

Abstract
Background
We conducted a phase II study of oral capecitabine rapidly disintegrating tablets given concurrently with radiation therapy (RT) to assess progression-free survival (PFS) in children with newly diagnosed diffuse intrinsic pontine gliomas (DIPG).
Patients and Methods
Children 3 to 17 years with newly diagnosed DIPG were eligible. Capecitabine, 650 mg/m2/dose BID (MTD in children with concurrent radiation) was administered for 9 weeks starting the first day of RT. Following a 2 week break, 3 courses of capecitabine, 1250 mg/m2/dose BID for 14 days followed by a 7 day rest, were administered. As prospectively designed, 10 evaluable patients treated at the MTD on the phase I trial were included in the phase II analyses. The design was based on comparison of the PFS distribution to a contemporary historical control (n=140) with 90% power to detect a 15% absolute improvement in the 1-year PFS with a Type-1 error rate, α=0.10.
Results
Forty-four patients were evaluable for the phase II objectives. Capecitabine and RT was well tolerated with low grade palmar plantar erythrodyesthesia, increased alanine aminotransferase, cytopenias, and vomiting the most commonly reported toxicities. Findings were significant for earlier progression with 1-year PFS of 7.21% (SE=3.47%) in the capecitabine treated cohort versus 15.59% (SE=3.05%) in the historical control (p=0.007), but there was no difference for overall survival distributions (p=0.30). Tumor enhancement at diagnosis was associated with shorter PFS and OS. Capecitabine was rapidly absorbed and converted to its metabolites.
Conclusion
Capecitabine did not improve the outcome for children with newly diagnosed DIPG.
Keywords: Malignant glioma, clinical trial, capecitabine, child, radiation, diffuse intrinsic pontine glioma (DIPG)
INTRODUCTION
While the overall survival (OS) for children with brain tumors has improved substantially over the last 30 years, the outcome for children with diffuse intrinsic pontine gliomas (DIPG) remains dismal. The diagnosis of DIPG is based on characteristic magnetic resonance imaging (MRI) findings and a clinical history of cranial nerve deficits and long-tract signs.1 Focal radiation therapy (RT) often provides symptomatic improvement and prolongs progression–free (PFS) and OS however, the 2-year OS remains less than 10%.2,3 Because DIPG remains a radiographic diagnosis, biologic data to guide therapeutic choices are limited and have largely been extrapolated from data in adults and children with non-brainstem high-grade gliomas. Unfortunately, numerous clinical trials that have coupled chemotherapy or biologic therapy with RT have failed to significantly impact survival in children with DIPG. 2,4–12
Capecitabine is an oral fluoropyrimidine carbamate prodrug which exploits high intratumoral concentrations of thymidine phosphorylase (TP) to preferentially generate high concentrations of 5-Fluororuacil (5-FU) in tumor tissue. Capecitabine is rapidly absorbed after oral administration, then metabolized through a three step enzymatic process with the final stage of this conversion mediated by TP.13 Many tumor cells, including gliomas, have been shown to express high levels of TP. Radiation also induces TP, with studies in U87 glioblastoma xenografts demonstrating a 70-fold increase in TP expression following a single dose of radiation.14 Thus the combination of radiation and capecitabine is hypothesized to result in higher concentrations of 5-FU in tumor cells with capecitabine alone. Furthermore, 5FU and capecitabine are radiation sensitizers, and may enhance the therapeutic effect of RT.15,16
Capecitabine plus concomitant radiotherapy has been well tolerated in both adults and children with newly-diagnosed gliomas.17 A prior Pediatric Brain Tumor Consortium (PBTC) trial demonstrated that the recommended phase II capecitabine dose during radiotherapy for children with newly diagnosed high-grade or brainstem gliomas was 650 mg/m2/dose administered twice daily.[18] Single agent capecitabine, 1250 mg/m2/dose, administered twice daily for 14 consecutive days of a 21-day course, was administered post radiotherapy for 3 courses. Of note, a new rapidly disintegrating tablet (RDT) formulation was created for the phase I and II trials to facilitate enrollment of children unable to swallow pills. Preliminary pharmacokinetic data from the phase I trial demonstrated that the disposition of the RDT in children was similar to that of adults receiving the standard capecitabine formulation.18
The primary objective of this study was to estimate the PFS distribution for children with newly diagnosed DIPG treated with the combination of capecitabine and RT and compare to a contemporary historical control. Secondary objectives were to: (i) estimate the OS; (ii) further characterize the safety profile of capecitabine plus concomitant radiotherapy; (iii) further characterize the pharmacokinetics of capecitabine RDTs; and (iv) describe radiographic changes including diffusion tensor imaging (DTI) variables in response to therapy. Since DIPG is a rare disease, this trial and the preceding phase I trial were prospectively designed to have the same inclusion/exclusion criteria so that evaluable patients treated at the MTD in the phase I trial could also be included in the phase II analyses.
