

ABSTRACT
Resolution of virus infections depends on the priming of virus-specific CD8+ T cells by dendritic cells (DC). While this process requires major histocompatibility complex (MHC) class I-restricted antigen presentation by DC, the relative contribution to CD8+ T cell priming by infected DC is less clear. We have addressed this question in the context of a peripheral infection with herpes simplex virus 1 (HSV). Assessing the endogenous, polyclonal HSV-specific CD8+ T cell response, we found that effective in vivo T cell priming depended on the presence of DC subsets specialized in cross-presentation, while Langerhans cells and plasmacytoid DC were dispensable. Utilizing a novel mouse model that allows for the in vivo elimination of infected DC, we also demonstrated in vivo that this requirement for cross-presenting DC was not related to their infection but instead reflected their capacity to cross-present HSV-derived antigen. Taking the results together, this study shows that infected DC are not required for effective CD8+ T cell priming during a peripheral virus infection.
IMPORTANCE The ability of some DC to present viral antigen to CD8+ T cells without being infected is thought to enable the host to induce killer T cells even when viruses evade or kill infected DC. However, direct experimental in vivo proof for this notion has remained elusive. The work described in this study characterizes the role that different DC play in the induction of virus-specific killer T cell responses and, critically, introduces a novel mouse model that allows for the selective elimination of infected DC in vivo. Our finding that HSV-specific CD8+ T cells can be fully primed in the absence of DC infection shows that cross-presentation by DC is indeed sufficient for effective CD8+ T cell priming during a peripheral virus infection.
KEYWORDS: CD8 T cells, dendritic cells, HSV-1, cross-presentation, direct presentation
INTRODUCTION
Protective immunity against virus infections relies on professional antigen-presenting cells (APC) driving the activation and effector differentiation of naive CD8+ T cells. Among the different types of APC, dendritic cells (DC) are the most effective initiators of T cell priming. DC comprise heterogeneous subsets that differ in location, phenotype, ontogenesis, and function (1, 2). XCR1-expressing DC subsets with developmental dependence on the transcription factors interferon-regulating factor 8 (Irf8), inhibitor of DNA binding 2 (Id2), and basic leucine zipper transcriptional factor ATF-like 3 (Batf3) can be further classified in mice into CD8+ DC and CD103+ DC (3, 4). CD8+ DC exist exclusively in lymphoid organs, whereas CD103+ DC reside primarily in peripheral tissues and enter lymphoid organs only upon migration from the tissue. CD11bhi DC require the transcription factor Irf4, consist largely of interstitial DC such as dermal DC (dDC), and are typically associated with efficient major histocompatibility complex class II (MHC-II) presentation. Plasmacytoid DC (pDC) are potent producers of alpha/beta interferon (IFN-α/β) and have been linked to CD8+ T cell priming in antiviral immunity, although this role remains controversial (5).
CD8+ T cell priming by DC requires presentation of peptides in the context of MHC class I molecules. Such peptides can originate from cytosolic and endosomal sources (6, 7). The presentation of endogenously synthesized, cytosolic antigen is referred to as direct presentation. It typically involves self-antigens and peptides derived from microbial proteins expressed by an infected cell. Cross-presentation, in contrast, enables the translocation of exogenous protein material from endosomes into the cytosolic MHC class I presentation machinery. XCR1+ DC are particularly effective at utilizing this pathway to display model antigens or antigens acquired in the context of tumors to CD8+ T cells (1, 3, 4). Requirements for XCR1+ DC in the induction of virus-specific CD8+ T cell immunity imply that cross-presentation might also be important for the display of viral antigen (8, 9). However, it has also been suggested that DC could be particularly susceptible to infection and therefore drive antiviral CD8+ T cell immunity through direct presentation (10).
We therefore set out to examine whether DC in the lymph nodes (LN) were infected in the context of a murine model of peripheral herpes simplex virus 1 (HSV) infection (11) and whether such infection was required for effective CD8+ T cell priming in vivo. Our data yielded no evidence to suggest that infectious virus translocated from the site of inoculation to the local draining LN, and we could not detect HSV-infected DC in the LN. Using mice in which infected DC can be eliminated, we found that in vivo priming of HSV-specific CD8+ T cell immunity was intact. Together with strict in vivo requirements for the presence of cross-presenting XCR1+ DC, this study argues that direct infection of DC is not required for induction of effective virus-specific CD8+ T cell immunity in vivo during epidermal HSV-1 infection.
RESULTS
In vivo priming of HSV-specific CD8+ T cells is impaired in the absence of Irf8.
Skin infection of mice with HSV-1 results in the stimulation of a CD8+ T cell response that is directed primarily against an MHC class I H2-Kb-restricted, immunodominant epitope from HSV glycoprotein B (gB498–505). These virus-specific effector CD8+ T cells contribute to the clearance of HSV from the skin and the nervous system (11, 12). Moreover, when present as tissue-resident memory cells at sites of virus inoculation (13) or virus reemergence (14), HSV-specific CD8+ T cells can also prevent HSV-1 infection. We began addressing whether cross-presentation drives the priming of these protective HSV-specific CD8+ T cell responses in vivo by examining whether the DC subsets known to have superior cross-presenting capacities, namely, CD8+ DC and CD103+ DC, were required for the priming of HSV-specific CD8+ T cells. For this, we employed Irf8−/− mice that have been described to lack CD8+ DC and CD103+ DC (15). We first validated that this deficiency also applied to the brachial LN (bLN), which drains the site of HSV skin infection and represents the site of T cell priming (16–18). For this, we enriched DC (CD11chi MHC class IIint-hi) from the bLN by depleting T cells, B cells, monocytes/macrophages, and granulocytes as previously described (17) and distinguished these further into DC subsets using phenotypic characteristics specific for CD8+ DC (CD205+ CD8+), CD103+ DC (CD8− CD205+ CD103+), dermal DC (CD8− CD103− CD11b+ CD326−), and Langerhans cells (LC) (CD8− CD103− CD11b− CD326+). As expected, CD8+ DC and CD103+ DC were missing in Irf8−/− mice, while dermal DC and LC were unaffected (Fig. 1A).
FIG 1.

Irf8-dependent cells, but not pDC, are required for HSV-specific CD8+ T cell priming. (A) Representative plots of DC and DC subsets enriched from the brachial LN of Irf8−/− and WT mice. (B to D) Absolute number of gB498–505-specifc CD8+ T cells (B), frequency of IFN-γ producing CD8+ T cells in the spleen (C), and virus titers in the skin (D) of Irf8−/− and WT mice infected with HSV-1 on flank skin 3 and 7 days earlier. All data are pooled results from at least two independent experiments (n = 5 per experiment) and are expressed as mean + standard error of the mean (SEM). (E) Representative plots of splenic pDC in BDCA2-DTR mice treated with or without DTX and absolute number of gB498–505-specific CD8+ T cells in the spleens of BDCA2-DTR mice treated with or without DTX that were infected with HSV-1 on flank skin 7 days earlier. Asterisks indicate statistically significant differences versus controls as assessed by the Student t test (***, P < 0.0001).
