

ABSTRACT
The papillomavirus (PV) E2 protein is a sequence-specific DNA binding protein that recruits cellular factors to its genome in infected epithelial cells. E2 also binds to and loads the viral E1 DNA helicase at the origin of replication. Posttranslational modifications (PTMs) of PV E2 have been identified as potential regulators of E2 functions. We recently reported lysine 111 (K111) as a target of p300 acetylation in bovine PV (BPV). The di-lysines at 111 and 112 are conserved in almost all papillomaviruses. We pursued a mutational approach to query the functional significance of lysine in human PV (HPV) E2. Amino acid substitutions that prevent acetylation, including arginine, were unable to stimulate transcription and E1-mediated DNA replication. The arginine K111 mutant retained E2 transcriptional repression, nuclear localization, DNA and chromatin binding, and association with E2 binding partners involved in PV transcription and replication. While the replication-defective E2-K111R mutant recruited E1 to the viral replication origin, surprisingly, unwinding of the duplex DNA did not occur. In contrast, the K111 glutamine (K111Q) mutant increased origin melting and stimulated replication compared to wild-type E2. These experiments reveal a novel activity of E2 necessary for denaturing the viral origin that likely depends on acetylation of highly conserved lysine 111.
IMPORTANCE HPV is one of the most common sexually transmitted infections in the United States. Over 200 HPVs have been described, and they manifest in a variety of ways; they can be asymptomatic or can result in benign lesions (papillomas) or progress to malignancy. Although 90% of infections are asymptomatic and resolve easily, HPV16 and -18 alone are responsible for 70% of all cervical cancers, which are almost entirely caused by HPV infection. Interestingly, 60 to 90% of other cancers have been linked to HPV. The goal of this research is to further elucidate the mechanisms that regulate and mediate viral replication.
KEYWORDS: acetylation, lysine, papillomavirus, viral replication
INTRODUCTION
Papillomaviruses (PVs) are a family of nonenveloped DNA viruses that are the major etiological agents of cervical and oropharyngeal cancers. The circular, double-stranded DNA genome is approximately 8 kb and is comprised of a long control region (LCR) (1) and eight open reading frames (2–5). The proteins expressed by the virus are conserved in both structure and function across the various papillomaviruses (6). These multifunctional proteins and splice variants regulate viral mechanisms and manipulate infected cells directly and through interactions with host proteins.
The viral replicative program can be separated into three defined stages: establishment, maintenance, and vegetative amplification (7). The initiation of a papillomavirus infection results in the establishment and maintenance of a low copy number of double-stranded viral episomes within infected basal keratinocytes. The papillomavirus replicative program is closely linked to the life cycle of keratinocyte host cells and is moderated by a combination of viral and host cell factors. During establishment, there is an initial amplification of replication to establish a low copy number of viral genomes in basal cells. During the maintenance stage, low copy numbers of viral episomes are maintained in the basal keratinocytes. This is followed by the vegetative amplification stage in differentiating cells of the upper epithelial strata, where the viral copy number is increased, late genes are expressed, and virions are packaged (3). There is a distinct correlation between the differentiation of keratinocytes and the switch from the maintenance to the amplification stage; however, the details of how the virus senses and progresses through these stages and how replication is regulated remain unknown.
The papillomavirus E2 protein is the major transcriptional regulator of viral gene expression and a viral replication initiation protein (8–11). The structure of E2 consists of an N-terminal transactivation domain (TAD) and a C-terminal DNA binding domain, held together by an unstructured linker region. E2 regulates transcription by recruiting factors to the viral genome that result in either the activation or repression of transcription (12), through steric hindrance or by blocking the binding of host transcription factors such as Sp1 and TATA binding protein (TBP) (1, 13, 14). These activities involve the recruitment of certain host factors that are involved in chromatin processing such as chromatin adaptor Brd4, AMF1/GPS2, Tip60 histone acetyltransferase (HAT) complex subunit EP400, and the demethylase JARDIC/SMCX (15–19). Transcriptional repression of E6 and E7 by E2 is important for the stable maintenance of the virus, and loss of this function and subsequent increases in the levels of E6 and E7 may lead to the progression of malignancy with high-risk papillomaviruses (20, 21).
Previous replication studies have identified the early proteins E1 and E2 as the only viral proteins necessary for transient replication of the viral genome (22–24). E1 is the major replication initiation protein and has both DNA binding and helicase activity but does not demonstrate high specificity in DNA recognition. The papillomavirus replication origin contains E1 binding sites, which are flanked by high-affinity E2 binding sites. E2 binds specifically to these sites at the origin and recruits E1 monomers. E2 is displaced in an ATP-dependent process, and E1 forms a double hexameric helicase complex that unwinds the DNA (25). Although the behavior of E1 and E2 during replication initiation has been established (26, 27), the exact dynamics at the replication origins and what host replication machinery is involved are still being investigated.
Higher levels of E2 protein are present in the upper layers of the epithelium, where the virus enters the amplification stage of its replicative program (28). Recent reports demonstrated that keratinocyte differentiation coincides with and is induced by expression of the acetyltransferase p300 (29). Posttranslational modifications (PTMs) that affect papillomavirus E2 function have been previously identified. Phosphorylations in the hinge region of bovine papillomavirus 1 (BPV1) E2 at serines 290, 298, and 301 result in higher copy numbers of viral genomes (30). We have identified conserved lysine residues in BPV1 E2 that were acetylated by p300 in vitro and regulate its transcriptional activation capability (31). These lysines at positions 111 and 112 (K111 and K112) are conserved in more than 90% of papillomaviruses, including BPV1 and cancer-associated “high-risk” HPV16, -18, and -31. Here we examined the role of these HPV31 E2 lysines in the initiation of DNA replication. We discovered that viral replication did not tolerate the conservative mutation of K111 to arginine (K111R). Unexpectedly, our data reveal that DNA unwinding by the E1 helicase depends on a PTM at lysine 111 in E2.
RESULTS
Mutation of K111 affects transient viral replication.
