

SUMMARY
Objective
Homeostatic synaptic plasticity (HSP) serves as a gain control mechanism at CNS synapses, including those between the dentate gyrus (DG) and CA3. Improper circuit control of DG-CA3 synapses is hypothesized to underlie epileptogenesis. Here, we sought to (1) identify compounds that preferentially modulate DG-CA3 synapses in primary neuronal culture and (2) determine if these compounds would delay or prevent epileptogenesis in vivo.
Methods
We previously developed and validated an in vitro assay to visualize the behavior of DG-CA3 synapses and predict functional changes. We used this ‘synapse-on-chip’ assay (quantification of synapse size, number, and type using immunocytochemical markers) to dissect the mechanisms of HSP at DG-CA3 synapses. Using DREADD constructs and pharmacological agents we determined the signaling cascades necessary for gain control at DG-CA3 synapses. Finally, we tested the implicated cascades (using kappa OR agonists and antagonists) in two models of epileptogenesis: electrical amygdala kindling in the mouse and chemical (pentylenetetrazole) kindling in the rat.
Results
In vitro, synapses between DG mossy fibers (MF) and CA3 neurons are the primary homeostatic responders during sustained periods of activity change. Kappa opioid receptor (OR) signaling is both necessary and sufficient for the homeostatic elaboration of DG-CA3 synapses, induced by presynaptic DG activity levels. Blocking kappa OR signaling in vivo attenuates the development of seizures in both mouse and rat models of epilepsy.
Keywords: Mossy fiber, dentate gyrus, CA3, kappa opioid receptors, homeostatic synaptic plasticity, temporal lobe epilepsy
INTRODUCTION
The most common form of adult acquired epilepsy – temporal lobe epilepsy (TLE) – is characterized by seizures originating in the temporal lobe, including hippocampus, amygdala and temporal cortex.1 Improper “gating” of input to the hippocampus, in particular the recurrent networks of the CA3 region, has been extensively implicated in temporal lobe seizures,2,3 particularly in mTLE.4–6 In the healthy hippocampus, input to the CA3 region arrives predominantly from the dentate gyrus (DG) via mossy fibers, the enormous specialized axon terminals of DG granule cells.7 DG neurons heavily gate input to CA3 both directly and via hilar interneurons. Several hypotheses have been presented to explain the loss of dentate-CA3 gating in TLE. Major theories include the selective loss of hilar interneurons and the pathological reorganization of mossy fiber axons.8–10 A more recent hypothesis is that a failure of circuit-level closed-loop control underlies hippocampal epileptogenesis.
Neurons in several areas of the nervous system recalibrate their connections in response to overall network activity: chronic inactivity often increases excitatory synaptic strength, while chronic hyperactivity decreases it.11–13 These compensatory negative-feedback mechanisms, collectively termed homeostatic synaptic plasticity (HSP), are thought to maintain appropriate levels of neuronal output, providing stability within dynamic neural networks.14,15 However, it has been difficult to test this idea as many factors involved in homeostatic plasticity are also important in general synaptic function.
Recently, we showed that homeostatic compensation in hippocampus occurs preferentially at synapses between dentate gyrus mossy fibers (MF) and the proximal dendrites of CA3 neurons16. Other excitatory hippocampal synapses were unchanged, both in vitro and in vivo, as assayed through structural and functional measures.16 The large, complex MF-CA3 synapse has therefore been suggested to act as a hippocampal “gain control,” adjusting the bandwidth of information entering the hippocampus.17 It has been hypothesized that improper gain control at the proposed MF-CA3 checkpoint may lead to excessive hippocampal inputs and potentially seizures.18
We have previously developed and validated an in vitro assay for MF-CA3 synapses.16 Here, we used this ‘synapse-on-chip’ approach to screen for compounds capable of selectively altering MF-CA3 synapses in vitro. We then proposed to test these compounds in vivo to determine whether improper homeostatic calibration at MF-CA3 synapses was responsible for the emergence of mTLE.
METHODS
All procedures were performed in accordance with Georgetown University’s Institutional Animal Care and Use Committee’s regulations.
Primary hippocampal cultures & in vitro pharmacology
Hippocampal neurons were prepared from E19 rat embryos as previously described19 (Supplementary Methods) and analyzed at >21 days in vitro (DIV). Neurons were treated for 24–48 hours as indicated with drugs from the following sources. Concentrations in bold were selected after dose-response tests for efficacy and toxicity. Tetrodotoxin [TTX; 1 μM] or picrotoxin [PTX; 100 μM] (both from Sigma, St. Louis, MO) were used to suppress neuronal firing (TTX) and increase activity (PTX), respectively. Opioid receptor (OR) targeting drugs were obtained from Tocris (Bristol, UK). We tested the mu receptor agonist, DAMGO [100 nM, 1 μM, 10 μM], the mu receptor antagonist CTOP [50 nM, 500 nM, 5 μM], the delta receptor agonist DPDPE [100 nM, 1 μM, 10 μM], the delta receptor antagonist ICI-174,864 [20 nM, 200 nM, 2 μM], the kappa receptor agonist U-50488 [250 nM, 2.5 μM, 25 μM], and the kappa receptor antagonist norBNI [1 μM, 10 μM, 100 μM]. We also tested the broad-spectrum opioid receptor antagonist, naloxone [1 μM, 10 μM, 100 μM] (Santa Cruz Biotechnology, Santa Cruz, CA). TTX (1 mM) and naloxone (100 mM) stock solutions were prepared in distilled water. PTX was freshly prepared in 0.1 M NaOH. Stock solutions of OR drugs were prepared in saline. Drugs were diluted in Neurobasal media to the final concentration from stock solutions.
Immunocytochemistry
Neurons were stained using the following primary antibodies at the indicated dilutions: PSD-95 (NeuroMab, Davis, CA) [1:200-400], SPO (Synaptic Systems, Goettingen, Germany) [1:400], MAP2 (Abcam, Cambridge, UK) [1:200], CTIP2 (Abcam) [1:200–1000]. See Supplementary Methods for full details.
Immunohistochemistry
After chemical kindling, brains were stained for Synaptoporin (Supplementary Fig. 5D). See Supplementary Methods for full details.
Image acquisition
Fluorescent microscopy: Images were acquired using a Zeiss Axiovert 200M epifluorescent microscope and analyzed using MetaMorph software (Molecular Devices, Sunnyvale, CA). Images were taken of neurons with separable dendritic arbors. Only dendrites that could be unambiguously traced for at least 100 μm from the soma were used for analysis. For computation of immunofluorescence intensity as a function of distance, dendrites were divided into 20 μm segments, starting from the cell body.16 The strongest three dendrites on each neuron were traced and 20 μm regions centered on the dendrite were automatically generated. PSD-95 or SPO immunoreactivity was quantified as total integrated intensity per 20 μm region after thresholding. Cellular averages were obtained and subsequently pooled into group averages. Immunoreactivity is expressed per 20 μm region or in proximal (<20 μm) and distal (>80 μm) subregions. Stimulated emission depletion (STED) microscopy: See Supplementary Methods for full details.
Cell type identification
For cell type identification (Figure 1A), we used established criteria that we and others have developed16,20: DG neurons are immunopositive for CTIP2 and have small oblong cell bodies with 2–3 primary neurites, along with mossy fiber axon terminals enriched for the vesicle-associated protein synaptoporin (SPO).20,21 CA1 neurons are immunopositive for CTIP2 and have large somata with >7 primary neurites. CA3 neurons are CTIP2-negative and have proximal dendrites with excitatory synapses that closely contact SPO-positive mossy fiber terminals (Fig. S1). Details of neurite number, thickness, and other morphological/biochemical/electrophysiological properties of these cells types can be found in16.
Figure 1. Excitatory cells of the hippocampus display distinct morphologies and homeostatic compensations to inactivity.

(A) Schematic (top) and representative image (bottom) of the major excitatory hippocampal cells types in vitro. CA3 neuron indicated with open triangle; DG neurons with arrows; CA1 neuron with filled triangle. (A-D) Neuronal morphology and excitatory synapses of mature (>21 DIV) primary hippocampal neurons were visualized with the dendritic marker MAP2 (green) and excitatory scaffolding protein PSD-95 (red), respectively. DG neurons (C, arrows in A) can be easily identified based on small soma and limited number of primary neurites. Both DG (C) and CA1 (D, filled triangle in A) neurons have nuclei which stain for CTIP2 (blue). CA3 neurons have medium soma size, moderate number of strong primary neurites, and no nuclear CTIP2 stain (circles in B). (E-G) Mature (>21 DIV) primary hippocampal neurons where grown for 1 or 7 days under normal activity conditions (NT) or inactivity (TTX). Quantification of PSD-95 immunoreactivity in dendrites of the distinct excitatory cell types as a function of distance from the soma. Data are expressed as mean±SEM from neuronal averages (N = 20 neurons/condition, 3 dendrites/neuron). ***P <0.001 v. NT, repeated-measures ANOVA and post hoc Tukey test. Scale bar = 20 μm.
For DG-CA3 synapse identification (Figure 2A, Fig S1), we identified GFP-filled DG neurons based on appearance (described above, Figure 1A) and traced the GFP-filled axons to terminals on proximal (<40 um) dendrites of a GFP-negative target cell. DG terminals are larger than normal synaptic terminals and show preference for proximal dendrites, making them easy to trace unambiguously. In our experience, almost 80% of SPO-positive DG terminals target CA3 neurons.16 We imaged GFP-filled presynaptic terminals and then captured the SPO content of the presynaptic terminals and the PSD-95 content of the nearby synapses blindly (using pre-established channel settings, Figure 2A, top row). Analysis of DG-CA3 synapses was done by placing circles of equal size around GFP-filled terminals within the proximal (<40 um) portion of a CA3 dendrite and quantifying the bouton volume (GFP area), amount of presynaptic SPO and amount of PSD-95 staining within that area (Figure 2A, bottom row).
Figure 2. Presynaptic DG inactivity drives elaboration of DG-CA3 synapses.