In the next step, we infected Irf8−/− mice and wild-type (WT) mice with the KOS strain of HSV-1 on the skin and determined the systemic HSV-specific CD8+ T cell response 7 days later in the spleen. We did this by measuring the number of polyclonal, endogenous CD8+ T cells using H2-Kb-gB498–505-restricted tetramers (Fig. 1B) and assessing the ability of CD8+ T cells to produce IFN-γ in response to restimulation with gB498–505 (Fig. 1C). Irf8−/− mice were dramatically impaired in mounting an HSV-specific CD8+ T cell response (Fig. 1B and C). They also had elevated viral titers in the skin on days 3 and 7 after infection compared to WT mice (Fig. 1D). The fact that Irf8−/− mice also lack plasmacytoid DC (pDC), which can be identified in LN suspensions depleted of T cells, B cells, and other mononuclear cells by expression of CCR9 and intermediate levels of CD11c (Fig. 1E), appeared to be inconsequential, as conditional depletion of pDC in BDCA2-DTR mice using diphtheria toxin (DTX) administration (19) did not impair the HSV-specific CD8+ T cell response following HSV-1 skin infection (Fig. 1E). These findings indicate that Irf8-dependent cells, such as CD8+ DC and CD103+ DC, but not pDC were required for optimal priming of HSV-specific CD8+ T cells in vivo.
XCR1+ DC are required for HSV-specific CD8+ T cell priming in vivo.
Irf8−/− mice are developmentally deficient in CD8+ DC, CD103+ DC, and pDC (2). These mice also have defects in monocytes, granulocytes, and mast cells (20) and with increasing age are prone to developing a syndrome similar to chronic myeloid leukemia (21). It was therefore important to validate whether HSV-specific CD8+ T cell priming in Irf8−/− mice was indeed defective because of the absence of cross-presenting DC. We considered Batf3−/− mice as a suitable alternative to assess the relative in vivo requirements for CD8+ DC and CD103+ DC in virus-specific CD8+ T cell priming after HSV skin infection (8). However, consistent with a recent report (22), we observed that skin-draining LN of Batf3−/− mice on a C57BL/6 background still harbored some CD8+ DC and CD103+ DC (data not shown) and therefore refrained from using these mice for further experiments. We turned our attention to Langerin-DTR mice, which express the human diphtheria toxin receptor (DTR) under the control of the langerin promoter (23). Langerin is a C-type lectin that was originally described in LC but is also expressed by CD8+ DC and CD103+ DC. DTX treatment of Langerin-DTR mice therefore enables the conditional depletion of all three types of DC (24). To facilitate detection of langerin-expressing cells, we used a strain of Langerin-DTR mice that also expressed enhanced green fluorescent protein (GFP) under the control of the langerin promoter (Lg-DTR/GFP). We first validated how effectively CD8+ DC, CD103+ DC, and LC could be depleted in the bLN and skin of these mice using flow cytometry. For this, we depleted T cells, B cells, monocytes/macrophages, and granulocytes from the bLN 2 days after the last DTX treatment and gated on DC using their characteristic expression of high levels of MHC-II and CD11c. The DC were first plotted based on expression of CD8 and GFP (langerin) to identify CD8+ DC (Fig. 2A). The CD8− fraction was then further divided into LC (GFP+ CD103−) and CD103+ DC (GFP+ CD103+). Figure 2B shows that DTX treatment effectively depleted CD8+ DC, CD103+ DC, and LC from the brachial LN. We also assessed LC in the epidermis. Whereas the epidermis of untreated control mice harbored a network of GFP-expressing cells with typical DC morphology, these GFP+ cells were absent from skin examined on day 2 after the last DTX treatment (Fig. 2B). Having thus validated that our DTX treatment regimen depleted CD8+ DC, CD103+ DC, and LC in vivo, we infected DTX-treated mice with HSV-1 on the skin and measured HSV-specific CD8+ T cell priming in the spleen 7 days later. This DTX treatment regimen (Fig. 2C) induced a dose-dependent reduction of the number of endogenous HSV-specific CD8+ T cells (Fig. 2C), highlighting that langerin-expressing DC were required for virus-specific CD8+ T cell priming after HSV-1 skin infection. Although LC share the expression of langerin with CD8+ DC and CD103+ DC, LC develop in situ from yolk sac-derived precursors and therefore replenish with very different kinetics after DTX treatment (24). This becomes obvious on day 21 after DTX treatment, when CD8+ DC and CD103+ DC were reconstituted but LC remained absent (Fig. 2A and B). This differential repletion kinetics enabled us to examine the relative contribution of LC to HSV-specific CD8+ T cell responses. The observation that Langerin-DTR mice treated 21 days earlier with DTX had numbers of HSV-specific CD8+ T cells similar to those of Langerin-DTR mice given phosphate-buffered saline (PBS) (Fig. 2D) further supports the conclusion that in vivo priming of HSV-specific CD8+ T cells following HSV skin infection depended on the presence of CD8+ DC and CD103+ DC but did not require LC or pDC.
FIG 2.
