

Abstract
L-Cystine bismorpholide (1a) and L-cystine bis(N′-methylpiperazide) (1b) were seven and twenty-four times more effective than L-cystine dimethyl ester (CDME) in increasing the metastable supersaturation range of L-cystine, respectively, effectively inhibiting L-cystine crystallization. This behavior can be attributed to inhibition of crystal growth at microscopic length scale, as revealed by atomic force microscopy. Both 1a and 1b are more stable than CDME, and 1b was effective in vivo in a knockout mouse model of cystinuria.
Graphical Abstract
INTRODUCTION
Cystinuria is an autosomal recessive disorder caused by mutations in gene SLC3A1 on chromosome 2 or an incompletely dominant disorder caused by mutations in gene SLC7A9 on chromosome 19. Each gene codes for a component of the major proximal renal tubular cystine and dibasic amino acid transporter.1–3 These mutations result in abnormal reabsorption of L-cystine and dibasic amino acids from the luminal fluid of the renal proximal tubule, leading to elevated concentrations of these amino acids in the urine of affected individuals. Defective expression in the small bowel leads to reduced absorption of these amino acids from the intestinal lumen. Because of its limited aqueous solubility, L-cystine crystallizes readily in urine and forms L-cystine stones in the kidney, ureter, and bladder. Although the incidence of L-cystine stones is much lower than that of calcium oxalate stones, L-cystine stones are larger, occur at a young age, recur more frequently, and are more likely to cause chronic kidney disease.4 Cystine stones account for 1% of all stones and as many as 7% of stones in children. Cystinuria is a chronic, lifelong condition, and patients with cystinuria have a >50% chance of stone formation during their lifetime, most experiencing onset and diagnosis between the ages of 2 and 40.5
Current clinical treatment of cystinuria has not changed over the last 30 years and is aimed at reducing the concentration of free L-cystine in urine and increasing its solubility.6,7 A high fluid intake of around 4–5 L a day and alkalinization of urine pH with citrate or bicarbonate salt can suppress but may not completely prevent stone formation. Drug therapy, based on disulfide exchange with D-penicillamine or α-mercaptopropionylglycine with L-cystine to generate more soluble mixed disulfides, has been used for severe cases.4,8 These drugs, however, have side effects that include loss of taste, fever, proteinuria, serum sickness-type reactions, and even frank nephritic syndrome.8
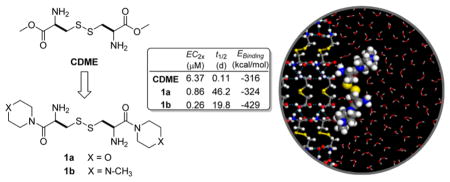
Recently, Ward and his co-workers reported an alternative approach to prevent cystinuria based on crystal growth inhibition, which is achieved through the binding of tailored crystal growth inhibitors (aka molecular imposters), L-cystine dimethyl ester (CDME) and L-cystine methyl ester (CME), to L-cystine crystal surfaces.9–12 Real-time in situ atomic force microscopy (AFM) revealed that CDME and CME dramatically reduce the growth velocity of six symmetry-equivalent {100} steps because of specific binding to crystal growth sites, which frustrates the further attachment of L-cystine molecules.9 CDME was found to significantly reduce stone burden in cystinuria mice compared with a water-treated group accompanied by the formation of smaller stones, but did not prevent stone formation.13 Herein, we report the discovery of two novel L-cystine diamides, L-cystine bismorpholide (CDMOR, LH707 1a) and L-cystine bis(N′-methylpiperazide) (CDNMP, LH708, 1b),14 which are more effective inhibitors of L-cystine crystallization and exhibit significantly better stability. Moreover, both 1a and 1b are orally bioavailable and 1b is effective in vivo in inhibiting L-cystine crystallization and stone formation.