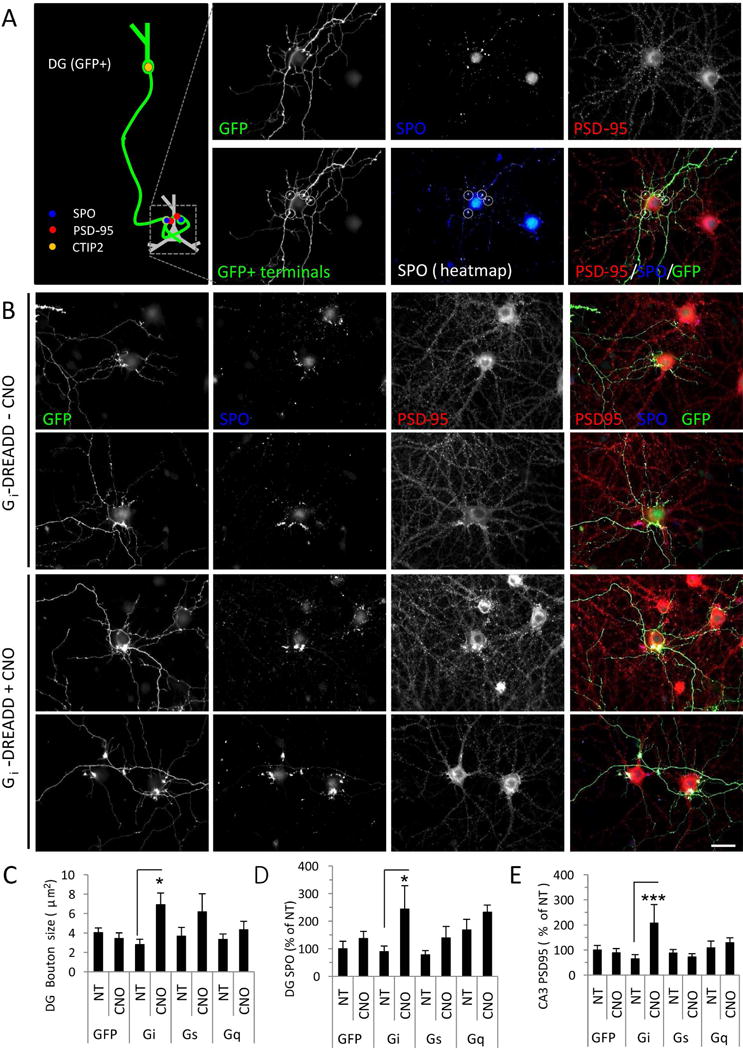
(A) Left: Schematic illustrating the in vitro DG-CA3 activity assay. The axon of a GFP-filled DG neuron is traced to the SPO-positive terminal on the proximal dendrites of the postsynaptic CA3 target (GFP negative). Right: Representative images of neurons immunostained for GFP (green) to reveal transfected DG neurons, synaptoporin (SPO, blue) to label presynaptic DG terminals, and PSD-95 (red) to label excitatory synapses. Bottom row shows selection of GFP+ terminals for analysis. (B) DG neurons were transfected with GFP and Gi-DREADD and left untreated (top) or activated by addition of CNO (bottom). (C-D) Quantification of bouton size (C) and presynaptic SPO accumulation (D) in axon boutons from DG neurons transfected with GFP vector control, or with Gi-, Gs-, or Gq- DREADD, in the absence (NT) or presence of CNO. (E) Quantification of postsynaptic PSD-95 accumulation in untransfected CA3 neurons opposite DG neurons transfected and treated as indicated. Data are expressed as mean±SEM from neuronal averages (N=10 neurons/condition, 2–5 boutons/neuron). *P<0.05, ***P<0.001 NT vs. CNO, ANOVA and post hoc Bonferroni multiple comparison test. Scale bars, 20 μm.
DREADD transfection & analysis
Cultured neurons (>21 DIV) were sparsely transfected using Lipofectamine 2000 (Invitrogen) with GFP alone (AAV5-CaMKIIa-HA-EGFP) or transfected with one of the following GFP-containing DREADD (Designer Receptors Exclusively Activated by Designer Drugs) constructs: Gq-DREADD (AAV5-CaMKIIa-HA-hM3D(Gq)-IRES-mCitrine), Gi-DREADD (AAV5-CaMKIIa-HA-hM4D(Gi)-IRES-mCitrine), or Gs-DREADD (AAV5-CaMKIIa-HA-rM3D(Gs)-IRES-mCitrine), all from UNC Vector Core, Chapel Hill, NC). Although these are AAV vectors, we used transfection to obtain sparse expression of the DREADDs (~0.05% efficiency). After 48 hours of transfection, neurons were incubated in the absence or presence of CNO [1 or 10 μM, Enzo Life Sciences] for 24–48 hours and then processed for immunocytochemistry. Randomly transfected cells were classified as DG neurons as described in the section above. See Supplementary Methods for full details.
Electrical kindling
Adult male C57Bl/6J mice were obtained from Charles River (Boston, MA) and a bipolar stimulating electrode was stereotaxically implanted into their left basolateral amygdalae. We waited at least 14 days after surgery to allow normalization of seizure threshold,22 after which mice were kindled daily at an individually-determined afterdischarge (AD) threshold. ADs were monitored electrographically and behavior was manually recorded by an observer blinded to the experimental condition. Seizures were scored using a modified Racine score (Table S3). Kindling continued daily until mice experienced four stage 5 seizures, a status defined as fully kindled. See Supplementary Methods for full details.
Chemoconvulsant kindling
Adult male Sprague Dawley rats were acquired from Charles River (Boston, MA). Rats were treated every two days with pentylenetetrazole (PTZ) (20–22.5 mg/kg, i.p.). This subconvulsant dose was selected to maximize sensitization to PTZ, as single injections of 20–22.5 mg/kg rarely evoked generalized seizures.23 Rats were observed for 15–30 minutes following injection of chemoconvulsant, and behavior was quantitatively scored using a modified Racine scale (Table S3). See Supplementary Methods for full details.
In vivo opioid receptor pharmacology
Each kindling session, animals (mice or rats) received intraperitoneal injections of either saline, U50488 (5 mg/kg, Tocris) or norBNI (5 mg/kg, Tocris), approximately 1.5 hours after kindling. Drugs were prepared fresh in sterile saline. U50488 at higher doses (10 mg/kg) and other kappa OR agonists24 are known to acutely mitigate chemoconvulsant-induced seizures when administered <2 hours before chemoconvulsant injection.25–27 Conversely, norBNI (10 mg/kg)24 and other kappa OR antagonists26 are known to block the effects of U50488. We selected drug concentrations 50% below the effective doses shown to acutely alter seizure severity and/or duration. We also administered drugs after daily kindling to reduce drug interference with kindling per se. With this dosing scheme, U50488 (half-life 2.60 hours)28 would not be present appreciably during subsequent electrical kindling sessions, although residual norBNI (half-life 18.94 hours)29 may remain.
Statistics
Data were analyzed using GraphPad Prism. Data are expressed as mean±SEM unless otherwise indicated. Experiments were performed and analyzed blind to treatments. Normally distributed group data were evaluated via one-way ANOVA and, if significant, by appropriate post hoc analysis, either Tukey tests (all pairs of groups) or Dunnett’s multiple comparison test (all groups vs. control). Non-normally distributed group data were analyzed using Kruskal-Wallis tests and post hoc Dunn’s comparisons. Full statistical methods are presented in Supporting Information. Table S1 provides a comprehensive list of statistical tests and results.
RESULTS
Various cell types in the nervous system recalibrate in response to sustained changes in overall activity, most commonly through compensatory changes in synaptic strength: prolonged inactivity increases excitatory synaptic strength, while prolonged hyperactivity decreases it.11,12 We have previously observed that excitatory synapses of excitatory hippocampal neurons do not respond uniformly to activity changes. Both in vitro and in vivo, synapses between mossy fibers (MF) of dentate gyrus (DG) granule cells and proximal dendrites of CA3 neurons demonstrate large bidirectional compensations to activity changes, while other synapses do not.16
Remarkably, the MF-CA3 synapse is recapitulated faithfully in dissociated hippocampal neuron cultures: in vitro MF-CA3 connections demonstrate the same striking size, unique morphology, proximal dendritic distribution, molecular components, pharmacological sensitivity, and activity-responsiveness as the in vivo synapse.16,20 Here, we use our validated ‘synapse-on-chip’ approach to dissect the mechanisms of regulation of the DG-CA3 proximal synapses (hereafter MF-CA3 synapses) in vitro, screen drugs which preferentially act on this synapse, and predict in vivo drug effects.
Homeostatic elaboration of DG-CA3 proximal synapses in vitro is determined by presynaptic activity
The various types of excitatory hippocampal neurons (DG granule and CA1/CA3 pyramidal cells) can be identified in dissociated cultures using a combination of previously established criteria16,20 including cell body size, number of neurites, and expression of the marker protein CTIP2 (Fig. 1A–D; see Methods for additional details). For example, DG granule cells are particularly easy to identify based on their relatively small and round somata, few (2–3) primary neurites, and expression of unique proteins including the transcription factor CTIP2 (Fig. 1A; Fig. 2A; Fig. S1).
Consistent with prior studies,16,30 we found that homeostatic compensations of excitatory hippocampal neurons following sustained inactivity (induced via application of the sodium channel antagonist TTX) occurred only at excitatory post-synapses of CA3 neurons (Fig. 1B,E); in contrast, DG (Fig. 1C,F) and CA1 (Fig. 1D,G) neurons did not demonstrate compensatory post-synaptic strengthening (N=20 neurons/condition, 3 dendrites/neuron; ***P<0.001 v. NT). Furthermore, the increases in accumulation of post-synaptic proteins was confined to the proximal (<40 μm) portion of CA3 dendrites (Fig. 1D,G).