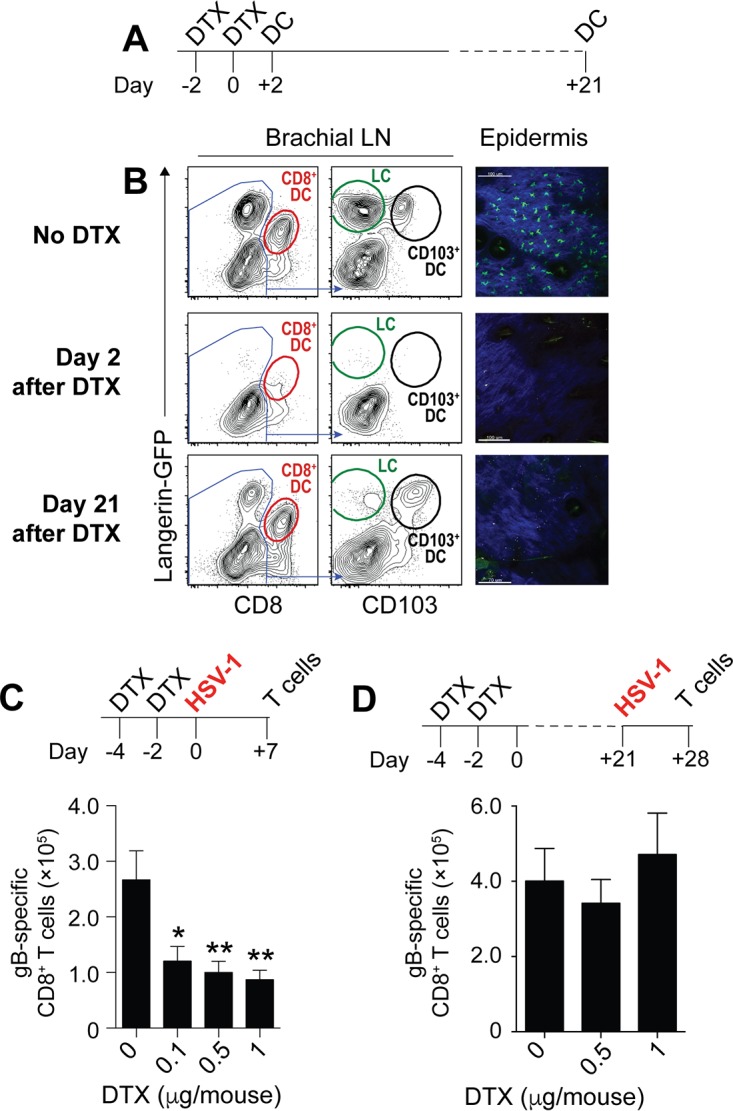
Langerin-positive DC, but not LC, are required for HSV-specific CD8+ T cell priming. (A) Schematic depicting the DTX treatment regimen for the assessment of DC depletion. Mice were treated with DTX on days −4 and −2. DC in the brachial LN and the epidermis were analyzed 2 days or 21 days after the last DTX treatment. (B) Representative plots of MHC-IIhi CD11chi cells enriched from the brachial LN on day 2 or day 21 after DTX treatment on two consecutive days. Cells were first plotted based on expression of CD8 and GFP (langerin) to identify CD8+ DC. The CD8− fraction was then further divided into LC (GFP+ CD103−) and CD103+ DC (GFP+ CD103+). (C and D) Top panels, schematics depicting the DTX treatment regimen to determine its impact on HSV-specific CD8+ T cell priming. Mice were treated with DTX on days −4 and −2. On day 0 (C) or day 21 (D) mice were infected on the skin with HSV-1, and the spleens were analyzed for virus-specific CD8 T cells 7 days later. Bottom panels, absolute numbers of gB498–505-specific CD8+ T cells in the spleen on day 7 after HSV-1 skin infection of Lg-DTR mice that last received DTX 2 days (C) or 21 days (D) earlier relative to those in PBS controls. All data are pooled results from at least two independent experiments (n = 3 to 5 per experiment) and are expressed as mean + SEM. Asterisks indicate statistically significant differences versus controls as assessed by one-way analysis of variance (ANOVA) (*, P < 0.05).
Infectious virus, viral RNA, or HSV-infected DC are not detectable in the LN draining the site of infection.
The above findings showed that HSV-specific CD8+ T cell priming required the presence of DC subsets with superior capacities to perform cross-presentation. These findings are consistent with an important role for cross-presentation in the type of HSV-specific T cell immunity under investigation. However, it was also possible that CD8+ DC and CD103+ DC were infected with HSV-1 and consequently drove T cell priming through direct presentation rather than cross-presentation. We therefore determined whether cross-presentation itself was required for HSV-specific CD8+ T cell priming. As direct presentation requires infection of DC, we first examined whether infectious virus was detectable in the bLN. We chose day 2 after infection for this analysis, as it represents the onset of HSV-derived MHC class I antigen presentation by CD8+ DC in the bLN (17) and reflects a time when viral replication in the skin is still increasing (11, 17). Although DC acquire HSV-derived antigen and present it to naive CD8+ T cells at this time point (17), we could not grow virus from bLN homogenates of mice infected 2 days earlier (Fig. 3A). When we enriched CD11chi MHC class IIhi cells from the bLN, we also could not find a clearly discernible population of DC that were positive for GFP in mice infected on the skin with a GFP-expressing HSV-1 KOS strain (HSV-GFP). To determine if this failure to detect virus or infected DC in the LN was due to a general inability of HSV-1 to reach the LN and infect DC, we repeated these experiments in mice deficient in the IFN-α/β receptor (Ifnar2−/− mice), which are unable to contain HSV-1 infections. Notwithstanding the fact that the mice also have higher viral titers upon HSV infection (25), the primary purpose for including these mice in the present study was to serve as positive controls for virus detection. Consistent with this, Ifnar2−/− mice infected epidermally with HSV-GFP indeed had readily detectable levels of HSV-1 in the bLN (Fig. 3A) and a clearly discernible population of GFP-expressing DC (Fig. 3B). We also investigated viral RNA expression in DC subsets isolated from the bLN of Ifnar2−/− and WT mice infected 2 days earlier with HSV-1 on the skin. We focused on several viral genes that are expressed throughout different phases of the infection (26). Rl2 is an immediate early gene, the product of which transactivates the next class of viral genes. Us12 is also an immediate early gene but is more robustly expressed throughout the infection. Ul27 and Us6 are late genes and were chosen because they encode relevant T cell antigens (glycoprotein B and glycoprotein D, respectively). While we detected mRNA coding for all these viral genes in CD8+ DC, CD103+ DC, dermal DC, and LC from Ifnar2−/− mice, these were absent from the same DC subsets isolated from WT mice (Fig. 3C). These observations indicate that HSV-1 could generally translocate from the site of infection into the draining LN, that it could infect different LN DC with comparable effectiveness, and that these events could be measured through the techniques employed here.
FIG 3.