Two lysine residues, K111 and K112, are very highly conserved among human and animal papillomaviruses (PVs) (Fig. 1A). We previously demonstrated the requirement of lysine 111 for BPV1 E2 transcriptional activation (31). While E2 is known to bind and localize the E1 helicase to high-affinity E2 binding sites at the viral origin, additional activities for E2 in DNA replication have not been elucidated. We began testing both BPV and HPV31 E2 lysine 111 and 112 mutants (E2-K111 and E2-K112) for stimulation of E1-dependent viral replication by cotransfection with the corresponding E1 gene and the PV origin-containing luciferase reporter construct. This di-lysine motif has been previously studied in BPV1, and mutations of each of these lysines resulted in decreased viral replication, with K111R causing a more severe reduction (32, 33). Compared to wild-type BPV E2, mutations of these lysines had deleterious effects on replication; K112R resulted in a 3-fold reduction, and K111R completely abrogated transient DNA replication, similarly to the replication-dead W145R mutant (Fig. 1B). The conservative arginine and alanine mutations were then tested in the context of HPV31 E2. The results indicate that mutations that prevent acetylation at K111 or mimic a deacetylated lysine—arginine and alanine, respectively—failed to induce E1-dependent replication (Fig. 1C). However, the mutation of HPV31 K111 to glutamine (K111Q), which resembles an acetylated lysine, stimulated replication to levels that surpassed wild-type E2. We ensured that the abrogation of replication from the K111R mutant HPV31 E2 protein was not due to reduced cell viability (Fig. 1C). These experiments also demonstrated that while replication is affected by K112 in the context of BPV1 (31), mutations at this position in HPV31 E2 did not exhibit a similar dependence (Fig. 1C).
FIG 1.

K111 is necessary for transient viral replication. Mutations of E2 lysine 111 (K111) alter transient replication. (A) Sequence alignment shows the highly conserved K111. (B) C33A cells were cotransfected with BPV1 E2-K111 mutants, pFLORI-BPV1, and pRRL and (C) C33A cells were cotransfected with HPV31 E2-K111 mutants. E2 mutants were cotransfected with pFLOri31. The luminescence was normalized to Renilla luciferase and the respective background E2 luminescence. Each of the samples was normalized to its respective control without E1 to cancel out any background effect. (Inset) The effect of K111R mutation on cell viability and cell cycle was determined by flow cytometry. (D) 293TT cells were cotransfected with HPV31 FLAG-E2 and HA-E1. E1 protein pulldown was conducted using 12CA5 antibody and blotted with HA7 and M2 antibodies. *, P < 0.05; **, P < 0.005.
An E2 mutation that reduces the interaction between E2 and E1 would likely alter viral replication, so we investigated E1-E2 association to determine if that was affected by the K111 mutations. Coimmunoprecipitation (co-IP) studies were conducted in 293TT cells that were cotransfected with hemagglutinin (HA)-tagged E1 and FLAG-tagged E2. E1 immunoprecipitation (with HA7 antibody) and Western blot analysis revealed that E1 interacted with both of the E2 mutants (Fig. 1D).
A possible explanation for this loss of activity is that K111 is located near the putative nuclear localization signal (NLS) within the E2 TAD. Deletion mutations in this region of BPV1 were reported to cause E2 to be diffused throughout the cell rather than nuclear (34). To determine if the mutants demonstrate the same effect in HPV31, we used immunofluorescence to observe the location of E2 and investigate whether the inability of the K111R mutant to initiate replication is due to the protein not being present within the nucleus. In transfected C33A cells and cells of a line of spontaneously immortalized keratinocytes (NIKs), E2 nuclear localization was unaffected by the K111 mutations (Fig. 2). These results clearly indicate that mutations at K111 do not inactivate the putative NLS and K111R remains predominantly nuclear.
FIG 2.
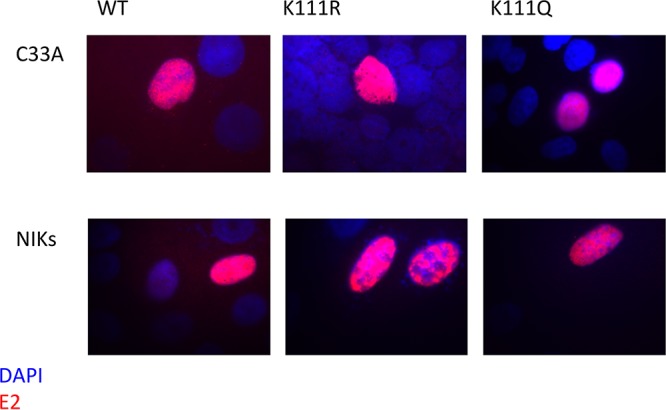
K111 is not required for nuclear localization of E2. C33A cells and NIKs were transfected with the HPV31 E2 wild type (wt) and the K111 mutants. The cells were stained with HA7 48 h posttransfection and analyzed by immunofluorescence for E2 (red) and DAPI (blue).
HPV31 E2 lysine 111 mutants repress transcription.
Although K111 is located in the transactivation domain of E2 and not the C-terminal DNA binding region, we still investigated the ability of the mutants to recognize the E2 binding sites. Chromatin immunoprecipitation (ChIP) was used to determine whether the DNA binding ability of E2 was impaired in the K111R mutant (Fig. 3A). Both HPV31 E2-K111R and E2-K111Q were able to bind near the origin using the HPV31 LCR4 primers, with no significant differences compared to wild-type E2.
FIG 3.

HPV31 E2 lysine 111 (K111) mutations repress transcription. C33A cells were cotransfected with 31-Fluc and HPV31 FLAG-tagged E2 wild type (wt) or K111 mutants. The amount of E2 at the origin was determined by ChIP using LCR4 primers (A). C33A cells were cotransfected with 31-Fluc and HPV31 FLAG-E2 wild type or K111 mutants. Repression was measured by luminometry and is shown as a fraction of baseline transcription (B). HeLa cells were transfected with HPV31 HA-E2 wild type and harvested after 48 h, and E6 transcripts were quantified by qPCR (C). *, P < 0.05.
Due to the similar consequences of the K111 mutants on replication in BPV1 and HPV31, we investigated the transcriptional activities of the K111 mutants. Unlike BPV E2 protein, which is a strong activator, the HPV31 E2 protein acts as an efficient repressor of viral gene transcription (35–37). To examine the effects of mutating K111 on E2 transcriptional repression, we used the HPV31 promoter-luciferase assay (Fig. 3B). C33A cells were transfected with plasmids expressing HPV31 E2 and a plasmid containing a portion of the HPV31 LCR containing the E2 binding sites and p97 promoter upstream of the firefly luciferase gene. Transfected amounts of wild-type E2 plasmid were first titrated to validate the concentrations of E2 used for the assay. K111R, K111Q, and K111A mutants repressed transcription at levels equivalent to those of wild-type E2 (Fig. 3B). This indicates that K111 is not necessary for HPV31 transcriptional repression. This conclusion was confirmed by an assay for repression of E6 transcription in HeLa cells. E2 represses transcription in the integrated HPV18 genomes present in these cells, resulting in a reduction of E6 mRNA. HeLa cells were transfected with either a vector control or HPV31 E2 plasmids. Compared to the negative control, HeLa cells transfected with wild-type E2 as well as the E2 mutants showed diminished transcription of the E6 gene (Fig. 3C). Together with the ChIP results, these experiments confirm that the K111 mutants are able to access E2 binding sites in the context of chromatin.