CA3 proximal dendrites are the territory of the large mossy fiber (MF) axon terminal boutons of DG granule cells (Fig. 2A); even in vitro, DG axons show a pronounced preference for proximal dendrites of their targets (Fig. S1B, Fig. 2A, Fig. S2). DG terminals are also extremely large and readily identifiable by immunocytochemistry because, unlike other presynaptic terminals, these compartments are enriched in the vesicle-associated protein synaptoporin (SPO)20,21 (Fig. S1E). We verified the presynaptic vesicular localization of SPO using super-resolution microscopy (Fig. S2). SPO was the presynaptic scaffolding protein, Bassoon, and showed strong co-localization with vGlut1, found on vesicles containing the excitatory neurotransmitter glutamate (Fig. S2D–E,G–H). SPO puncta were directly opposed to PSD-95 puncta on proximal dendrites, as previously observed (Fig. S2B–C,F).16
Presynaptic SPO accumulation is necessary and sufficient for the functional homeostatic strengthening of DG-CA3 proximal synapses,16 suggesting that presynaptic mechanisms may drive compensatory changes at this synapse. To determine the locus of compensation, we transfected neurons with DREADDs, G protein-coupled receptors engineered to be activated exclusively by an exogenous, otherwise inert agent, clozapine N-oxide (CNO).31,32 CNO silences cells expressing Gi-DREADD, activates those expressing Gq-DREADD, and increases cyclic AMP (cAMP) levels via Gs-DREADD,31,32 while leaving untransfected neurons unaffected.
We examined the consequences of DREADD expression on DG neurons and their postsynaptic targets. We identified GFP-positive, DREADD-transfected DG granule cells (Fig. S1, Fig. 2A) and traced their GFP-positive axons to their untransfected CA3 targets. We then quantified the area and SPO content of the bouton, as well as PSD-95 accumulation in nearby proximal dendrites (Fig. 2A). Chronic inactivation of Gi-DREADD-expressing DG neurons provoked a large (~250%) increase in presynaptic bouton size (in μm2: Gi+NT 2.8±0.6, Gi+CNO 6.9±0.1.2) (Fig. 2B–C) and a similar (~270%) increase in the presynaptic accumulation of SPO within these boutons (SPO immunoreactivity (as % of NT): Gi+NT 89.6±20.6, Gi+CNO 243.4±84.9) (Fig. 2B,D). DG inactivation with CNO-induced Gi signaling also tripled the postsynaptic accumulation of PSD-95 in untransfected target CA3 neuron proximal dendrites opposing the SPO-positive terminals (PSD-95 immunoreactivity (as % of NT): Gi+NT 64.9±17.1, Gi+CNO 207.2±73.9) (Fig. 2B,E). In contrast, neither GFP transfection alone nor Gq-/Gs-DREADD activation had significant effects on bouton size, SPO accumulation, or PSD-95 accumulation (Fig. 2C–E; Fig. S3). These results suggest that presynaptic mechanisms drive the concerted pre- and postsynaptic expansion of DG-CA3 proximal synapses.
Opioid receptor signaling collectively constrains DG-CA3 synapses
Suppression of activity in presynaptic DG neurons induced DG-CA3 proximal synapse elaboration in vitro. We next aimed to determine the signaling mechanisms responsible. Unlike other presynaptic terminals in the hippocampus, MF terminals are highly enriched with endogenous opioid neuropeptides as well as mu, kappa, and delta opioid receptors (ORs).33–37 Interestingly, MF opioid content – like MF synaptic strength – is inversely regulated by activity. Under basal conditions, MFs contain high levels of dynorphin and low levels of enkephalin.35,37 However, following hilar lesion-induced seizures, MF enkephalin levels are elevated, while dynorphin is depleted.37
To determine if opioid receptor (OR) signaling contributes to DG-CA3 HSP, we incubated hippocampal neurons for 24 hours under conditions of normal activity, inactivity (action potential blockade with TTX), or hyperactivity (disinhibition with the GABAA receptor antagonist, picrotoxin (PTX)) in the absence or presence of naloxone, a broad-spectrum OR antagonist (Fig. 3A). Chronic inactivity significantly increased the postsynaptic accumulation of PSD-95 (TTX: 211.2±22.7% of NT; P<0.01) (Fig. 3A–C) and the presynaptic accumulation of SPO (Fig. 3A,B,D; Fig. S4) (TTX: 338.2±47.9% of NT) along proximal dendrites of CA3 neurons. Hyperactivity had no effect on SPO levels (PTX: 104.6±21.5% of NT, PTX+1 μM naloxone: 136.5±15.1%), and decreased proximal dendritic PSD-95 accumulation (PTX: 34.1±4.7% of NT) (Fig. 3A–C), as has been previously observed.16 No significant differences in PSD-95 levels were observed in distal dendrites under any condition (TTX: 136.0±15.3% of NT; PTX: 87.6±10.0% of NT; P>0.05) (Fig. 3A–C, Fig. S4), supporting previous findings that homeostatic synaptic compensations in vitro occurs preferentially at proximal DG-CA3 synapses16,30.
Figure 3. Opioid receptor (OR) signaling collectively constrains DG-CA3 proximal synapses.

(A) Cultured hippocampal neurons (>21 DIV) incubated for 24 hours under normal activity conditions (NT), hyperactivity (PTX) or inactivity (TTX) in the absence (top) or presence (bottom) of the broad-spectrum OR antagonist, naloxone. DG axon terminals and excitatory synapses were visualized with SPO (blue) and PSD-95 (red), respectively. (B) Representative proximal (<20 μm from soma) and distal (>80 μm) CA3 dendrites immunostained with SPO (blue) and PSD-95 (red). (C) Quantification of PSD-95 immunoreactivity in proximal (<20 μm from soma) and distal (>80 μm) CA3 dendrites. **P<0.01, ***P <0.001, one-way ANOVA and post hoc Dunnett’s comparison vs. NT. NS (P>0.05) vs. conditions indicated. (D) Quantification of SPO accumulation in presynaptic DG terminals opposite proximal (<20 μm from soma) and distal (>80 μm) CA3 dendrites. **P<0.01, ***P <0.001, ++P<0.01, one-way ANOVA and post hoc Bonferroni multiple comparison test. Data are expressed as mean±SEM from neuronal averages (n=10–20 neurons/group, 3 dendrites/neuron). Scale bar, 20 μm.
Blockade of opioid receptors with naloxone caused a dose-dependent increase in proximal dendritic PSD-95 accumulation in CA3 neurons (1 μM naloxone: 94.1±10.9% of NT; 100 μM naloxone: 261.5±35.0% of NT; P<0.001, Fig. 3A–B) and presynaptic SPO accumulation in opposing terminals (1 μM naloxone: 78.5±25.3% of NT; 100 μM naloxone: 418.5±60.2% of NT, Fig. 3A,C). Concurrent treatment with TTX and naloxone caused no additional increase in PSD-95 upregulation over TTX or naloxone treatment alone (TTX+100 μM naloxone: 239.7±23.9% of NT), suggesting that the two pathways may be mechanistically overlapping. Naloxone did, however, enhance the inactivity-driven accumulation of SPO (TTX: 338.2±47.9% of NT; TTX+naloxone: 525.8±51.2%). Interestingly, low doses of naloxone (1 μM) that had no effect alone were sufficient to counter the effects of PTX treatment on proximal PSD-95 (Fig. 3A–B) (PTX: 34.1±4.7% of NT; PTX+1 μM naloxone: 95.5±13.6%). These results suggest that OR signaling collectively constrains DG-CA3 proximal synapses under basal conditions, and that a loss of OR signaling permits unchecked DG-CA3 synapse expansion.
Kappa ORs mediate homeostatic upregulation of DG-CA3 synapses
To determine which opioid receptors (ORs) regulated DG-CA3 synapses, we incubated neurons for 24 hours in the presence or absence of selective agonists and antagonists of the kappa, delta, and mu ORs. Kappa OR activation with the agonist U50488 caused a dose-dependent increase in SPO opposite CA3 proximal dendrites (250 nM U50488: 133.7±18.6% of NT; 2.5 μM U50488: 246.9±41.3; P<0.001 vs. NT) (Fig. 4A–C) and a dose-dependent increase in proximal dendritic PSD-95 in CA3 neurons (250 nM U50488: 98.7±20.7% of NT; 2.5 μM U50488: 225.9±45.2; P<0.05 vs. NT) (Fig. 4A–B,D). Conversely, chronic blockade of kappa ORs with the antagonist norBNI decreased proximal PSD-95 (1 μM norBNI: 72.0±8.9; P<0.05 vs. NT) (Fig. 4E). These results suggest that kappa OR signaling is sufficient to induce trans-synaptic DG-CA3 synapse elaboration.
Figure 4. Effect of opioid receptor drugs on DG-CA3 synapses.

(A–B) Cultured hippocampal CA3 neurons (>21 DIV) incubated for 24 hours under normal activity (NT), inactivity (TTX), or in the presence of kappa (U50488), delta (DPDPE), or mu (DAMGO) opioid receptor agonists at concentrations indicated. MAP2 (blue, dendritic morphology), PSD-95 (red, excitatory postsynapses), SPO (green, presynaptic MF terminals). (C) Quantification of SPO accumulation in presynaptic DG terminals opposite proximal CA3 dendrites under conditions indicated in D. (D-E) Quantification of PSD-95 immunoreactivity in proximal and distal dendrites of CA3 neurons (>21 DIV) incubated for 24 hrs under normal activity (NT), inactivity (TTX), or in the presence of kappa, delta, or mu OR agonists (D) or antagonists (E) as indicated. Data are mean±SEM from neuronal averages (N=3 dendrites/neuron, 10–15 neurons/condition), normalized to control. *P<0.05, ***P<0.001, one-way ANOVA and post hoc Dunnett’s comparison. Scale bars, 20 μm.
In contrast, activation of mu ORs with the agonist DAMGO caused a mild dose-dependent decrease in presynaptic SPO (100 nM DAMGO: 93.6±20.0 % of NT; 1 μM DAMGO: 58.1±9.6) (Fig. 4A–C) and proximal PSD-95 (100 nM DAMGO: 127.6±22.5% of NT; 1 μM DAMGO: 63.8±12.5) (Fig. 4D), though these did not reach significance. Conversely, mu OR blockade with the antagonist CTOP increased proximal PSD-95 (50 nM CTOP: 304.2±33.7% of NT; P<0.001 vs. NT) (Fig. 4E). These results suggest that mu OR signaling negatively constrains DG-CA3 synapses.
