

ABSTRACT
Epstein-Barr virus (EBV) expresses several mRNAs produced from intronless genes that could potentially be unfavorably translated compared to cellular spliced mRNAs. To overcome this situation, the virus encodes an RNA-binding protein (RBP) called EB2, which was previously found to both facilitate the export of nuclear mRNAs and increase their translational yield. Here, we show that EB2 binds both nuclear and cytoplasmic cap-binding complexes (CBC and eukaryotic initiation factor 4F [eIF4F], respectively) as well as the poly(A)-binding protein (PABP) to enhance translation initiation of a given messenger ribonucleoparticle (mRNP). Interestingly, such an effect can be obtained only if EB2 is initially bound to the native mRNPs in the nucleus. We also demonstrate that the EB2-eIF4F-PABP association renders translation of these mRNPs less sensitive to translation initiation inhibitors. Taken together, our data suggest that EB2 binds and stabilizes cap-binding complexes in order to increase mRNP translation and furthermore demonstrate the importance of the mRNP assembly process in the nucleus to promote protein synthesis in the cytoplasm.
IMPORTANCE Most herpesvirus early and late genes are devoid of introns. However, it is now well documented that mRNA splicing facilitates recruitment on the mRNAs of cellular factors involved in nuclear mRNA export and translation efficiency. To overcome the absence of splicing of herpesvirus mRNAs, a viral protein, EB2 in the case of Epstein-Barr virus, is produced to facilitate the cytoplasmic accumulation of viral mRNAs. Although we previously showed that EB2 also specifically enhances translation of its target mRNAs, the mechanism was unknown. Here, we show that EB2 first is recruited to the mRNA cap structure in the nucleus and then interacts with the proteins eIF4G and PABP to enhance the initiation step of translation.
KEYWORDS: Epstein-Barr virus, EB2, BMLF1, translation, mRNP, CBC, PABP, eIF4G
INTRODUCTION
Epstein-Barr virus (EBV) is a ubiquitous human gammaherpesvirus that is the causative agent of infectious mononucleosis. It has been associated with the development of B-cell and epithelial malignancies such as Burkitt's lymphoma, Hodgkin's disease, undifferentiated nasopharyngeal carcinoma, a subtype of gastric carcinoma, and posttransplant lymphoproliferative disorders (1). After primary infection, EBV persists in a lifelong latent state in the memory B lymphocytes of infected individuals, with intermittent viral production from differentiated oropharyngeal epithelial cells, B-cell receptor-activated B cells, and plasma cells (2–5). These phases of viral reactivation are considered to be a risk factor for the emergence of EBV-associated malignancies (6).
In contrast with higher eukaryote genes, most herpesviruses early and late genes are intronless. Since splicing plays a very important role in mRNA export and translation (7, 8), efficient expression of intronless herpesvirus genes requires that a specific mechanism is used for their mRNA export and translation. In the case of EBV, nuclear export of intronless viral mRNAs is facilitated by the essential virus-encoded RNA-binding protein (RBP) EB2, also called SM, Mta, or BMLF1 (9–12). EB2 shuttles between the nucleus and the cytoplasm (13) and directly interacts with both the export factor NXF1 and the export adapter Aly/REF (13, 14) to promote the nuclear export of intronless viral mRNAs (15–18).
Every human herpesvirus expresses an EB2 homolog: ICP27 for herpes simplex virus 1 (HSV-1), ORF57 for Kaposi's sarcoma-associated herpesvirus (KSHV), and UL69 for human cytomegalovirus (HCMV). All members of this family of regulatory proteins share an ability to bind RNA and have been implicated in various aspects of RNA processing, including mRNA stabilization, splicing regulation, polyadenylation, and export (19–21). However, their functions are not interchangeable (11, 19). Some of these proteins may also play a role in viral gene transcriptional activation (22, 23). In addition, these proteins also have important functions in mRNA translation. However, they appear to use different mechanisms. ORF57 from KSHV is able to enhance the translation of an intronless KSHV mRNA by recruiting the cellular factor PYM. PYM's depletion results in a marked reduction of ORF57's association with components of the translational machinery (24). Those authors thus proposed a model suggesting that the ORF57-PYM complex makes a bridge between the 40S ribosomal subunit and initiation factors. UL69 from HCMV stimulates viral mRNA translation through its interactions with the translation initiation factor eukaryotic initiation factor 4A (eIF4A) and the poly(A)-binding protein (PABP) (25). Several studies have shown that ICP27 from HSV-1 also enhances translation of unspliced viral mRNAs (26, 27), and a recent report showed that ICP27 targets PABP and eIF4G to promote translation initiation (28). Regarding EB2 from EBV, it has been previously demonstrated that besides its role as a nuclear export factor, it also strongly stimulates translation of unspliced mRNAs without affecting overall cellular translation (29). Moreover, EB2 associates with translating ribosomes and increases the proportion of its target RNAs in the polyribosomal fraction (29). However, whether EB2 enhances intronless mRNA translation through mechanisms similar to those used by the ORF57, UL69, or ICP27 protein remained completely unknown.
In eukaryotic cells, mRNAs interact with numerous RNA-binding proteins (RBPs). Together, the RNA and its associated RBPs form messenger ribonucleoparticles (mRNPs) (30, 31). The composition of mRNPs is highly dynamic, and understanding how the RBPs influence each step of mRNA biogenesis is an important question. The first RBP loaded onto the mRNA during transcription is the cap-binding complex (CBC). This complex is very important for the subsequent recruitment of other complexes, such as the spliceosome (32, 33), the transcription/export (TREX) complex (34), or the RNA decay machinery (35). In addition, after maturation of the mRNA, CBC translocates to the cytoplasm in association with the mRNP and localizes to the polysomes, where it supports the first round of translation (36–38). During this step, there is an extensive remodeling of mRNP's composition, starting with the replacement of CBC by the eIF4E cap-binding protein (38, 39).
Translation initiation is the most highly regulated phase of the translation cycle (40, 41). Most eukaryotic mRNAs are tagged by the cap structure and a poly(A) tail at their 5′ and 3′ ends, respectively. The assembly of the translation machinery requires at least 11 eukaryotic initiation factors (eIFs) and is enhanced by the cytoplasmic poly(A)-binding protein (PABPC1 or PABP). In the cytoplasm, PABP binds to the mRNA 3′ poly(A) tails through its RNA recognition motifs (RRMs) and interacts with the N terminus of the eIF4G protein. The interaction of PABP with both the mRNA and eIF4G causes the mRNA to form a closed-loop intermediate, thereby stabilizing the mRNA and promoting ribosome recruitment and translation initiation (42, 43). The eIF4F complex composed of eIF4E (a small cap-binding protein), eIF4A (a DEAD box RNA helicase), and the central factor eIF4G (a large scaffold protein) anchors the 43S preinitiation complex (consisting of eIF3, eIF1, eIF1A, eIF2-GTP-tRNAiMet, eIF5, and the 40S subunit of the ribosome) to the 5′ end of the mRNA through the interaction between eIF3 and eIF4G. Once assembled at the cap, the helicase activity of the eIF4F complex unwinds secondary structures in the 5′ untranslated region (5′UTR) to facilitate ribosomal scanning until it encounters an AUG start codon. At this point, the preinitiation complex is joined by the 60S ribosomal subunit, and translation elongation of the mRNA begins (44, 45).
Several observations suggest that mRNA splicing enhances translation due to the deposition in the nucleus of the exon junction complex (EJC) on the mRNA (46–50). As discussed above, most early and late herpesvirus genes are intronless, and the translation of their mRNAs is enhanced by a specific viral factor such as the EB2 protein from EBV (29). However, how EB2 contributes to the translation of mRNAs transcribed from intronless genes has been unknown. Here, we demonstrate that EB2 associates with both the nuclear cap-binding complex (CBC) and the cytoplasmic eIF4F-PABP complex. EB2 interacts with both PABP and the N-terminal part of eIF4G, via its C-terminal region. The resulting complex loaded onto the 5′ cap structure of the mRNA promotes translation initiation. Moreover, by using a recently published in vitro translation method that we have developed (51), we show that EB2 needs to be associated with its target mRNPs in the nucleus to stimulate their translation in the cytoplasm and that EB2-associated mRNPs are resistant to translation initiation inhibitors. Importantly, this work provides the first mechanistic model explaining how EB2 ensures the efficient translation of intronless mRNAs.
RESULTS
EB2 is associated with both nuclear and cytoplasmic CBCs.