HPV31 E2-K111 mutants maintain interactions with cellular factors.
E2 interacts with a large number of host factors, some of which have adverse impact on replication when binding to E2 is abrogated. Brd4 is a cellular transcription factor that recognizes acetyl-lysines. In the BPV1 system, interaction between E2 and Brd4 has been shown to be critical for its transcriptional activation function. The HPV16 E2 Brd4 binding-deficient R37A I73A mutant is also defective in viral replication (38). Brd4 is known to bind to the E2 N-terminal transcriptional activation domain and localizes to distinct foci during the initiation of viral replication before being displaced from actively replicating origins (38, 39). Three specific regions of Brd4 were reported to potentially interact with E2 (40, 41): the C-terminal motif (CTM), basic residue-enriched interaction domain (BID), and phosphorylation-dependent interaction domain (PDID). The interaction of HPV31 E2 was investigated by coimmunoprecipitation experiments with each of the Brd4 binding regions (Fig. 4A). Neither E2-K111R nor E2-K111Q was found to be deficient for Brd4 binding.
FIG 4.

HPV31 E2-K111 mutants maintain interactions with cellular factors. (A) 293TT cells were cotransfected with HPV31 FLAG-E2, and plasmids expressing BRD4 fragments CTM, BID, and PDID. BRD4 proteins were pulled down using glutathione S-transferase (GST) antibody and blotted with GST and M2 antibodies. (B) 293TT cells were cotransfected with HPV31 FLAG-E2 and HA-GPS2. HPV31 E2 protein pulldown was executed with M2 antibody and blotted with HA7 and M2 antibodies. Relative GPS2 binding was calculated by densitometry and presented graphically. (C) 293TT cells were transfected with HPV31 FLAG-E2 followed by E2 protein immunoprecipitation with M2 antibody. The amounts of endogenous TopBP1 coimmunoprecipitated were determined by blotting with anti-TopBP1 and anti-M2 antibodies, and relative binding was calculated by densitometry and represented graphically.
The GPS2 (AMF1) protein is another factor known to interact with E2 proteins (16). GPS2 is a positive regulator of E2 transcriptional activation in BPV1 (17). A binding-deficient mutant of BPV1 E2, E2-W145R, is also transcription and replication defective (16, 17). We investigated the interaction of the HPV31 E2 mutants with GPS2 by coimmunoprecipitation and found that the K111 mutants interacted with GPS2 (Fig. 4B). Interestingly, we also found that the E2 mutants differentially affected the expression of GPS2 from the transfected plasmid. The relative interactions of the E2 proteins and GPS2 were analyzed by densitometry, and we found that K111Q-transfected cells showed higher levels of GPS2, while K111R cells exhibited reduced levels compared to cells transfected with wild-type E2.
Topoisomerase II-binding protein 1 (TopBP1) is involved in DNA damage repair and DNA replication (42). TopBP1 influences viral replication (43) and colocalizes with HPV E2 at viral replication origins, proposed to occur as part of a DNA damage response (44, 45). TopBP1 complexes with HPV E2 and is important for establishing the copy number of viral genomes (46), and it also increases replication and transcriptional activities of E2. We found that both HPV31 E2-K111R and E2-K111Q maintained TopBP1 interaction in a similar manner to the wild type (Fig. 4C). Blots were analyzed by densitometric analysis to quantify the association of TopBP1 and the mutants relative to the wild type.
E1 protein complexes with and is recruited to the replication origin by E2-K111 mutants.
During replication initiation, E2 binds and recruits E1 to the replication origin. Although we have already established that the E2 mutants bind both DNA and E1, we investigated the efficiency with which the K111 mutants translocated E1 to the viral origin using ChIP assays. C33A cells were cotransfected with E1, E2, and PFLORI (Fig. 5B), and HeLa cells were cotransfected with HPV31 E1 and E2 (Fig. 5B). We found that the K111R and K111Q mutants both effectively recruited E1 to the viral replication origin in both models.
FIG 5.
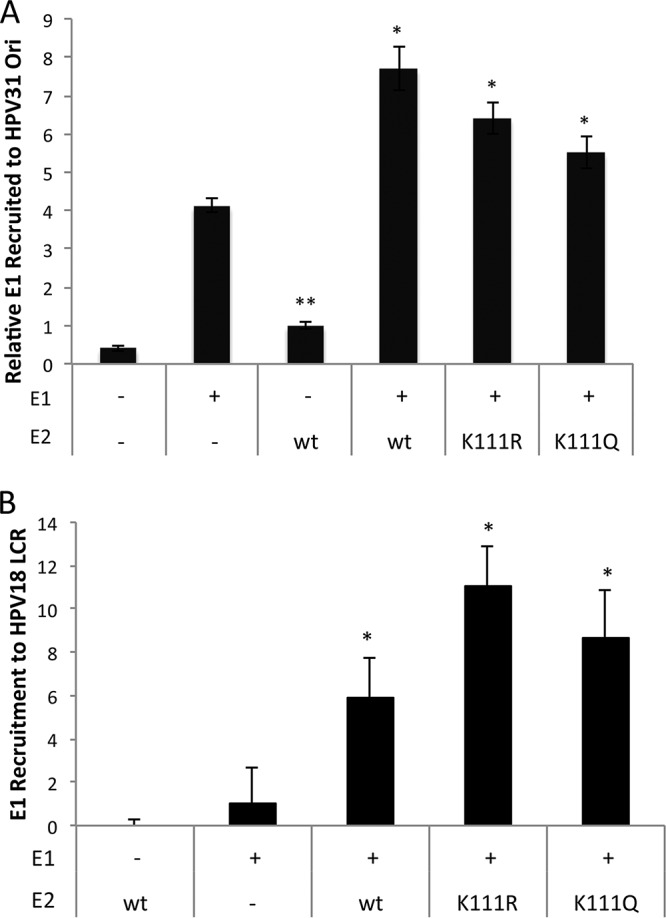
E1 protein complexes with and is recruited to the replication origin by the E2-K111 mutants. (A) C33A cells were cotransfected with PFLORI31, HPV31 HA-E2, and FLAG-E1. ChIP for E1 was conducted with M2 antibody and LCR4 primers. (B) HeLa cells were transfected with HPV31 FLAG-E1 and HA-E2, and the presence of E2 at the HeLa origin was determined by ChIP. *, P < 0.05; **, P < 0.005.