Activation of delta ORs with DPDPE did not significantly alter SPO expression (1 μM DPDPE: 77.1±18.2% of NT; 10 μM DPDPE: 65.7±9.1) (Fig. 4A–C) or proximal PSD-95 levels (1 μM DPDPE: 108.9±13.4% of NT; 10 μM DPDPE: 122.8±13.1) (Fig. 4D). Blockade of delta ORs with ICI-174,864 caused only a slight dose-dependent increase in proximal PSD-95 levels (20 nM ICI-174,864: 101.6±9.0% of NT; 2 μM ICI-174,864: 155.0±16.4) (Fig. 4E), which did not reach significance.
These results suggest that mu OR signaling (and possibly delta OR signaling) constrains DG-CA3 synapses. Loss of delta/mu signaling allows DG-CA3 synaptic elaboration, likely accounting for the effects of naloxone (Fig. 3). In contrast, kappa OR signaling is sufficient to induce the elaboration of DG-CA3 synapses. Under no condition did we observe significant changes in distal PSD-95 levels (P=0.274, ANOVA) (Fig. 4–5; Fig. S4). Distinct OR signaling pathways therefore bidirectionally and selectively mediate DG-CA3 plasticity in vitro.
Figure 5. Kappa ORs are necessary and sufficient for homeostatic upregulation of DG-CA3 synapses.
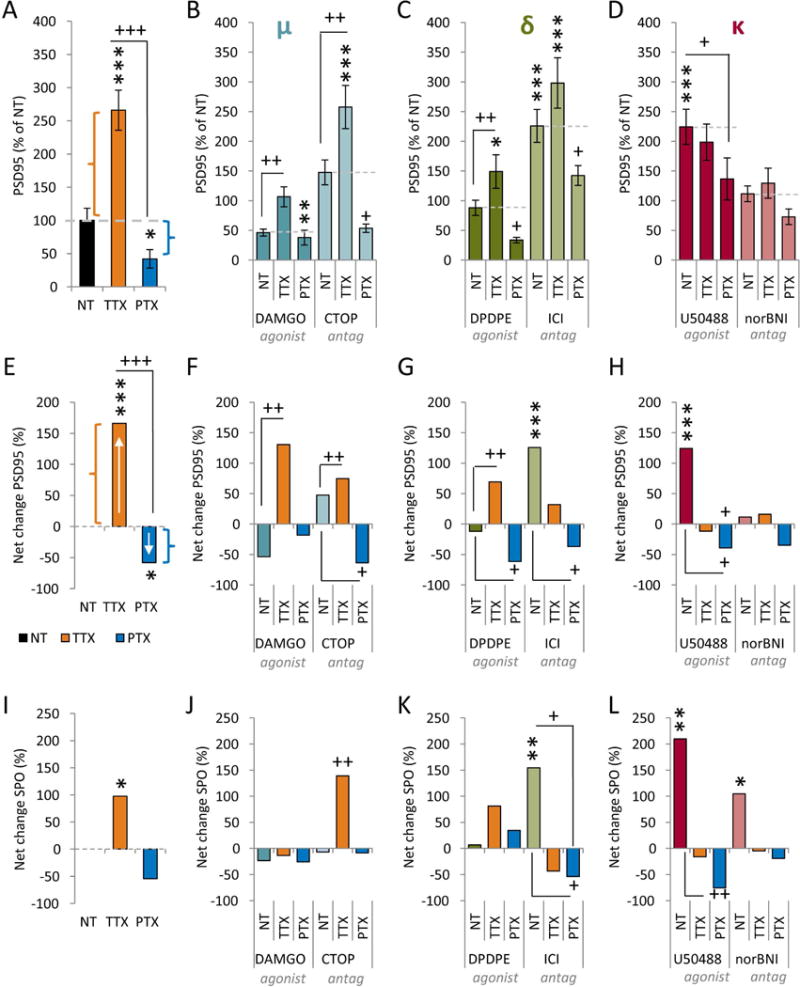
(A–D) Quantification of PSD-95 immunoreactivity in proximal CA3 dendrites after 24 hrs of normal activity (NT), inactivity (TTX), or hyperactivity (PTX), in the absence (A) or presence (B-D) of agonists (left) or antagonists (right) of the mu (B), delta (C), or kappa (D) ORs. Data are mean±SEM (N=20 neurons/condition, 3 dendrites/neuron), normalized to control. *P<0.05, **P<0.01, ***P<0.001 v. untreated control (black bar); +P<0.05, ++P<0.01, +++P <0.001 versus drug control (e.g., DAMGO NT). Significance determined by one-way ANOVA and post hoc Tukey test. (E-L) Net changes in PSD-95 (E-H) or SPO (I-L) with activity and OR drugs. The basal effect of each OR drug was calculated v. untreated control (black bar in A, 100% in B-D). The net effects of TTX (orange) and PTX (dark blue) was calculated as (OR drug+TTX)/(OR drug alone) and (OR drug+PTX)/(OR drug alone). Dotted lines in A-D indicate x-axes in E-H.
To determine whether OR signaling is a component of the activity-dependent homeostatic adjustment of DG-CA3 synapses, we modulated the activity of mature hippocampal neurons in the presence or absence of either agonists or antagonists of the mu, delta, and kappa ORs (Fig. 5, Table S2). With intact OR signaling, inactivity increased, while hyperactivity decreased, proximal dendritic PSD-95 in CA3 neurons (Fig. 5A) (TTX: 266.0±30.1% of NT; PTX: 42.0±13.9% of NT; P <0.0001) and SPO along proximal dendrites (Fig. 5I). To better visualize activity-dependent responses, regardless of basal drug effects, we plotted the net changes in Fig. 5E–L.
We then determined whether OR signaling influenced the ability to respond to inactivity and/or hyperactivity. As before, manipulation of mu or delta OR signaling had basal effects on DG-CA3 synapses (NT columns in Fig. 5B–C, F–G). However, these basal drug effects did not prevent homeostatic compensations. In the presence of mu OR agonist or antagonist (Fig. 5B, F), TTX still induced significant increases in PSD-95 levels (DAMGO+TTX: 130.6% increase over DAMGO, CTOP+TTX: 74.6% increase over CTOP; P<0.001, Bonferroni test vs. CTOP alone) (orange bars in Fig. 5F) while PTX decreased PSD-95 levels (Fig. 5B, and dark blue bars in Fig. 5F). Manipulation of delta OR signaling gave largely similar results (Fig. 5C, G). These observations suggest that homeostatic synaptic elaboration in response to inactivity (orange bars in Fig. 5E–G show net positive change) and contraction in response to hyperactivity (dark blue bars) are both intact in the presence or absence of mu and delta OR signaling.
The kappa OR agonist U50488 again increased proximal PSD-95 and SPO levels but, in marked contrast to our findings with mu and delta targeted drugs, this occluded the effects of TTX (U50488: 224.3±29.5% of NT; U50488+TTX: 198.5±30.5% of NT) (Fig. 5D,H,L). These results suggest that TTX-induced homeostatic upregulation proceeds through kappa OR signaling. Indeed, the kappa OR antagonist norBNI blocked inactivity-induced homeostatic upregulation of PSD-95 (norBNI: 111.6±13.3% of NT; norBNI+TTX: 126.4±25.3% of NT) (Fig. 5D,H). Taken together, these data reveal that kappa OR signaling is necessary and sufficient to mediate the homeostatic elaboration of DG-CA3 proximal synapses in response to inactivity.
Kappa ORs influence temporal lobe seizure progression
It has been hypothesized that MF-CA3 synapses serve as gain control for hippocampal circuits, regulating the bandwidth of information arriving from the cortex via the dentate gyrus.17 Inappropriate gain control at MF-CA3 synapses has been suggested to underlie temporal lobe epileptogenesis.18 If this were true, blocking homeostatic elaboration of DG synapses would prevent epileptogenesis, while promoting homeostatic elaboration would exacerbate it. Given the effects of kappa OR signaling on DG-CA3 synaptic homeostasis in vitro, we hypothesized that these drugs would bidirectionally modulate epileptogenesis in vivo.
We tested this hypothesis using the amygdala kindling model, which generates increasingly severe temporal lobe seizures.38–40 Each day, mice were kindled followed by injections of either saline, the kappa OR agonist U50488 (5 mg/kg/day, i.p.) or the kappa OR antagonist norBNI (5 mg/kg/day, i.p.) (Fig. 6B). The kappa OR agonist U50488 and antagonist norBNI are known to have acute anti- and pro-convulsant effects, respectively, when administered before injection of a chemoconvulsant.26,27,41 However, the action of these drugs at DG-CA3 synapses in vitro predicts the opposite effect of the drugs on the gradual emergence of seizures if chronically administered during epileptogenesis. As we are interested in long-term homeostatic adaptations induced by the drugs, not acute effects, drugs were administered after daily electrical stimulation and at doses below those shown to have acute effects on seizure severity (see Methods).
Fig. 6. Chronic blockade of kappa OR signaling counteracts epileptogenesis in mice and rats.