We have previously shown that EB2 can stimulate nuclear export and translation of viral mRNAs produced from intronless genes (13, 18, 29). To understand how EB2 can stimulate translation, a Flag-tagged EB2 protein (Flag-EB2) was purified, by two sequential immunoprecipitations in the presence of RNase, from an EBV-infected cell line (HEK293BMLF1-KO cells) (11) transfected with a Flag-EB2 expression plasmid following induction of the viral productive cycle via ectopic expression of the viral transcription factor EB1. EB2 cellular partners were then identified by mass spectrometry (MS). MS data analysis identified numerous EB2 cellular partners, many of which are involved in RNA metabolism (Table 1). Some of the proteins identified were already known to interact with EB2, such as the splicing factors SRSF2, SRSF3, and SRSF7 (18, 52) and the RBM15 protein (53). Interestingly, among the newly identified proteins was the cytoplasmic poly(A)-binding protein PABPC1 (referred to as PABP in this study) (Table 1). As the purification was performed in the presence of RNase, this suggests that the interaction is not mediated by RNA. Since PABP is known to play an essential role in translation due to its association with the eIF4F cap-binding complex (CBC), the interaction of EB2 with this protein suggests that EB2 could be an active component of the translation initiation machinery.
TABLE 1.
Cellular proteins interacting with EB2a
| Category and UniProt accession no. | Protein | Description | Coverage (%) | Reference |
|---|---|---|---|---|
| RNA metabolism | ||||
| Q0VCY7 | SRSF1 | Serine/arginine-rich splicing factor 1 | 17.34 | 18 |
| Q3SZR8 | SRSF3 | Serine/arginine-rich splicing factor 3 | 23.17 | 18 |
| Q3TWW8 | SRSF6 | Serine/arginine-rich splicing factor 6 | 9.14 | |
| Q99020 | hnRNPA/B | Heterogeneous nuclear ribonucleoprotein A/B | 12.98 | |
| P52597 | hnRNPF | Heterogeneous nuclear ribonucleoprotein F | 18.31 | |
| P31943 | hnRNPH | Heterogeneous nuclear ribonucleoprotein H | 43.43 | |
| P02057 | hnRNPK | Heterogeneous nuclear ribonucleoprotein K | 25.29 | |
| P52272 | hnRNPM | Heterogeneous nuclear ribonucleoprotein M | 8.77 | |
| Q00839 | hnRNPU | Heterogeneous nuclear ribonucleoprotein U | 21.09 | |
| Q4R7L5 | DDX1 | ATP-dependent RNA helicase DDX1 | 10.41 | |
| O00571 | DDX3X | ATP-dependent RNA helicase DDX3X | 32.78 | |
| P17844 | DDX5 | Probable ATP-dependent RNA helicase DDX5 | 22.31 | |
| Q501J6 | DDX17 | Probable ATP-dependent RNA helicase DDX17 | 24.15 | |
| O75533 | SF3B1 | Splicing factor 3B subunit 1 | 3.74 | |
| Q13435 | SF3B2 | Splicing factor 3B subunit 2 | 2.68 | |
| A1A4K8 | U2AF35 | Splicing factor U2AF 35-kDa subunit | 29.11 | |
| Q96T37 | RBM15 | Putative RNA-binding protein 15 | 3.99 | 53 |
| P29341 | PABPC1 | Polyadenylate-binding protein 1 | 11.64 | |
| Other | ||||
| P08775 | RBP1 | DNA-directed RNA polymerase II subunit RPB1 | 24.20 | |
| Q8TAQ2 | SMARCC2 | SWI/SNF complex subunit SMARCC2 | 18.4 | |
| P35658 | Nup214 | Nuclear pore complex protein Nup214 | 1.67 |
Coimmunoprecipitation experiments were conducted with Flag-EB2 expressed in HEK293BMLF1-KO cells. After a first immunoprecipitation using an anti-Flag antibody resin, all samples were treated with RNase A. They were then subjected to a second immunoprecipitation using a specific antibody against EB2. Protein complexes recovered after this second immunoprecipitation were analyzed by MS.
To validate this hypothesis, S7 nuclease-treated cytoplasmic extracts from HeLa cells previously transfected with a Flag-tagged EB2 expression vector were passed through an m7GTP Sepharose affinity matrix, and after extensive washes, proteins bound to the matrix were eluted and analyzed by Western blotting. Interestingly, Flag-EB2 was retained on the m7GTP Sepharose affinity matrix together with the eIF4F components eIF4E and eIF4G (Fig. 1A). The interaction between Flag-EB2 and the m7GTP matrix was likely to be mediated by eIF4F, since preincubation of the cell extract with the m7GpppG cap analog to titrate eIF4E also titrated EB2 (Fig. 1A, lane 6). To completely exclude the possibility that EB2 directly binds to m7GTP, the protein was purified from a bacterial extract and incubated with an m7GTP Sepharose affinity matrix. The amount of EB2 retained on the m7GTP was then analyzed by Western blotting. The results show that all the protein charged on the m7GTP Sepharose affinity matrix was recovered in the flowthrough, confirming that there is no direct interaction between EB2 and m7GTP (Fig. 1A, bottom panel).
FIG 1.

EB2 is an mRNP cap-associated protein. (A) Top, 100 μg of S7 nuclease-treated cytoplasmic HeLa cell extracts were incubated with an m7GTP cap affinity matrix. Cell extracts were incubated in the absence or presence of m7GpppG cap analog (lanes 3 to 6), and proteins retained on the column were analyzed by Western blotting. Input (lanes 1 and 2) corresponds to 50 μg of cytoplasmic cell extract. Bands shown in each panel correspond to the same Western blot but were cropped for simplicity. Bottom, EB2 protein purified from bacteria was incubated with an m7GTP cap affinity matrix, and proteins bound to the column (lane 4) or from the flowthrough (lane 6) were analyzed by Western blotting. (B and C) HeLa total cell extracts expressing Flag-tagged EB2 were subjected to immunoprecipitation (IP) using an anti-Flag M2 affinity gel in the absence (lanes 3 and 4) or presence (lanes 5 and 6) of RNase A, and associated proteins were analyzed by Western blotting. Input (lanes 1 and 2) corresponds to 1/10 of cell extracts.
The interaction between EB2 and several translation initiation factors, including the eIF4F components eIF4E and eIF4G, was then tested by coimmunoprecipitation assays using HeLa cells expressing Flag-tagged EB2 (Fig. 1B). As expected from the MS analysis of EB2-associated proteins, PABP interacted with EB2. Similarly, eIF4G interacted with EB2, but none of the other initiation factors (eIF2, eIF3, eIF4A, eIF4B, or eIF4E) or the ribosomal proteins (rpS6 for the 40S subunit and rpL11 for the 60S subunit) were found to be associated with EB2. Although RNase treatment slightly decreased the amount of proteins coimmunoprecipitated with Flag-EB2, these interactions appear to be mostly resistant to RNase treatment, showing that they are not mediated by RNA. Quantitation of the bands in the gel shows that after RNase A treatment, 66% of the PABP- and 57% of the eIF4G-interacting complexes are resistant to the treatment. This shows that although RNase treatment can destabilize some of the EB2-PABP or EB2-eIF4G complexes, most of the interaction is preserved. The RNase used in this experiment was efficient, since the interaction between EB2 and NXF1, previously shown to be RNA dependent (14), was clearly sensitive to RNase treatment. Quantitation of the bands in the gel shows that after RNase A treatment, 17% of the NXF1-EB2 complex is still visible (in accordance with our previous published results). These results suggest that EB2 interacts with the PABP and eIF4G components of the cytoplasmic cap-binding complex.
Whether EB2 could also interact with the nuclear cap-binding complex (CBC) is of interest since EB2 is mostly a nuclear protein with important functions in pre-mRNA splicing and mRNA export (13). This was tested by coimmunoprecipitation experiments using HeLa cells expressing Flag-tagged EB2. As shown in Fig. 1C, endogenous CBP80 was efficiently coimmunoprecipitated with EB2, and again, this interaction was resistant to RNase treatment. Taken together, these results indicate that EB2 can associate with both the nuclear and cytoplasmic cap-binding complexes (CBC and eIF4F, respectively).
The C-terminal domain of the EB2 protein interacts in vitro with the translation initiation factors eIF4G and PABP.
EB2 has been previously found to be associated with cellular polysomes and to enhance the translation of intronless mRNAs (29). The association of EB2 with the cap-binding complexes suggests that these interactions could play an important role in EB2-dependent stimulation of mRNA translation. To confirm that EB2 interacts directly with both PABP and eIF4G and to identify the domain of EB2 involved in these interactions, we performed in vitro binding assays using either glutathione S-transferase (GST)–EB2 or truncated GST-EB2 fusion proteins produced in bacteria (Fig. 2A), together with [35S]methionine-labeled PABP or eIF4G, produced in vitro in a coupled transcription-translation rabbit reticulocyte system.
FIG 2.

EB2 interacts in vitro with the eIF4G and PABP initiation factors through its C-terminal domain. (A) Schematic representation of both recombinant GST-EB2 fusion proteins and the eIF4G plus eIF4G deletion mutants used. (B) GST pulldown assays were performed using GST, GST-EB2, GST-EB2Nter, or GST-EB2Cter and [35S]methionine-radiolabeled proteins eIF4GNter, eIF4Gp100, PABP, or eIF4E, synthesized in vitro in a rabbit reticulocyte lysate (RRL), under untreated or RNase-treated conditions as indicated. (C) GST pulldown assays were also performed with GST or GST-PABP fusion proteins and [35S]methionine-radiolabeled proteins EB2 (full length), EB2Nter, or EB2Cter, synthesized in vitro in RRL, under untreated or RNase-treated conditions. Eluates were subjected to SDS-PAGE and analyzed by autoradiography. In lane 1, the equivalent of 1/10 of the total RRL used in each assay was loaded onto the gel as an input control.
