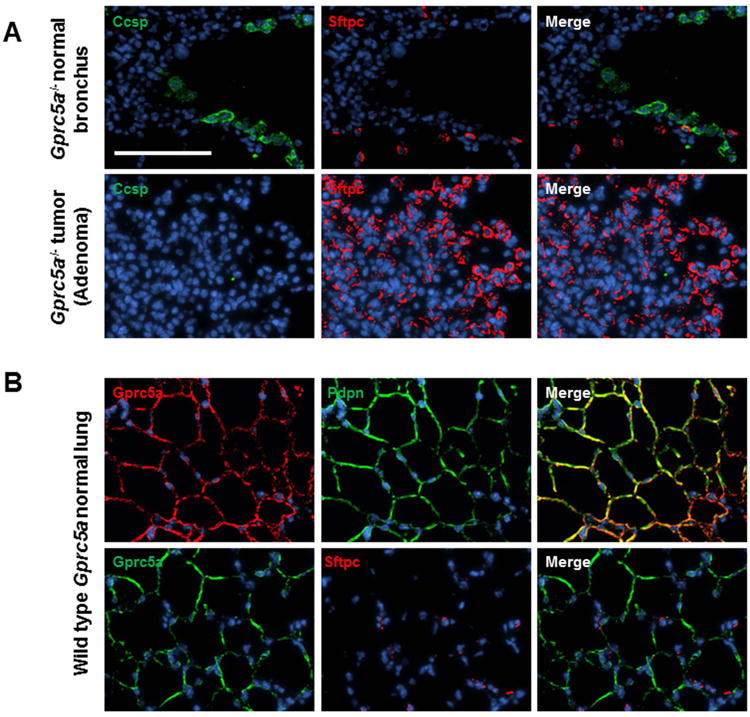
Immunofluorescence (IF) analysis of airway lineage markers of Clara (Ccsp) and alveolar type 2 (AT2) (Sftpc) cells was performed in Gprc5a-/- lesions and adjacent normal regions as described in Materials and Methods. B. IF analysis of Gprc5a and the AT1 cell marker Pdpn was performed as described in Materials and Methods. All images were captured using a BX61 immunofluorescent system (Olympus) and merged using the CytoVision workstation (Leica biosystems Inc.) at a magnification of 200× (scale bar denoting 100 μm).
Mutational landscape of tobacco carcinogen exposed Gprc5a-/- LUADs
We then sought to understand genome-wide alterations in the pathogenesis of the LUADs in tobacco carcinogen exposed Gprc5a-/- mice. Using the Illumina HiSeq 2500 platform, we performed WES of seven Gprc5a-/- LUADs; two spontaneously developing LUADs from two saline treated mice (>16 months after saline) and five tumors (from five mice) that developed five to seven months after NNK treatment. In accordance with previously reported criteria on classification of proliferative mouse lung lesions 24, all seven LUADs were confirmed to have either nuclear atypia (Figure S6; white arrows) or both nuclear atypia and evidence of cellular mitosis (Figure S6; red arrows). We then inferred somatic contrasts, in the manner described previously 25, by also sequencing three tail veins from one WT and two Gprc5a-/- mice as well as two Gprc5a-/- normal lung tissues at one month following saline or NNK. Targeted bases were sequenced to a mean depth of 215× (Table S1). Across samples, 99%, 97% and 86% of targeted bases were sequenced to at least 10×, 20× and 50× coverage, respectively (Table S1). There were no significant differences in depth of coverage or proportion of regions covered to 20× among samples (Table S1). In these seven LUADs, we identified 5,223 exonic single nucleotide variants (SNVs). Of note, LUADs that developed in NNK-exposed Gprc5a-/- mice exhibited substantially higher mutation burdens (Figure S7, left) and different base substitution spectra (Figure S7, right) compared with spontaneously developing tumors, consistent with what is known about smoking-induced human LUADs 3, 7, 8, 10.