The K111R mutant abrogates unwinding of the replication origin.
These results with the K111R E2 protein were intriguing. Our current understanding of HPV replication suggests that since the K111R mutant binds chromatin and recruits E1 to the origin, replication initiation should be unaffected; however, that was not the case. During initiation, the assembly of the E1 double hexamer follows the recruitment of E1 to the origin by E2. To determine whether the E1 helicase was active, a FAIRE (formaldehyde-assisted isolation of regulatory elements) assay was modified to infer DNA unwinding at the replication origin. FAIRE assays are typically used to identify active promoters by detecting sites in chromatin that are nucleosome free (47–49). This principle was applied to investigate the viral replication origin. Compared to E1 alone, E1 cotransfected with the wild type resulted in 16-fold unwinding at the origin. The K111R mutant yielded low levels of sheared origin DNA indicating that E1 helicase activity is restricted by this mutation (Fig. 6A). The K111Q mutant also yielded interesting results as it produced 21-fold more naked origin DNA than E1 alone, which is significantly higher than that produced by the wild type. These data imply that the E2-K111R change did not allow unwinding of the replication origin while the K111Q mutation enhanced it.
FIG 6.
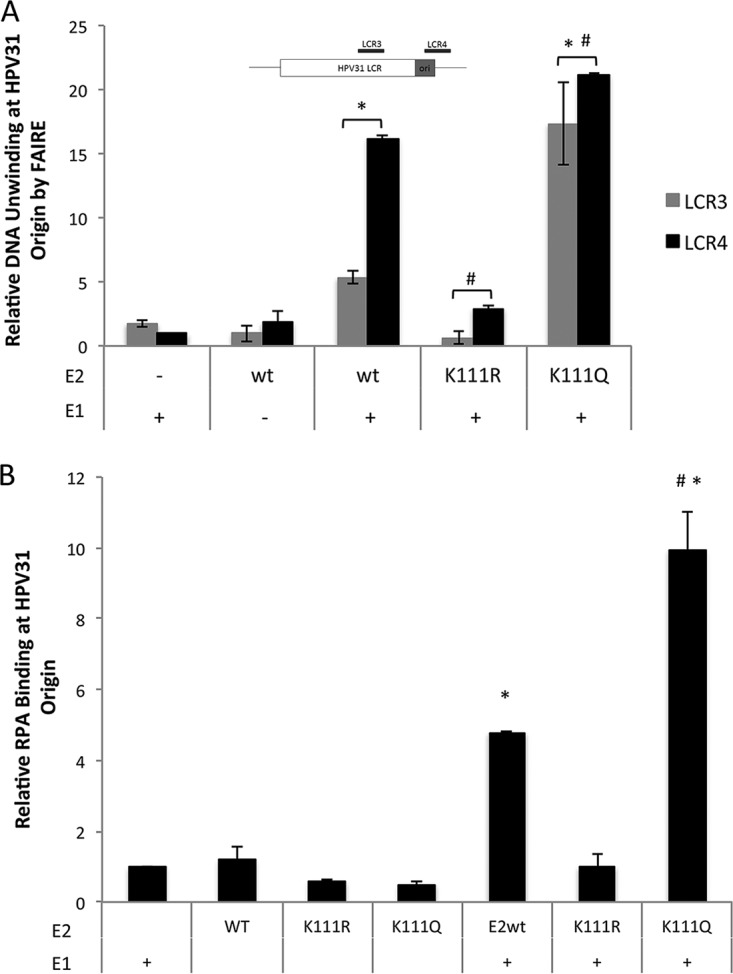
The K111R mutant reduces unwinding of the replication origin. C33A cells were transfected with FLAG-E1, FLAG-E2, or PFLORI31. Unwinding at origin DNA was analyzed by FAIRE assay using HPV31 LCR primers LCR3 and LCR4 (A). (B) ChIP for RPA protein at the LCR used anti-RPA70 antibody and LCR4 primers. *, P < 0.05 by one-way ANOVA compared to the wild type in the absence of E1; #, P < 0.05 by one-way ANOVA compared to wild-type E2 plus E1.
We then corroborated the FAIRE results with a second assay. Replication protein A (RPA) is a cellular factor that engages single-stranded DNA (ssDNA) at the viral origin (50). We performed a ChIP assay for endogenous RPA (51) in cells cotransfected with HPV31 E1 and E2 (Fig. 6B). In cells expressing E1 and E2-K111R, recruitment of RPA to the HPV31 origin was not observed. Interestingly, the K111Q mutant showed an ∼3-fold increase in unwound DNA compared to the wild type. These results support the findings of the FAIRE assay and demonstrate that both E1 and E2 are required for the recruitment of RPA and that origin melting is the event in replication initiation that is dependent upon acetylation of K111.
DISCUSSION
The role of papillomavirus E2 in replication initiation has been extensively studied; however, the mechanisms by which replication and copy number are regulated and the specific involvement of E2 beyond E1 recruitment remain undefined. Activation of the early promoter (p97) leads to the expression of proteins E1, E2, E6, and E7 (9, 23, 33). The E1 protein is the primary viral replication protein. It has been reported that in vitro, E1 is capable of initiating viral replication from a histone-free template in the absence of E2; however, the binding of E1 to DNA is relatively nonspecific and E2 is required in vivo (50). For viral genome replication, E2 binds to DNA in a sequence-specific manner at sites that form the origin of replication and recruits E1 monomers to its preferred adjacent sites. After the first pair of E1 molecules arrives at the replication origin, E2 is released from its binding sites through a process of ATP hydrolysis (52). More E1 molecules are recruited and form a double hexameric complex at the origin that is similar to the eukaryotic ORC-MCM helicase. This E1 complex then acts as a helicase and unwinds the DNA to allow cellular DNA polymerases and associated factors to be recruited. Without E2 serving as a viral origin recognition factor, the inability of E1 to bind specifically to the replication origin would cause inefficient and inconsistent firing.