In a first series of experiments, 35S-labeled eIF4GNter or eIF4Gp100 (Fig. 2B) was incubated with the GST, GST-EB2, GST-EB2Nter, or GST-EB2Cter protein bound to glutathione-Sepharose beads, with or without RNase treatment. As shown in Fig. 2B, eIF4GNter interacted with GST-EB2 and GST-EB2Cter but not with GST or GST-EB2Nter, while eIF4Gp100 did not significantly bind any of these proteins. RNase treatment did not change the interaction between EB2 and eIF4GNter, demonstrating that the interaction was not dependent on RNA. From these results, we conclude that the main domains involved in the interaction between EB2 and eIF4G are located in the C-terminal half of EB2 and the N-terminal part of eIF4G, respectively.
In a second series of experiments, 35S-labeled PABP or eIF4E was incubated with the GST-EB2 series of proteins bound to glutathione-Sepharose beads (Fig. 2B). Complementary experiments using a GST-PABP fusion protein produced in bacteria and in vitro-translated 35S-labeled EB2, EB2Nter, or EB2Cter were also performed (Fig. 2C). Interestingly, as was the case in our coimmunoprecipitation assays, eIF4E did not interact in vitro with the GST-EB2 proteins in the pulldown assays (Fig. 2B), whereas PABP interacted with both EB2 and EB2Cter, but not EB2Nter, in an RNase-resistant manner. This result argues strongly in favor of a direct EB2 interaction with both eIF4G and PABP.
Taken together, our data suggest that by directly interacting with eIF4G on one hand and PABP on the other, EB2 could stabilize the PABP-eIF4F complex, thereby promoting more efficient translation initiation.
EB2 stimulates in vitro translation of its associated mRNPs.
We have shown previously that EB2 stimulates translation of reporter genes driven by different heterologous promoters (26). To definitively prove that the effect of EB2 on translation is not dependent on regulatory sequences present in the promoter used, the Renilla luciferase (Luc) reporter gene was cloned under the control of two different viral promoters: the promoter of the BMRF1 early gene (pBMRF1), an EBV gene which is not affected by EB2, and the promoter of the BDLF1 late gene (pBDLF1), an EBV gene which is highly dependent on EB2 for its cytoplasmic RNA accumulation and translation (15, 29) (Fig. 3A). First, cytoplasmic mRNA was extracted from HEK293BMLF1-KO cells transfected with an EB1 expression vector together with various quantities of an EB2 expression vector, and the relative amounts of BMRF1 or BDLF1 mRNA were quantified by reverse transcription-quantitative PCR (RT-qPCR). As expected, the amounts of endogenous cytoplasmic BMRF1 mRNA and protein were not affected by EB2 (Fig. 3B), whereas the amount of endogenous cytoplasmic BDLF1 mRNA was dependent on the amount of EB2 expressed (Fig. 3B). Unfortunately, as we have already published, it is very difficult, if even possible, to detect the expression of endogenous EB2-dependent proteins in the absence of EB2 (15). These results confirm that EB2 has no impact on the expression or cytoplasmic accumulation of BMRF1 mRNA, whereas it increases the level of BDLF1 mRNA accumulated in the cytoplasm. In contrast, when cells were cotransfected with the pBMRF1-Luciferase or pBDLF1-Luciferase reporter plasmid, we observed that the cytoplasmic accumulation of Renilla luciferase mRNA (Fig. 3C and D, panels a) and, as a consequence, the luciferase activity (Fig. 3C and D, panels b) were both enhanced by EB2, independently of the promoter used to direct transcription of the reporter gene. Next, the relative luciferase activity was compared with the amount of luciferase mRNA expressed in the cytoplasm of the cells in order to evaluate translation efficiency (Fig. 3C and D, panels c). It is clear that in both cases, EB2 stimulates translation of the luciferase mRNA, regardless of the promoter used to drive luciferase expression. Taken together, these results demonstrate that the effect of EB2 on the level of protein expressed is not dependent on the promoter controlling the expression of the gene.
FIG 3.

Stimulation of mRNA translation is independent of the promoter used to control the expression of the reporter mRNA. (A) Schematic representation of the reporter constructs used. The reporter plasmids carry the Renilla luciferase-coding sequence preceded by the β-globin 5′UTR (glo-RLuc) under the control of the viral BMRF1 or BDLF1 promoter. (B) Quantification by RT-qPCR of the amount of cytoplasmic endogenous BMRF1 and BDLF1 mRNA expressed in HEK293BMLF1-KO cells transfected with an EB1 expression vector together with different amounts of an EB2 expression vector. The Western blot shows the expression of the EB1 and EB2 proteins after transfection of the corresponding expression vectors in HEK293BMLF1-KO cells. Expression of the endogenous BMRF1 protein is also shown. (C and D) Panels a, mRNA quantification by RT-qPCR of the cytoplasmic luciferase-encoding mRNA expressed from the pBMRF1-RLuc (C) and pBDLF1-RLuc (D) reporter plasmids (using GAPDH as an internal control); panels b, total luciferase activity measured at 24 h posttransfection; panels c, translational efficiency calculated by normalizing the total luciferase activity by reference to the amount of cytoplasmic luciferase mRNA. The results are expressed as mean ± standard deviation (SD) from three independent experiments (n = 3). *, P < 0.05; **, P < 0.01 (two-tailed paired t test).
In order to gain insights into the molecular mechanisms by which EB2 promotes translation, we used a modified in vitro translation system, called the hybrid system, which utilizes rabbit reticulocyte lysate (RRL) deprived of endogenous ribosomes but complemented with a riboproteome isolated from heterogeneous cells (Fig. 4A) (51). We recently demonstrated that this hybrid system allows in vitro translation of purified mRNPs and that translation efficiency is sensitive to mRNA splicing and the mRNP composition (50). Since EB2 protein pellets predominantly with the ribosomal fraction (Fig. 4B), we took advantage of this new protocol to use EB2-containing riboproteomes and challenge our hypothesis in vitro. Briefly, HeLa cells were first transfected with the glo-RLuc reporter plasmid, together or not with an EB2 expression vector (Fig. 4A). Since the amount of Luc mRNA is known to be strongly increased in the presence of EB2, we used conditions under which similar cytoplasmic mRNA concentrations of Luc mRNA were accumulated in cells. These conditions were obtained when cells expressing EB2 were transfected with 0.5 μg of the glo-RLuc reporter plasmid and when cells not expressing EB2 were transfected with 6 μg of the reporter (Fig. 4C). Based on these conditions, mRNA expressed from HeLa cells previously transfected with the glo-RLuc reporter plasmid was purified and then added to the in vitro hybrid system reconstituted with a HeLa cell riboproteome containing EB2 (RH-EB2) or not (RH). The results clearly show that the mRNA purified from HeLa cells (or, as a control, in vitro-transcribed glo-RLuc mRNA) is translated with similar efficiency in both lysates (Fig. 4D).
FIG 4.

The RNA-binding protein EB2 stimulates translation of its associated mRNPs in a hybrid system. (A) Schematic representation of the reporter constructs used and the protocol followed. The reporter plasmid carries the Renilla luciferase gene driven by the β-globin 5′UTR (glo-RLuc) under the control of the CMV promoter. HeLa cells were initially transfected or not by the reporter plasmid. The cellular ribosomal fraction was then pelleted by ultracentrifugation at 36 h posttransfection. Part of the ribosomal fraction purified from HeLa cells that had been mock transfected (RH) or transfected with an EB2 expression plasmid (RH-EB2) was transferred to the supernatant of rabbit reticulocyte lysate (RRL) that had been depleted of its ribosomes to generate the in vitro translation hybrid system (51). The other part of the ribosomal fraction was used to extract total mRNA that was quantified by RT-qPCR. This purified naked mRNA fraction (containing the glo-RLuc mRNA) was transferred to the hybrid system, which was supplemented with cellular ribosomes RH or RH-EB2. The mRNP (glo-RLuc mRNP) containing the ribosome fraction was used to directly supplement the RRL depleted of its ribosomes. For both mRNP and mRNA, translation was then carried out for 30 min at 30°C before determination of Renilla activity. (B) EB2 detection by Western blotting using either the HeLa total cytoplasmic fraction (S10) or the ribosomal fraction (RH) or supernatant (S100), both obtained after ultracentrifugation of HeLa S10 through a sucrose cushion. (C) Different amounts (0.5 or 6 μg per 107 cells) of the glo-RLuc plasmid were transfected in HeLa cells in the absence or presence of EB2. At 36 h after transfection, cytoplasmic mRNA was purified and quantified by RT-qPCR. (D) In vitro-transcribed or cytoplasmic mRNA purified from HeLa cells transfected with the glo-RLuc plasmid was translated in the hybrid system containing a HeLa ribosomal fraction either in the absence (RH) or in the presence (RH-EB2) of EB2. (E) Ribosomal pellets containing mRNPs obtained from 6 μg of glo-RLuc or 0.5 μg of glo-RLuc plus EB2 were collected and translated directly in the hybrid system. For panels C to E, results are presented as mean ± SD from three independent experiments (n = 3). **, P < 0.01 (two-tailed paired t test).