RESULTS AND DISCUSSION
Design Principle
CDME inhibits the crystallization of L-cystine in water at micromolar concentrations, which is below the concentrations needed to produce toxic effects in cells and animals.9 CDME was shown to be effective in reducing stone size and overall mass in a knockout mouse model of cystinuria.13 Its in vivo effectiveness after oral administration may be compromised by esterase-mediated hydrolysis; indeed, esters are commonly used prodrug forms and are readily converted in vivo to their precursor carboxylic acids.15,16 This prompted us to synthesize and evaluate more stable analogues of L-cystine that may serve as inhibitors of L-cystine crystallization. Out of a panel of Nα-methyl and diamide derivatives of L-cystine, 1a and 1b were selected and shown to be substantially more potent than CDME with respect to inhibiting bulk crystallization. They also are more stable than CDME, and preliminary in vivo studies in a knockout mouse model of cystinuria indicate that both 1a and 1b are orally bioavailable and 1b is effective in inhibiting L-cystine crystallization and stone formation.
Chemical Synthesis
As illustrated in Scheme 1, L-cystine diamides 1a and 1b were readily synthesized through the amidation of Boc-protected L-cystine 2 using HOBt-activated ester followed by deprotection of the Boc groups using 4 equiv of 4 N HCl in dioxane. Amidation using activated esters afforded higher yields and fewer side products than other coupling methods. The overall yields of the straightforward three-step synthesis were 40–50%. All compounds were characterized fully using 1H, 13C NMR and HRMS and were >95% pure based on NMR and HPLC.
Scheme 1. Synthesis of 1a and 1b from Boc-L-cystinea.

aReaction conditions: (i) HOAt, EDC, DIEA, (ii) morpholine or N-methylpiperazine, (iii) 4 N HCl/dioxane
Determination of L-Cystine Solubility Using a Fluorescence Assay after OPA/NBC Derivatization
A fluorescence-based assay using O-phthaldialdehyde (OPA) and N-Boc-cysteine (NBC) was used to measure the solubility of L-cystine in water based on a similar protocol reported previously for the analysis and resolution of L- and D-amino acids.17 OPA/NBC derivatization is fast and simple, but the reaction of OPA with cystine and NBC yielded derivatives with decreased fluorescence. In the protocol of Scheme 2, the disulfide bond in L-cystine (3) was reduced to L-cysteine (4) and the sulfhydryl group was then alkylated with iodoacetic acid to form S-carboxymethyl L-cysteine (5), which afforded the OPA/NBS derivative 6 with normal fluorescence.18
Scheme 2. Fluorescence-Based Assay for L-Cystine Concentration Measurementa.

aReaction conditions: (i) DTT, rt, 10 min, (ii) iodoacetic acid, rt, 15 min, (iii) O-phthaldialdehyde (OPA)/N-Boc-cysteine (NBC), rt, 3 min
Effect on the Metastable Supersaturation Range of L-Cystine
A highly supersaturated solution of L-cystine was prepared in Millipore water according to the literature method.9 Solutions of 1a, 1b, or CDME were then added to the supersaturated solution of L-cystine in water to give supersaturated solutions of L-cystine containing various concentrations of 1a, 1b, or CDME. After incubation of the mixtures at 25 °C for 72 h, the solubility of L-cystine in the presence of a given concentration of each test compound was determined using the aforementioned fluorescence assay.
As illustrated in Figure 1, the concentration of L-cystine in the presence of 1a, 1b, and CDME is greater than the equilibrium solubility of L-cystine. This is a consequence of these crystal growth inhibitors increasing the metastable supersaturation range of L-cystine. In the absence of inhibitor, the concentration of L-cystine in the supernatant is approximately 1 mM, somewhat greater than the reported equilibrium concentration of 0.7 mM,19 and identical to the value measured as part of a kinetic study of L-cystine crystal growth.11 The higher-than-expected concentration can be attributed to the limited time of incubation for crystallization and/or unknown adventitious impurities that inhibit crystallization, which is not unusual. At low inhibitor concentrations, the L-cystine concentration slightly exceeds the equilibrium concentration, corresponding to minimal inhibition. At high inhibitor concentration, the L-cystine concentration reaches a plateau at or near the initial L-cystine concentration in the supersaturated solution. The data reveal that 1a and 1b inhibit bulk crystallization more effectively than CDME. The concentration of an inhibitor that elevates the level of L-cystine supersaturation (aka L-cystine concentration in our assay) to twice the concentration of the lower plateaus, denoted as EC2x, is a useful metric for comparing the effectiveness of the crystallization inhibitors. The EC2x values for 1a and 1b were measured to be 0.86 and 0.26 μM, respectively, which are seven and twenty-four times that observed for CDME (EC2x = 6.37 μM). The upper plateau for 1a is lower than those for 1b and CDME, but this can be attributed to a lower L-cystine concentration in the initial supersaturated solution used to test 1a.