Lysine acetylation is a reversible PTM that is essential for activation/deactivation of many pathways; for example, the acetylation of histones by acetyl transferases is necessary for transcriptional activation. We first identified this E2 lysine modification through a p300 in vitro acetylation assay (31). It has been reported that p300 levels in basal keratinocytes are very low, while differentiating cells show high levels (29). This difference in p300 levels coincides with the switch of viral genomes from maintenance to vegetative amplification in infected cells. To explore the importance of this acetylation, we designed mutants that represent deacetylated and acetylated K111 (K111R and K111Q, respectively). The acetylation-deficient mutant E2-K111R was observed to abrogate replication in both BPV1 (31) and HPV31, as reported here. As shown in Fig. 1C, the acetylation mimetic E2-K111Q demonstrated significantly higher levels of replication than wild-type E2. This supports the hypothesis that acetylation at the 111 residue is a necessary step in the initiation of replication. Furthermore, the K111Q result suggests that acetylation at K111 may be an integral regulatory step in switching the viral replicative program from maintenance to vegetative amplification (53).
To determine how K111 affects replication, we further characterized the K111R and K111Q mutants. Since K111 is located in the putative NLS, we investigated the localization of E2 and found that like wild-type E2, the mutants were also nuclear (Fig. 2). Since the K111 mutations did not affect nuclear localization, we enquired whether other functions of E2 were compromised. Knowing that a DNA-binding-deficient mutant would be detrimental to viral replication, we conducted a ChIP assay of E2 at the origin that allowed direct comparisons of the K111 mutants to wild-type E2. We observed no differences among the E2 proteins bound to the viral replicon (Fig. 5), confirming that the C-terminal DNA binding domain was not influenced by the K111 mutations. HPV31 E2 serves as a strong repressor of transcription, and according to our results from two corroborating experiments (Fig. 3), this function is maintained at levels similar to wild type for both mutants. The ability of HPV31 E2 to repress transcription results from binding at sites located on the TATA box of the p97 promoter as well as interaction with host cell transcription factors (1, 12, 54–56). The K111 mutants allow transcriptional repression, which indirectly indicates that DNA binding and recruitment of any essential cofactors are unaffected.
Several cellular factors that interact with E2 have been reported to directly affect E2 activities in viral transcription and genome replication. We investigated the ability of the K111 mutants to interact with a subset of these proteins and found that the K111R mutant was not binding deficient for any of them. An interesting observation was that the E2 mutants exhibited differential expression levels (Fig. 4). The K111R mutant showed lower expression than the wild type, and the K111Q mutant was expressed at extremely low levels compared to the wild type or K111R mutant. Attempts to increase the protein levels of the K111Q mutant by increasing the plasmid quantity transfected as well as using proteasome inhibitor MG132 were ineffective. The coimmunoprecipitation experiments with the fragments of BRD4 were an exception. The CTM of BRD4 stabilizes E2 (57), and that effect was clearly demonstrated, and the BID and PDID fragments also produced E2 stabilization to a lesser degree. While the expression of K111Q was low, it remained highly active in all of the experiments, indicating that despite low levels of expression, the acetylation mimic K111Q mutant is active and results in enhanced viral replication.
K111 is located in the TAD, which is responsible for E1 binding (58). We found that E1 binds to both K111 mutants, and the E2 mutants bound DNA and subsequently recruited E1 to the replication origin at levels comparable to the wild type (Fig. 1D and 5). This interaction also demonstrates that the TAD of E2 is functional and therefore not misfolded in the mutants. For the assembly of the E1 double hexamer helicase, E2 should not remain bound to E1 at the origin. One possible explanation for the lack of replication is that the K111R mutant binds strongly to E1 and is unable to be released, thereby hindering formation of the helicase complex and preventing unwinding at the origin. We investigated the formation of the helicase complex by determining if the origin becomes exposed under replication conditions by FAIRE assay. The FAIRE assay was modified to measure the open chromatin at the origin under replication conditions. Our results confirm that K111R is not capable of facilitating E1-dependent unwinding of the replication origin (Fig. 6A).
To corroborate the results from the FAIRE assay, we performed a ChIP for RPA. Our data reveal reduced RPA at the replication origin with K111R and a significant increase with K111Q (Fig. 6B), consistent with a direct effect of E2 on the E1 recruitment of RPA to the origin. Previous reports suggest that RPA is recruited to single-stranded DNA (ssDNA) by interacting directly with E1 and this interaction is inhibited by ssDNA (50, 59). RPA was reported to interact with E2 using in vitro-expressed partially purified proteins (60); however, this has not been established by in vivo coimmunoprecipitation, and our attempts to visualize such an interaction have been unsuccessful. Since RPA is necessary for DNA replication, it was proposed that E1 loads RPA onto ssDNA during unwinding (50). The role of RPA in viral replication has been more extensively studied in simian virus 40 (SV40). SV40 large T-antigen (TAg) has been reported to interact with RPA, and during replication, the SV40 TAg-RPA interaction is essential for assembly of the primosome (61). This specific function has not been determined for the E1-RPA interaction, but a similar mechanism could explain how a lack of RPA loading at the origin inhibits replication. Our data amend this proposed mechanism by adding the requirement of E2 for the recruitment of RPA, since neither E1 nor E2 independently recruited RPA to the origin (Fig. 6B).
This study characterized an acetylation site on HPV E2 that directly impacts viral replication. Manipulation of K111 left the E2 localization, repression, DNA binding, and E1 recruitment intact while only affecting viral replication. K111R was replication defective, while the acetylation-mimetic K111Q enhanced viral replication, which lends credence to the idea that it is an acetylation at this site that regulates viral replication. We found that K111R abrogates replication by preventing the helicase activity of E1. It is possible that E2 blocks the formation of the E1 helicase complex or that another factor involved in episome unwinding is not recruited by the K111R mutant. Further investigation of the interactions of HPV E2-K111 with factors directly involved in replication initiation and DNA unwinding will be necessary to further our understanding of how the K111 acetylation regulates replication mechanistically. An interesting observation made here is that K111Q not only rescued the deficiencies of K111R, but this mutant outperformed wild-type E2. We are currently studying K111 E2 mutants in the context of the viral genome for establishment and replication in keratinocytes. This possible acetylation presents a novel regulatory step in the PV replication program that may be necessary for the transition from maintenance stage replication to vegetative amplification.
MATERIALS AND METHODS
Cell culture, plasmids, and reagents.
C33A and HeLa (ATCC), HEK 293TT (J. Schiller, C. Buck), and J23T3 (H. Green) cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Atlas Biologicals) and 100 U/ml penicillin-streptomycin (Life Technologies). Cells of a spontaneously immortalized keratinocyte cell line (NIKs) were cocultured in F medium (62) with mitomycin C-treated J23T3 cells.