Next we sought to discern potential somatic driver mutations in the LUADs by prioritization of exonic and nonsynonymous variants. We compiled exonic nonsynonymous variants that occurred in genes described by Vogelstein and colleagues 27 as known to contain bona fide driver mutations in cancer (see Supplementary Methods). We also included mutations in Trp53 as well as in Mtus1 based on the report by Westcott and colleagues 25. Notably, this analysis revealed that all LUADs exhibited somatic activating mutations in Kras (p.G12D or P.Q61R) (Figure 4, Table S2), the same variants purported to act as drivers of human LUAD in smokers 3, 11. We also confirmed these Kras p.G12D and p.Q61R mutations by deep (> 1,000×) targeted sequencing (Ion Torrent) and digital PCR; both platforms revealed similar variant allele frequencies (VAFs) for the SNVs (Figures S8A-D). The NNK-exposed LUADs exhibited additional driver genes that were mutated in at least one LUAD including the cell cycle kinase Atm and the epigenetic modulator and methyltransferase Kmt2d as well as the tumor suppressors Apc, Nf1 and Trp53 (Figure 4), of which a subset was confirmed by deep targeted sequencing (> 1,000×) (Figure S9). The spontaneous LUADs, albeit few, exhibited very few mutated driver genes besides Kras. Of note, one NNK-exposed LUAD, unlike the other four similar carcinogen derived tumors, exhibited a low somatic mutational burden concomitant with a mutation in p.Q61R (Figure 4). Of note, we found no significant differences in GPRC5A expression among human LUADs based on the type of KRAS codon 12 variant (Figure S10A) or between KRAS-mutant LUADs with and without TP53 mutations (Figure S10B). We next surveyed genome-wide copy number variations (CNVs) in the seven LUADs (see Materials and Methods) and found disparate global CNV patterns between NNK-exposed and spontaneous Gprc5a-/- LUADs (Figure 5). Four out of the five NNK-exposed LUADs, which we had found to exhibit high mutational burdens and specifically a p.G12D Kras mutation, shared gains in broad regions within chromosome 6 (Figure 5). These shared CNVs, along with other copy number gains not found in spontaneous LUAD, included the oncogenes Kras, Met, Braf and Ezh2. Our findings highlight genomic alterations that may play crucial roles in accelerated development and progression of Kras mutant LUADs in presence of tobacco carcinogen exposure.
Figure 4. Whole-exome sequencing analysis of spontaneous and NNK-derived Gprc5a-/- LUADs.
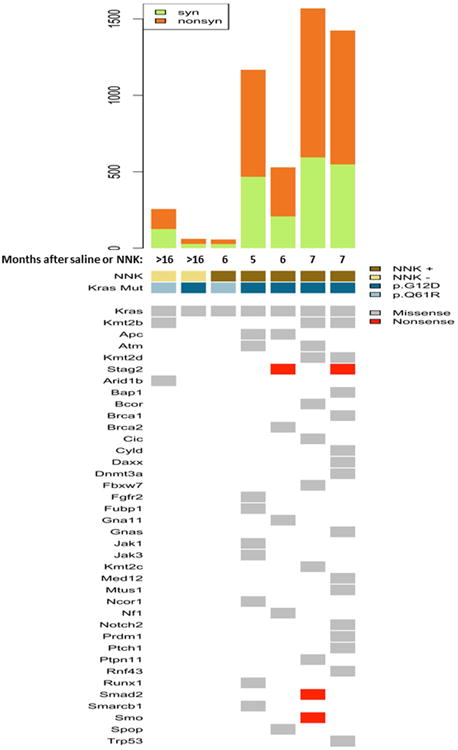
Two spontaneous (> 16 months following saline) LUADs from two Gprc5a-/- mice as well as five LUADs that developed in different mice by five to seven months following NNK treatment were analyzed by whole-exome sequencing (WES) as described in Materials and Methods. Somatic calls were contrasted against whole-exomes from three tail veins (from two Gprc5a-/- and one WT mice) and Gprc5a-/- normal lung tissues at one month following saline (see Methods). Single nucleotide variants (SNVs) were prioritized based on variants occurring in bona fide driver genes 27. The top panel depicts total number of silent (syn) and non-silent (nonsyn) exonic mutations per LUAD. In the lower panel, columns represent LUADs and rows represent mutated driver genes. Arrow indicates somatic Kras (p.G12D or p.Q61R) mutations.