METHODS
Eligibility
Children 3 to 17 years of age with newly diagnosed DIPG were eligible. Histologic diagnosis was not required. No prior therapy other than surgery or corticosteroid therapy was allowed. Patients were also required to have: a Karnofsky (patients > 16 years) or Lansky (patients ≤ 16 years) performance status of ≥ 50; adequate hematologic function (absolute neutrophil count ≥ 1,000/mm3, platelets ≥ 100,000/mm3, hemoglobin ≥ 8 g/dl); age appropriate renal function; and adequate hepatic function (bilirubin ≤ 1.5× and SGPT ≤ 5× the institutional upper limit of normal for age). Patients were excluded if they were receiving other anti-cancer or experimental therapy; had uncontrolled infection or significant systemic disease; had a known hypersensitivity to capecitabine components; were receiving warfarin, sorivudine or related analogues, or were pregnant or lactating. If applicable, patients were required to agree to use medically acceptable forms of birth control. The institutional review boards of each PBTC institution approved the protocol prior to patient enrollment and continuing approval was maintained throughout the study. Patients or their legal guardians signed written informed consent and assent was obtained as appropriate.at the time of enrollment.
Treatment Plan
This study was designed with an identical treatment plan as in the preceding phase I study. In both trials, the dose and schedule of capecitabine were different during the radiation and post-radiation phases, with the goal to maximize capecitabine exposure during and for a period following radiation based on preclinical data that demonstrated a selective increase in intratumoral TP following radiation. Additionally, capecitabine is known to be a radiosensitizing agent.15,16
Drug Administration and Radiation Therapy
Capecitabine was provided as 125, 175, 250 and 350 mg strawberry flavored film coated RDTs by Hoffmann-La Roche (Nutley, NJ). Tablets could be swallowed whole or after complete dispersion in room temperature water. Administered doses did not deviate from the dose based on body surface area by more than 10%.
Capecitabine therapy began within 24 hours of the initiation of RT and was administered at 650 mg/m2/dose twice daily without interruption for 9 weeks, followed by a 2-week break. During the post-radiation phase, the dose was 1250 mg/m2/dose twice daily for 14 consecutive days followed by a 7 day break, for 3 courses. The entire duration of protocol treatment was 20 weeks. If a patient developed protocol defined unacceptable toxicities, the dose was reduced to 500 mg/m2/dose during the radiotherapy phase or 900 mg/m2/dose during the post-radiation phase. Patients were taken off therapy due to: completion of therapy, progressive disease, development of unacceptable toxicity or illness that required permanent drug cessation, refusal by the patient, parent or legal guardian to continue therapy, pregnancy, or non-compliance to protocol guidelines.
Conventional or conformal RT was administered to all patients once daily five days a week in 180 cGy/day fractions to a total dose of 5580 cGy. Treatment was directed to the maximal tumor dimensions demonstrated on MRI (typically on T2 weighted images) plus a 1.5 cm anatomic margin (clinical target volume- CTV). The planning target volume was defined as an additional 3 to 5 mm margin to the CTV depending on the type of head immobilization and institutional preference.
Monitoring
Baseline clinical assessment for all patients included a history, physical exam and laboratory studies, including a pregnancy test for females of childbearing potential. Testing for dihydropyrimidine dehydrogenase (DPD) deficiency was not required, but recommended if a patient developed significant toxicity during therapy. During therapy, clinical assessments, including history, physical exam and laboratory studies, were completed approximately every 3 weeks with complete blood counts performed weekly. Neuro-imaging (MRI including T1, T2, FLAIR and gradient sequences 19) was performed at diagnosis, week 11, end of therapy (week 20) and then every 3 months until disease progression. Diffusion imaging was performed pre-treatment and at week 11. Patients were followed after the end of therapy for resolution of any treatment-related adverse events and then for progression and survival for up to 3 years.