Figure 1.

Effect of CDME and L-cystine diamides 1a and 1b on the aqueous concentration of L-cystine. The errors bars represents the standard deviation calculated from the triplicate measurements.
Atomic Force Microscopy (AFM) Study of L-Cystine Crystal Growth Inhibition
In situ AFM measurements of step velocities, which are known to correspond with crystal growth rates, were performed for hexagonal L-cystine crystals in aqueous solutions containing 2 mM L-cystine. The average step velocity of L-cystine in 2 mM L-cystine solution, without additives, was 9.44 ± 0.17 nm/s. The effect of additives was expressed by the quantity V/V0, where V is the step velocity measured in the presence of additives and V0 is the velocity measured initially in the absence of additives (a lower V/V0 corresponds to greater inhibition). Like CDME, the V/V0 measured in the presence of 1a or 1b decreased with increasing additive concentration (Table 1). This is accompanied by a reduction in crystal mass yield as well as changes in morphology from plates to rods (in vitro). Step roughening was observed in the presence of 1a and 1b, like that reported previously for CDME.9 The extent of step roughening increases with additive concentration (Figure 2), consistent with the Cabrera–Vermilyea mechanism, in which adsorbed impurity particles block step propagation.20,21 The data reveal that the inhibition efficacies for 1a, 1b, and CDME, as deduced solely from the reduction of the {100} step velocities on the (001) basal plane, are comparable. The values of V/V0 are best represented as the range that is indicative of crystal growth inhibition. The range of V/V0 values corresponds to measurements made on different L-cystine crystals in different 2 mM L-cystine solutions by several operators. The observation of a range of V/V0 values reflects inherent uncertainty in the actual L-cystine concentrations when working with metastable supersaturated L-cystine solutions as well as small uncertainties in the very low inhibitor concentration. Moreover, measurement of V/V0 becomes more difficult when the step advancement rate becomes very slow due to higher inhibitor concentrations. Nonetheless, the V/V0 values for CDME, 1a, and 1b are comparable, supporting a common mechanism for crystal growth inhibition.
Table 1.
Effects of L-Cystine Diamides on L-Cystine Crystallization in Comparison to CDME
| Compound | Structure | Effect on L-cystine supersaturation | Effect on AFM step velocity | Chemical stabilityd | in vivo activitye | EBinding (kcal/mol)g to L-cystine crystal surface | |
|---|---|---|---|---|---|---|---|
|
|
|
|
|
||||
| EC2xa (μM) | Ratiob | V/V0c @ 15/30/45μM | t1/2 (days) | fraction of mice with stones | |||
| CDME |
|
6.37 | 1.0 | 0.4–0.65/0.27–0.54/0.21–0.48 | 0.11 | 7/14f | −316 |
|
| |||||||
| 1a |
|
0.86 | 7.4 | 0.44–0.65/0.34–0.58/0.29–0.53 | 46.2 | 6/6 | −324 |
|
| |||||||
| 1b |
|
0.26 | 24.5 | 0.52–0.66/0.32–0.50/0.30–0.40 | 19.8 | 1/7 | −429 |
EC2x refers to the concentration required to double L-cystine concentration in solution without observable crystallization.
Ratio refers to the improvement in potency over the control CDME.
The range of normalized step velocity (V/V0) as obtained at three different inhibitor concentrations in aqueous 2 mM L-cystine.
Chemical stability of each compound was measured using LCMS after incubation of test compounds in pH 7.4 phosphate buffered saline at 37 °C.
The in vivo activity was measured in Slc3a1 knockout male mice. In the parallel water control group, 5/7 mice formed stones.