C33A, and HeLa cells were transfected with Lipofectamine 2000. 293TT cells were transfected with 2 μg/ml polyethylenimine (PEI) at a ratio of 1:2, and NIKs were transfected using X-tremeGene HP DNA transfection reagent (Roche) at a ratio of 1:2 DNA to transfection reagent. HPV31 E2 mutant plasmids were constructed by site-directed mutagenesis (Agilent; Quick-Change II) and verified by sequencing of the entire E2 gene. HA-tagged HPV31 E2 was obtained from P. Howley (63), codon-optimized FLAG HPV31 E2 was supplied by A. McBride (64), and the HPV31 E1, pCI-RLuc, and PFLORI31 constructs were obtained from J. Archambault (65). Codon-optimized HA-31E1 was cloned into the NheI and ApaI sites of PCDNA3.
Antibodies.
Antibodies used include anti-TopBP1 (Bethyl), mouse anti-FLAG M2 (Sigma-Aldrich), and mouse anti-HA (HA7; Sigma-Aldrich, 12CA5). Anti-RPA70 antibody (Abcam), goat anti-mouse 594, and goat anti-rabbit 488-horseradish peroxidase (HRP) (Jackson ImmunoResearch) and Alexa Fluor (Thermo Fisher) antibodies were purchased from commercial sources. Glutathione- and M2-conjugated beads were obtained from Sigma-Aldrich.
Viral replication assay.
The luciferase-based replication assay was performed as previously reported (65). Ten nanograms each of BPV1/HPV31 E1, E2, and PFLORI31 constructs was cotransfected into C33A cells in a 96-well plate. The pCI-RLuc construct (2.5 ng) was included to normalize for transcriptional activation and transfection efficiency. Cells were incubated in transfection medium overnight, and the medium was replaced. Seventy-two hours posttransfection, cells were lysed in Dual-Glo luciferase reagent (Promega), and both firefly and Renilla luciferase levels were determined by luminometry on a PHERAStar FS (BMG Labtech).
Flow cytometry.
C33A cells at 50% confluence were transfected with 2 μg of HPV31 HA-E2 in 60-cm dishes. Forty-eight hours after transfection, the cells were harvested with 0.1% trypsin-EDTA, spun down, and fixed in 90% ethanol. The samples were washed in PBS and treated with 50 μg/μl each of propidium iodide and RNase A for 20 min. The samples were analyzed using a FACSCalibur instrument and the FlowJo program.
Immunofluorescence.
C33A cells and NIKs grown on coverslips were transfected with 2.5 ng of the indicated HPV31 E2 plasmid. Cells were fixed 48 h posttransfection for 15 min in 4% formaldehyde solution in PBS, washed three times with PBS, and permeabilized in PBT (0.2% Triton X–PBS) for 10 min. The cells were blocked for 1 h in 10% normal goat serum–PBT and incubated with HA7 primary antibody (1:5,000 in 10% normal goat serum–PBT) overnight. The cells were washed, incubated with secondary antibodies (1:5,000 in 10% normal goat serum–PBT) for 1 h, and mounted with Vectashield antifade mounting medium with DAPI (4′,6-diamidino-2-phenylindole [Vector Laboratories]). Cells were analyzed using a Nikon microscope and QCapture Pro software.
Transcriptional repression luciferase assay.
Twenty-five nanograms of HPV31 HA-E2 (WT or mutant) or empty vector control (pCI) and 100 ng of HPV31-luc constructs were cotransfected into C33A cells in each well of a 12-well plate. Forty-eight hours posttransfection, cells were lysed and processed with the Dual-Glo luciferase kit (Promega). Luminescence was measured using the PHERAStar FS (BMG Labtech).
HeLa repression assay.
HeLa cells in 6-well dishes were transfected at 50% confluence with 1 μg HPV31 HA-E2 (WT or mutants). Forty-eight hours posttransfection, the cells were rinsed with cold PBS and lysed directly in plates using 1 ml TRIzol. The cells were collected by scraping and homogenized by several passes through an 18-gauge syringe. RNA was isolated by phase separation and resuspended in diethyl pyrocarbonate (DEPC)-treated water. cDNA synthesis was conducted with the SuperScript III first-strand synthesis system (Thermo Fisher Scientific), and cDNA was analyzed by quantitative PCR using the following HPV18 E6 and actin primers: SKH 18E6 F01 (5′-GGTGCCAGAAACCGTTGAAT-3′) and SKH 18E6 R01 (5′-CGAATGGCACTGGCCTCTAT-3′) and HeLabActin F01 (5′-GGACATCCGCAAAGACCTGT-3′) and HeLabActin R01 (5′-CCAGGGCAGTGATCTCCTTC-3′).
Coimmunoprecipitation and chromatin immunoprecipitation.
293TT cells were cotransfected with plasmids expressing a codon-optimized FLAG-tagged HPV31 E2 and the protein of interest. The cells were harvested 48 h posttransfection and lysed in 1 ml NP-40 buffer (0.5% NP-40, 50 mM Tris, 150 μM NaCl) for 10 min on ice. One hundred units of Benzonase endonuclease was added to each sample for 30 min. Samples were centrifuged, and supernatants were collected in fresh tubes for immunoprecipitations and inputs. For co-IP, the samples were rotated overnight at 4°C with either antibody-conjugated beads (glutathione or M2), or Sepharose A/G beads and ∼1 μg of antibody. The beads were washed five times with NP-40 lysis buffer, resuspended, and boiled in SDS sample buffer. The samples and their respective inputs were then analyzed by Western blotting.
For ChIP, C33A cells were transfected with plasmids (1:1:1) expressing HPV31 E1, E2 and the HPV31 origin of replication (PFLORI). Forty-eight hours posttransfection, the cells were cross-linked in 1% paraformaldehyde for 10 min at room temperature. The cells were rinsed with cold PBS after the cross-linking was stopped by addition of 0.125 M glycine, and then the cells were collected by scraping. These samples were processed using the ChIP-It enzymatic shearing kit (Active Motif). The DNA from the immunoprecipitations and their inputs were analyzed by quantitative PCR using primer pairs designed against the HPV18 or -31 LCR.
FAIRE assay.