Figure 5. Genome-wide copy number variation in spontaneous and NNK-derived Gprc5a-/- LUADs.
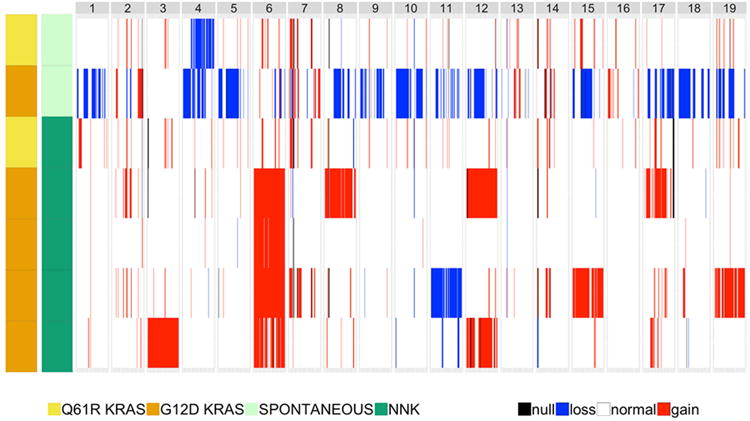
Copy number variations (CNVs) were assessed from sequencing data and read depth using FREEC and Sequenza as described in the Supplementary Methods. Read depth was analyzed in LUADs relative to depth in normal samples to estimate copy number in 8 kb windows followed by segmentation via a LASSO-based algorithm. Genome-wide CNAs across all LUADs were then plotted. Loss, blue; gain, red; not discernable, black.
Discussion
In this study we sought to further understand the role of Gprc5a expression in LUAD pathogenesis. First we surveyed publicly available expression datasets and found that human GPRC5A was largely preferentially expressed in normal lung tissues relative to other organ-specific normal samples and was also consistently suppressed in LUADs. We found that Gprc5a-/- mice exposed to a relatively chronic NNK treatment regimen resulted in development of LUADs (within 5 months after exposure) as opposed to the latent development (> 16 months following saline treatment) in control treated mice. Whole-exome sequencing of latent (from saline treated mice) and NNK-associated Gprc5a-/- LUADs, revealed activating somatic activating Kras mutations in all tumors. Additional (besides Kras) driver mutations and copy number alterations were also found in NNK-associated (and not in latent) Gprc5a-/- LUADs that may underlie the observed accelerated Kras mutant LUAD development by tobacco carcinogen exposure. Our findings pinpoint genome-wide alterations that may be implicated in the progression of Kras mutant LUADs (by smoking).
The G-protein coupled receptor, Gprc5a, is a retinoid-inducible gene that was shown to be specifically expressed in murine normal lung tissues 21. Gprc5a-/- mice were demonstrated to develop late onset LUADs spontaneously 22, 23, suggesting that Gprc5a may function as a lung lineage-specific tumor suppressor. We sought to comprehensively probe the expression patterns of the gene in human normal and malignant tissues. We found that GPRC5A was largely expressed in normal lung and airway tissues relative to other organ-specific tissues and was consistently (across all datasets examined) suppressed in LUADs. We further studied the expression of GPRC5A in a dataset that we previously reported and that constituted lung tumors and normal-appearing airway cells in the adjacent cancerization field 31. This analysis demonstrated that GPRC5A is down-regulated in both LUADs and the surrounding normal-appearing airway field of cancerization relative to distant normal lung tissues. It is worthwhile to mention that the expression patterns of GPRC5A in premalignant precursors (e.g. atypical adenomatous hyperplasia/AAH) to LUAD development is not known. Due to the paucity of profiled AAH lesions we could not probe GPRC5A expression in premalignant phases of LUAD development. Analysis of GPRC5A expression in human lesions (AAH) that are precursors to LUAD development would shed light on the implication of this gene in the early pathogenesis of human LUAD. Of note, we previously reported that GPRC5A protein was significantly down-regulated in normal-appearing airways of lung cancer-free smokers relative to phenotypically healthy smokers 30. Our data suggest that GPRC5A exhibits an expression profile typical of an airway lineage-specific suppressor 33. These findings suggest that Gprc5a, a plausible airway lineage-specific gene may be suppressed in early (“normal”) phases of lung oncogenesis.