These data were taken from published results and are comparable to those from its parallel water control group.13
The binding energies in kcal/mol for the binding of test compound to cystine crystal surface {100} were computed using the COMPASS force field in BIOVIA’s Materials Studio after Monte Carlo searches of the configurational space for possible adsorption configuration in the presence of explicit water molecules (see Experimental Section for details).
Figure 2.

AFM images of crystal growth on the (001) basal plane in the presence of 1a, (A) 15 μM, (B) 30 μM, and (C) 45 μM, and 1b, (D) 15 μM, (E) 30 μM, and (F) 45 μM. All images were taken ~25 min after injection of inhibitor. The roughening of the {100} steps in the presence of 1a and 1b is evident.
Chemical Stability
The chemical stabilities of 1a and 1b were determined in pH 7.4 phosphate buffered saline at 37 °C using LC-MS by following the disappearance of the test compounds (Figure 3). The half-life for CDME is shorter (t1/2 = ~2.7 h) than that measured for 1a and 1b (t1/2 = 19.8 and 46.2 days, respectively), indicating that these L-cystine diamides are >170 times more stable than CDME under physiologically simulated conditions.
Figure 3.

Chemical stability of L-cystine diamides 1a and 1b in PBS at 37°C in comparison to CDME.
In Vivo Activity in a Genetic Mouse Model of Cystinuria
Slc3a1 knockout male mice were used to test the effectiveness of L-cystine diamides for the treatment of cystinuria. Two groups of six or seven mice were treated with either 1a or 1b at 29.3 μmol/kg through daily gavage for 4 weeks, and a third control group of seven mice received water only. Five out of the seven mice in the control group formed stones. All six mice treated with 1a formed stones, but only one out of the group of seven mice treated with 1b formed stones. These results indicate that 1b is effective while 1a is ineffective in preventing stone formation in cystinuria mice. In a previous study, CDME was shown to be effective in reducing stone size and stone burden in a knockout mouse model of cystinuria but had no effect on the number of mice that formed stones.13 Our preliminary in vivo study here suggests that 1b is a better candidate for stone prevention than CDME.
L-Cystine Diamides in Mouse Urine after Oral Dosing
Two groups of 4–5 wild-type male mice and two groups of Slc3a1 knockout male mice were treated with either 1a or 1b at 29.3 μmol/kg through daily gavage for 1 week and the urine of each mouse was collected individually during the 4 h period immediately after the last dosing. The amounts of L-cystine diamides in the urine samples were determined using LC-MS/ MS and shown in Figure 4. While no test compounds were found in urine samples collected prior to oral dosing (data not shown), micromolar concentrations of 1a and 1b were found in the urine of each mouse after dosing, suggesting that these compounds are orally bioavailable. Interestingly, significantly higher concentrations of 1b were found in urine samples collected from the Slc3a1 knockout cystinuria mouse group than those from the normal mouse group (7.59 ± 1.34 μM vs 2.10 ± 0.57 μM for 1b). Significantly higher concentrations of 1b were found in mice with cystinuria as compared to 1a at equivalent oral doses (7.59 ± 1.34 μM for 1b vs 2.09 ± 0.62 μM for 1a). These unexpected results may indicate that the activities of other transporters are elevated after knocking out the Slc3a1 gene which worked in our favor in the case of 1b but not in the case of 1a.
Figure 4.

Drug concentration in mouse urine after 7 daily oral dosing of L-cystine diamides 1a and 1b.