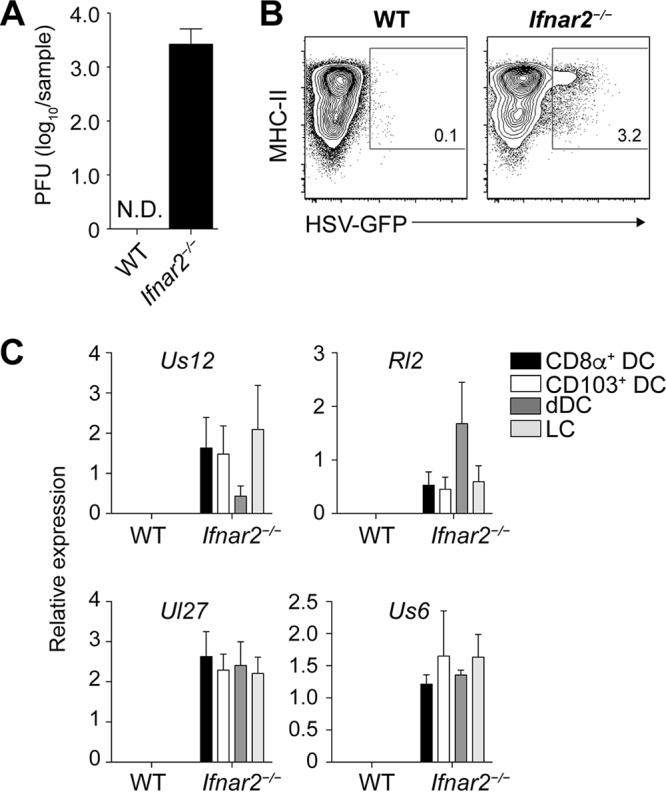
No evidence for virus infection of DC in the LN draining the site of infection in wild-type mice. (A) Viral titers in homogenates of the brachial LN measured 2 days after epidermal infection of Ifnar2−/− and WT mice with HSV-1. (B) Representative plots of MHC-IIhi CD11chi DC enriched from the brachial LN of Ifnar2−/− and WT mice infected 2 days earlier with a GFP-expressing HSV-1 strain. (C) Relative expression of viral mRNA in DC subsets isolated from the brachial LN of Ifnar2−/− and WT mice infected with HSV-1 on the skin 2 days earlier. Data are pooled results from two independent experiments (n = 5 per experiment) and are expressed as mean + SEM. N.D., not detectable.
Lack of viral recrudescence in HSV-Cre-infected R-DTA mice.
The above findings are consistent with the interpretation that virus and HSV-infected DC were absent from the bLN following epidermal infection of wild-type mice. We sought to validate these findings through an alternative approach in which potentially infected DC can be eliminated from the LN. For this, we utilized mice in which a loxP stop cassette prevents the ROSA26 promoter-driven expression of diphtheria toxin in the germ line configuration (R-DTA mice) (27). Exposure of cells from R-DTA mice to the recombinase Cre removes the stop cassette, induces diphtheria toxin expression, and therefore kills cells containing Cre (28). We reasoned that infecting R-DTA mice with a Cre-expressing HSV-1 KOS strain (HSV-Cre) should eliminate productively infected cells. Before we used this system to examine how HSV-specific CD8+ T cell priming was affected in the absence of direct presentation by infected DC, we tested whether infection of R-DTA mice with HSV-Cre led to the expected depletion of productively infected cells. In this regard, it is important to note that epidermal inoculation of mice with HSV-1 results in two distinct phases of virus replication (11). The virus undergoes a first round of replication at the site of skin infection (referred to as the “primary site” in Fig. 4A). It then enters local sensory nerves and travels in a retroaxonal manner to the dorsal root ganglia (DRG). Here it replicates again, which upon infection of neighboring nerve cells allows the virus to gain access to their axons and directs new virions back to the skin. The consequence of this replication-dependent recrudescence at the “secondary” site is a zosteriform skin lesion covering the entire innervated dermatome (Fig. 4A). The dependence of viral recrudescence on productive infection in the DRG is demonstrated through the use of a thymidine kinase-deficient HSV-1 KOS strain (HSV-ΔTK), which does not undergo the second round of replication in the DRG (29) and therefore fails to generate a secondary-site lesion (Fig. 4A). R-DTA mice infected with HSV-Cre had slightly reduced viral titers at the primary site 2 days after infection (Fig. 4B). More prominently, these mice failed to generate a secondary-site lesion, as demonstrated clinically (Fig. 4A) and through viral titer measurements at the secondary site (Fig. 4C). HSV-Cre-induced prevention of viral recrudescence thus supported the notion that HSV-infected cells were eliminated in R-DTA mice.
FIG 4.

R-DTA mice inoculated with HSV-Cre eliminate infected cells in the DRG and DC in the LN. (A to C) Photograph of the primary- and secondary-site viral lesions (A) and viral titers (B and C) in wild-type and R-DTA mice infected on the skin with a Cre-expressing HSV strain compared to infection of WT mice with a TK-deficient HSV strain. (D) Percentage of GFP+ DC in the brachial LN of WT, Ifnar2−/−, R-DTA × Ifnar2−/−, and R-DTA mice infected 2 days earlier on the skin with HSV-Cre-GFP. Data are pooled from two independent experiments (n = 5 per experiment) and are expressed as mean + SEM. N.D., not detectable.
Elimination of infected DC in HSV-Cre-infected R-DTA mice.
As different cell types can differ in susceptibility to virus infection, we investigated whether this system allowed for the elimination not only of HSV-infected keratinocytes or neurons but also of DC infected by HSV. Given that infected DC could not be detected in the bLN of wild-type mice (Fig. 3A and B), we harnessed our observation that a proportion of LN DC were infected in Ifnar2−/− mice to determine whether HSV-Cre-infected DC could be eliminated in R-DTA mice. We first analyzed the different DC subsets from the bLN of Ifnar2−/− mice infected on the skin with an HSV-1 KOS strain that expressed both Cre and GFP (HSV-Cre-GFP). GFP+ cells were represented at similar frequencies among CD8+ DC, CD103+ DC, dermal DC, and LC (Fig. 4D). Notably, when we crossed the Ifnar2−/− mice onto the R-DTA background and reexamined GFP expression in HSV-Cre-GFP-infected mice, we could no longer detect GFP+ DC subsets (Fig. 4D). These observations highlight that HSV-infected DC could also be efficiently eliminated in R-DTA mice.
HSV-specific CD8+ T cell priming is not affected by the absence of infected DC.