The nicotine-specific nitrosamine ketone NNK is present in tobacco and found to be causally linked to lung cancer in human smokers 34, suggesting that NNK exposure is a viable experimental method to mimic carcinogenic effects of cigarette smoking 35, 36. We previously showed that Gprc5a-/- mice developed late onset LUADs even after a single dose of the tobacco carcinogen NNK (104 mg/kg body weight) 22. In order to emulate smoking-associated lung cancer in humans -- particularly in light of the fact that the majority of diagnosed lung cancers such as LUADs occur in lifetime smokers 3, 4 -- we determined to better delineate tobacco carcinogen-mediated LUAD development in Gprc5a-/- mice, through relatively more chronic NNK exposure. Indeed, we found that this more chronic exposure (50 mg/kg per body weight three times per week for eight weeks) markedly accelerated LUAD development. Despite the fact that the Gprc5a-/- mice are of the C57BL/6 × sv129 strain resistant to lung tumorigenesis by carcinogens such as NNK 35, 36, all mice developed LUADs by six months following NNK exposure. It is reasonable to surmise that knockout of Gprc5a in the airway may render the lung more susceptible to tumorigenesis by tobacco carcinogen. It is worthwhile to mention that histopathological analysis revealed the malignant features of the LUADs exemplified by high nuclear crowding and cytological atypia 24 and demonstrated that the lesions comprised a mixed papillary/solid histology. Of note, this histopathological pattern is commonly observed in human LUADs 37 and further augments the suggestion of this model and our findings as motivation for interrogating these molecular alterations as targets for personalized prevention. More generally, our data suggest that the tobacco carcinogen-exposed Gprc5a-/- may be a viable model to understand numerous aspects of human LUAD development in smokers.
The cell-of-origin of Kras mutant LUADs is still not well established. Studies in mice demonstrated that Kras-mutant LUADs can develop from stem cells within the bronchioalveolar junction 38 or from multiple cell lineages (Sftpc-expressing AT2 cells or Ccsp-positive clara cells) 39. In our study interrogating tobacco carcinogen-exposed Gprc5a-/- mice, we found lesions lacked Ccsp and exclusively expressed Sftpc suggesting that they developed from AT2 cells. Our observations are in accordance with recent reports interrogating in vivo the cell-of-origin of LUADs. A study by Mainardi and colleagues revealed that malignant adenocarcinoma only progressed from Sftpc-expressing alveolar hyperplasias in mice engineered to express mutant Kras (G12V) 40. Moreover, AT2 cells were found to act as alveolar progenitor/stem cells leading to clonal foci of lesions and, subsequently, to adenocarcinomas 41. Interestingly, we noted in WT normal lung tissues that expression of Gprc5a protein itself was restricted to AT1 cells. These observations are in accordance with the report by Treutlein and colleagues 42 demonstrating, via single-cell RNA-sequencing of mouse lung tissues, that Gprc5a mRNA was highly correlated with expression of Pdpn and with dynamic generation of AT1 cells from bipotent progenitor cells (found in the supplement of the reported study). It is important to note that various injury model studies have suggested potential pathophysiological links between both alveolar cell types and self-renewal signaling cues in AT2 cells. Earlier pneumonectomy experiments demonstrated that most AT2 cells possess latent regenerative capacity 43. Also, AT2 cell ablation was found to induce self-renewal (duplication) of remaining AT2 cells 44. The recent report by Desai and colleagues further demonstrated that injury (by increased oxygen tension) to AT1 cells activated stem cell renewal properties in AT2 cells and mutations of Kras in the latter drove constitutive self-renewal and subsequent oncogenesis 41. It is reasonable to speculate that Gprc5a knockout may induce “molecular injury” to AT1 cells resulting in self-duplication of AT2 cells which in turn is further exacerbated by tobacco carcinogen exposure (and Kras mutations). Additional studies are warranted to support this plausible supposition.