This result suggests that EB2 may stimulate translation only if it is incorporated within mRNPs. To test this hypothesis, the in vitro hybrid system was reconstituted with similar amounts of luciferase mRNPs associated with ribosomes purified from HeLa cells expressing EB2 or not. It is clear that luciferase mRNPs purified from cells expressing EB2 (RH-EB2) are more efficiently translated than luciferase mRNPs purified from cells which do not express EB2 (RH) (Fig. 4E).
Taken together, these data show that EB2 stimulates translation only when it is associated with mRNPs. By virtue of its association with the CBC, the loading of EB2 onto the mRNPs probably takes place in the cell nucleus.
EB2-associated mRNPs are refractory to translation initiation inhibitors.
Our data strongly suggest that EB2 enhances translation of its target mRNAs by acting at the initiation step. In order to confirm this hypothesis, different translation initiation inhibitors were added to the hybrid system. We used either the m7GpppG cap analog, which titrates the initiation factor eIF4E, the drug hippuristanol, which inactivates the DEAD box RNA helicase eIF4A (54, 55), or the L protease from foot-and-mouth disease virus (FMDV), which cleaves the eIF4G initiation factor (56). All are strong inhibitors of cap-dependent, but not internal ribosome entry site (IRES)-driven, translation initiation. We also used two translation elongation inhibitors: puromycin, which induces ribosome dislocation from the mRNPs, and cycloheximide, which binds to the ribosome and inhibits its translocation.
In vitro translation of mRNAs purified from HeLa cells expressing a capped and polyadenylated Renilla luciferase reporter (glo-RLuc) was performed (Fig. 5A). In addition, we included two control mRNAs containing an IRES to monitor the specificity of the translation initiation inhibitors (Fig. 5A). One control mRNA (EMCV-RLuc), which is driven by the encephalomyocarditis virus (EMCV) IRES, is uncapped but polyadenylated and can initiate translation independently of eIF4E. The other mRNA (HCV-RLuc), which contains the hepatitis C virus (HCV) IRES, is uncapped and nonpolyadenylated and can initiate translation without any members of the eIF4F complex.
FIG 5.

EB2 strongly reduces the sensitivity of its associated mRNPs to translation initiation inhibitors. (A) Schematic representation of mRNA expressed from the constructs containing the Renilla luciferase reporter gene driven by either the 5′UTR of β-globin or the EMCV or HCV IRES. These were in vitro transcribed with a cap and a poly(A) tail in the case of the glo-RLuc construct, without a cap but with a poly(A) tail in the case of the EMCV-RLuc construct, or without a cap and a poly(A) tail for the HCV-RLuc construct. (B) For mRNAs, luciferase activity from 2.7 nM glo-RLuc mRNA translated for 30 min in the hybrid system containing ribosomes prepared from mock-transfected HeLa cells (RH) or cells transfected with an EB2 expression vector (RH-EB2) in the presence of 100 μM cap analog (m7GpppG) (panel a), 10 μM hippuristanol (panel b), or 0.2 μl of RRL expressing L protease (panel c) is shown. The EMCV-RLuc construct was used as a control in panels a and c, while in panel b the HCV-RLuc construct was used as a control. For mRNPs, luciferase activity from mRNPs associated with the ribosome pellet prepared from HeLa cells transfected with 6 μg or 0.5 μg of expression vector for the glo-RLuc reporter, respectively, without (RH) or with (RH-EB2) an expression vector for EB2 is shown. The translation was carried out for 30 min in the hybrid system in the presence of 100 μM cap analog (m7GpppG) (panel a), 10 μM hippuristanol (panel b), or 0.2 μl of RRL expressing L protease (panel c). The efficiency of cleavage of eIF4G by the L protease in both the RH and RH-EB2 lysates was verified by Western blotting (bottom panel). (C) Luciferase activity from mRNP associated with the ribosome pellet prepared from HeLa cells transfected with 6 μg or 0.5 μg of glo-RLuc expression vector, respectively, without (RH) or with (RH-EB2) an expression vector for EB2. Translation was carried out for 30 min in the hybrid system in the presence of different concentrations of puromycin (panel a) or cycloheximide (panel b). The results are expressed relative to the control, which was set to 100%, and they are presented as mean ± SD from three independent experiments (n = 3). *, P < 0.05; **, P < 0.01 (two-tailed paired t test).
Under the experimental conditions used, which were identical to those described above, translation of the mRNA glo-RLuc reporter was strongly inhibited by a high concentration of cap analog m7GpppG, whereas this treatment had no effect on the control EMCV-RLuc or HCV-RLuc mRNA, as can be expected for cap-independent translation targets (Fig. 5B, panel a). No difference was observed for the translation of glo-RLuc mRNA purified from lysates containing EB2 (RH-EB2) or not (RH). In contrast, the same concentration of m7GpppG cap analog had significantly less effect on mRNPs issuing from cells containing EB2 (RH-EB2) rather than their counterparts devoid of EB2 (RH) (Fig. 5B, panel a). It is important to note that, as described previously (50), translation inhibition is more important for mRNAs than for mRNPs, regardless of the presence of EB2. These results suggested that EB2-containing mRNPs are, to a certain extent, protected from m7GpppG cap analog inhibition. These results were confirmed by the use of the alternative translation initiation inhibitors hippuristanol (Fig. 5B, panel b) and FMDV L protease (Fig. 5B, panel c), which gave similar results. It should be noted (Fig. 5B, panel c) that luciferase translation from the EMCV IRES is enhanced in response to eIF4G cleavage, as expected since it has been shown that active eIF4E functions as a negative modulator of IRES-mediated translation by increasing competition from capped mRNAs for the eIF4F complex (57).
Drugs inhibiting the translation elongation step affected the rate of translation of the mRNPs containing EB2 (RH-EB2) or not (RH) to the same level (Fig. 5C), showing that the protective effect seen in the presence of the viral protein EB2 is specific for the initiation step. Taken together, these results demonstrate that the viral protein EB2, loaded onto the mRNPs, has a specific positive impact on translation initiation.
DISCUSSION
The role of the EB2 protein in nuclear export of unspliced mRNAs has been extensively studied (9, 11, 13–15, 18, 52, 53, 58, 59) and its effect on translation demonstrated (29), although the mechanism involved in the latter process has not been identified. We previously showed that EB2 interacts with intronless mRNAs and facilitates their accumulation in the cytoplasm, where EB2 is found associated with polysomes (18, 29). Here, we show that EB2 interacts with the nuclear cap-binding complex (CBC) and is present in the cytoplasm in a complex formed between the eIF4F cap-binding complex and PABP. This association appears to protect translation initiation from several inhibitors, such as the L protease, which cleaves the eIF4G protein, hippuristanol, which specifically inhibits eIF4A, and the cap analog, which titrates eIF4E and thus prevents its association with the capped mRNA. The effect of EB2 is specific for translation initiation, since the presence of the viral protein had no protective effect against treatments with either puromycin, which causes premature chain termination during translation, or cycloheximide, which exerts its effect by interfering with the translocation step in protein synthesis, thereby blocking translational elongation.
In an attempt to understand how EB2 stimulates mRNA translation, we first identified PABP by coimmunoprecipitation of EB2-associated partners and mass spectrometry analysis. PABP is known to enhance the recruitment of the small ribosomal subunit through its interaction with the eIF4G-eIF4E complex (40). The results of GST pulldown assays using purified and in vitro-translated proteins suggest that the interaction between EB2 and PABP is direct. In addition, we also found an interaction between EB2 and eIF4G, both in a coimmunoprecipitation assay and in a GST pulldown assay. Although both interactions appear to be mostly resistant to RNase treatment, we observed a slight decrease in the amount of coimmunoprecipitated proteins in the assay using HeLa cell extracts. It is thus possible that the presence of RNA stabilizes these interactions. RNA-protein interactions may modify the conformation of one of the proteins involved in the complex and hence stabilize the complex. Noticeably, the interaction observed between EB2 and eIF4G in the GST pulldown assay appears to be less efficient than that between EB2 and PABP, whereas both eIF4G and PABP appear to be very efficiently coimmunoprecipitated with EB2 from HeLa cell extracts. This suggests that PABP could stabilize the interaction between EB2 and eIF4G. In a recent publication, Smith et al. have reported an interaction between ICP27, the EB2 homologous protein from HSV-1, and PABP (28). Moreover, they have shown that although eIF4G and ICP27 do not seem to interact directly, eIF4G associates with ICP27 in the presence of PABP. Thus, both EB2 and ICP27 appear to associate with a PABP-eIF4G complex to enhance translation initiation.