We finally used the R-DTA system to interrogate whether infected DC were required for optimal HSV-specific CD8+ T cell priming in vivo. We first compared the capacities of bLN DC from R-DTA and WT mice to present HSV-derived antigen ex vivo to T cell receptor (TCR)-transgenic HSV-specific CD8+ T cells (gBT-I). For this, we sorted CD8+ DC, CD103+ DC, dermal DC, and LC 2 days after epidermal HSV-Cre infection, coincubated them in vitro with naive gBT-I, and determined cell trace violet dilution after 60 h by flow cytometry as a readout for proliferation (16, 17). Reflecting the situation in HSV-infected WT mice, CD8+ DC were the only DC subset that induced proliferation of gBT-I in R-DTA mice (Fig. 5A and B). Given the well-documented capacity of CD8+ DC to cross-present and the above considerations on the R-DTA system, these findings are consistent with the conclusion that the gBT-I response in this experiment was driven by cross-presentation. In the next step, we enumerated the magnitude of the HSV-specific CD8+ T response in the spleens of R-DTA mice and WT mice 7 days after epidermal HSV infection and assessed their capacity to produce cytokines upon ex vivo peptide restimulation. The magnitude of the HSV-specific CD8+ T cell response in R-DTA mice was not different from that in WT mice infected with the same strain of virus (Fig. 5C) or HSV-ΔTK. The proportion of CD8+ T cells responding in vitro to gB498–505 restimulation with secretion of IFN-γ showed a slight reduction, but this was not statistically significant (Fig. 5D). We also observed no significant differences in further functional analyses, such as frequency of tumor necrosis factor alpha-positive (TNF-α+) CD8+ T cells or intensity of binding to the H2-Kb-gB498–505-restricted tetramer (data not shown). Consistent with these measures of intact functionality, R-DTA mice infected with HSV-Cre cleared the virus as efficiently as WT mice from the primary site of infection by day 7 (Fig. 5E). We also examined HSV-specific CD8+ T cell priming in R-DTA × Ifnar2−/− mice (Fig. 5C and D). Although the amount of HSV-specific CD8+ T cell priming was significantly reduced in these mice relative to WT controls (Fig. 5C), this reduction was unlikely to due to the elimination of infected DC, as Ifnar2−/− mice were equally impaired in HSV-specific CD8+ T cell priming (30), and these mice harbor multiple infected DC subtypes (Fig. 3 and 4). Taking the results together, we conclude that mice lacking infected DC from the LN were fully capable of mounting protective HSV-specific CD8+ T cell responses.
FIG 5.

Ex vivo CD8+ DC presentation of HSV-derived antigen and HSV-specific CD8+ T cell priming does not require directly infected DC. (A and B) Absolute numbers of proliferated, CTV-labeled gBT-I upon coincubation for 60 h with serial dilutions of LN DC subsets isolated 2 days after skin HSV-Cre infection of WT (A) and R-DTA (B) mice. (C and D) Absolute number of gB498–505-specific CD8+ T cells (C) and frequency of IFN-γ-producing CD8+ T cells (D) in the spleens of R-DTA (open bars), WT (black bars), and R-DTA × Ifnar2−/− mice (gray bars) infected with HSV-Cre or HSV-ΔTK on flank skin 7 days earlier. Data are pooled from three independent experiments (n = 5 to 10 per group and experiment) and are expressed as mean + SEM.
DISCUSSION
The relative contributions of cross-presented and directly presented antigens to the induction of antiviral CD8+ T cell immunity by DC have remained controversial (8, 31–33). Little doubt exists that DC located at the inoculation site can be infected with virus (34). However, whether the DC responsible for CD8+ T cell priming in the LN are infected during epidermal virus infections has not been resolved. Our analysis of the LN draining the site of epidermal HSV-1 infection in wild-type mice revealed no infectious virus, no viral RNA, and no discernible population of infected DC. This was not related to a general inability to identify virus translocating from the epidermis to the LN and/or to infect LN DC, as we readily detected replicative virus and HSV-infected DC in the bLN of Ifnar2−/− mice. This analysis was done on day 2 after infection, when viral replication in the skin is still increasing (17) and MHC class I-restricted antigen presentation by DC in the LN commences (16, 17). While we cannot rule out that some infected DC might have reached the LN prior to this time point, this is unlikely to make any meaningful contribution to HSV-specific CD8 T cell priming, as the earliest signs of in vivo T cell receptor stimulation (CD69 upregulation) do not occur before about 48 h after infection (35).
Concluding that infected DC were very rare in, if not absent from, the LN draining the site of HSV-1 infection hinges on the sensitivity of the assays utilized to measure the virus and therefore warranted further validation. For this we established a new mouse model in which virus-infected DC could be eliminated. By infecting R-DTA mice with a Cre-expressing strain of HSV-1, we reasoned that infected cells would produce Cre, transcribe DTX, and consequently die. This has previously been shown to occur when R-DTA mice are crossed onto backgrounds that express Cre in certain cell types, such as DC (27, 28). Two lines of evidence indicated that virus-mediated delivery of Cre also worked to excise the lox P stop cassette in R-DTA mice. First, HSV could not reemerge at the secondary site in the skin of R-DTA mice, a process that is well known to require viral replication and productive infection of cells in the DRG (29). This lack of viral recrudescence in HSV-Cre-infected R-DTA mice is likely a combinatorial effect of reduced viral input into the DRG from the site of inoculation and the Cre-induced elimination of infected cells in the DRG. As it takes about 96 h for viral titers in the DRG to reach levels comparable to those in the inoculation site (11), these observations also imply that Cre-induced death of host cells in R-DTA mice could be most effective at preventing spread to uninfected cells when viral loads are comparatively low. The second line of evidence harnessed our observation that HSV-infected DC can be identified in the LN in the absence of IFN-α/β responses. Crossing Ifnar2−/− mice onto the R-DTA background and infecting these epidermally with HSV-Cre demonstrated that infected DC could also be eliminated from the LN in R-DTA mice. We therefore conclude that infecting R-DTA mice with a Cre-expressing virus provides a novel and effective in vivo model to specifically study the contribution of infected cells to antiviral immunity. This system is an important advance over previous studies, which so far have examined the role of cross-presentation in antiviral T cell immunity through the use of mice lacking the DC subtypes specialized in cross-presentation (8) or in models where mice were infected through routes that deposit virus within the priming lymphoid organ (36).
We showed that R-DTA mice lacking infected DC in the LN can effectively initiate and prime HSV-specific CD8+ T cell priming. Combined with our observation that mice lacking DC specialized in cross-presentation failed to mount HSV-specific CD8+ T cell responses, these findings argue that cross-presentation by DC in vivo is sufficient to induce effector differentiation of virus-specific CD8+ T cells following epidermal HSV infection. The very small reduction in the ex vivo capacity of CD8+ DC from R-DTA mice to induce proliferation of gBT-I cells and the ability of endogenous HSV-specific CD8+ T cells from R-DTA mice infected with HSV-Cre to mount ex vivo IFN-γ recall responses is likely related to reduced viral replication at the site of infection and the absence of viral recrudescence. This conclusion is based on the decrease in viral titers at the inoculation site in HSV-Cre-infected R-DTA mice and our previous demonstration that CD8+ DC isolated from mice infected epidermally with HSV-ΔTK, which does not cause viral recrudescence, have slightly reduced capacitates to drive the proliferation of gBT-I ex vivo (17). Even if these minor reductions were due to the elimination of a few HSV-infected DC that escaped our analysis of the draining LN, our data still show that effective CD8+ T cell responses do not require infected DC.