(A–F) Adult male mice (N=7 NT, 8 norBNI, 5 U50488) were kindled once daily and seizure behavior was visually scored in blinded manner using a modified Racine scale (Table S3). 1 to 3 hours after kindling, mice received daily i.p. injections of either saline, kappa OR agonist U50488 (5 mg/kg/day) or kappa OR antagonist norBNI (5 mg/kg/day). (A–B) Days until initial first generalized seizure (A, defined as first stage 5 seizure, P=0.034 NT vs. norBNI, ++P=0.009 all curves, Mantel-Cox test) or until full kindling (B, defined as 4 stage 5 seizures, P=0.017 NT vs. norBNI, ++P=0.008 all curves, Mantel-Cox test). (C-D) Median seizure duration (C) and severity (D) between groups; 3 day averages for each animal were pooled for group analysis. +P=0.027, +++P<0.001, treatment effect, two-way ANOVA (time v. treatment). (E-F) Lack of relationship between average seizure severity and either initial (E, R2=0.139) or final after-discharge threshold (ADT) (F, R2=0.117). (G-I) Adult male rats were kindled every other day via PTZ injection and seizure behavior visually scored in blinded manner using a modified Racine scale (Table S3). Approximately 1 to 3 hours after each kindling, mice received i.p. injections of saline or kappa OR antagonist norBNI (5 mg/kg). (G) Median seizure severity per group for each kindling session (N=6 per group; **P<0.001, treatment effect, aligned rank transform test). Essentially no seizure behavior was observed in norBNI animals. (H) Mean seizure score ± SD overlaid with individual animal scores on last day of kindling (Mann Whitney; *P=0.015). (I) Mean and individual maximum seizure score attained during kindling (Mann Whitney; **P=0.0043).
Administration of the kappa OR agonist, U50488 (5 mg/kg/day) resulted in faster kindling compared to control animals (Fig. 6A–B). Conversely, mice receiving daily injections of the kappa OR antagonist norBNI (5 mg/kg/day) kindled significantly more slowly (Fig. 6A–B) and less severely (Fig. 6D). NorBNI increased, while U50488 decreased, both time to first generalized seizure (Fig. 6A) (median time to first stage 5 seizure (in days): NT 10, norBNI 15, U50488 9; P=0.009, Mantel-Cox test of survival curves) and time to full kindling (Fig. 6B) (median time to 4 stage 5 seizures (in days): NT 16, norBNI >21, U50488 11; P=0.008, Mantel-Cox test). Indeed, after the 21 day protocol, norBNI animals remained incompletely kindled (fully kindled by day 21: NT 7/7 animals, norBNI 3/8, U50488 5/5) and some norBNI mice failed to reach even a single stage 5 seizure (Fig. 6A). NorBNI decreased both seizure duration (Fig. 6C) and severity (Fig. 6D), while U50488 increased these parameters (seizure duration: P<0.001; seizure severity: P=0.027 ANOVA).
We observed no significant difference in after-discharge duration at the start of the study (mean±SD (in sec): NT 7.8±9.8, norBNI 10.1±8.6, U50488 13.3±11.5; P=0.226, Kruskal-Wallis test), suggesting that intrinsic excitability did not differ between groups at baseline. We also observed no significant changes in after-discharge threshold (ADT) (P=0.170, Kruskal-Wallis test), a measure of kindling stimulation efficacy, over the course of the study. Lastly, we observed no strong relationship between average seizure severity and initial or final ADT (Fig. 6E–F). Stimulation intensity was therefore not considered a factor in the findings.
The in vivo effects of chronic kappa OR treatment predicted by our ‘synapse-on-chip’ assay are precisely the opposite pattern that has been reported in response to acute, pre-seizure administration of these drugs. These findings demonstrate that the chronic norBNI treatment paradigm, which prevents DG-CA3 elaboration in vitro, suppresses the emergence of temporal lobe seizures in vivo.
To generalize our findings to another species and seizure model, we investigated chemically-induced kindling in rats (Fig. 6G–I; Fig. S5). We selected pentylenetetrazole (PTZ), a GABAA receptor antagonist, as the chemoconvulsant since PTZ-kindling is a widely used animal model for screening novel antiepileptic compounds.42 Rats received an initially subconvulsant dose of PTZ (20–22.5 mg/kg, i.p.)23 every other day for 11 sessions (Fig. 6G). After each kindling session, rats were given either saline (NT) or norBNI (5 mg/kg i.p., a concentration 50% below that shown to acutely affect seizure severity and EEG patterns in rats). Delivery of norBNI was delayed 1–3 hrs after PTZ to minimize acute interaction effects with the chemoconvulsant (Schematic in Fig. 6G).
Saline-treated rats developed progressively stronger behavioral seizures over the course of the kindling sessions (main effect of kindling day, P<0.0001; aligned-rank-transform test) (Fig. 6G). In contrast, the norBNI-treated group displayed essentially no seizures (Fig. 6G) (main effect of treatment, P=0.0059; treatment by day interaction effect, P<0.0001; aligned-rank-transform test). On the last kindling day, we observed a significant difference between groups in mean seizure severity (P=0.015, Mann Whitney test) (Fig. 6H) and in maximal seizure severity scores (P=0.0043, Mann Whitney test) (Fig. 6I). We also observed a significant difference in the proportion of animals displaying any limbic motor response (score of 2 or greater): the majority (5/6) of saline-treated, but no norBNI-treated, animals attained this response (P=0.0152, Fisher’s Exact test).
To determine if norBNI’s impact on chemoconvulsant kindling was due to acute (not the intended chronic) effects, we pre-treated a group of rats (n=6) with norBNI or saline 46.5 hours prior to a single challenge dose of PTZ; this timing was selected to match the interval between drug treatment and PTZ administration in our chronic studies. We found that both groups displayed a median seizure severity of 0.5, and did not differ significantly from each other (Mann Whitney, U=16, P=0.999). Thus, acute effects of norBNI do not account for the anti-epileptogenic effects we demonstrate here.
To assess if norBNI-dependent changes in seizure progression correlated with alterations in SPO levels in the brains of protected animals, we repeated the treatment in an independent cohort of saline or norBNI-treated animals (n=5/group). We again observed that norBNI was protective against epileptogenesis: norBNI seizure burden was significantly less throughout the study (Fig. S5A–C). However, in these animals, we observed no significant changes in SPO expression in the granule cell layer of the dentate gyrus (Fig. S5D–H). CA3 expression of SPO was mildly correlated with seizure burden (R2=0.256), while hilar SPO expression was moderately correlated with seizure burden (R2=0.530) (Fig. S5I–K).
Taken together, we concluded that the kappa OR antagonist is capable of suppressing seizure development in vivo in two rodent species and two epilepsy paradigms. The kappa OR agonist U-50488 has the opposite effect, exacerbating seizure progression. The direction of action of the kappa OR drugs during chronic treatment while seizures emerge is opposite their reported acute effects. However, the direction of action of the kappa OR drugs is predicted by their chronic effects at DG-CA3 synapses in vitro, as determined by our ‘synapse-on-chip’ assay. We therefore suggest that DG-CA3 synapse elaboration, a critical component of hippocampal homeostatic plasticity which is mediated by kappa OR signaling, is a necessary component of temporal lobe epileptogenesis and a viable target for therapeutic interventions.
DISCUSSION
In this study, we investigated whether inappropriate hippocampal gain control could contribute to epileptogenesis and whether harnessing endogenous neuronal calibration mechanisms could inhibit development of seizures. Our previous studies implicated the MF-CA3 synapse as a major mediator of homeostatic compensation in the hippocampus. Here, we found that presynaptic activity of DG granule cells dictates coordinated pre- and postsynaptic homeostatic changes across DG-CA3 proximal synapses in vitro. Because DG axon terminals are highly and distinctively enriched with endogenous opioid neuropeptides, we examined the role of ORs in HSP. The pharmacological experiments using complete blockade of ORs by naloxone suggested that ORs collectively restrain DG-CA3 synapses. Interestingly, distinct OR types appear to act in opposing ways: mu and delta ORs basally constrain DG-CA3 synapses, while kappa ORs promote their elaboration (Table S2). Moreover, while mu and delta OR signaling appear dispensable for homeostatic adaptation, kappa OR signaling is both necessary and sufficient for homeostatic growth of DG-CA3 synapses in vitro (Table S2).
Based on these properties, we suggest that presynaptic opioid neuropeptides are major coordinators of homeostatic plasticity at MF-CA3 synapses, providing closed-loop activity-dependent control of CA3 networks18 (Fig. S6). A common strategy in man-made closed-loop systems is proportional-integral control: the appropriate compensation to a deviation from the expected course is calculated based on both the current magnitude (proportional) and overall persistence of the problem (integral, i.e., cumulative deviation over time). We propose that presynaptic opioid peptides could provide such proportional-integral control. The relative accumulation of the opioid peptides would encode the activity history of DG neurons, while the release of the opioids would encode the current activity. Opioid signaling could thus coordinate trans-synaptic changes at the MF-CA3 synaptic ‘gate’ that are appropriate to the duration and magnitude of a change in neural activity. Upregulation of this gate in principle could involve increased number/size of synapses or the recruitment of synaptic components including PSD-95. The integrity of the opioid signaling system may therefore be crucial to maintaining the DG “gate” to the CA3 network (Fig. S6).
We hypothesized that improper strengthening of MF-CA3 synapses in vivo promotes the development of seizures. We thus sought to exert a “homeostatic brake,” combating epileptogenesis by weakening these synapses via their endogenous control mechanism. We propose that KOR signaling regulates the homeostatic gain control program at MF-CA3 synapses. Without KOR signaling, MF-CA3 synapses cannot undergo inactivity-induced expansion. This expansion may contribute to epileptogenesis, therefore blocking the runaway elaboration of MF-CA3 synapses would be protective. Mu and delta antagonists, like the kappa agonist U50488, act on MF-CA3 synapses but promote elaboration in vitro. We would therefore expect chronic treatment with mu and delta antagonists to exacerbate seizure progression, similar to U50488. These results provided the rationale for the focus on KORs in our in vivo pharmacology studies. Indeed, we showed that kappa OR signaling bidirectionally alters the emergence of severe seizures in the mouse electrical kindling model, and that suppression of kappa ORs dramatically inhibited chemical kindling in rats. These effects are generally consistent with the in vitro finding that kappa OR activity is necessary and sufficient for DG-CA3 synapse homeostatic upregulation.