Although viruses encode numerous proteins with a wide variety of functions, they remain completely dependent on the host cell translational machinery to synthesize viral polypeptides essential for their replication and propagation. Consequently, since viruses utilize host ribosomes to translate their mRNAs, they use different strategies to promote viral mRNA translation and to diminish cellular mRNA translation (60). Although nearly every step of the translation process can be targeted by virally encoded functions, viruses mostly target the initial step. Many RNA viruses, in particular picornaviruses, induce host translation shutoff by targeting eukaryotic initiation factors. For example, the L protease of FMDV cleaves eIF4G, and the poliovirus-encoded 3C protease cleaves PABP. In order to maintain viral mRNA translation after these proteolytic events, viral mRNAs possess a cis-regulatory element called the internal ribosome entry site (IRES) that allows their cap-independent translation initiation (61).
We show that in the case of DNA viruses such as herpesviruses, the strategy to ensure efficient translation of viral mRNAs during the productive cycle is different. Most viral mRNAs are produced from intronless genes. However, it is now clear that cellular pre-mRNA splicing and export factors are important modulators of spliced mRNA translation (7, 47). Consequently, all herpesviruses encode an RNA-binding protein (EB2, ICP27, ORF57, or UL69) that rescues viral intronless mRNAs for nuclear export and translation (13, 16, 62–64). All these factors are able to shuttle between the nucleus and the cytoplasm, where they localize within polysomes and enhance translation of their target mRNAs. However, the means by which they stimulate translation differ: ORF57 interacts with PYM to enhance the recruitment of the 40S ribosomal subunit (24), and UL69 interacts with both the RNA helicase eIF4A and PABP (25). Here, we show that EB2 interacts with the cap-binding complexes (CBC in the nucleus and eIF4F in the cytoplasm) and PABP to stabilize the capped structure of the mRNA. Interestingly, the same type of mechanism has recently been proposed for ICP27 (28), suggesting that these two particular viral proteins target the PABP-eIF4G complex to enhance translation initiation from intronless viral mRNAs. Since EB2 is also able to interact with the nuclear cap-binding complex, we can speculate that the nuclear loading of EB2 onto the target mRNA is required in order to facilitate the subsequent recruitment of PABP onto the cytoplasmic eIF4F cap-binding complex.
In contrast to ORF57 from KSHV, which is able to enhance translation of a viral intronless mRNA in vitro in a rabbit reticulocyte lysate (24), EB2 has no effect in this system. However, by using a new hybrid system to translate mRNPs in vitro (51), we have been able to show that EB2 needs to be loaded onto the mRNPs in the cell nucleus to exert a stimulatory effect on translation.
Taken together, these results have prompted us to propose a model (Fig. 6) in which the EB2 protein is first loaded onto the nascent mRNPs during transcription via an interaction with CBC. EB2 then promotes nuclear mRNP export through interaction with NXF1 (14). Finally, EB2 remains associated with the cytoplasmic mRNPs, probably through a transfer from CBC to eIF4G and PABP. Consequently, the translation initiation complex would be more stably associated with the mRNPs, leading to translation stimulation. The direct interactions mapped between eIF4G, PABP, and the C-terminal region of EB2 could render EB2-associated mRNPs less sensitive to translation initiation inhibitors.
FIG 6.
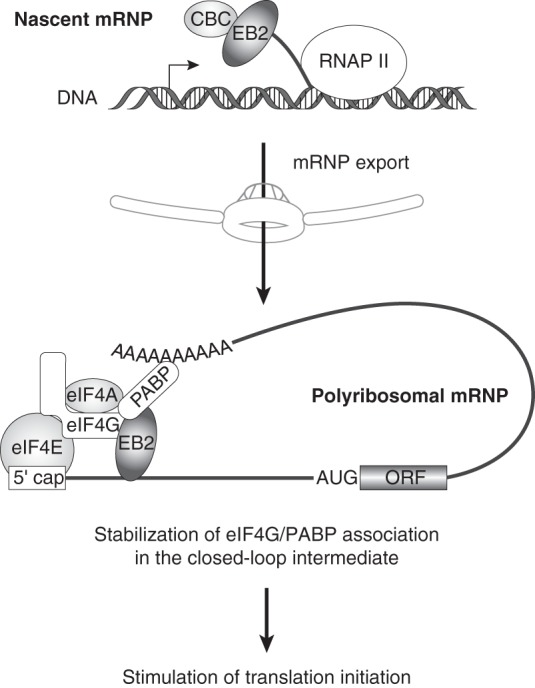
Model for translation initiation of EB2-associated mRNPs. EB2 protein is loaded onto the mRNP during the nuclear steps through its interaction with the CBC. In the cytoplasm, EB2 stabilizes the interaction between eIF4G and PABP and hence contributes to the stabilization of the closed-loop mRNA organization by binding both proteins via its C-terminal domain.
From a dynamic point of view, mRNPs are remodeled extensively following their synthesis in the nucleus to their decay in the cytoplasm, and the impact of mRNP composition on translation is difficult to access. It has been demonstrated that pre-mRNA splicing enhances mRNA translation through the deposition of the EJC complex, but the mechanisms are not fully understood (47). In a previous study we have shown a similar effect in vitro in the hybrid system, in which we observed that spliced transcripts were more efficiently translated than their unspliced counterparts (51). In this study, we show that EB2-associated mRNPs are more efficiently translated in vitro. This is a prime example that nuclear events that modify mRNP composition can strongly influence their translation efficiency.
MATERIALS AND METHODS
DNA constructs and in vitro transcription.
pcDNA-RLuc, pCMV-EB1, pCI-Flag-EB2, pGEX-EB2, pGEX-EB2Nter, pGEX-EB2Cter, pGEX-eIF4GNter pGEX-eIF4Gp100, and pGEX-PABP have been described previously (15, 17, 29, 65) as, have EMCV-RLuc and HCV-RLuc (50). The pBMRF1-RLuc and pBDLF1-RLuc constructs were generated by cloning a PCR fragment containing the BMRF1 and the BDLF1 viral promoters, respectively, in the pcDNA-RLuc expression vector digested by NruI and BamHI. mRNA was synthesized in vitro by using 1 μg of linear DNA template, 20 U of T7 RNA polymerase (Promega), 1.6 mM each ribonucleoside triphosphate (except for rGTP used at 0.32 mM), and 1.28 mM m7GpppG cap analog (New England BioLabs) in transcription buffer (40 mM Tris-HCl [pH 7.9], 6 mM MgCl2, 10 mM NaCl, 3 mM dithiothreitol [DTT], 2 mM spermidine), and RNasin (Promega). Transcription reactions were carried out at 37°C for 2 h and the mRNA precipitated with 2.5 M ammonium acetate after 1 h of treatment with 1 U of RQ1 DNase (Promega). The mRNA pellet was then resuspended in 30 μl RNase-free water and the mRNA concentration determined by absorbance using NanoDrop technology. mRNA integrity was checked by electrophoresis on nondenaturing 1% agarose gels.
Cell culture and transfection.
HeLa and HEK293BMLF1-KO (11) cells were grown at 37°C in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum and 1% penicillin-streptomycin plus 100 μg/ml hygromycin B in the case of HEK293BMLF1-KO. Plasmid transfections were performed using cationic polymers (JetPEI from Polyplus Transfection) as specified by the manufacturer. Cells were collected at 24 or 48 h after DNA transfection for further analysis.
HeLa cell ribosomal fraction isolation and ribosome depletion from reticulocyte lysates.
The detailed method has previously been described (51). Briefly, HeLa cells were pelleted by centrifugation at 1,000 × g for 5 min, resuspended in the same volume of hypotonic cell lysis buffer R [10 mM HEPES, 10 mM CH3CO2K, 1 mM (CH3CO2)2Mg, and 1 mM DTT] and lysed using a Potter-Elvehjem homogenizer. Cell lysates were centrifuged at 16,000 × g for 10 min to yield the S10 supernatant extract. Three hundred microliters of each S10 preparation was centrifuged through a 1-ml sucrose cushion for 2 h 15 min at 240,000 × g. The resulting pellets were gently rinsed once in buffer R and resuspended in R2 buffer (20 mM HEPES, 10 mM NaCl, 25 mM KCl, 1.1 mM MgCl2, and 7 mM β-mercaptoethanol). The resuspended solutions were quantified by reading the absorbance at 260 nm. Bradford quantification was also performed to estimate the ribosomal fraction concentration before storage at −80°C.
In parallel, untreated rabbit reticulocyte lysate (uRRL) (Promega) preparation was performed as previously described (66). After centrifugation of 1 ml of uRRL for 2 h 15 min at 240,000 × g, 900 μl of postribosomal supernatant (Su) was collected, frozen, and stored at −80°C. The extent of ribosome depletion was measured by translating 27 nM in vitro-transcribed, capped, and polyadenylated glo-RLuc mRNA in the Su and validated when no luciferase activity over background level could be detected.