In this context, it is also worthwhile to come back to the observation that several DC were infected in the bLN of Ifnar2−/− mice. Although one could expect that infected DC would directly present HSV-derived antigen and therefore should stimulate HSV-specific CD8+ T cells, any such contribution does not seem to be sufficient, as Ifnar2−/− mice failed to mount effective HSV-specific CD8+ T cell responses (30). This defect in HSV-specific CD8+ T cell priming was related to CD8+ DC requiring stimulation by IFN-α/β to provide interleukin 15 (IL-15) to the CD8+ T cells recognizing HSV-derived antigen on the DC (30). With HSV-infected R-DTA × Ifnar2−/− mice comparably impaired in HSV-specific CD8+ T cell priming as Ifnar2−/− mice, it appears unlikely that this complex regulation was simply required because some CD8+ DC were infected in Ifnar2−/− mice. Rather, these observations suggest that infected DC alone, at least in the absence of appropriate activation cues, are not sufficient to drive the priming of HSV-specific CD8+ T cells in vivo.
Our previous work showed that CD8+ DC were the only DC subset capable of presenting antigen to naive HSV-specific CD8+ T cells over the first 4 days of the infection (17). Together with the demonstration that in vivo interactions between XCR1+ DC and naive HSV-specific CD8+ T cells occur largely within the first 48 h after the infection (35) and that HSV-specific CD8+ T cell priming also takes place in vivo if the site of viral inoculation is excised after 8 h (16), CD8+ DC appear to play the dominant role in the initiation of the CD8+ T cell response in this model. Interestingly, as the virus bounces back to the skin, a second wave of MHC class I-restricted antigen presentation can be detected from day 5 after infection, which now also involves a strong contribution by CD103+ DC (17). It is conceivable that CD103+ DC add a “second” antigenic hit to HSV-specific CD8+ T cells initially primed by CD8+ DC as they egress from the LN. This could be relevant for migrational imprinting, consolidation of effector function, and/or their potential to form memory. However, disentangling such potential roles for CD103+ DC in vivo is currently not possible, as identical transcriptional and developmental requirements of CD8+ DC and CD103+ DC (37) render all available mouse models, including the ones used in this study, deficient in both DC subtypes. The exclusion of an important contribution by LC to HSV-specific CD8+ T cell priming in response to this epidermal infection also shows that LC are not the migratory DC subset required in our model to deliver antigen to the LN for cross-presentation by CD8+ DC (16, 38). With dermal DC being the most numerous DC subset migrating from the site of inoculation to the draining LN (2, 17, 39), they are the most likely source of antigen that is engulfed by CD8+ DC in the LN and subsequently cross-presented to naive HSV-specific CD8+ T cell priming. The redundancy of pDC in the response studied here aligns with an earlier report (19) arguing that pDC matter more for viral immunity against systemic infections.
Taken together, this study reveals that cross-presentation by CD8+ DC is sufficient to elicit effective in vivo HSV-specific CD8+ T cell priming in response to a peripheral infection. While in previous studies a requirement for direct presentation in response to cutaneous orthopoxvirus infection has been observed and suggested (40, 41), our results highlight that this is not the case for all viruses. Here we show that cross-presentation provides an important avenue for effective in vivo CD8+ T cell priming against a cytolytic virus that largely evades direct priming.
MATERIALS AND METHODS
Mice.
C57BL/6, Ifnar2−/− (42), R-DTA (27), R-DTA × Ifnar2−/−, Irf8−/− (21), BDCA2-DTR (19), Lg-DTR/EGFP (23), and gBT-I (17) mice were crossed and bred at the Department of Microbiology and Immunology, The Peter Doherty Institute, The University of Melbourne, Australia, under specific-pathogen-free conditions. All animal protocols were carried out under the authority of Australian ethics project licenses (number 1112345 and 1513504) and approved by the University of Melbourne Animal Ethics Committee in strict accordance with Australian governmental regulations (Australian Code of Practice for the Care and Use of Animals for Scientific Purpose). To deplete DC in BDCA2-DTR and Lg-DTR mice, diphtheria toxin (DTX) (Sigma-Aldrich) were injected intraperitoneally (i.p.) at 200 ng/mouse or as indicated at days −4 and −2 prior to infection.
Virus, viral mRNA, and titer determination.
Mice were infected with 1 × 106 PFU of HSV-KOS (HSV-1), thymidine kinase-deficient HSV (HSV-TK), Cre-expressing HSV (HSV-Cre; KOS0152) (29), GFP/Cre-expressing (HSV-GFP; KOS/pCMV/eGC) (26) by employing the flank scarification model. As previously described (11), anesthetized mice were shaved and excess hair was removed using commercially available depilation cream. A small area of epidermis at the left flank was then abraded for virus inoculation, and mice were bandaged thereafter for 2 days. To determine virus titers in skin or lymph nodes, mice were euthanized and samples were removed, homogenized, clarified by centrifugation, and then assayed for infectious virus by plaque assay on Vero cells (CSL, Parkville, Australia). For quantitative real-time PCR analysis of viral RNA, total RNA from sorted cells was DNase treated and extracted using a Direct-Zol RNA MicroPrep kit (Zymo Research). Samples were reverse transcribed using a SuperScript VILO cDNA synthesis kit (Invitrogen). Controls without reverse transcription yielded no viral transcripts, which excludes viral genomic DNA as a source for the transcripts measured. Real-time PCR was performed with TaqMan universal PCR master mix (Life Technologies) with primers and probes for HSV-1 genes Rl2 (coding for infected-cell polypeptide 0 [ICP0]) (43) Us12 (coding for ICP47) (26), Ul27 (coding for glycoprotein B [gB]) (26), and Us6 (coding for gD) (26) and cellular housekeeping genes obtained from Life Technologies: pgk1 (Mm00435617_m1), tbp (Mm00446973_m1), hmbs (Mm00660260_m1), gapdh (Mm99999915_g1), b2m (Mm00437762_m1), and hprt (Mm00446968_m1). mRNA expression was normalized to all cellular housekeeping genes, and relative expression (RE) was determined (RE = 2−ΔΔCT).
Flow cytometry, cell staining and intracellular cytokine staining.