Molecular Modeling
Crystal morphology and adsorption/docking calculations were performed using BIOVIA’s Materials Studio software suite. Bravais–Friedel Donnay–Harker (BFDH) calculations provided a plausible explanation for the hexagonal plate habit observed experimentally,9 with a large (001) basal face and six small {100} faces, which have been identified as the fast-growing faces (i.e, fast growth normal to the {100} plane). Crystal growth inhibition will be most effective for additives that slow the advance of the {100} steps, as demonstrated previously.9
Crystal surfaces, such as those observed for L-cystine (Figure 2), are complex, decorated with steps and kinks that serve as sites for binding of solute molecules during crystal growth.20 One approach to screening prospective crystal growth inhibitors is to calculate binding energies associated with adsorption to morphologically important crystal surfaces. Binding energies of the L-cystine diamides onto the fast growing {100} surface of L-cystine in an explicitly solvated environment (Figure 5) are listed in Table 1. Compounds 1a and 1b have binding energies greater in magnitude than L-cystine (−85.8 kcal/mol). The magnitude of the binding energy for 1b was greater than those of 1a and CDME, which is consistent with smaller EC2x observed for 1b (Figure 1).
Figure 5.

Structure configurations of L-cystine (A), CDME (B), and 1b (C) when adsorbed onto the {100} surface of L-cystine crystal (in ball-and-stick presentation). Cystine and its derivatives are in space-filling representation at 60% of vdW radii; solvent (H2O) molecules are in line representation. Dashed blue lines represents selected hydrogen bonding between molecules.
CONCLUSIONS
In summary, L-cystine diamides 1a and 1b are potent inhibitors of L-cystine crystallization. These compounds reduce the {100} step velocities to an extent comparable to CDME but are more effective than CDME with respect to sustaining higher concentrations of L-cystine in solution, which is tantamount to inhibition of crystal growth. The inhibition of L-cystine crystallization in vitro by these two L-cystine diamides occurs at submicromolar concentrations, which are seven and twenty-four times lower than that of CDME. In situ AFM studies indicate that 1a, 1b, and CDME reduce step velocities by comparable amounts, accompanied by step roughening that is consistent with a step-pinning mechanism due to binding of the additives to {100} step sites on the (001) basal face. The observation that 1a and 1b are significantly more effective than CDME in inhibiting the bulk crystallization of L-cystine despite comparable step velocity reduction on the (001) face suggests that 1a and 1b exert much stronger inhibition of growth on the {100} faces, which is supported by binding energy calculations on the flat {100} surface. The small size of the (100) faces has precluded AFM studies of the (100) faces and characterization of the active growth steps on the (100) faces. A reduction in the (100) step velocities on the (001) basal plane is an indicator of a good inhibitor, but we have found recently (using a combination of SEM and optical microscopy) that certain inhibitors, including 1a and 1b, are more effective with respect to inhibiting growth on the (100) face than CDME, which can account for the greater effectiveness of 1a and 1b. Furthermore, both 1a and 1b are more stable than CDME at physiological pH and temperature and are expected to be more resistant to proteolytic degradation. 1b effectively inhibited L-cystine stone formation in vivo in a genetic mouse model of cystinuria. At present, we have no data to indicate that there is disulfide metathesis that might possibly affect their in vivo activity. Both 1a and 1b were found in micromolar concentrations in mouse urine after seven daily oral gavage, indicating that they are orally bioavailable and can reach their site of action in the urine. L-Cystine bis(N′-methylpiperazide), 1b, seems to be better absorbed in Slc3a1 knockout cystinuria mice than in normal mice. These results suggest that the L-cystine diamides may have utility as oral therapy for the prevention of L-cystine stones in people with cystinuria.
EXPERIMENTAL SECTION
Synthesis of L-Cystine Diamides 1a–1b
L-Cystine bis(N′-methylpiperazide) (1b): To a solution of N,N′-bis(tert-butoxycarbonyl)-L-cystine (8.81 g, 20 mmol) in DMF (80 mL) were added HOAt (8.01 g, 52.0 mmol), EDC (9.97 g, 52.0 mmol), 1-methylpiperazine (4.01 g, 40.0 mmol), and DIEA (17.47 mL, 100 mmol). The reaction mixture was stirred at room temperature overnight. Then 200 mL of cold water was added (no precipitate formed). The solution was extracted with dichloromethane three times. The combined organic extracts were washed with brine, dried over MgSO4, filtered, concentrated, and purified by ISCO flash silica gel chromatography (DMF was removed by blowing nitrogen before column), eluted with dichloromethane–6%MeOH in dichloromethane containing 0.5 M NH3 to give 7.21 g (60% yield) of the desired intermediate N,N′-bis(tert-butoxycarbonyl)-L-cystine bis(N′-methylpiperazide).