RNA extraction and RT-qPCR.
One milliliter of TRIzol reagent (Life Technologies) was added to 100 μg of pelleted ribosomes, and RNA was extracted as indicated by the manufacturer. Extracted cytoplasmic RNA (1 μg) was treated with RQ1 DNase (Promega) before being reversed transcribed using the High Capacity RNA-to-cDNA master mix (Life Technologies). Quantitative PCR (qPCR) was then performed as described previously (29) using endogenous glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA as an internal control for relative quantification. Absolute quantification of mRNA was obtained by performing reverse transcription-quantitative PCR (RT-qPCR) on in vitro-transcribed Renilla luciferase mRNA with amounts ranging from 1 pg to 1 ng. The relative copy number of Renilla luciferase cDNA was compared to that of GAPDH cDNA, using x−ΔCT (where x corresponds to the experimentally calculated amplification efficiency of each primer couple). The sequences of the primers used have been described previously (18, 29).
In vitro translation assays.
The hybrid system was generated either with a combination of 5 μl of Su and 1 μg of pelleted ribosomes purified from cells, as previously described (50), or with mRNA which had been extracted from 1 μg of pelleted ribosomes. For the experiments with the results shown in Fig. 5B (left panel), translation reactions were performed using in vitro-transcribed RNA at 2.7 nM in a final volume of 10 μl of the hybrid system (5 μl of Su mixed with 1 μg of pelleted ribosomes) supplemented with 75 mM KCl, 0.75 mM MgCl2, and a 20 μM amino acid mix. In all in vitro translation assays, in order to ensure that Renilla activity detected in vitro from de novo translation was not biased by contaminating luciferase protein which could have been carried over during polysome purification, we performed the same experiments using a fraction of the purified ribosomes in the presence of the translation inhibitor cycloheximide (100 μM). This set the baseline for luciferase activity, which could then be subtracted from total Renilla activity. All translation reactions were performed for 30 min at 30°C before the reaction was stopped by the addition of Renilla lysis buffer (Promega).
Renilla activity.
Renilla activity was measured using the Renilla luciferase assay system (Promega) in a Mithras apparatus (Berthold Technologies) with 50 μl substrate injection and 10 s of signal integration program.
Coimmunoprecipitation and Western blotting.
HeLa cells were transfected in 100-mm plates with 0.5 μg of pCI-Flag-EB2 vector as indicated above. Transfected cells were lysed in 400 μl of hypotonic lysis buffer (10 mM Tris-HCl [pH 7.5], 10 mM NaCl, 10 mM EDTA, 0.5% Triton X-100) and protease inhibitors (complete EDTA-free cocktail; Roche). After 10 min on ice, NaCl was adjusted to 150 mM and the lysate cleared by centrifugation at 10,000 × g for 10 min at 4°C. The supernatant was incubated with 30 μl of anti-Flag M2 affinity gel (Sigma-Aldrich) for 4 h at 4°C. RNase treatment was performed by adding 2 μl of RNase A (10 mg/ml) to the incubation mixture when indicated. Recovered complexes were washed extensively with NET-2 buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% NP-40) and resuspended in Laemmli SDS loading buffer. Immunopurified proteins were then analyzed by Western blotting using anti-Flag (Sigma-Aldrich), anti-PABPC1 (Sigma-Aldrich), anti-eIF4A (Cell Signaling Technology), anti-eIF4GI, anti-eIF4B, and anti-eIF4E (kindly provided by Simon Morley, University of Sussex, UK), anti-eIF2α (Cell Signaling Technology), anti-eIF3 (kindly provided by Pierre Jalinot, LBMC-ENS de Lyon, France), anti-rpS6 (Cell Signaling Technology), anti-BMRF1 (Cell Signaling Technology), anti-rpL11 (Cell Signaling Technology), anti-NXF1, and anti-CBP80 (kindly provided by Elisa Izaurralde, Max Planck Institute for Developmental Biology, Germany).
m7GTP cap affinity matrix.
Cytoplasmic extracts from HeLa cells (100 μg of total protein) previously treated with S7 nuclease (Roche) were incubated for 10 min at 37°C in cap binding buffer (50 mM Tris-HCl [pH 7.5], 30 mM NaCl, 1 mM DTT, 2.5 mM MgCl2, 0.5% glycerol) in the absence or presence of 5 mM m7GpppG cap analog (New England BioLabs) as a control. Extracts were incubated overnight at 4°C with 20 μl of m7GTP-Sepharose 4B (GE Healthcare Life Sciences) under gentle shaking. Sepharose beads were washed three times with cap washing buffer (50 mM HEPES [pH 7.5], 40 mM NaCl, 2 mM EDTA, 0.1% Triton), and then bound proteins were eluted by boiling in Laemmli SDS loading buffer and analyzed by Western blotting as indicated above.
In vitro GST pulldown assays.
GST and GST fusion proteins were expressed in Escherichia coli BL21(DE3) and bound to glutathione-Sepharose 4 Fast Flow beads (GE Healthcare Life Sciences). Beads carrying GST or the GST fusion proteins were equilibrated in TNTB buffer (10 mM Tris-HCl [pH 8], 250 mM NaCl, 0.1% NP-40, and 2 mg/ml bovine serum albumin [BSA]) in the presence of protease inhibitors (complete EDTA-free cocktail; Roche) and incubated with radiolabeled proteins synthesized in vitro in the presence of [35S]methionine using the TNT-coupled transcription/translation system (Promega) in TNTB buffer for 4 h at 4°C. RNase treatment was performed by adding 2 μl of RNase A (10 mg/ml) in the incubation reaction mixture when indicated. Beads were washed five times in TNTB buffer without BSA, and bound proteins were fractionated by SDS-PAGE and analyzed by autoradiography on a Fuji FLA-5100 phosphorimager.
Mass spectrometric (MS) identification of EB2-associated proteins.
HEK293BMLF1-KO cells were transfected with pCMV-EB1 (in order to activate the EBV productive cycle) and pCI-Flag-EB2. At 48 h after transfection, cells (around 75 × 106) were washed with phosphate-buffered saline (PBS), recovered by centrifugation at 500 × g and 4°C, and lysed in 4 ml hypotonic lysis buffer (20 mM Tris-HCl [pH 7.5], 15 mM NaCl, 10 mM EDTA, 0.5% NP-40, 0.1% Triton X-100) and protease inhibitors (complete EDTA-free cocktail; Roche) for 10 min on ice. NaCl was adjusted to 150 mM and the lysate cleared by centrifugation at 10,000 × g for 10 min at 4°C. The cleared lysate was diluted to 10 ml in the above-described lysis buffer with 150 mM NaCl. The diluted lysate was incubated for 4 h at 4°C with 500 μl of anti-Flag M2 affinity gel (Sigma-Aldrich) prewashed twice with isotonic wash buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% NP-40) and 2 mg/ml BSA. Protein complexes captured on beads were washed six times with ice-cold NET-2 buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.05% NP-40) and then incubated 10 min at room temperature with RNase A at a final concentration of 200 μg/ml. Flag epitope-containing complexes were affinity eluted from the beads in one volume of isotonic wash buffer containing 150 μg/ml 3× Flag peptide (Sigma-Aldrich). The eluate was diluted in 10 ml hypotonic lysis buffer with 150 mM NaCl and incubated with rabbit polyclonal anti-EB2 antibody that was precoupled to 50 μl of PureProteome protein A/G Mix magnetic beads (Millipore), according to the manufacturer's instructions. Immunoprecipitation was carried out at 4°C for 4 h. Captured protein complexes were sequentially washed six times with ice-cold NET-2 buffer and twice with ice-cold 10 mM Tris-HCl (pH 7.5) buffer. Samples were completely dried by vacuum evaporation and resuspended in 15 μl of 0.1% SDS and 10 mM DTT. Samples were heated at 95°C for 5 min to denature proteins and reduce disulfides and then cooled down to room temperature. The reduced thiol groups were alkylated by incubating with 0.8 μl of freshly prepared 1 M iodoacetamide at room temperature for 45 min in the dark. The resulting samples were mixed with 15.8 μl of 2× Laemmli SDS loading buffer (Bio-Rad) and loaded onto a 4 to 15% Mini-Protean TGX gel (Bio-Rad). Samples were migrated until the dye-front had run ∼1 cm into the gel from the bottom of the well. The gel was washed three times with ∼200 ml high-pressure liquid chromatography (HPLC)-grade water for 5 min each and stained with the Thermo Scientific Pierce silver stain kit as indicated by the manufacturer.
MS analyses.
Each well from the SDS-PAGE gels was cut into four bands. Each band was reduced with 10 mM dithiothreitol (DTT) for 45 min at 60°C, alkylated with 100 mM iodoacetamide for 30 min at room temperature, and digested overnight at 37°C with sequencing-grade trypsin (Promega) at a 1:20 trypsin/protein mass ratio.