Our WES analysis demonstrated that Gprc5a-/- LUADs from NNK-treated mice overall exhibited increased total somatic mutation burdens compared to spontaneous LUADs. These data suggest that tobacco carcinogen exposure induces mutational signatures that may be involved in accelerated LUAD development. These findings are in accordance with the report by Westcott and colleagues demonstrating increased mutation burdens in tumors from mice exposed to various carcinogens (e.g. urethane) relative to tumors arising spontaneously from genetically engineered Kras mutant mice 25. Notably, our WES analysis revealed that all seven Gprc5a-/- LUADs exhibited somatic activation mutations in Kras despite normal lungs from the mice being WT for the oncogene. These observed mutations (p.G12D and p.Q61R) are the same variants thought to act as drivers of human LUAD in smokers 3. Our analysis of human LUADs (TCGA dataset) showed that there were no significant differences in GPRC5A expression based on the type of Kras codon 12 variant (i.e., p.G12A/C/D/V). It cannot be neglected that it is difficult to determine the natural order and causal relationship between GPRC5A suppression and KRAS (p.G12D) mutation in human LUADs that had already developed. Of note, one NNK-exposed LUAD, unlike the other four similar carcinogen derived tumors, exhibited a low somatic mutational burden concomitant with a mutation in p.Q61R. It is reasonable to speculate that different Kras variants may be implicated in disparate levels of mutational heterogeneity, a supposition that indeed has been previously interrogated 25. Further analysis in our study revealed additional (besides Kras) mutated driver 27 genes in Gprc5a-/- LUADs from NNK-treated mice and in at least one of these LUADs. In contrast very few, if any, additional driver genes were mutated in the two spontaneous (from saline treated mice) LUADs. It cannot be neglected that we studied very few spontaneous/latent Gprc5a-/- LUADs, largely due to the long latency of these lesions. Yet, our data in the Gprc5a-/- carcinogen-exposed mouse are in line with previous reports surveying genomes of human smoker LUADs and underscoring the causal link of tobacco carcinogen exposure to increased mutational heterogeneity 25, 26.
Our whole-exome sequencing analysis revealed additional co-occurring mutated driver genes in the tobacco carcinogen-exposed LUADs, such as Trp53, Kmt2d, Nf1 and Kmt2c. We also noted amplification of the Ezh2 oncogene in the LUADs. Our findings in the tobacco carcinogen-exposed Gprc5a-knockout mouse are in close concordance with previously reported alterations in human LUADs. In agreement with previous studies of human Kras mutant LUAD 45, it is also plausible that the mutated (e.g. Trp53 and Mll2/Kmt2d) and copy number altered (e.g. Ezh2 gain) driver genes that we accentuate here may cooperate with Kras to accelerate LUAD development in the tobacco carcinogen-exposed Gprc5a-/- mouse. Nonetheless, particularly in light of the fact that therapies targeting Kras have been challenging 13, the co-occurring mutations and CNVs described here may constitute viable targets for early treatment and prevention of Kras mutant LUAD. It is noteworthy, that we found various co-occurring mutated or copy number altered genes that are epigenetic regulators (e.g. Kmt2d, Kmt2c and Ezh2) suggesting that epigenetic deregulation may be an important mechanism in development and progression of smoke-associated Kras mutant LUAD. It is plausible that drugs that reprogram the epigenome (e.g. histone deacetylase and DNA methyltransferase inhibitors) may be viable agents, alone or in combination, for (chemo)prevention of this malignancy.