Data were collected on an LSR Fortessa or FACSAria III instrument (both from BD Biosciences) and analyzed using FlowJo software (Tree Star). The staining protocols used combinations of antibodies listed below: from BD Bioscience, anti-CD8 (53-6.7), anti-IFN-γ (XMG1.2), anti-CD11c (N418), anti-CD16/CD32 (2.4G2, Fc block), anti-CD44 (IM70), and anti-Vα2+ (B20.1); from eBioscience, anti-CD11b (M1/70) and anti-CD205 (205yekta); from BioLegend, anti-CD19 (6D5), anti-CD103 (2E7), anti-CD326 (EpCAM, G8.8), anti-CD62L (53–6.70), and anti-GzmB (GB11); and from R&D Systems, anti-CD199 (CCR9, 242503). Single-cell suspensions of spleen and skin draining/nondraining lymph nodes were surface stained directly ex vivo in the presence anti-CD16/CD32 to block nonspecific binding. Enumeration of HSV-1-specific T cells was achieved using either HSV-specific H-2Kb-gB498–505 tetramers (Andrew Brooks, The University of Melbourne) or following incubation with 1 μM HSV-1 gB498–505 for 1 h followed by an additional 4 h in the presence of 10 μg/ml brefeldin A (Sigma, USA). Cells from HSV-1-infected mice were then stained with anti-CD8 and anti-CD44 and fixed with BD Cytofix/Cytoperm (BD Biosciences, USA) for 20 min at room temperature. Cells were washed twice with fluorescence-activated cell sorter (FACS) buffer (PBS, 1% fetal calf serum [FCS], 2 mM EDTA), stained with anti-IFN-γ in BD Perm/Wash buffer (BD Biosciences, USA), and then analyzed. The in vivo determination of cells infected with HSV-1 was performed using HSV-GFP virus during primary infection, with expression of cellular GFP indicating infected cells.
DC isolation and sorting strategy.
Brachial lymph nodes from mice at 2 days after HSV-1 infection were subjected to enzymatic digestion in RPMI 1640 supplemented with glutamine, penicillin, and streptomycin (all from Gibco), collagenase type IV (200U/ml; Worthington), and DNase 1 (0.2 mg/ml; Roche) for 20 min, followed by 5 min of incubation with 0.01 M (final concentration) EDTA. Single-cell suspensions were enriched for conventional DC (excluding plasmacytoid DC) using magnetic bead depletion as previously described (17). Cells were stained with anti-CD11c, anti-CD8, anti-CD205, anti-CD326, anti-CD103, anti-CD11b, and anti-CD19. DC subtypes from propidium iodide-negative CD11c+ events were sorted by flow cytometry according to strategies described previously (17); briefly, CD8α+ DC (CD19− CD11c+ CD205int CD8α+), CD103+ DC (CD19− CD11c+ CD205hi CD8α− CD103+), dermal DC (dDC) (CD19− CD11c+ CD205int CD8α− CD103− CD326− CD11b+), and Langerhans cells (LC) (CD19− CD11c+ CD205int CD8α− CD103− CD326+) were used. The purity of sorted subsets was routinely over 94%.
Preparation of T cells.
gBT-I T cells were isolated from pooled LN of gBT-I mice and enriched using goat anti-rat IgG-coupled magnetic bead (Qiagen, Australia) depletion with antibodies against Mac-1 (M1/70), F4/80 (F4/80), erythrocytes (TER-119), GR-1 (RB6-8C5), I-A/E (M5114), and CD4 (GK1.5). Cells were routinely 90 to 95% pure as determined by flow cytometry. T cells were labeled with 5 μM CellTrace violet (CTV) (Molecular Probes, USA) as per the manufacturer's protocol. Labeled cells (5 × 104) were cocultured with various concentrations of purified DC subtypes from HSV-1 infected for 60 h as previously described (17). Proliferation was measured by CTV dilution of CD8+, Vα2+, and CD44+ cells using a flow cytometer.
Microscopy.
Lg-DTR/EGFP mice were anesthetized, shaved on the left flank, and hair depilated for flank skin imaging as described elsewhere (44). Imaging was performed with an upright LSM710 NLO multiphoton microscope (Carl Zeiss, Jena, Germany) with a 20×/1.0 numerical aperture (NA) water immersion objective enclosed in an environmental chamber that was maintained at 35°C with heated air. Fluorescence excitation was provided by a Chameleon Vision II Ti:Sapphire laser (Coherent Inc., USA) with dispersion correction and fluorescence emission detected using external nondescanned photomultiplier tubes. GFP and second-harmonic generation were excited at 920 nm. Three-dimensional image stacks were acquired at a resolution of 512 by 512. Raw imaging data were then processed with Imaris 8 (Bitplane).
ACKNOWLEDGMENTS
We thank R. Locksley, M. Colonna, K. Murphy, and B. Malissen for mice. We gratefully acknowledge the excellent assistance by Amanda Turner.
Our research is supported by the National Health and Medical Research Council of Australia. P. Whitney is supported by an Overseas Biomedical Fellowship (NHMRC) and a MDHS Faculty Fellowship (University of Melbourne). T. Gebhardt is supported by a fellowship from the Sylvia and Charles Viertel Charitable Foundation. D. Tscharke is supported by a Senior Research Fellowship (NHMRC).