Peptide sequences were determined by mass spectrometry performed using an LTQ Velos instrument (dual-pressure linear ion trap) equipped with a nanospray source (Thermo Fisher Scientific) and coupled to a U3000 nano-liquid chromatography (nanoLC) system (Thermo Fisher Scientific). An MS survey scan was acquired over the m/z range 400 to 1,600 in enhanced-resolution mode. The MS/MS scans were acquired in normal resolution mode over a dynamic m/z range for the 20 most intense MS ions with a charge of 2 or more and with a collision energy set to 35 eV. The spectra were recorded using dynamic exclusion of previously analyzed ions for 0.5 min with 50 millimass units (mmu) of mass tolerance. Peptide separation was made on a C18 PepMapmicro precolumn (5 μm; 100 Å; 300 μm by 5 mm; Dionex) and a C18 PepMap nanocolumn (3 μm; 100 Å; 75 μm by 200 mm; Dionex) using a linear 90-min gradient from 0 to 40% of solvent B, where solvent A was 0.1% HCOOH in H2O-CH3CN (95:5) and solvent B was 0.1% HCOOH in H2O-CH3CN (20:80), at a 300-nl/min flow rate.
Protein identification was performed using the MASCOT 2.4.1 algorithm through the Proteome Discoverer software v1.4 (Thermo Fisher Scientific) against the Swiss-Prot database (release 2014_11). The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/) via the PRIDE partner repository (67).
ACKNOWLEDGMENTS
We thank Simon Morley (University of Sussex, UK), Pierre Jalinot (LBMC-ENS de Lyon, France), and Elisa Izaurralde (Max Planck Institute for Developmental Biology, Germany) for kindly providing antibodies. We thank Robin Buckland and Emiliano P. Ricci for critical reading of the manuscript. We also acknowledge the contribution of the AniRA-Analyze Génétique platform of the SFR BioSciences (UMS3444/US8).
The Oncogenic Herpesviruses team was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Agence Nationale pour la Recherche (ANR) (no. RPV09056CSA), and the Ligue Régionale Contre le Cancer. The proteomic analysis was financially supported by SFR BioSciences (UMS3444/US8).
We declare that we have no conflict of interest.
REFERENCES
- 1.Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700. In Knipe, DM, Howley, PM, Griffin, DE, Martin, MA, Lamb, RA, Roizman, B, Straus, SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol 79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niedobitek G, Young LS, Lau R, Brooks L, Greenspan D, Greenspan JS, Rickinson AB. 1991. Epstein-Barr virus infection in oral hairy leukoplakia: virus replication in the absence of a detectable latent phase. J Gen Virol 72:3035–3046. doi: 10.1099/0022-1317-72-12-3035. [DOI] [PubMed] [Google Scholar]
- 4.Temple RM, Zhu J, Budgeon L, Christensen ND, Meyers C, Sample CE. 2014. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc Natl Acad Sci U S A 111:16544–16549. doi: 10.1073/pnas.1400818111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Young LS, Lau R, Rowe M, Niedobitek G, Packham G, Shanahan F, Rowe DT, Greenspan D, Greenspan JS, Rickinson AB, et al. 1991. Differentiation-associated expression of the Epstein-Barr virus BZLF1 transactivator protein in oral hairy leukoplakia. J Virol 65:2868–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kenney SC, Mertz JE. 2014. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin Cancer Biol 28:60–68. doi: 10.1016/j.semcancer.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Hir H, Nott A, Moore MJ. 2003. How introns influence and enhance eukaryotic gene expression. Trends Biochem Sci 28:215–220. doi: 10.1016/S0968-0004(03)00052-5. [DOI] [PubMed] [Google Scholar]
- 8.Luo M, Reed R. 1999. Splicing is required for rapid and efficient mRNA export in metazoans. Proc Natl Acad Sci U S A 96:14937–14942. doi: 10.1073/pnas.96.26.14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyer JL, Swaminathan S, Silverstein SJ. 2002. The Epstein-Barr virus SM protein is functionally similar to ICP27 from herpes simplex virus in viral infections. J Virol 76:9420–9433. doi: 10.1128/JVI.76.18.9420-9433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farjot G, Buisson M, Duc Dodon M, Gazzolo L, Sergeant A, Mikaelian I. 2000. Epstein-Barr virus EB2 protein exports unspliced RNA via a crm-1-independent pathway. J Virol 74:6068–6076. doi: 10.1128/JVI.74.13.6068-6076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gruffat H, Batisse J, Pich D, Neuhierl B, Manet E, Hammerschmidt W, Sergeant A. 2002. Epstein-Barr virus mRNA export factor EB2 is essential for production of infectious virus. J Virol 76:9635–9644. doi: 10.1128/JVI.76.19.9635-9644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruvolo V, Gupta AK, Swaminathan S. 2001. Epstein-Barr virus SM protein interacts with mRNA in vivo and mediates a gene-specific increase in cytoplasmic mRNA. J Virol 75:6033–6041. doi: 10.1128/JVI.75.13.6033-6041.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hiriart E, Farjot G, Gruffat H, Nguyen MVC, Sergeant A, Manet E. 2003. A novel nuclear export signal and a REF interaction domain both promote mRNA export by the Epstein-Barr virus EB2 protein. J Biol Chem 278:335–342. doi: 10.1074/jbc.M208656200. [DOI] [PubMed] [Google Scholar]
- 14.Juillard F, Hiriart E, Sergeant N, Vingtdeux-Didier V, Drobecq H, Sergeant A, Manet E, Gruffat H. 2009. Epstein-Barr virus protein EB2 contains an N-terminal transferable nuclear export signal that promotes nucleocytoplasmic export by directly binding TAP/NXF1. J Virol 83:12759–12768. doi: 10.1128/JVI.01276-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batisse J, Manet E, Middeldorp J, Sergeant A, Gruffat H. 2005. Epstein-Barr virus mRNA export factor EB2 is essential for intranuclear capsid assembly and production of gp350. J Virol 79:14102–14111. doi: 10.1128/JVI.79.22.14102-14111.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han Z, Marendy E, Wang YD, Yuan J, Sample JT, Swaminathan S. 2007. Multiple roles of Epstein-Barr virus SM protein in lytic replication. J Virol 81:4058–4069. doi: 10.1128/JVI.02665-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiriart E, Bardouillet L, Manet E, Gruffat H, Penin F, Montserret R, Farjot G, Sergeant A. 2003. A region of the Epstein-Barr virus (EBV) mRNA export factor EB2 containing an arginine-rich motif mediates direct binding to RNA. J Biol Chem 278:37790–37798. doi: 10.1074/jbc.M305925200. [DOI] [PubMed] [Google Scholar]
- 18.Juillard F, Bazot Q, Mure F, Tafforeau L, Macri C, Rabourdin-Combe C, Lotteau V, Manet E, Gruffat H. 2012. Epstein-Barr virus protein EB2 stimulates cytoplasmic mRNA accumulation by counteracting the deleterious effects of SRp20 on viral mRNAs. Nucleic Acids Res 40:6834–6849. doi: 10.1093/nar/gks319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han Z, Swaminathan S. 2006. Kaposi's sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J Virol 80:5251–5260. doi: 10.1128/JVI.02570-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandri-Goldin RM. 2011. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol 6:1261–1277. doi: 10.2217/fmb.11.119. [DOI] [PubMed] [Google Scholar]
- 21.Winkler M, Rice SA, Stamminger T. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J Virol 68:3943–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai-Ju JQ, Li L, Johnson LA, Sandri-Goldin RM. 2006. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J Virol 80:3567–3581. doi: 10.1128/JVI.80.7.3567-3581.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palmeri D, Spadavecchia S, Carroll KD, Lukac DM. 2007. Promoter- and cell-specific transcriptional transactivation by the Kaposi's sarcoma-associated herpesvirus ORF57/Mta protein. J Virol 81:13299–13314. doi: 10.1128/JVI.00732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyne JR, Jackson BR, Taylor A, Macnab SA, Whitehouse A. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J 29:1851–1864. doi: 10.1038/emboj.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoyagi M, Shenk TE, Gaspar M. 2010. Human cytomegalovirus UL69 protein facilitates translation by associating with the mRNA cap-binding complex and excluding 4EBP1. Proc Natl Acad Sci U S A 107:2640–2645. doi: 10.1073/pnas.0914856107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontaine-Rodriguez EC, Knipe DM. 2008. Herpes simplex virus ICP27 increases translation of a subset of viral late mRNAs. J Virol 82:3538–3545. doi: 10.1128/JVI.02395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larralde O, Smith RWP, Wilkie GS, Malik P, Gray NK, Clements JB. 2006. Direct stimulation of translation by the multifunctional herpesvirus ICP27 protein. J Virol 80:1588–1591. doi: 10.1128/JVI.80.3.1588-1591.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith RWP, Anderson RC, Larralde O, Smith JWS, Gorgoni B, Richardson WA, Malik P, Graham SV, Gray NK. 2017. Viral and cellular mRNA-specific activators harness PABP and eIF4G to promote translation initiation downstream of cap binding. Proc Natl Acad Sci U S A 114:6310–6315. doi: 10.1073/pnas.1610417114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricci EP, Mure F, Gruffat H, Decimo D, Medina-Palazon C, Ohlmann T, Manet E. 2009. Translation of intronless RNAs is strongly stimulated by the Epstein-Barr virus mRNA export factor EB2. Nucleic Acids Res 37:4932–4943. doi: 10.1093/nar/gkp497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller-McNicoll M, Neugebauer KM. 2013. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat Rev Genet 14:275–287. doi: 10.1038/nrg3434. [DOI] [PubMed] [Google Scholar]