Plasmids expressing HPV31 E2 and E1, along with a plasmid containing the LCR of HPV31 (PFLORI), were transfected into C33A cells. Cells were harvested 48 h posttransfection. One replicate of each sample was harvested immediately by scraping into cold PBS, and the other was formaldehyde cross-linked with 1% paraformaldehyde for 10 min at room temperature. The cross-linking was stopped with 0.125 M glycine for 1 min, and after washing with cold PBS, the samples were collected by scraping. After shearing with the ChIP-It enzymatic shearing kit (Active Motif), the chromatin was extracted by phenol-chloroform extraction followed by ethanol precipitation and resuspended in Tris-EDTA (TE) buffer. The cross-linked and respective uncross-linked control samples were analyzed by quantitative PCR with primer sets designed for the HPV31 replication origin (66) LCR4: LCR3 F (5′-GTTCTGCGGTTTTTGGTTTC-3′) and LCR3 R (5′-TGTTGGCAAGGTGTGTTAGG-3′) and LCR4 F (5′-AAAGTGGTGAACCGAAAACG-3′) and LCR4 R (5′-CATGCAATTTCCGAGGTCTT-3′).
Statistical analyses.
Mean values are reported ± the standard error of the mean (SEM). Two-way t tests and one-way analysis of variance (ANOVA) were used for analyses (*, P ≤ 0.05).
ACKNOWLEDGMENTS
We appreciate the generosity of Alison McBride (NIAID/NIH) for the codon-optimized HPV31 E2 and Jacques Archambault (McGill University) for HPV31 and BPV1 luciferase replicons.
This work was supported by National Cancer Institute/National Institutes of Health grant R01CA58376.
REFERENCES
- 1.Hou SY, Wu SY, Zhou T, Thomas MC, Chiang CM. 2000. Alleviation of human papillomavirus E2-mediated transcriptional repression via formation of a TATA binding protein (or TFIID)-TFIIB-RNA polymerase II-TFIIF preinitiation complex. Mol Cell Biol 20:113–125. doi: 10.1128/MCB.20.1.113-125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadaja M, Silla T, Ustav E, Ustav M. 2009. Papillomavirus DNA replication—from initiation to genomic instability. Virology 384:360–368. doi: 10.1016/j.virol.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 3.Hebner CM, Laimins LA. 2006. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol 16:83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 4.Soeda E, Ferran MC, Baker CC, McBride AA. 2006. Repression of HPV16 early region transcription by the E2 protein. Virology 351:29–41. doi: 10.1016/j.virol.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 5.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 6.Conway MJ, Meyers C. 2009. Replication and assembly of human papillomaviruses. J Dent Res 88:307–317. doi: 10.1177/0022034509333446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. The biology and life-cycle of human papillomaviruses. Vaccine 30(Suppl 5):F55–F70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- 8.Stenlund A. 2003. Initiation of DNA replication: lessons from viral initiator proteins. Nat Rev Mol Cell Biol 4:777–785. doi: 10.1038/nrm1226. [DOI] [PubMed] [Google Scholar]
- 9.Sedman J, Stenlund A. 1995. Co-operative interaction between the initiator E1 and the transcriptional activator E2 is required for replicator specific DNA replication of bovine papillomavirus in vivo and in vitro. EMBO J 14:6218–6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melendy T, Sedman J, Stenlund A. 1995. Cellular factors required for papillomavirus DNA replication. J Virol 69:7857–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Androphy EJ, Lowy DR, Schiller JT. 1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 325:70–73. doi: 10.1038/325070a0. [DOI] [PubMed] [Google Scholar]
- 12.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stubenrauch F, Pfister H. 1994. Low-affinity E2-binding site mediates downmodulation of E2 transactivation of the human papillomavirus type 8 late promoter. J Virol 68:6959–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stubenrauch F, Leigh IM, Pfister H. 1996. E2 represses the late gene promoter of human papillomavirus type 8 at high concentrations by interfering with cellular factors. J Virol 70:119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith JA, White EA, Sowa ME, Powell ML, Ottinger M, Harper JW, Howley PM. 2010. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc Natl Acad Sci U S A 107:3752–3757. doi: 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breiding DE, Sverdrup F, Grossel MJ, Moscufo N, Boonchai W, Androphy EJ. 1997. Functional interaction of a novel cellular protein with the papillomavirus E2 transactivation domain. Mol Cell Biol 17:7208–7219. doi: 10.1128/MCB.17.12.7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng YC, Breiding DE, Sverdrup F, Richard J, Androphy EJ. 2000. AMF-1/Gps2 binds p300 and enhances its interaction with papillomavirus E2 proteins. J Virol 74:5872–5879. doi: 10.1128/JVI.74.13.5872-5879.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev 20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee AY, Chiang CM. 2009. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J Biol Chem 284:2778–2786. doi: 10.1074/jbc.M805835200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Francis DA, Schmid SI, Howley PM. 2000. Repression of the integrated papillomavirus E6/E7 promoter is required for growth suppression of cervical cancer cells. J Virol 74:2679–2686. doi: 10.1128/JVI.74.6.2679-2686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. 1989. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol 63:4417–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders CM, Stenlund A. 2001. Mechanism and requirements for bovine papillomavirus, type 1, E1 initiator complex assembly promoted by the E2 transcription factor bound to distal sites. J Biol Chem 276:23689–23699. doi: 10.1074/jbc.M101861200. [DOI] [PubMed] [Google Scholar]
- 23.Ustav M, Stenlund A. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J 10:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ustav M, Ustav E, Szymanski P, Stenlund A. 1991. Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. EMBO J 10:4321–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanders CM, Stenlund A. 1998. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. EMBO J 17:7044–7055. doi: 10.1093/emboj/17.23.7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L, Mohr I, Fouts E, Lim DA, Nohaile M, Botchan M. 1993. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proc Natl Acad Sci U S A 90:5086–5090. doi: 10.1073/pnas.90.11.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berg M, Stenlund A. 1997. Functional interactions between papillomavirus E1 and E2 proteins. J Virol 71:3853–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klumpp DJ, Laimins LA. 1999. Differentiation-induced changes in promoter usage for transcripts encoding the human papillomavirus type 31 replication protein E1. Virology 257:239–246. doi: 10.1006/viro.1999.9636. [DOI] [PubMed] [Google Scholar]