Trial Design
The Phase II design required 44 evaluable patients to detect with 90% power and 10% type-1 error rate an absolute improvement of 15% in the 1-year PFS distribution as compared to the PBTC DIPG historical control cohort (1-year PFS of 15.9% in the historical cohort versus 30.9% hypothesized for this study). The primary efficacy endpoint was PFS, defined as the time from initiation of treatment to the earliest date of failure (disease progression, or death from any cause). Progression was determined at the local institution and defined as worsening neurologic status not explained by causes unrelated to tumor progression or a greater than 25% increase in the bi-dimensional tumor measurements or the appearance of a new lesion. Overall survival was a secondary endpoint, defined as the time from initiation of treatment to death from any cause.
Patients who did not experience an event for PFS or OS were censored at their off-study dates. Analyses were based on an intent-to-treat hypothesis. The Kaplan Meier estimate of the PFS and OS distributions were compared using Log-rank test to the PBTC historical control which consists of 140 patients with newly diagnosed DIPG treated on three previously published PBTC phase I and II treatment studies (PBTC006, PBTC007, PBTC014) with similar eligibility criteria, disease assessments and definition of progression. An interim analysis for futility was conducted after the 21st failure on study and the threshold for stopping the trial was not reached. Toxicity data is reported descriptively. Cox proportional hazards models were used to investigate the association of predefined neuroimaging variables of interest with PFS and OS 19.
Pharmacokinetics
Patients consenting to participate in the pharmacokinetic (PK) substudy were assigned to 1 of 3 sampling schedules, to maximize the information obtained from limited sampling. Initial patients were assigned to schedules A and B, which were utilized in the phase I study.18 Subsequently the sampling was further reduced to encourage participation and in schedule C samples were obtained on days 1 and 14 at predose, and 0.5, 1, 3 and 6 hours after dosing as well as 1–4 hours after the morning dose on days 7 and 21. Determination of plasma concentrations of capecitabine and its metabolites (5′-deoxy-5-fluorocytidine [DFCR], 5′-deoxy-5-fluorouridine [DFUR], 5-FU, and α-fluoro-β-alanine [FBAL]) was done using a validated liquid chromatography (LC) method with mass spectrometry detection.18 Pharmacokinetic parameters (Cmax, tmax, AUC6h and t1/2) of capecitabine and its metabolites were estimated individually on days 1 and 14 of the RT phase using non-compartmental methods using WinNonlin version 6.2 software.
In addition to the non-compartmental approach, a population analysis was performed to describe the PK time course profile as well as to obtain PK parameter estimates in children from the pooled PK data. A multi-response population PK model, previously developed in adults for the main capecitabine metabolites (5’-DFUR, 5-FU, and FBAL) was used to compare the adult exposure with the exposures from the pediatric studies.20 This model was used with a Bayesian feedback analysis using NONMEM. Pediatric PK parameters and exposure estimates were subsequently compared with the expected exposure in an adult population at the same dose level.
STUDY RESULTS
Thirty-five children with DIPG, all eligible, were enrolled from February 2010 to May 2011. Thirty-four patients received at least one dose of capecitabine and thus were evaluable for the survival and toxicity endpoints. Ten patients treated at the MTD of capecitabine on the phase I study also met the eligibility criteria for this study, bringing the total sample size to 44 evaluable patients. Patient characteristics are presented in Table 1.
TABLE 1.
Patient Characteristics (n=44)
| Characteristic | Capecitabine Cohort | DIPG Historical cohort |
|---|---|---|
|
| ||
| Total eligible patients enrolled in Phase II | 35 | 140 |
| Evaluable | 34 | 140 |
| Inevaluable | 1 | |
| Evaluable Patients Contributing from Phase I | 10 | |
|
| ||
| Analysis Cohort |
Patient Number N (NPhase-1+NPhase-II) |
Patient Number N |
|
| ||
| Sex | ||
| Male | 22 (1+21) | 57 |
| Female | 22 (9+13) | 83 |
|
| ||
| Age at diagnosis (years) | ||
| Median (Range) | 7.2 (3.4–16.2) | 6.6 (3.0–18.7) |
|
| ||
| Time from diagnosis to start of therapy (days) | ||
| Median (Inter quartile Range) | 11.5 (8–15) | 13 (9–19) |
|
| ||
| Baseline Lansky/Karnofsky | ||
| Mean (Range) | 90 (50–100) | 80 (50–100) |
|
| ||
| Race | ||
| Black | 6 (2+4) | 16 |
| More than one race | 1 (0+1) | 0 |
| Native American | 1 (0+1) | 1 |
| White, Non-Hispanic | 32 (5+27) | 106 |
| Pacific Islander | 1 (1+0) | 1 |
| Asian | 1 (1+0) | 8 |
| Unknown | 2 (1+1) | 8 |
|
| ||
| Baseline Tumor Volume | ||
| Median (Range) | 40.8 (13.9–86.2 | 31.5 (6.6–80.2) |
Twenty-five patients completed the entire planned protocol therapy. In five patients treatment was stopped prior to completion due to adverse events and three withdrew from treatment; two early in course 1 due to difficulty taking study medication, and one after the first post-RT course in the context of wound dehiscence and neutropenia. Eleven patients developed clinical (n=3) or radiographic (n=8, 4 local and 4 distant) progression during therapy. These results are summarized in Supplementary Table S1.