To a solution of N,N′-bis(tert-butoxycarbonyl)-L-cystine bis(N′-methylpiperazide) (5.2 g, 8.60 mmol) in 50 mL of dichloromethane at 0 °C was added 4 M HCl in 1,4-dioxane (12.90 mL, 51.6 mmol). Solid precipitate formed. The reaction mixture was stirred at room temperature for 2 h. The solid was collected by filtration, rinsed with cold ether, and dried in a vacuum oven at 50 °C to give 2.45 g (71% yield) of the desired product 1b. 1H NMR (500 MHz, DMSO-d6, 100 °C) δ 2.80 (s, 6H), 3.31 (b, 4H), 3.31–3.46 (m, 8H), 3.98 (b, 8H), 4.7 (t, 2H), 9.5 (b, 6H). 13C NMR (500 MHz, CD3OD) δ 167.36, 68.20, 54.88, 53.92, 51.01, 43.73, 40.66. LC-MS (ESI+) m/z 404.9 [M + H]+. HRMS (ESI+) m/z calculated for C16H33N6O2S2+ [M + H]+ 405.2101, found 405.2095.
L-Cystine bismorpholide (1a) was synthesized following the same procedure as that for 1b above. 1H NMR (400 MHz, CD3OD, δ): 3.26 (dd, 2H), 3.37 (dd, 2H), 3.55–3.76 (m, 8H), 3.72–3.85 (m, 8H), 4.78 (dd, 2H). 13C NMR (500 MHz, CD3OD) δ 166.97, 68.21, 67.63, 67.53, 50.70, 47.50, 44.13, 39.45. LC-MS (ESI+) m/z 378.8 [M + H]+. HRMS (ESI+) m/z calculated for C14H27N4O4S2+ [M + H]+ 379.1468, found, 379.1480.
Fluorescence Assay for the Inhibition of L-Cystine Crystal Formation
First, 5 μL of each solution was added to 500 μL of L-cystine supersaturated solution as previously reported.9 The mixtures were allowed to stand at 25 °C for 72 h. At the end of incubation, the mixtures were centrifuged at 10000 rpm for 4 min and the supernatants were diluted 2-fold for concentration measurement. Each diluted mixture (10 μL), 0.1 M dibasic sodium phosphate solution (90 μL), and 10 μL of DTT solution (12.5 mM) were mixed at room temperature for 10 min before the addition of 10 μL of iodoacetic acid (100 mM) and continued incubation at room temperature for an additional 15 min. Derivatization was performed by the addition of 10 μL of OPA (100 mM in methanol) and 10 μL of NBC (100 mM in methanol) for 3 min. Then 40 μL of the derivatized mixture was plated in a 384-well plate and fluorescence was read at Ex 355 nm/Em 460 nm to derive the concentrations of the original mixtures. More details can be found in Supporting Information.
Molecular Modeling
Adsorption Locator in BIOVIA’s Materials Studio was used in docking the inhibitors onto the target surface and computing the sorption energies as Ebinding = Etotal interaction − Eslab.solvent − Esolvent.sorbate, where slab and sorbate refer to the surface slab of cystine crystal and the candidate inhibitor molecule, respectively. Possible adsorption configurations were identified by performing Monte Carlo searches of the configurational space through a simulated annealing procedure.22,23 The COMPASS force field24 was used in the evaluation of interaction energies. Binding energies without and with the presence of solvent (water) were computed and, in the latter case, water molecules were introduced explicitly into the model for binding energy calculations, with a nominal density of 1.0 g/cm3. The Bravais–Friedel Donnay–Harker (BFDH) method25 was used in computing the crystal morphology and identifying fast growing crystal faces.
Supplementary Material
Acknowledgments
We acknowledge Rutgers University Foundation for partial support of this work. This work was also supported in part by the Rutgers Supercomputing Cluster (NIH 1S10OD012346), by the MRSEC Program of the National Science Foundation under award no. DMR-1420073 and by the Rare Kidney Stone Consortium (U54KD083908), which is a part of the NIH Rare Diseases Clinical Research Network, supported through collaboration between the NIH Office of Rare Diseases Research at the National Center for Advancing Translational Sciences and National Institute of Diabetes and Digestive and Kidney Disease.