In conclusion, we demonstrate that GPRC5A is predominantly expressed in the human lung and is commonly suppressed in human LUADs. We find that relatively chronic NNK tobacco carcinogen treatment accelerates LUAD development in Gprc5a-/- mice that otherwise develop late onset spontaneous tumors. Following WES analysis, we report that Gprc5a-/- LUADs display activating mutations in the Kras oncogene. Our sequencing analysis also underscores co-occurring mutations and CNVs in additional driver genes that may cooperate with Kras in LUAD pathogenesis. Lastly, our findings insinuate that the tobacco carcinogen exposed Gprc5a-/- model recapitulates carcinogen-induced Kras mutant LUAD; this offers a unique opportunity to map the temporal evolution of this malignancy.
Supplementary Material
Novelty And Impact.
The development of Kras-mutant lung adenocarcinoma (LUAD), the most common molecular lung cancer subtype, is poorly understood. Using in vivo carcinogenesis models and whole-exome sequencing, we report that Gprc5a-/- mice develop spontaneous LUADs with somatic driver Kras mutations. We also demonstrate that tobacco carcinogen-exposed Gprc5a-/- mice exhibit accelerated development of LUADs harboring co-occurring mutations in additional drivers that may cooperate with Kras in LUAD pathogenesis and, thus, constitute targets for early treatment of Kras-mutant LUAD.
Acknowledgments
Grant support: Supported by the University Cancer Foundation via the Institutional Research Grant program at MD Anderson Cancer Center (to H.K.), the Lung Cancer Discovery Award from the American Lung Association (to H.K.), National Cancer Institute (NCI) grant 1R01CA205608-01A1 (to P.S. and H.K.) and by NCI cancer center support grant CA16672. D.E. was supported by the Medical Research Volunteer Program (MRVP) at the American University of Beirut.
Footnotes
The authors report no conflicts of interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends--An Update. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2016;25:16–27. doi: 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 3.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kadara H, Scheet P, Wistuba II, Spira AE. Early Events in the Molecular Pathogenesis of Lung Cancer. Cancer Prev Res (Phila) 2016;9:518–27. doi: 10.1158/1940-6207.CAPR-15-0400. [DOI] [PubMed] [Google Scholar]
- 5.Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc Natl Acad Sci U S A. 2004;101:10143–8. doi: 10.1073/pnas.0401422101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steiling K, Ryan J, Brody JS, Spira A. The field of tissue injury in the lung and airway. Cancer Prev Res (Phila Pa) 2008;1:396–403. doi: 10.1158/1940-6207.CAPR-08-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, Chen K, Walker J, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–34. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, Zhao Z, Behrens C, Fujimoto J, Chow C, Yoo Y, Kalhor N, et al. Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Annals of oncology : official journal of the European Society for Medical Oncology. 2016 doi: 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi J, Hua X, Zhu B, Ravichandran S, Wang M, Nguyen C, Brodie SA, Palleschi A, Alloisio M, Pariscenti G, Jones K, Zhou W, et al. Somatic Genomics and Clinical Features of Lung Adenocarcinoma: A Retrospective Study. PLoS medicine. 2016;13:e1002162. doi: 10.1371/journal.pmed.1002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstraw P, Ball D, Jett JR, Le Chevalier T, Lim E, Nicholson AG, Shepherd FA. Non-small-cell lung cancer. Lancet. 2011;378:1727–40. doi: 10.1016/S0140-6736(10)62101-0. [DOI] [PubMed] [Google Scholar]