Adverse Events
The study therapy was well tolerated in the majority of patients. Adverse events resulted in cessation of therapy in 1 patient during the radiation phase (grade 4 neutropenia), and 4 patients during the post radiation phase [grade 2 CNS necrosis (n=2), grade 4 neutropenia that did not resolve within 7 days (n=1), and persistent toe infection (n=1)]. Five additional patients required protocol directed dose reductions during therapy, two during the radiation phase for grade 4 neutropenia, and the remainder during the post radiation phase for grade 2 palmar plantar erythrodyesthesia (n=2) or grade 3 elevation of alanine aminotransferase (ALT) (n=1). The most commonly reported toxicities on treatment were grade 2 or lower and expected, including palmar plantar erythrodyesthesia (hand-foot syndrome), increase in ALT, cytopenias, and vomiting. Grade 2 and higher treatment related toxicities are presented in Table 2. CNS necrosis grades 2-3 was reported as an adverse event in 4 patients after RT with capecitabine. Two patients were taken off treatment early due to this toxicity and associated symptoms.
TABLE 2.
Capecitabine Related Toxicities during treatment (n=44)
| Toxicity | RT Phase (Course 1–3) |
Maintenance (Course 4–6) |
||||
|---|---|---|---|---|---|---|
| Gr-2 | Gr-3 | Gr-4 | Gr-2 | Gr-3 | Gr-4 | |
|
Hematologic Toxicities Lymphocyte count decreased |
12 | 12 | 2 | 8 | 7 | 2 |
| White blood cell decreased | 10 | 3 | 4 | 5 | 1 | |
| Neutrophil count decreased | 7 | 3 | 2 | 3 | 2 | 2 |
| Lymphopenia | 3 | 5 | 2 | 2 | ||
| Anemia | 5 | 3 | 1 | |||
| Platelet count decreased | 2 | |||||
| Non-hematologic Toxicities | ||||||
| Palmar-plantar erythrodysesthesia syndrome | 5 | 7 | ||||
| Vomiting | 4 | 1 | ||||
| Diarrhea | 1 | 3 | 2 | |||
| Anorexia | 2 | 2 | ||||
| Somnolence | 1 | |||||
| Central nervous system necrosis | 3 | 1 | ||||
| Constipation | 2 | 1 | ||||
| Alanine aminotransferase increased | 5 | 1 | 2 | 1 | ||
| Hypocalcemia | 1 | 1 | ||||
| Blood bilirubin increased | 2 | 3 | ||||
| Mucositis oral | . | 2 | ||||
| Enterocolitis | 1 | |||||
Outcome
Figures 1 and 2 demonstrate the PFS and OS, respectively, for patients on study compared to those in the PBTC historical cohort (n=140). Findings were significant for earlier progression (p=0.007) with the study treatment, with 6 and 12 month PFS of 33.7% (SE=7.1%) and 7.2% (SE=3.5%) versus 54.7% (SE=4.2%) and 15.6% (SE=3.1%) in the historical control, respectively. However, there was no difference in the OS for patients treated with capecitabine and RT when compared to the historical control (p=0.30).
Figure 1.

Progression Free Survival: Kaplan-Meier curve of progression-free survival for patients on PBTC030 compared to aggregate data from prior PBTC DIPG studies
Figure 2.

Overall Survival: Kaplan-Meier curve of overall survival for patients on PBTC030 compared to aggregate data from prior PBTC DIPG studies.