- 29.Wong PP, Pickard A, McCance DJ. 2010. p300 alters keratinocyte cell growth and differentiation through regulation of p21(Waf1/CIP1). PLoS One 5:e8369. doi: 10.1371/journal.pone.0008369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McBride AA, Bolen JB, Howley PM. 1989. Phosphorylation sites of the E2 transcriptional regulatory proteins of bovine papillomavirus type 1. J Virol 63:5076–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinlan EJ, Culleton SP, Wu SY, Chiang CM, Androphy EJ. 2012. Acetylation of conserved lysines in bovine papillomavirus E2 by p300. J Virol 87:1497–1507. doi: 10.1128/JVI.02771-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brokaw JL, Blanco M, McBride AA. 1996. Amino acids critical for the functions of the bovine papillomavirus type 1 E2 transactivator. J Virol 70:23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abroi A, Kurg R, Ustav M. 1996. Transcriptional and replicational activation functions in the bovine papillomavirus type 1 E2 protein are encoded by different structural determinants. J Virol 70:6169–6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skiadopoulos MH, McBride AA. 1996. The bovine papillomavirus type 1 E2 transactivator and repressor proteins use different nuclear localization signals. J Virol 70:1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernard BA, Bailly C, Lenoir MC, Darmon M, Thierry F, Yaniv M. 1989. The human papillomavirus type 18 (HPV18) E2 gene product is a repressor of the HPV18 regulatory region in human keratinocytes. J Virol 63:4317–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romanczuk H, Thierry F, Howley PM. 1990. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P97 and type 18 P105 promoters. J Virol 64:2849–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thierry F, Howley PM. 1991. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol 3:90–100. [PubMed] [Google Scholar]
- 38.Wang X, Helfer CM, Pancholi N, Bradner JE, You J. 2013. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J Virol 87:3871–3884. doi: 10.1128/JVI.03068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakakibara N, Chen D, Jang MK, Kang DW, Luecke HF, Wu SY, Chiang CM, McBride AA. 2013. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog 9:e1003777. doi: 10.1371/journal.ppat.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McBride AA, Jang MK. 2013. Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses 5:1374–1394. doi: 10.3390/v5061374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. 2013. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49:843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Makiniemi M, Hillukkala T, Tuusa J, Reini K, Vaara M, Huang D, Pospiech H, Majuri I, Westerling T, Makela TP, Syvaoja JE. 2001. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J Biol Chem 276:30399–30406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- 43.Kanginakudru S, DeSmet M, Thomas Y, Morgan IM, Androphy EJ. 2015. Levels of the E2 interacting protein TopBP1 modulate papillomavirus maintenance stage replication. Virology 478:129–135. doi: 10.1016/j.virol.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reinson T, Toots M, Kadaja M, Pipitch R, Allik M, Ustav E, Ustav M. 2013. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J Virol 87:951–964. doi: 10.1128/JVI.01943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boner W, Taylor ER, Tsirimonaki E, Yamane K, Campo MS, Morgan IM. 2002. A functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1. J Biol Chem 277:22297–22303. doi: 10.1074/jbc.M202163200. [DOI] [PubMed] [Google Scholar]
- 46.Donaldson MM, Mackintosh LJ, Bodily JM, Dornan ES, Laimins LA, Morgan IM. 2012. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J Virol 86:12806–12815. doi: 10.1128/JVI.01002-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hogan GJ, Lee CK, Lieb JD. 2006. Cell cycle-specified fluctuation of nucleosome occupancy at gene promoters. PLoS Genet 2:e158. doi: 10.1371/journal.pgen.0020158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. 2007. FAIRE (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res 17:877–885. doi: 10.1101/gr.5533506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsompana M, Buck MJ. 2014. Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7:33. doi: 10.1186/1756-8935-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loo YM, Melendy T. 2004. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J Virol 78:1605–1615. doi: 10.1128/JVI.78.4.1605-1615.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuck S, Stenlund A. 2015. A conserved regulatory module at the C terminus of the papillomavirus E1 helicase domain controls E1 helicase assembly. J Virol 89:1129–1142. doi: 10.1128/JVI.01903-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen G, Stenlund A. 2002. Sequential and ordered assembly of E1 initiator complexes on the papillomavirus origin of DNA replication generates progressive structural changes related to melting. Mol Cell Biol 22:7712–7720. doi: 10.1128/MCB.22.21.7712-7720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reinson T, Henno L, Toots M, Ustav M Jr, Ustav M. 2015. The cell cycle timing of human papillomavirus DNA replication. PLoS One 10:e0131675. doi: 10.1371/journal.pone.0131675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Demeret C, Desaintes C, Yaniv M, Thierry F. 1997. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J Virol 71:9343–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schweiger MR, Ottinger M, You J, Howley PM. 2007. Brd4-independent transcriptional repression function of the papillomavirus E2 proteins. J Virol 81:9612–9622. doi: 10.1128/JVI.00447-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dostatni N, Lambert PF, Sousa R, Ham J, Howley PM, Yaniv M. 1991. The functional BPV-1 E2 trans-activating protein can act as a repressor by preventing formation of the initiation complex. Genes Dev 5:1657–1671. doi: 10.1101/gad.5.9.1657. [DOI] [PubMed] [Google Scholar]
- 57.Zheng G, Schweiger MR, Martinez-Noel G, Zheng L, Smith JA, Harper JW, Howley PM. 2009. Brd4 regulation of papillomavirus protein E2 stability. J Virol 83:8683–8692. doi: 10.1128/JVI.00674-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abbate EA, Berger JM, Botchan MR. 2004. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2. Genes Dev 18:1981–1996. doi: 10.1101/gad.1220104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han Y, Loo YM, Militello KT, Melendy T. 1999. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A. J Virol 73:4899–4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li R, Botchan MR. 1993. The acidic transcriptional activation domains of VP16 and p53 bind the cellular replication protein A and stimulate in vitro BPV-1 DNA replication. Cell 73:1207–1221. doi: 10.1016/0092-8674(93)90649-B. [DOI] [PubMed] [Google Scholar]
- 61.Melendy T, Stillman B. 1993. An interaction between replication protein A and SV40 T antigen appears essential for primosome assembly during SV40 DNA replication. J Biol Chem 268:3389–3395. [PubMed] [Google Scholar]
- 62.Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, O'Connor SL. 2000. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J Investig Dermatol 114:444–455. doi: 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- 63.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. 2010. NCoR1 mediates papillomavirus E8;E2C transcriptional repression. J Virol 84:4451–4460. doi: 10.1128/JVI.02390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oliveira JG, Colf LA, McBride AA. 2006. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc Natl Acad Sci U S A 103:1047–1052. doi: 10.1073/pnas.0507624103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeSmet M, Kanginakudru S, Rietz A, Wu WH, Roden R, Androphy EJ. 2016. The replicative consequences of papillomavirus E2 protein binding to the origin replication factor ORC2. PLoS Pathog 12:e1005934. doi: 10.1371/journal.ppat.1005934. [DOI] [PMC free article] [PubMed] [Google Scholar]