- 12.Kadota K, Sima CS, Arcila ME, Hedvat C, Kris MG, Jones DR, Adusumilli PS, Travis WD. KRAS Mutation Is a Significant Prognostic Factor in Early-stage Lung Adenocarcinoma. The American journal of surgical pathology. 2016;40:1579–90. doi: 10.1097/PAS.0000000000000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomasini P, Walia P, Labbe C, Jao K, Leighl NB. Targeting the KRAS Pathway in Non-Small Cell Lung Cancer. The oncologist. 2016;21:1450–60. doi: 10.1634/theoncologist.2015-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gontan C, de Munck A, Vermeij M, Grosveld F, Tibboel D, Rottier R. Sox2 is important for two crucial processes in lung development: branching morphogenesis and epithelial cell differentiation. Dev Biol. 2008;317:296–309. doi: 10.1016/j.ydbio.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 15.Kadara H, Wistuba II. Field cancerization in non-small cell lung cancer: implications in disease pathogenesis. Proceedings of the American Thoracic Society. 2012;9:38–42. doi: 10.1513/pats.201201-004MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nature genetics. 2009;41:1238–42. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan P, Kadara H, Behrens C, Tang X, Woods D, Solis LM, Huang J, Spinola M, Dong W, Yin G, Fujimoto J, Kim E, et al. Sex determining region Y-Box 2 (SOX2) is a potential cell-lineage gene highly expressed in the pathogenesis of squamous cell carcinomas of the lung. PloS one. 2010;5:e9112. doi: 10.1371/journal.pone.0009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan B, Li C, Kimura S, Engelhardt RT, Smith BR, Minoo P. Inhibition of distal lung morphogenesis in Nkx2.1(-/-) embryos. Developmental dynamics : an official publication of the American Association of Anatomists. 2000;217:180–90. doi: 10.1002/(SICI)1097-0177(200002)217:2<180::AID-DVDY5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka H, Yanagisawa K, Shinjo K, Taguchi A, Maeno K, Tomida S, Shimada Y, Osada H, Kosaka T, Matsubara H, Mitsudomi T, Sekido Y, et al. Lineage-specific dependency of lung adenocarcinomas on the lung development regulator TTF-1. Cancer Res. 2007;67:6007–11. doi: 10.1158/0008-5472.CAN-06-4774. [DOI] [PubMed] [Google Scholar]
- 21.Cheng Y, Lotan R. Molecular cloning and characterization of a novel retinoic acid-inducible gene that encodes a putative G protein-coupled receptor. The Journal of biological chemistry. 1998;273:35008–15. doi: 10.1074/jbc.273.52.35008. [DOI] [PubMed] [Google Scholar]
- 22.Fujimoto J, Kadara H, Men T, van Pelt C, Lotan D, Lotan R. Comparative functional genomics analysis of NNK tobacco-carcinogen induced lung adenocarcinoma development in Gprc5a-knockout mice. PloS one. 2010;5:e11847. doi: 10.1371/journal.pone.0011847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tao Q, Fujimoto J, Men T, Ye X, Deng J, Lacroix L, Clifford JL, Mao L, Van Pelt CS, Lee JJ, Lotan D, Lotan R. Identification of the retinoic acid-inducible Gprc5a as a new lung tumor suppressor gene. J Natl Cancer Inst. 2007;99:1668–82. doi: 10.1093/jnci/djm208. [DOI] [PubMed] [Google Scholar]
- 24.Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, Fraire AE, Gabrielson EW, Gunning WT, Haines DC, Kaufman MH, Linnoila RI, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–16. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 25.Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, Fredlund E, Quigley DA, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–92. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boeva V, Zinovyev A, Bleakley K, Vert JP, Janoueix-Lerosey I, Delattre O, Barillot E. Control-free calling of copy number alterations in deep-sequencing data using GC-content normalization. Bioinformatics. 2011;27:268–9. doi: 10.1093/bioinformatics/btq635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, Eklund AC. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Annals of oncology : official journal of the European Society for Medical Oncology. 2015;26:64–70. doi: 10.1093/annonc/mdu479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujimoto J, Kadara H, Garcia MM, Kabbout M, Behrens C, Liu DD, Lee JJ, Solis LM, Kim ES, Kalhor N, Moran C, Sharafkhaneh A, et al. G-protein coupled receptor family C, group 5, member A (GPRC5A) expression is decreased in the adjacent field and normal bronchial epithelia of patients with chronic obstructive pulmonary disease and non-small-cell lung cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012;7:1747–54. doi: 10.1097/JTO.0b013e31826bb1ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadara H, Fujimoto J, Yoo SY, Maki Y, Gower AC, Kabbout M, Garcia MM, Chow CW, Chu Z, Mendoza G, Shen L, Kalhor N, et al. Transcriptomic architecture of the adjacent airway field cancerization in non-small cell lung cancer. J Natl Cancer Inst. 2014;106:dju004. doi: 10.1093/jnci/dju004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 33.Perdomo C, Campbell JD, Gerrein J, Tellez CS, Garrison CB, Walser TC, Drizik E, Si H, Gower AC, Vick J, Anderlind C, Jackson GR, et al. MicroRNA 4423 is a primate-specific regulator of airway epithelial cell differentiation and lung carcinogenesis. Proc Natl Acad Sci U S A. 2013;110:18946–51. doi: 10.1073/pnas.1220319110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hecht SS. Approaches to chemoprevention of lung cancer based on carcinogens in tobacco smoke. Environmental health perspectives. 