Neuroimaging
The 44 evaluable patients had a total of 173 neuroimaging assessments. Forty-three patients had standard diffusion imaging with 16 having specific diffusion tensor imaging performed. Baseline enhancement as well as the change in enhancement after RT were significantly associated with an increased risk of progression (p-values = 0.028 and 0.035 respectively) and patients with baseline volume of enhancement above the median had a significantly shorter PFS (p=0.04) and OS (p=0.042; figure not shown) as demonstrated in Figure 3. Patients with tumor enhancement at baseline had significantly lower diffusion values (p=0.007) and a lower decrease in their diffusion ratio measurements after RT (p=0.005) compared to patients with non-enhancing tumor at baseline (data not shown). The specific regional diffusion tensor metrics assessed were not associated with PFS due to small sample size; however, patients with higher 2D mean apparent diffusion coefficient (ADC) had a relatively shorter PFS. Although the presence of necrosis or cystic enhancement after radiation based on imaging assessment at day 70 was similar in patients on this study compared to the historical control (27% vs 21%, p=0.42), there was a suggestion that the median volume of necrosis was increased (2.6 vs 0.8, p=0.07).
Figure 3.
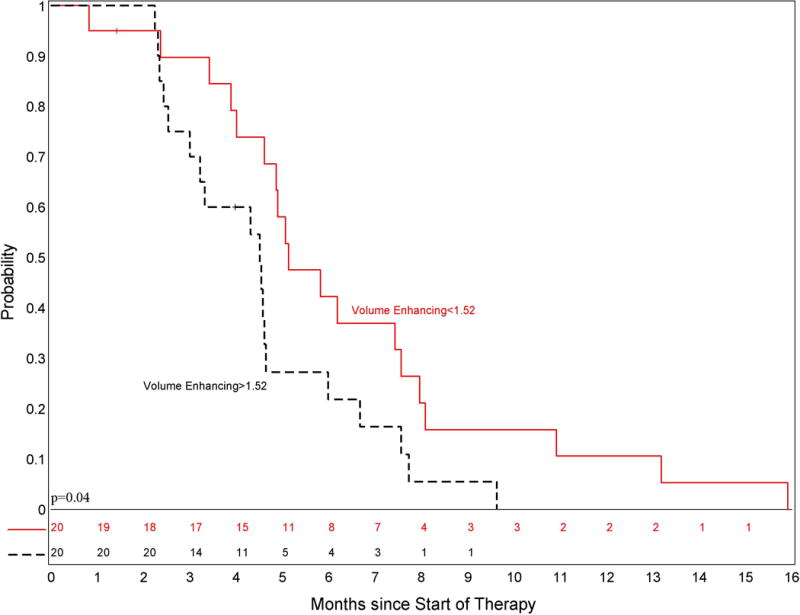
PFS by Volume Enhancing categorization; Kaplan-Meier curve of progression-free survival for patients stratified by enhancement. Patients were stratified based on volume of enhancement above the median
Pharmacokinetics
Plasma concentrations for capecitabine and the metabolites (5′-DFCR, 5′-DFUR, 5-FU, and FBAL) were available from 15 patients, who participated in the PK portion of this study. Following oral administration of capecitabine, data indicated that capecitabine was rapidly absorbed and converted to its metabolites, 5′-DFCR, 5′-DFUR, FBAL and the pharmacologically active 5-FU. Plasma concentrations of capecitabine and 5′-DFCR, 5′-DFUR, and 5-FU achieved median maximum levels between 0.5 and 1 hour after dosing on days 1 and 14. PK parameters are summarized in Table 3. A large inter-subject variability was observed for the PK parameters and due to the limited number of samples collected, individual observed pharmacokinetic parameters could not be calculated for all patients and should be interpreted with caution.
TABLE 3.