An interesting paradox is that the effects of kappa signaling observed here during the emergence of epilepsy are opposite to those expected from the acute in vivo effects of U50488 and norBNI on chemically and electrically induced seizures. Acute pre-treatment with norBNI is known to be pro-convulsant, while acute U50488 is anti-convulsant.26,27,41 We observed the opposite effects of the drugs when chronically administered after seizures during kindling. Moreover, with the dose and timing we employed, we observed no acute effects of the drugs on seizures in control experiments. We therefore do not attribute the effects of chronic drug administration to acute action, or to accumulation or residual presence of the drugs, but rather to engagement of the homeostatic negative feedback mechanism that acts counter to the applied acute manipulation. We propose that repetitive, low-level administration of norBNI, which blocks the elaboration of MF-CA3 synapses in vitro, prevents the maladaptive elaboration of MF-CA3 synapses in vivo, thereby slowing or arresting seizure progression (Supplementary Fig. 6). Conversely, chronic administration of U50488 mimics the homeostatic expansion of MF-CA3 synapses, exacerbating seizure progression.
These effects contrast with a previous report that prodynorphin knockout mice display accelerated kindling26. Dynorphins are high affinity agonists for kappa OR. However, developmental compensation or cross-reactivities of various dynorphin peptides to different ORs may obscure the interpretation of these data. Moreover, prodynorphin knockout mice (which have a developmental depletion of this peptide) display decreased seizure threshold, and thus kindling stimulation may be more severe in the knockouts than wild-type controls, influencing the rate of kindling. By contrast, our pulsatile blockade of kappa OR avoids effects on acute seizures and rather selectively and transiently decreases kappa OR signaling in the post-seizure period. The dissociation between the genetic studies and our present findings may underscore the importance of a temporal window for intervention after an epileptogenic insult in defining the net effect.
The classical opioid receptors are G-protein coupled receptors that all link to Gαi small GTPases, leading to various well-established effects such as inhibition of cAMP production, activation of G-protein gated inward rectifying potassium channels, and reduction in calcium channel conductances.43 In view of this shared G protein second messenger, how can ORs produce differential effects in homeostatic regulation of DG-CA3 synapses? ORs are known to exist in large protein complexes, and distinct scaffold protein networks may link each receptor to a specific microdomain of signaling proteins and downstream effectors. For instance, ORs bind to arrestin proteins, and different arrestin family members could underlie some of the distinct receptor effects via downstream targets including MAP kinases.44 Furthermore, it is possible that differential receptor localization may play a role in the specific effects of KORs. An attractive hypothesis is that KORs are more highly enriched at the DG-CA3 synapse itself and thus have a more potent effect on this system.
In this regard, the observation that Gi-DREADD could mimic the effects of KOR in DG neurons suggests that it is the neuronal activity state that drives homeostatic plasticity. Thus, changes induced by KOR signaling in our studies would be due to the effects of its Gi-signaling on neuronal activity, and its putative localization at the DG-CA3 synapse. Given the nature of Gi receptor signaling, we expect that this effect may in fact be achievable with other Gi-coupled receptors as well, and we cannot exclude the possibility that mu/delta receptors may also play a role in these mechanisms in vivo. However, although other endogenous Gi-receptors may have similar signaling activity, these may not be sufficiently expressed in the DG-CA3 synapse. By this model, any Gi-receptor expressed in the right location would have an analogous effect, as would any chronic activity manipulation that selectively engages homeostatic adaptation at this synapse. Indeed, this interpretation is supported by our data with Gi-DREADDs in vitro. Because the only apparent commonality between KOR and Gi-DREADD is the Gi signaling cascade, it is possible that blocking KOR Gi-signaling would also prevent DREADD function in our assays. Future studies will be needed to dissect the precise signaling cascades and expression patterns of the KORs that may be involved in modulating hippocampal homeostatic plasticity and seizure progression.
The MF-CA3 synapse contains specific molecular constituents (e.g., opioid peptides and synaptoporin) that are not widely or strongly expressed at other types of hippocampal synapses. Both the present study, and prior genetic ablations have globally modulated the function of the kappa OR system. Local genetic manipulation of molecular targets specifically at MF-CA3 synapses (e.g., knockdown of synaptoporin or kappa ORs in MF terminals) may provide more selective manipulation of endogenous HSP mechanisms. These studies will be key to resolving the role of intra-hippocampal kappa OR signaling on seizures and epileptogenesis. Although we did not find a significant change in SPO levels in the hippocampus of protected animals following norBNI treatment, it is possible that the localization of SPO is changing, rather than the total amount, or that the change is only affecting a subset of synapses that is difficult to detect against the background of unaffected synapses. Further experiments are necessary to conclusively demonstrate that endogenous homeostatic mechanisms are responsible for epileptogenesis or its prevention. It will be important to test this approach in animal models that exhibit spontaneous seizures to determine whether this method can alter or prevent disease pathogenesis.
While single-synapse gain control is unlikely to represent a cure for all forms of temporal lobe epilepsy, our present findings are consistent with a long-standing literature involving the “dentate gate.” Transient suppression of the dentate in mice with spontaneous seizures can suppress seizure activity, whereas driving activation of the dentate can trigger seizures.2 Here, we have proposed a synaptic mechanism for dentate gate regulation and provided evidence that systemic pharmacological manipulation of a signaling platform enriched in the MF-CA3 pathway can bidirectionally modulate epileptogenesis. Utilizing endogenous mechanisms of homeostatic compensation may be particularly helpful as it can be utilized after an epileptogenic perturbation, and induced on fairly slow timescales. Exploiting endogenous homeostatic mechanisms may therefore expand the timeframe for intervention after injury. This raises the possibility that intervention after insult, but before spontaneous seizures emerge, can modify disease trajectory. This feature is therapeutically attractive as treatment does not require foreknowledge of when an insult would occur, and could potentially be administered effectively even after some delay.
Supplementary Material
SupplementaryFigure 1. In vitro identification of DG-CA3 proximal synapses. (A) Schematic of in vivo hippocampal connections. Note the large size of the DG terminals targeting the proximal dendrites of CA3 neurons. (B) Representative image of a GFP-transfected DG neuron. Note how the DG axon wraps around the soma of nearby untransfected neurons (red triangles). (C-E) Schematic (C) and representative examples (D-E) of experimental protocol: Mature (>21 DIV) primary hippocampal neurons are transfected with GFP. DG neurons (D) can be easily identified by small soma size and few, short dendrites (as well as nuclear staining with CTIP2). DG axons can be easily traced to the proximal dendrites of target cells (E) and unambiguously identified by staining with the presynaptic vesicle protein synaptoporin (SPO). PSD-95 accumulation on the opposing postsynaptic cell can be readily quantified. Scale bars = 20 μm.
Supplementary Figure 2. Synaptoporin is localized to presynaptic glutamatergic vesicles in vitro. (A) Schematic of images visualized in panels B–H. The proximal dendrites of CA3 neurons in vitro were imaged using conventional fluorescence microscopy (B) or stimulated emission depletion (STED) super-resolution microscopy (C-H). Boxes in A indicate the relative sizes of the subsequent panels. (B) Cultured hippocampal neurons immunostained with synaptoporin (SPO) and PSD-95. A representative CA3 neuron is shown with soma indicated by dotted circle. (C–H) Super-resolution images of the proximal dendrites of CA3 neurons in vitro. Soma indicated with dotted white line. To determine the intracellular localization of SPO, neurons were immunolabeled with SPO and either the postsynaptic scaffolding protein PSD-95 to label all excitatory postsynaptic terminals (C,F); the presynaptic scaffolding protein Bassoon to label all presynaptic terminals (D,G); or the vesicular glutamate transporter 1 (vGlut1) to label all presynaptic vesicles containing the excitatory neurotransmitter glutamate (E,H). (F–H) Magnified images of the images in C–E. Note the precise co-localization of SPO with vGlut1 on glutamatergic vesicles. Scale bars = 20 μm (B), 2 μm (C–E), 1 μm (F–H).
Supplementary Figure 3. Presynaptic DG inactivity drives elaboration of DG-CA3 synapses. Mature (>21 DIV) primary hippocampal neurons are transfected with various constructs – namely GFP control (A), hM3Dq (Gq) DREADD-GFP (B), hM4Di (Gi) DREADD-GFP (C), or Gs DREADD-GFP (D) constructs. Neurons were then left untreated (NT, top in A–D) or treated with CNO to activate the relevant G-protein cascade (bottom in A–D). For analysis, we identified DG neurons transfected with GFP-DREADDs and traced these GFP-filled terminals to their un-transfected target neurons. (A–D) Representative images of the un-transfected targets of the DREADD-transfected DG neurons. Neurons were immunostained for GFP (green) to reveal transfected DG neurons, synaptoporin (SPO, blue) to label presynaptic DG terminals, and PSD-95 (red) to label excitatory synapses. Scale bars, 20 μm.
Supplementary Figure 4. Opioid receptor (OR) signaling collectively constrains DG-CA3 proximal synapses. (A–C) Representative images of CA3 neurons (from >21 DIV hippocampal cultures) incubated for 24 hours under normal activity conditions (NT), hyperactivity (PTX) or inactivity (TTX) in the absence (top) or presence (bottom) of the broad-spectrum OR antagonist, naloxone. DG axon terminals and excitatory synapses were visualized with SPO (blue) and PSD-95 (red), shown in overlay. (B-C) Representative linearized dendrites of CA3 hippocampal neurons from conditions indicated. (C) Representative examples of proximal (<20 μm from soma) and distal (>60 μm) CA3 dendrites used for analysis in Figures 4–6.