1997;105(4):955–63. doi: 10.1289/ehp.97105s4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hollander MC, Balogh AR, Liwanag J, Han W, Linnoila RI, Anver MR, Dennis PA. Strain-specific spontaneous and NNK-mediated tumorigenesis in Pten+/- mice. Neoplasia. 2008;10:866–72. doi: 10.1593/neo.08406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng HC, Takano Y. NNK-Induced Lung Tumors: A Review of Animal Model. Journal of oncology. 2011;2011:635379. doi: 10.1155/2011/635379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solis LM, Behrens C, Raso MG, Lin HY, Kadara H, Yuan P, Galindo H, Tang X, Lee JJ, Kalhor N, Wistuba II, Moran CA. Histologic patterns and molecular characteristics of lung adenocarcinoma associated with clinical outcome. Cancer. 2012;118:2889–99. doi: 10.1002/cncr.26584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–35. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 39.Sutherland KD, Song JY, Kwon MC, Proost N, Zevenhoven J, Berns A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111:4952–7. doi: 10.1073/pnas.1319963111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mainardi S, Mijimolle N, Francoz S, Vicente-Duenas C, Sanchez-Garcia I, Barbacid M. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111:255–60. doi: 10.1073/pnas.1320383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature. 2014;507:190–4. doi: 10.1038/nature12930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–5. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brody JS, Burki R, Kaplan N. Deoxyribonucleic acid synthesis in lung cells during compensatory lung growth after pneumonectomy. The American review of respiratory disease. 1978;117:307–16. doi: 10.1164/arrd.1978.117.2.307. [DOI] [PubMed] [Google Scholar]
- 44.Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, Hogan BL. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–36. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, Zhang J, Giri U, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer discovery. 2015;5:860–77. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, Loda M, Weber G, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A. 2001;98:13790–5. doi: 10.1073/pnas.191502998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, Grosveld F, Philipsen S. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PloS one. 2010;5:e10312. doi: 10.1371/journal.pone.0010312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Yang P, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PloS one. 2008;3:e1651. doi: 10.1371/journal.pone.0001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–11. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 50.Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, Lam S, Gazdar AF, et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome research. 2012;22:1197–211. doi: 10.1101/gr.132662.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC, Lin CH, Whang-Peng J, Hsu SL, Chen CH, Huang CY. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics. 2007;8:140. doi: 10.1186/1471-2164-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei TY, Juan CC, Hisa JY, Su LJ, Lee YC, Chou HY, Chen JM, Wu YC, Chiu SC, Hsu CP, Liu KL, Yu CT. Protein arginine methyltransferase 5 is a potential oncoprotein that upregulates G1 cyclins/cyclin-dependent kinases and the phosphoinositide 3-kinase/AKT signaling cascade. Cancer science. 2012;103:1640–50. doi: 10.1111/j.1349-7006.2012.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabbout M, Garcia MM, Fujimoto J, Liu DD, Woods D, Chow CW, Mendoza G, Momin AA, James BP, Solis L, Behrens C, Lee JJ, et al. ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin Cancer Res. 2013;19:3383–95. doi: 10.1158/1078-0432.CCR-13-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.