ABBREVIATIONS USED
- AFM
atomic force microscopy
- BFDH
Bravais–Friedel Donnay–Harker
- CDME
L-cystine dimethyl ester
- CDMOR
L-cystine bismorpholide
- CDNMP
L-cystine bis(N′-methylpiperazide)
- CME
L-cystine methyl ester
- LC-MS
liquid chromatography mass spectrometry
- MRM
multiple reaction monitoring
- NBC
N-Boc-cysteine
- NHS
N-hydroxysuccimide
- OPA
O-phthaldialdehyde
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.6b00647.
Experimental data for fluorescence assay for the inhibition of L-cystine crystal formation, AFM crystal growth experiments, chemical stability measurement, in vivo activity in mouse model of cystinuria, and measurement of compound concentration in urine; 1H NMR and 13C NMR spectroscopic data for new compounds 1a and 1b (PDF)
Molecular formula strings (CSV)
References
- 1.Calonge MT, Gasparini P, Chillaron J, Chillon M, Gallucci M, Rousaud F, Zelante L, Testar X, Dallapiccola B, Disilverio F, Barcelo P, Estivill X, Zorzano A, Nunes V, Palacin M. Cystinuria caused by mutations in rbat, a gene involved in the transport of cystine. Nat Genet. 1994;6:420–425. doi: 10.1038/ng0494-420. [DOI] [PubMed] [Google Scholar]
- 2.Feliubadalo L, Font M, Purroy J, Rousaud F, Estivill X, Nunes V, Golomb E, Centola M, Aksentijevich I, Kreiss Y, Goldman B, Pras M, Kastner DL, Pras E, Gasparini P, Bisceglia L, Beccia E, Gallucci M, de Sanctis L, Ponzone A, Rizzoni GF, Zelante L, Bassi MT, George AL, Jr, Manzoni M, De Grandi A, Riboni M, Endsley JK, Ballabio A, Borsani G, Reig N, Fernandez E, Estevez R, Pineda M, Torrents D, Camps M, Lloberas J, Zorzano A, Palacin M. Non-type I cystinuria caused by mutations in SLC7A9 encoding a subunit (b(o +)at) of rBAT. Nat Genet. 1999;23:52–57. doi: 10.1038/12652. [DOI] [PubMed] [Google Scholar]
- 3.Chillaron J, Font-Llitjos M, Fort J, Zorzano A, Goldfarb DS, Nunes V, Palacin M. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol. 2010;6:424–34. doi: 10.1038/nrneph.2010.69. [DOI] [PubMed] [Google Scholar]
- 4.Moe OW. Kidney stones: Pathophysiology and medical management. Lancet. 2006;367:333–344. doi: 10.1016/S0140-6736(06)68071-9. [DOI] [PubMed] [Google Scholar]
- 5.Claes D, Jackson E. Cystinuria: Mechanisms and management. Pediatr Nephrol. 2012;27:2031–2038. doi: 10.1007/s00467-011-2092-6. [DOI] [PubMed] [Google Scholar]
- 6.Mattoo A, Goldfarb DS. Cystinuria. Semin Nephrol. 2008;28:181–191. doi: 10.1016/j.semnephrol.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 7.Andreassen KH, Pedersen KV, Osther SS, Jung HU, Lildal SK, Osther PJS. How should patients with cystine stone disease be evaluated and treated in the twenty-first century? Urolithiasis. 2016;44:65–76. doi: 10.1007/s00240-015-0841-x. [DOI] [PubMed] [Google Scholar]
- 8.Becker G. Cystine stones. Nephrology. 2007;12:S4–S10. doi: 10.1111/j.1440-1797.2006.00722.x. [DOI] [PubMed] [Google Scholar]
- 9.Rimer JD, An Z, Zhu Z, Lee MH, Goldfarb DS, Wesson JA, Ward MD. Crystal growth inhibitors for the prevention of l-cystine kidney stones through molecular design. Science. 2010;330:337–341. doi: 10.1126/science.1191968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mandal T, Shtukenberg AG, Yu AC, Zhong X, Ward MD. Effect of urinary macromolecules on l-cystine crystal growth and crystal surface adhesion. Cryst Growth Des. 2016;16:423–431. [Google Scholar]