Supplementary Figure 5. Chronic norBNI treatment blocks epileptogenesis in rats, but does not alter SPO expression. Adult male mice (N=5/group) were kindled once daily. 1 to 3 hours after kindling, mice received daily i.p. injections of either saline or kappa OR antagonist norBNI (5 mg/kg/day). At the end of the study, animals were perfused and brains were immunostained to visualize SPO expression in the hippocampus (D). (A-C) Seizure behavior was visually scored in blinded manner using a modified Racine scale (Table S3). (A) Mean seizure score ± SD between groups. 2 session averages for each animal were pooled for group analysis. Time: P <0.0001, Treatment: P=0.0584, Interaction +P=0.0226, two-way ANOVA (time v. treatment). **P<0.01, Bonferroni post-test v. NT. (B) Mean seizure score ± SD overlaid with individual animal scores on last day of kindling (Mann Whitney; * P = 0.0350). (C) Mean seizure burden (total seizure score) ± SD overlaid with individual animal scores on last day of kindling (Mann Whitney; * P = 0.0356). (D–J) After kindling, this cohort of animals was perfused and brains were stained for synaptoporin (SPO, green) to visualize MF terminals and DAPI (blue) to visualize cell nuclei. (D–E) Representative images of hippocampi and hippocampal regions. Regions in E are indicated as boxes in D. Scale bars = 100 um (D), 25 um (E). (F–H) Quantification of SPO integrated intensity (IR, quantified as arbitrary units (a.u.) as percent of background IR). SPO immunostaining was quantified in the granule cell layer and inner molecular layer of the dentate gyrus (DG, F), the hilus (G), and the stratum pyramidale of the CA3 region (H). Dotted lines delineate regions used for quantification. ns = P>0.05, Mann Whitney t-test. (I–K) Within-animal analysis of seizure burden (total seizure score over all kindling sessions) versus SPO immunoreactivity in the dentate gyrus (I), hilus (J), and CA3 (K). We observed no significant relationship between SPO immunoreactivity and seizure burden in either the DG (P=0.619) or CA3 (P=0.165). However, SPO immunoreactivity in the hilus was negatively correlated with seizure burden: the best-fit linear approximation (blue) had a slope that was significantly non-zero (P=0.026) and reasonably fit the data (R2 = 0.053) upon linear regression. Dotted lines in I–K indicate 95% confidence intervals.
Supplementary Figure 6. Proposed model of opioid receptor regulation of MF-CA3 synapses. Schematic model of opioids on the putative MF-CA3 “gain control” synapse. The DG-CA3 synapse is represented as a “gate” that controls activity flow (lightning bolts) through the hippocampus. Under basal activity conditions (middle), mu and delta receptors oppose gate opening, while kappa receptors promote it. Sustained bouts of inactivity in the dentate gyrus (left) increase the accumulation of presynaptic opioids which promote kappa OR signaling, leading to homeostatic upregulation of DG-CA3 synapses. Conversely, high DG activity (right) leads to the accumulation of presynaptic opioids which promote mu and delta OR activity, thus downregulating this synapse. We propose that in some forms of mTLE, the endogenous upregulation program becomes uncoupled from network activity (bottom panels = Pathological). Thus, when network activity returns to normal, the DG-CA3 “gate” remains open, allowing for runaway excitation. Antagonizing the kappa OR (e.g., with norBNI) during this period prevents this inappropriate program of DG-CA3 strengthening, thus limiting the emergence of seizures. Meanwhile, enhancing kappa OR signaling (with U50488) exacerbates the maladaptive opening of the DG-CA3 gate.
Key point.
Presynaptic DG inactivity induces elaboration of synapses between DG mossy fibers and CA3 thorny excrescences in hippocampal cultures.
Kappa opioid signaling is necessary and sufficient for elaboration of hippocampal DG-CA3 synapses in vitro.
Pharmacological inhibition of kappa opioid receptors attenuates epileptogenesis in two independent in vivo kindling models.
Significance.
This study elucidates mechanisms by which synapses between DG granule cells and CA3 pyramidal neurons undergo activity-dependent homeostatic compensation, via OR signaling in vitro. Modulation of kappa OR signaling in vivo alters seizure progression, suggesting that breakdown of homeostatic closed-loop control at DG-CA3 synapses contributes to seizures, and that targeting endogenous homeostatic mechanisms at DG-CA3 synapses may prove useful in combating epileptogenesis.
Acknowledgments
Funding sources are as follows: F31-NS080462 (BNQ), R56-NS075278 (DTSP) and KL2-TR001432 (PAF) from the National Institutes of Health; a pilot grant from the Georgetown University Medical Center Dean for Research (DTSP, PAF); and the Georgetown Hughes Scholars program & Sigma Xi GIAR program (RLD).
Footnotes
Disclosure of Conflicts of Interest
None of the authors has any conflict of interest to disclose.
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Chang BS, Lowenstein DH. Epilepsy. New Engl J Med. 2003;349:1257–66. doi: 10.1056/NEJMra022308. [DOI] [PubMed] [Google Scholar]
- 2.Krook-Magnuson E, Armstrong C, Oijala M, et al. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Comm. 2013;4:1376. doi: 10.1038/ncomms2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinemann U, Beck H, Dreier JP, et al. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res. 1992;7:273–80. [PubMed] [Google Scholar]
- 4.Tallent MK, Madamba SG, Siggins GR. Nociceptin reduces epileptiform events in CA3 hippocampus via presynaptic and postsynaptic mechanisms. J Neurosci. 2001;21(17):6940–8. doi: 10.1523/JNEUROSCI.21-17-06940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boison D. Is Intrinsic Hyperexcitability in CA3 the Culprit for Seizures in Rett Syndrome? Epilepsy Curr. 2012;12(1):13–4. doi: 10.5698/1535-7511-12.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang S, Sun H, Fang Q, et al. Low-frequency stimulation of the hippocampal CA3 subfield is anti-epileptogenic and anti-ictogenic in rat amygdaloid kindling model of epilepsy. Neurosci Lett. 2009;455(1):51–5. doi: 10.1016/j.neulet.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 7.Henze DA, Urban N, Barrionuevo G. The multifarious hippocampal mossy fiber pathway: a review. Neuroscience. 2000;98(3):407–27. doi: 10.1016/s0306-4522(00)00146-9. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W, Huguenard JR, Buckmaster PS. Increased excitatory synaptic input to granule cells from hilar and CA3 regions in a rat model of temporal lobe epilepsy. J Neurosci. 2012;32(4):1183–96. doi: 10.1523/JNEUROSCI.5342-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5(4):1016–22. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235(4784):73–6. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- 11.Turrigiano GG, Leslie KR, Desai NS, et al. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391(6670):892–6. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- 12.O’Brien RJ, Kamboj S, Ehlers MD, et al. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21(5):1067–78. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- 13.Queenan BN, Lee KJ, Pak DTS. Wherefore art thou, homeo(stasis)? Functional diversity in homeostatic synaptic plasticity. Neural Plast. 2012;2012:1–12. doi: 10.1155/2012/718203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135(3):422–35. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson SB, Turrigiano GG. Strength through diversity. Neuron. 2008;60(3):477–82. doi: 10.1016/j.neuron.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KJ, Queenan BN, Rozeboom AM, et al. Mossy fiber-CA3 synapses mediate homeostatic plasticity in mature hippocampal neurons. Neuron. 2013;77(1):99–114. doi: 10.1016/j.neuron.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chater TE, Goda Y. CA3 mossy fiber connections: giant synapses that gain control. Neuron. 2013;77(1):4–6. doi: 10.1016/j.neuron.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 18.Queenan BN, Pak DT. Homeostatic synaptic plasticity in the hippocampus: therapeutic prospects for seizure control? Future Neurol. 2013;8(4):361–3. [Google Scholar]
- 19.Sala C, Piëch V, Wilson NR, et al. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31(1):115–30. doi: 10.1016/s0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- 20.Williams ME, Wilke SA, Daggett A, et al. Cadherin-9 Regulates Synapse-Specific Differentiation in the Developing Hippocampus. Neuron. 2011;71(4):640–55. doi: 10.1016/j.neuron.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singec I, Knoth R, Ditter M, et al. Synaptic vesicle protein synaptoporin is differently expressed by subpopulations of mouse hippocampal neurons. J Comp Neurol. 2002;452(2):139–52. doi: 10.1002/cne.10371. [DOI] [PubMed] [Google Scholar]
- 22.Forcelli PA, Kalikhman D, Gale K. Delayed effect of craniotomy on experimental seizures in rats. PLoS One. 2013;8(12):e81401. doi: 10.1371/journal.pone.0081401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rocha L, Cano A, Cruz C, et al. Opioid peptide systems following a subconvulsant dose of pentylenetetrazol in rats. Epilepsy Res. 1999;37(2):141–50. doi: 10.1016/s0920-1211(99)00056-x. [DOI] [PubMed] [Google Scholar]
- 24.Przewłocka B, Machelska H, Lasoń W. Kappa opioid receptor agonists inhibit the pilocarpine-induced seizures and toxicity in the mouse. Eur Neuropsychopharmacol. 1994;4(4):527–33. doi: 10.1016/0924-977x(94)90302-6. [DOI] [PubMed] [Google Scholar]
- 25.Yajima Y, Narita M, Takahashi-Nakano Y, et al. Effects of differential modulation of mu-, delta- and kappa-opioid systems on bicuculline-induced convulsions in the mouse. Brain Res. 2000;862(1–2):120–6. doi: 10.1016/s0006-8993(00)02096-5. [DOI] [PubMed] [Google Scholar]
- 26.Loacker S, Sayyah M, Wittmann W, et al. Endogenous dynorphin in epileptogenesis and epilepsy: anticonvulsant net effect via kappa opioid receptors. Brain. 2007;130(Pt 4):1017–28. doi: 10.1093/brain/awl384. [DOI] [PubMed] [Google Scholar]
- 27.Tortella FC, Robles L, Holaday JW. U50,488, a highly selective kappa opioid: Anticonvulsant profile in rats. J Pharmacol Exp Ther. 1986;237(1):49–53. [PubMed] [Google Scholar]
- 28.Jones D, Hallyburton I, Stojanovski L, et al. Identification of a κ-opioid agonist as a potent and selective lead for drug development against human African trypanosomiasis. Biochem Pharmacol. 2010 doi: 10.1016/j.bcp.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kishioka S, Kiguchi N, Kobayashi Y, et al. Pharmacokinetic evidence for the long-lasting effect of nor-binaltorphimine, a potent kappa opioid receptor antagonist, in mice. Neurosci Lett. 2013;552:98–102. doi: 10.1016/j.neulet.2013.07.040. [DOI] [PubMed] [Google Scholar]