- 31.Singh G, Pratt G, Yeo GW, Moore MJ. 2015. The clothes make the mRNA: past and present trends in mRNP fashion. Annu Rev Biochem 84:325–354. doi: 10.1146/annurev-biochem-080111-092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis JD, Izaurralde E, Jarmolowski A, McGuigan C, Mataj IW. 1996. A nuclear cap-binding complex facilitates association of U1snRNP with the cap proximal 5′ splice site. Genes Dev 10:1683–1698. doi: 10.1101/gad.10.13.1683. [DOI] [PubMed] [Google Scholar]
- 33.Pabis M, Neufeld N, Steiner MC, Bojic T, Shav-Tal Y, Neugebauer KM. 2013. The nuclear cap-binding complex interacts with the U4/U6·U5 tri-snRNP and promotes spliceosome assembly in mammalian cells. RNA 19:1054–1063. doi: 10.1261/rna.037069.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng H, Dufu K, Lee C-S, Hsu JL, Dias A, Reed R. 2006. Human mRNA export machinery recruited to the 5′ end of mRNA. Cell 127:1389–1400. doi: 10.1016/j.cell.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 35.Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, Sandberg R, Hyman A, LaCava J, Rout MP, Andersen JS, Bertrand E, Jensen TH. 2013. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol 20:1367–1376. doi: 10.1038/nsmb.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiu S-Y, Lejeune F, Ranganathan AC, Maquat LE. 2004. The pioneer translation initiation complex is functionally distinct from but structurally overlaps with the steady-state translation initiation complex. Genes Dev 18:745–754. doi: 10.1101/gad.1170204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lejeune F, Ranganathan AC, Maquat LE. 2004. eIF4G is required for the pioneer round of translation in mammalian cells. Nat Struct Mol Biol 11:992–1000. doi: 10.1038/nsmb824. [DOI] [PubMed] [Google Scholar]
- 38.Sato H, Maquat LE. 2009. Remodeling of the pioneer translation initiation complex involves translation and the karyopherin importin β. Genes Dev 23:2537–2550. doi: 10.1101/gad.1817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maquat LE, Tarn W-Y, Isken O. 2010. The pioneer round of translation: features and functions. Cell 142:368–374. doi: 10.1016/j.cell.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson RJ, Hellen CUT, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amrani N, Ghosh S, Mangus DA, Jacobson A. 2008. Translation factors promote the formation of two states of the closed-loop mRNP. Nature 453:1276–1280. doi: 10.1038/nature06974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kühn U, Wahle E. 2004. Structure and function of poly(A) binding proteins. Biochim Biophys Acta 1678:67–84. doi: 10.1016/j.bbaexp.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Aitken CE, Lorsch JR. 2012. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 19:568–576. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- 45.Hinnebusch AG. 2014. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 83:779–812. doi: 10.1146/annurev-biochem-060713-035802. [DOI] [PubMed] [Google Scholar]
- 46.Gehring NH, Lamprinaki S, Kulozik AE, Hentze MW. 2009. Disassembly of exon junction complexes by PYM. Cell 137:536–548. doi: 10.1016/j.cell.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 47.Hir HL, Saulière J, Wang Z. 2016. The exon junction complex as a node of post-transcriptional networks. Nat Rev Mol Cell Biol 17:41–54. doi: 10.1038/nrm.2015.7. [DOI] [PubMed] [Google Scholar]
- 48.Le Hir H, Séraphin B. 2008. EJCs at the heart of translational control. Cell 133:213–216. doi: 10.1016/j.cell.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Ma XM, Yoon S-O, Richardson CJ, Jülich K, Blenis J. 2008. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 133:303–313. doi: 10.1016/j.cell.2008.02.031. [DOI] [PubMed] [Google Scholar]
- 50.Panthu B, Mure F, Gruffat H, Ohlmann T. 2015. In vitro translation of mRNAs that are in their native ribonucleoprotein complexes. Biochem J 472:111–119. doi: 10.1042/BJ20150772. [DOI] [PubMed] [Google Scholar]
- 51.Panthu B, Décimo D, Balvay L, Ohlmann T. 2015. In vitro translation in a hybrid cell free lysate with exogenous cellular ribosomes. Biochem J 467:387–398. doi: 10.1042/BJ20141498. [DOI] [PubMed] [Google Scholar]
- 52.Verma D, Bais S, Gaillard M, Swaminathan S. 2010. Epstein-Barr Virus SM protein utilizes cellular splicing factor SRp20 to mediate alternative splicing. J Virol 84:11781–11789. doi: 10.1128/JVI.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hiriart E, Gruffat H, Buisson M, Mikaelian I, Keppler S, Meresse P, Mercher T, Bernard OA, Sergeant A, Manet E. 2005. Interaction of the Epstein-Barr virus mRNA export factor EB2 with human Spen proteins SHARP, OTT1, and a novel member of the family, OTT3, links Spen proteins with splicing regulation and mRNA export. J Biol Chem 280:36935–36945. doi: 10.1074/jbc.M501725200. [DOI] [PubMed] [Google Scholar]
- 54.Bordeleau M-E, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, Pelletier J. 2006. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol 2:213–220. doi: 10.1038/nchembio776. [DOI] [PubMed] [Google Scholar]
- 55.Lindqvist L, Oberer M, Reibarkh M, Cencic R, Bordeleau M-E, Vogt E, Marintchev A, Tanaka J, Fagotto F, Altmann M, Wagner G, Pelletier J. 2008. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS One 3:e1583. doi: 10.1371/journal.pone.0001583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohlmann T, Rau M, Morley SJ, Pain VM. 1995. Proteolytic cleavage of initiation factor eIF-4 gamma in the reticulocyte lysate inhibits translation of capped mRNAs but enhances that of uncapped mRNAs. Nucleic Acids Res 23:334–340. doi: 10.1093/nar/23.3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Svitkin YV, Herdy B, Costa-Mattioli M, Gingras A-C, Raught B, Sonenberg N. 2005. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol Cell Biol 25:10556–10565. doi: 10.1128/MCB.25.23.10556-10565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thompson J, Verma D, Li D, Mosbruger T, Swaminathan S. 2015. Identification and characterization of the physiological gene targets of the essential lytic replicative Epstein-Barr virus SM protein. J Virol 90:1206–1221. doi: 10.1128/JVI.02393-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verma D, Kim EA, Swaminathan S. 2013. A cell-based screening assay for antiviral compounds targeting the ability of herpesvirus posttranscriptional regulatory proteins to stabilize viral mRNAs. J Virol 87:10742–10751. doi: 10.1128/JVI.01644-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walsh D, Mohr I. 2011. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol 9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balvay L, Soto Rifo R, Ricci EP, Decimo D, Ohlmann T. 2009. Structural and functional diversity of viral IRESes. Biochim Biophys Acta 1789:542–557. doi: 10.1016/j.bbagrm.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 62.Soliman TM, Sandri-Goldin RM, Silverstein SJ. 1997. Shuttling of the herpes simplex virus type 1 regulatory protein ICP27 between the nucleus and cytoplasm mediates the expression of late proteins. J Virol 71:9188–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Swaminathan S. 2005. Post-transcriptional gene regulation by gamma herpesviruses. J Cell Biochem 95:698–711. doi: 10.1002/jcb.20465. [DOI] [PubMed] [Google Scholar]
- 64.Toth Z, Stamminger T. 2008. The human cytomegalovirus regulatory protein UL69 and its effect on mRNA export. Front Biosci 13:2939–2949. doi: 10.2741/2899. [DOI] [PubMed] [Google Scholar]
- 65.Soto-Rifo R, Rubilar PS, Limousin T, de Breyne S, Décimo D, Ohlmann T. 2012. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J 31:3745–3756. doi: 10.1038/emboj.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soto Rifo R, Ricci EP, Décimo D, Moncorgé O, Ohlmann T. 2007. Back to basics: the untreated rabbit reticulocyte lysate as a competitive system to recapitulate cap/poly(A) synergy and the selective advantage of IRES-driven translation. Nucleic Acids Res 35:e121. doi: 10.1093/nar/gkm682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu Q-W, Wang R, Hermjakob H. 2016. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]