- 11.Shtukenberg AG, Poloni LN, Zhu Z, An Z, Bhandari M, Song P, Rohl AL, Kahr B, Ward MD. Dislocation-actuated growth and inhibition of hexagonal l-cystine crystallization at the molecular level. Cryst Growth Des. 2015;15:921–934. [Google Scholar]
- 12.Shtukenberg AG, Zhu Z, An Z, Bhandari M, Song P, Kahr B, Ward MD. Illusory spirals and loops in crystal growth. Proc Natl Acad Sci U S A. 2013;110:17195–17198. doi: 10.1073/pnas.1311637110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahota A, Parihar JS, Capaccione KM, Yang M, Noll K, Gordon D, Reimer D, Yang I, Buckley BT, Polunas M, Reuhl KR, Lewis MR, Ward MD, Goldfarb DS, Tischfield JA. Novel cystine ester mimics for the treatment of cystinuria-induced urolithiasis in a knockout mouse model. Urology. 2014;84:1249.e9–1249.e15. doi: 10.1016/j.urology.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu L, Sahota A. US20140187546. Cystine diamide analogs for the prevention of cystine stone formation in cystinuria. 2014
- 15.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Prodrugs: Design and clinical applications. Nat Rev Drug Discovery. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 16.Hu L. Prodrug approaches to drug delivery. In: Wang B, Hu L, Siahaan TJ, editors. Drug Delivery: Principles and Applications. 2. Wiley & Sons; New York: 2016. pp. 227–271. [Google Scholar]
- 17.Wu X, Hu L. Efficient amidation from carboxylic acids and azides via selenocarboxylates: Application to the coupling of amino acids and peptides with azides. J Org Chem. 2007;72:765–774. doi: 10.1021/jo061703n. [DOI] [PubMed] [Google Scholar]
- 18.Birwé H, Hesse A. High-performance liquid chromatographic determination of urinary cysteine and cystine. Clin Chim Acta. 1991;199:33–42. doi: 10.1016/0009-8981(91)90006-x. [DOI] [PubMed] [Google Scholar]
- 19.Carta R, Tola G. Solubilities of l-cystine, l-tyrosine, l-leucine, and glycine in aqueous solutions at various phs and nacl concentrations. J Chem Eng Data. 1996;41:414–417. [Google Scholar]
- 20.De Yoreo JJ, Vekilov PG. Principles of crystal nucleation and growth. In: Dove PM, De Yoreo JJ, Weiner S, editors. Biomineralization. Mineralogical Society of America Geochemical Society; Washington, DC: 2003. pp. 57–93. [Google Scholar]
- 21.Cabrera N, Vermilyea DA. The growth of crystals from solution. In: Doremus RH, Roberts BW, Turnbull D, editors. Growth and Perfection of Crystals (Proceedings of the International Conference on Crystal Growth); Cooperstown, NY. August 27–29, 1958; New York: Wiley; 1958. pp. 393–410. [Google Scholar]
- 22.Cerný V. A thermodynamical approach to the travelling salesman problem: An efficient simulation algorithm. J Optim Theor Appl. 1985;45:41–51. [Google Scholar]
- 23.Kirkpatrick S, Gelatt CD, Vecchi MP. Optimization by simulated annealing. Science. 1983;220:671–680. doi: 10.1126/science.220.4598.671. [DOI] [PubMed] [Google Scholar]
- 24.Sun H. Compass: An ab initio forcefield optimized for condensed-phase applications - overview with details on alkane and benzene compounds. J Phys Chem B. 1998;102:7338–7364. [Google Scholar]
- 25.Donnay JDH, Harker D. A new law of crystal morphology extending the law of bravais. Am Mineral. 1937;22:446–467. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.