- 30.Queenan BN, Lee KJ, Tan H, et al. Mapping homeostatic synaptic plasticity using cable properties of dendrites. Neuroscience. 2016;315:206–16. doi: 10.1016/j.neuroscience.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armbruster BN, Li X, Pausch MH, et al. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Nat Acad Sci. 2007;104(12):5163–8. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander GM, Rogan SC, Abbas AI, et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63(1):27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weber E, Roth KA, Barchas JD. Immunohistochemical distribution of alpha-neo-endorphin/dynorphin neuronal systems in rat brain: evidence for colocalization. Proc Nat Acad Sci. 1982;79(9):3062–6. doi: 10.1073/pnas.79.9.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chavkin C, Bakhit C, Weber E, et al. Relative contents and concomitant release of prodynorphin/neoendorphin-derived peptides in rat hippocampaus. Proc Nat Acad Sci. 1983 Dec;80:7669–73. doi: 10.1073/pnas.80.24.7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGinty JF, Henriksen SJ, Goldstein A, et al. Dynorphin is contained within hippocampal mossy fibers: immunochemical alterations after kainic acid administration and colchicine-induced neurotoxicity. Proc Nat Acad Sci. 1983;80(2):589–93. doi: 10.1073/pnas.80.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yakovleva T, Bazov I, Cebers G, et al. Prodynorphin storage and processing in axon terminals and dendrites. FASEB J. 2006;20(12):2124–6. doi: 10.1096/fj.06-6174fje. [DOI] [PubMed] [Google Scholar]
- 37.Gall C. Seizures induce dramatic and distinctly different changes in enkephalin, dynorphin, and CCK immunoreactivities in mouse hippocampal mossy fibers. J Neurosci. 1988;8(6):1852–62. doi: 10.1523/JNEUROSCI.08-06-01852.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goddard G. Development of epileptic seizures through brain stimulation at low intensity. Nature. 1967:1020–1. doi: 10.1038/2141020a0. [DOI] [PubMed] [Google Scholar]
- 39.Racine RJ. Kindling: the first decade. Neurosurgery. 1978;3(2):234–52. doi: 10.1227/00006123-197809000-00018. [DOI] [PubMed] [Google Scholar]
- 40.Morimoto K, Fahnestock M, Racine RJ. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol. 2004;73(1):1–60. doi: 10.1016/j.pneurobio.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 41.Manocha A, Mediratta PK, Sharma KK. Studies on the anticonvulsant effect of U50488H on maximal electroshock seizure in mice. Pharmacol Biochem Behav. 2003;76(1):111–7. doi: 10.1016/s0091-3057(03)00218-1. [DOI] [PubMed] [Google Scholar]
- 42.Löscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20(5):359–68. doi: 10.1016/j.seizure.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Al-Hasani R, Bruchas MR. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology. 2011;115(6):1. doi: 10.1097/ALN.0b013e318238bba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lefkowitz RJ, Shenoy SK. Transduction of Receptor Signals by beta-Arrestins. Science. 2005;308(5721):512–7. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SupplementaryFigure 1. In vitro identification of DG-CA3 proximal synapses. (A) Schematic of in vivo hippocampal connections. Note the large size of the DG terminals targeting the proximal dendrites of CA3 neurons. (B) Representative image of a GFP-transfected DG neuron. Note how the DG axon wraps around the soma of nearby untransfected neurons (red triangles). (C-E) Schematic (C) and representative examples (D-E) of experimental protocol: Mature (>21 DIV) primary hippocampal neurons are transfected with GFP. DG neurons (D) can be easily identified by small soma size and few, short dendrites (as well as nuclear staining with CTIP2). DG axons can be easily traced to the proximal dendrites of target cells (E) and unambiguously identified by staining with the presynaptic vesicle protein synaptoporin (SPO). PSD-95 accumulation on the opposing postsynaptic cell can be readily quantified. Scale bars = 20 μm.
Supplementary Figure 2. Synaptoporin is localized to presynaptic glutamatergic vesicles in vitro. (A) Schematic of images visualized in panels B–H. The proximal dendrites of CA3 neurons in vitro were imaged using conventional fluorescence microscopy (B) or stimulated emission depletion (STED) super-resolution microscopy (C-H). Boxes in A indicate the relative sizes of the subsequent panels. (B) Cultured hippocampal neurons immunostained with synaptoporin (SPO) and PSD-95. A representative CA3 neuron is shown with soma indicated by dotted circle. (C–H) Super-resolution images of the proximal dendrites of CA3 neurons in vitro. Soma indicated with dotted white line. To determine the intracellular localization of SPO, neurons were immunolabeled with SPO and either the postsynaptic scaffolding protein PSD-95 to label all excitatory postsynaptic terminals (C,F); the presynaptic scaffolding protein Bassoon to label all presynaptic terminals (D,G); or the vesicular glutamate transporter 1 (vGlut1) to label all presynaptic vesicles containing the excitatory neurotransmitter glutamate (E,H). (F–H) Magnified images of the images in C–E. Note the precise co-localization of SPO with vGlut1 on glutamatergic vesicles. Scale bars = 20 μm (B), 2 μm (C–E), 1 μm (F–H).
Supplementary Figure 3. Presynaptic DG inactivity drives elaboration of DG-CA3 synapses. Mature (>21 DIV) primary hippocampal neurons are transfected with various constructs – namely GFP control (A), hM3Dq (Gq) DREADD-GFP (B), hM4Di (Gi) DREADD-GFP (C), or Gs DREADD-GFP (D) constructs. Neurons were then left untreated (NT, top in A–D) or treated with CNO to activate the relevant G-protein cascade (bottom in A–D). For analysis, we identified DG neurons transfected with GFP-DREADDs and traced these GFP-filled terminals to their un-transfected target neurons. (A–D) Representative images of the un-transfected targets of the DREADD-transfected DG neurons. Neurons were immunostained for GFP (green) to reveal transfected DG neurons, synaptoporin (SPO, blue) to label presynaptic DG terminals, and PSD-95 (red) to label excitatory synapses. Scale bars, 20 μm.
Supplementary Figure 4. Opioid receptor (OR) signaling collectively constrains DG-CA3 proximal synapses. (A–C) Representative images of CA3 neurons (from >21 DIV hippocampal cultures) incubated for 24 hours under normal activity conditions (NT), hyperactivity (PTX) or inactivity (TTX) in the absence (top) or presence (bottom) of the broad-spectrum OR antagonist, naloxone. DG axon terminals and excitatory synapses were visualized with SPO (blue) and PSD-95 (red), shown in overlay. (B-C) Representative linearized dendrites of CA3 hippocampal neurons from conditions indicated. (C) Representative examples of proximal (<20 μm from soma) and distal (>60 μm) CA3 dendrites used for analysis in Figures 4–6.
Supplementary Figure 5. Chronic norBNI treatment blocks epileptogenesis in rats, but does not alter SPO expression. Adult male mice (N=5/group) were kindled once daily. 1 to 3 hours after kindling, mice received daily i.p. injections of either saline or kappa OR antagonist norBNI (5 mg/kg/day). At the end of the study, animals were perfused and brains were immunostained to visualize SPO expression in the hippocampus (D). (A-C) Seizure behavior was visually scored in blinded manner using a modified Racine scale (Table S3). (A) Mean seizure score ± SD between groups. 2 session averages for each animal were pooled for group analysis. Time: P <0.0001, Treatment: P=0.0584, Interaction +P=0.0226, two-way ANOVA (time v. treatment). **P<0.01, Bonferroni post-test v. NT. (B) Mean seizure score ± SD overlaid with individual animal scores on last day of kindling (Mann Whitney; * P = 0.0350). (C) Mean seizure burden (total seizure score) ± SD overlaid with individual animal scores on last day of kindling (Mann Whitney; * P = 0.0356). (D–J) After kindling, this cohort of animals was perfused and brains were stained for synaptoporin (SPO, green) to visualize MF terminals and DAPI (blue) to visualize cell nuclei. (D–E) Representative images of hippocampi and hippocampal regions. Regions in E are indicated as boxes in D. Scale bars = 100 um (D), 25 um (E). (F–H) Quantification of SPO integrated intensity (IR, quantified as arbitrary units (a.u.) as percent of background IR). SPO immunostaining was quantified in the granule cell layer and inner molecular layer of the dentate gyrus (DG, F), the hilus (G), and the stratum pyramidale of the CA3 region (H). Dotted lines delineate regions used for quantification. ns = P>0.05, Mann Whitney t-test. (I–K) Within-animal analysis of seizure burden (total seizure score over all kindling sessions) versus SPO immunoreactivity in the dentate gyrus (I), hilus (J), and CA3 (K). We observed no significant relationship between SPO immunoreactivity and seizure burden in either the DG (P=0.619) or CA3 (P=0.165). However, SPO immunoreactivity in the hilus was negatively correlated with seizure burden: the best-fit linear approximation (blue) had a slope that was significantly non-zero (P=0.026) and reasonably fit the data (R2 = 0.053) upon linear regression. Dotted lines in I–K indicate 95% confidence intervals.
Supplementary Figure 6. Proposed model of opioid receptor regulation of MF-CA3 synapses. Schematic model of opioids on the putative MF-CA3 “gain control” synapse. The DG-CA3 synapse is represented as a “gate” that controls activity flow (lightning bolts) through the hippocampus. Under basal activity conditions (middle), mu and delta receptors oppose gate opening, while kappa receptors promote it. Sustained bouts of inactivity in the dentate gyrus (left) increase the accumulation of presynaptic opioids which promote kappa OR signaling, leading to homeostatic upregulation of DG-CA3 synapses. Conversely, high DG activity (right) leads to the accumulation of presynaptic opioids which promote mu and delta OR activity, thus downregulating this synapse. We propose that in some forms of mTLE, the endogenous upregulation program becomes uncoupled from network activity (bottom panels = Pathological). Thus, when network activity returns to normal, the DG-CA3 “gate” remains open, allowing for runaway excitation. Antagonizing the kappa OR (e.g., with norBNI) during this period prevents this inappropriate program of DG-CA3 strengthening, thus limiting the emergence of seizures. Meanwhile, enhancing kappa OR signaling (with U50488) exacerbates the maladaptive opening of the DG-CA3 gate.