Capecitabine and metabolites plasma PK parameters [Mean (%CV)] following administration of capecitabine 650mg/m2
| Day | n | Cmax (ng/ml) |
tmax
a (hours) |
n | AUC 0–8h (ng/ml) |
n | t ½ (hours) |
|
|---|---|---|---|---|---|---|---|---|
| Capecitabine | 1 | 15 | 5290 (95) | 0.5 (0.2–1.3) | 10 | 6180 (67) | 10 | 1.8 (97) |
| 14 | 14 | 3030 (64) | 0.7 (0.2–3.5) | 10 | 6340 (72) | 6 | 1.1 (29) | |
| 5’-DFCR | 1 | 15 | 2830 (62) | 0.5 (0.3–1.3) | 10 | 5260 (74) | 10 | 1.6 (67) |
| 14 | 14 | 2450 (57) | 1.0 (0.3–3.5) | 11 | 5120 (54) | 4 | 1.1 (33) | |
| 5’-DFUR | 1 | 15 | 3750 (53) | 0.5 (.3–1.3) | 8 | 5920 (54) | 8 | 1.8 (59) |
| 14 | 14 | 3370 (80) | 1.0 (0.3–3.5) | 8 | 5450 (44) | 2 | 1.3 (48) | |
| 5-FU | 1 | 15 | 122 (114) | 0.9 (0.3–3.0) | 6 | 121 (34) | 5 | 1.7 (59) |
| 14 | 14 | 138 (122) | 0.9 (0.2–3.5) | 9 | 178 (40) | 4 | 1.3 (48) | |
| FBAL | 1 | 15 | 1580 (34) | 2.4 (0.8–6.0) | 11 | 5600 (36) | 3 | 2.3 (42) |
| 14 | 14 | 1900 (37) | 3.0 (0.8–6.0)) | 12 | 6840 (32) | 3 | 1.9 (5.7) |
Median (min-max)
A total of 10 patients from the phase I and fifteen patients from the phase II trial were included in the Bayesian feedback analysis, for a total of 561 observations. Comparison of Cmax and AUC of parent drug and its metabolites revealed that the pediatric PK parameters appear to be comparable to the adult data, with a slightly lower than expected exposure compared to an adult population treated at 650mg/m2.20,21
DISCUSSION
Our study sought to enhance the efficacy of RT in children with DIPG through combination therapy with capecitabine via two hypothesized mechanisms: radiation sensitization and chemosensitization. The chemosensitization was hypothesized to occur as a result of increased intratumoral levels of TP from radiation leading to increased conversion of capecitabine to 5FU in the tumor versus the normal adjacent brain. The combination therapy was well tolerated in most children, with a toxicity profile consistent with other capecitabine trials. CNS necrosis was reported as an adverse event in 4/34 patients, and cystic/necrotic areas were present on imaging in 27% of patients post radiation. This is within the expected frequency for children with newly diagnosed DIPG receiving RT based on prior publications.22 However, the volume of necrosis was noted to be higher in this study compared to the historical control and two patients discontinued therapy due to symptomatic necrosis. It is unclear if this finding contributed to the shorter progression free survival seen on this study.
Unfortunately, similar to previous trials in children with DIPG, the combination therapy failed to improve the outcome compared with the aggregate data from prior PBTC studies. It is not known whether the shorter PFS in this study in the context of unchanged OS is related to larger baseline tumor volume, suggests increased pseudoprogression in response to therapy, clinical symptoms related to the increased volume of necrosis or cystic enhancement, or represents differences in treatment following progression.
As noted in prior PBTC studies, patients with tumor enhancement at baseline and over time had shorter survival [19]. In addition, as noted in our previous work, in the subgroup of patients with tumoral enhancement, there were significantly lower diffusion values consistent with increased cellularity within the tumor, lower decreases in diffusion values after RT, and shorter PFS and OS, compared to tumors without enhancement.
Analysis of additional pharmacokinetic data obtained in this phase II trial supported the findings from the phase I study, that pharmacokinetic parameters for capecitabine and its metabolites were similar or slightly lower than in adults at equivalent doses.
Because of the dismal prognosis for children with DIPG, offering treatment on a clinical trial is considered to be standard of care. This study was prospectively designed as a single arm phase II study with an identical treatment plan to the phase I study, such that children treated at the MTD could be included in the final survival analysis to maximize the use of available data from children with this rare and devastating disease. Although there are limitations to single arm phase II studies, based on a robust historical control of children with newly diagnosed DIPG treated on prior PBTC trials and the lack of change in survival over time, this design remains the standard for evaluating new agents in children with newly diagnosed DIPG. In contrast to recent studies in adult GBM that suggest a shift in survival over time, leading to a recommendation for randomized phase II studies in that patient population, the outcomes in children with DIPG, unfortunately, have not changed and the survival curves for the multiple studies in the PBTC historical control are virtually superimposable. Any single arm phase II study that demonstrated improved survival compared to the historical control would readily become the standard of care.
In conclusion, similar to prior studies, the combination of capecitabine and radiation therapy failed to improve survival for children with DIPG. However, our knowledge about potential therapeutic targets for DIPG, and other CNS tumors, is rapidly evolving. There may be trials when a tumor biopsy is an integral component of trial eligibility to assess the presence or absence of the putative target. Such an approach may lead to a more accurate understanding of the trial results, however, sampling differences with a small biopsy due to tumor heterogeneity as well as delivery of the targeted therapy to the tumor at adequate exposures will continue to be major challenges in the development of effective therapies for this challenging tumor.