REFERENCES
- 1.Heath WR, Carbone FR. 2009. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol 10:1237–1244. doi: 10.1038/ni.1822. [DOI] [PubMed] [Google Scholar]
- 2.Malissen B, Tamoutounour S, Henri S. 2014. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol 14:417–428. doi: 10.1038/nri3683. [DOI] [PubMed] [Google Scholar]
- 3.Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW, Henn V, Kloetzel PM, Gurka S, Kroczek RA. 2010. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 207:1273–1281. doi: 10.1084/jem.20100348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M. 2010. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med 207:1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swiecki M, Colonna M. 2015. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15:471–485. doi: 10.1038/nri3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joffre OP, Segura E, Savina A, Amigorena S. 2012. Cross-presentation by dendritic cells. Nat Rev Immunol 12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 7.Ackerman AL, Cresswell P. 2004. Cellular mechanisms governing cross-presentation of exogenous antigens. Nat Immunol 5:678–684. [DOI] [PubMed] [Google Scholar]
- 8.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. 2008. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh R, Cresswell P. 2010. Defective cross-presentation of viral antigens in GILT-free mice. Science 328:1394–1398. doi: 10.1126/science.1189176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freigang S, Probst HC, van den Broek M. 2005. DC infection promotes antiviral CTL priming: the ‘Winkelried’ strategy. Trends Immunol 26:13–18. doi: 10.1016/j.it.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 11.van Lint A, Ayers M, Brooks AG, Coles RM, Heath WR, Carbone FR. 2004. Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol 172:392–397. doi: 10.4049/jimmunol.172.1.392. [DOI] [PubMed] [Google Scholar]
- 12.Simmons A, Tscharke DC. 1992. Anti-CD8 impairs clearance of herpes simplex virus from the nervous system: implications for the fate of virally infected neurons. J Exp Med 175:1337–1344. doi: 10.1084/jem.175.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T. 2012. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109:7037–7042. doi: 10.1073/pnas.1202288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, Jin L, Diem K, Koelle DM, Wald A, Robins H, Corey L. 2013. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature 497:494–497. doi: 10.1038/nature12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC III, Belardelli F, Gabriele L. 2002. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med 196:1415–1425. doi: 10.1084/jem.20021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, Heath WR, Carbone FR. 2003. Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science 301:1925–1928. doi: 10.1126/science.1087576. [DOI] [PubMed] [Google Scholar]
- 17.Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, Brooks AG, Heath WR. 2009. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 10:488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- 18.Eidsmo L, Stock AT, Heath WR, Bedoui S, Carbone FR. 2012. Reactive murine lymph nodes uniquely permit parenchymal access for T cells that enter via the afferent lymphatics. J Pathol 226:806–813. doi: 10.1002/path.3975. [DOI] [PubMed] [Google Scholar]
- 19.Swiecki M, Wang Y, Gilfillan S, Colonna M. 2013. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog 9:e1003728. doi: 10.1371/journal.ppat.1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yanez A, Goodridge HS. 2016. Interferon regulatory factor 8 and the regulation of neutrophil, monocyte, and dendritic cell production. Curr Opin Hematol 23:11–17. doi: 10.1097/MOH.0000000000000196. [DOI] [PubMed] [Google Scholar]
- 21.Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, Lou J, Knobeloch KP, Gabriele L, Waring JF, Bachmann MF, Zinkernagel RM, Morse HC 3rd, Ozato K, Horak I. 1996. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 87:307–317. doi: 10.1016/S0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 22.Seillet C, Jackson JT, Markey KA, Brady HJ, Hill GR, Macdonald KP, Nutt SL, Belz GT. 2013. CD8alpha+ DCs can be induced in the absence of transcription factors Id2, Nfil3, and Batf3. Blood 121:1574–1583. doi: 10.1182/blood-2012-07-445650. [DOI] [PubMed] [Google Scholar]
- 23.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, Saeland S, Davoust J, Malissen B. 2005. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B. 2007. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med 204:3119–3131. doi: 10.1084/jem.20071724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med 189:663–672. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC. 2014. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response. PLoS Pathog 10:e1004237. doi: 10.1371/journal.ppat.1004237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Voehringer D, Liang HE, Locksley RM. 2008. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J Immunol 180:4742–4753. doi: 10.4049/jimmunol.180.7.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D. 2009. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 206:549–559. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakim LM, Jones CM, Gebhardt T, Preston CM, Carbone FR. 2008. CD8(+) T-cell attenuation of cutaneous herpes simplex virus infection reduces the average viral copy number of the ensuing latent infection. Immunol Cell Biol 86:666–675. doi: 10.1038/icb.2008.47. [DOI] [PubMed] [Google Scholar]
- 30.Greyer M, Whitney PG, Stock AT, Davey GM, Tebartz C, Bachem A, Mintern JD, Strugnell RA, Turner SJ, Gebhardt T, O'Keeffe M, Heath WR, Bedoui S. 2016. T cell help amplifies innate signals in CD8(+) DCs for optimal CD8(+) T cell priming. Cell Rep 14:586–597. doi: 10.1016/j.celrep.2015.12.058. [DOI] [PubMed] [Google Scholar]
- 31.Sei JJ, Haskett S, Kaminsky LW, Lin E, Truckenmiller ME, Bellone CJ, Buller RM, Norbury CC. 2015. Peptide-MHC-I from endogenous antigen outnumber those from exogenous antigen, irrespective of APC phenotype or activation. PLoS Pathog 11:e1004941. doi: 10.1371/journal.ppat.1004941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson NS, Behrens GM, Lundie RJ, Smith CM, Waithman J, Young L, Forehan SP, Mount A, Steptoe RJ, Shortman KD, de Koning-Ward TF, Belz GT, Carbone FR, Crabb BS, Heath WR, Villadangos JA. 2006. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat Immunol 7:165–172. [DOI] [PubMed] [Google Scholar]
- 33.Zinkernagel RM. 2014. On the role of dendritic cells versus other cells in inducing protective CD8+ T cell responses. Front Immunol 5:30. doi: 10.3389/fimmu.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Macleod BL, Bedoui S, Hor JL, Mueller SN, Russell TA, Hollett NA, Heath WR, Tscharke DC, Brooks AG, Gebhardt T. 2014. Distinct APC subtypes drive spatially segregated CD4+ and CD8+ T-cell effector activity during skin infection with HSV-1. PLoS Pathog 10:e1004303. doi: 10.1371/journal.ppat.1004303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. 2015. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity 43:554–565. doi: 10.1016/j.immuni.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 36.Nopora K, Bernhard CA, Ried C, Castello AA, Murphy KM, Marconi P, Koszinowski U, Brocker T. 2012. MHC class I cross-presentation by dendritic cells counteracts viral immune evasion. Front Immunol 3:348. doi: 10.3389/fimmu.2012.00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy KM. 2013. Transcriptional control of dendritic cell development. Adv Immunol 120:239–267. doi: 10.1016/B978-0-12-417028-5.00009-0. [DOI] [PubMed] [Google Scholar]
- 38.Kim M, Truong NR, James V, Bosnjak L, Sandgren KJ, Harman AN, Nasr N, Bertram KM, Olbourne N, Sawleshwarkar S, McKinnon K, Cohen RC, Cunningham AL. 2015. Relay of herpes simplex virus between Langerhans cells and dermal dendritic cells in human skin. PLoS Pathog 11:e1004812. doi: 10.1371/journal.ppat.1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eidsmo L, Allan R, Caminschi I, van Rooijen N, Heath WR, Carbone FR. 2009. Differential migration of epidermal and dermal dendritic cells during skin infection. J Immunol 182:3165–3172. doi: 10.4049/jimmunol.0802950. [DOI] [PubMed] [Google Scholar]
- 40.Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, Barber GN, Bennink JR, Yewdell JW. 2008. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol 9:155–165. doi: 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- 41.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. 2002. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol 3:265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 42.de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff SJ, Zaker-Tabrizi L, Fung KY, Forster SC, Beddoe T, Reid HH, Rossjohn J, Hertzog PJ. 2013. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nat Immunol 14:901–907. doi: 10.1038/ni.2667. [DOI] [PubMed] [Google Scholar]
- 43.Mott KR, Underhill D, Wechsler SL, Ghiasi H. 2008. Lymphoid-related CD11c+ CD8alpha+ dendritic cells are involved in enhancing herpes simplex virus type 1 latency. J Virol 82:9870–9879. doi: 10.1128/JVI.00566-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, Mueller SN. 2011. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477:216–219. doi: 10.1038/nature10339. [DOI] [PubMed] [Google Scholar]