Supplementary Material
Supplementary Table S1: Summary of the off treatment reason for all patients in the phase II analysis.
Acknowledgments
The authors and the PBTC acknowledge the clinical research assistant support of Ms. Emily Carps and Mr. Joyson Pekkattil
Funding: This study was sponsored by F. Hoffmann-La Roche Ltd. The study was also supported in part by: NIH grant U01CA081457, NIH, Grant K12 CA090433-06
Abbreviations
- RT
Radiation
- PFS
progression-free survival
- DIPG
diffuse intrinsic pontine glioma
- MTD
maximum tolerated dose
- SE
standard error
- OS
overall survival
- TP
thymidine phosphorylase
- 5-FU
5-Fluororuacil
- PBTC
Pediatric Brain Tumor Consortium
- RDT
rapidly disintegrating tablets
- DTI
diffusion tensor imaging
- MRI
magnetic resonance imaging
- cGy
centigray
- CTV
clinical target volume
- PK
pharmacokinetic
- DFCR
5′-deoxy-5-fluorocytidine
- DFUR
5′-deoxy-5-fluorouridine
- FBAL
α-fluoro-β-alanine
- LC
liquid chromatography
- Cmax
maximum concentration
- AUC
area under the concentration time curve
- tmax
time to maximum concentration
- t1/2
half-life
- ALT
alanine aminotransferase
- CNS
central nervous system
- 2D
two dimensional
- ADC
apparent diffusion coefficient
Footnotes
Conflicts of Interest: No authors report any conflicts of interest related to this manuscript. Several authors are employees of Roche as noted above.
References
- 1.Albright AL, Packer RJ, Zimmerman R, Rorke LB, Boyett J, Hammond GD. Magnetic resonance scans should replace biopsies for the diagnosis of diffuse brain stem gliomas: a report from the Children’s Cancer Group. Neurosurgery. 1993;33(6):1026–1029. doi: 10.1227/00006123-199312000-00010. discussion 1029-1030. [DOI] [PubMed] [Google Scholar]
- 2.Cohen KJ, Heideman RL, Zhou T, et al. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: a report from the Children’s Oncology Group. Neuro Oncol. 13(4):410–416. doi: 10.1093/neuonc/noq205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gajjar A, Packer RJ, Foreman NK, Cohen K, Haas-Kogan D, Merchant TE. Children’s Oncology Group’s 2013 blueprint for research: central nervous system tumors. Pediatr Blood Cancer. 60(6):1022–1026. doi: 10.1002/pbc.24427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey S, Howman A, Wheatley K, et al. Diffuse intrinsic pontine glioma treated with prolonged temozolomide and radiotherapy – Results of a United Kingdom phase II trial (CNS 2007 04) Eur J Cancer. doi: 10.1016/j.ejca.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradley KA, Zhou T, McNall-Knapp RY, et al. Motexafin-gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: a children’s oncology group phase 2 study. Int J Radiat Oncol Biol Phys. 85(1):e55–60. doi: 10.1016/j.ijrobp.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Korones DN, Fisher PG, Kretschmar C, et al. Treatment of children with diffuse intrinsic brain stem glioma with radiotherapy, vincristine and oral VP-16: a Children’s Oncology Group phase II study. Pediatr Blood Cancer. 2008;50(2):227–230. doi: 10.1002/pbc.21154. [DOI] [PubMed] [Google Scholar]
- 7.Bernier-Chastagner V, Grill J, Doz F, et al. Topotecan as a radiosensitizer in the treatment of children with malignant diffuse brainstem gliomas: results of a French Society of Paediatric Oncology Phase II Study. Cancer. 2005;104(12):2792–2797. doi: 10.1002/cncr.21534. [DOI] [PubMed] [Google Scholar]
- 8.Allen J, Siffert J, Donahue B, et al. A phase I/II study of carboplatin combined with hyperfractionated radiotherapy for brainstem gliomas. Cancer. 1999;86(6):1064–1069. doi: 10.1002/(sici)1097-0142(19990915)86:6<1064::aid-cncr24>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 9.Walter AW, Gajjar A, Ochs JS, et al. Carboplatin and etoposide with hyperfractionated radiotherapy in children with newly diagnosed diffuse pontine gliomas: a phase I/II study. Med Pediatr Oncol. 1998;30(1):28–33. doi: 10.1002/(sici)1096-911x(199801)30:1<28::aid-mpo9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Broniscer A, Iacono L, Chintagumpala M, et al. Role of temozolomide after radiotherapy for newly diagnosed diffuse brainstem glioma in children: results of a multiinstitutional study (SJHG-98) Cancer. 2005;103(1):133–139. doi: 10.1002/cncr.20741. [DOI] [PubMed] [Google Scholar]
- 11.Fouladi M, Nicholson HS, Zhou T, et al. A phase II study of the farnesyl transferase inhibitor, tipifarnib, in children with recurrent or progressive high-grade glioma, medulloblastoma/primitive neuroectodermal tumor, or brainstem glioma: a Children’s Oncology Group study. Cancer. 2007;110(11):2535–2541. doi: 10.1002/cncr.23078. [DOI] [PubMed] [Google Scholar]
- 12.Turner CD, Chi S, Marcus KJ, et al. Phase II study of thalidomide and radiation in children with newly diagnosed brain stem gliomas and glioblastoma multiforme. J Neurooncol. 2007;82(1):95–101. doi: 10.1007/s11060-006-9251-9. [DOI] [PubMed] [Google Scholar]
- 13.Reigner B, Blesch K, Weidekamm E. Clinical pharmacokinetics of capecitabine. Clin Pharmacokinet. 2001;40(2):85–104. doi: 10.2165/00003088-200140020-00002. [DOI] [PubMed] [Google Scholar]
- 14.Blanquicett C, Gillespie GY, Nabors LB, et al. Induction of thymidine phosphorylase in both irradiated and shielded, contralateral human U87MG glioma xenografts: implications for a dual modality treatment using capecitabine and irradiation. Mol Cancer Ther. 2002;1(12):1139–1145. [PubMed] [Google Scholar]
- 15.Vermund H, Gollin FF. Mechanisms of action of radiotherapy and chemotherapeutic adjuvants. A review. Cancer. 1968;21(1):58–76. doi: 10.1002/1097-0142(196801)21:1<58::aid-cncr2820210110>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Zhu AX, Willett CG. Chemotherapeutic and biologic agents as radiosensitizers in rectal cancer. Semin Radiat Oncol. 2003;13(4):454–468. doi: 10.1016/S1053-4296(03)00048-1. [DOI] [PubMed] [Google Scholar]
- 17.Grunda JM, Fiveash J, Palmer CA, et al. Rationally designed pharmacogenomic treatment using concurrent capecitabine and radiotherapy for glioblastoma; gene expression profiles associated with outcome. Clin Cancer Res. 16(10):2890–2898. doi: 10.1158/1078-0432.CCR-09-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilburn LB, Kocak M, Schaedeli Stark F, et al. Phase I trial of capecitabine rapidly disintegrating tablets and concomitant radiation therapy in children with newly diagnosed brainstem gliomas and high-grade gliomas. Neuro Oncol. 15(6):759–766. doi: 10.1093/neuonc/nos315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poussaint TY, Kocak M, Vajapeyam S, et al. MRI as a central component of clinical trials analysis in brainstem glioma: a report from the Pediatric Brain Tumor Consortium (PBTC) Neuro Oncol. 2011;13(4):417–427. doi: 10.1093/neuonc/noq200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gieschke R, Burger HU, Reigner B, Blesch KS, Steimer JL. Population pharmacokinetics and concentration-effect relationships of capecitabine metabolites in colorectal cancer patients. Br J Clin Pharmacol. 2003;55(3):252–263. doi: 10.1046/j.1365-2125.2003.01765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gieschke R, Reigner B, Blesch KS, Steimer JL. Population pharmacokinetic analysis of the major metabolites of capecitabine. J Pharmacokinet Pharmacodyn. 2002;29(1):25–47. doi: 10.1023/a:1015716617967. [DOI] [PubMed] [Google Scholar]
- 22.Pier DB, Levine D, Kataoka ML, et al. Magnetic resonance volumetric assessments of brains in fetuses with ventriculomegaly correlated to outcomes. J Ultrasound Med. 2011;30(5):595–603. doi: 10.7863/jum.2011.30.5.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124(3):439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21(7):827. doi: 10.1038/nm0715-827a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: Summary of the off treatment reason for all patients in the phase II analysis.