

ABSTRACT
Previous studies demonstrated that a single intramuscular (i.m.) dose of an attenuated recombinant vesicular stomatitis virus (rVSV) vector (VesiculoVax vector platform; rVSV-N4CT1) expressing the glycoprotein (GP) from the Mayinga strain of Zaire ebolavirus (EBOV) protected nonhuman primates (NHPs) from lethal challenge with EBOV strains Kikwit and Makona. Here, we studied the immunogenicities of an expanded range of attenuated rVSV vectors expressing filovirus GP in mice. Based on data from those studies, an optimal attenuated trivalent rVSV vector formulation was identified that included rVSV vectors expressing EBOV, Sudan ebolavirus (SUDV), and the Angola strain of Marburg marburgvirus (MARV) GPs. NHPs were vaccinated with a single dose of the trivalent formulation, followed by lethal challenge 28 days later with each of the three corresponding filoviruses. At day 14 postvaccination, a serum IgG response specific for all three GPs was detected in all the vaccinated macaques. A modest and balanced cell-mediated immune response specific for each GP was also detected in a majority of the vaccinated macaques. No matter the level of total GP-specific immune response detected postvaccination, all the vaccinated macaques were protected from disease and death following lethal challenge with each of the three filoviruses. These findings indicate that vaccination with a single dose of attenuated rVSV-N4CT1 vectors each expressing a single filovirus GP may provide protection against the filoviruses most commonly responsible for outbreaks of hemorrhagic fever in sub-Saharan Africa.
IMPORTANCE The West African Ebola virus Zaire outbreak in 2013 showed that the disease was not only a regional concern, but a worldwide problem, and highlighted the need for a safe and efficacious vaccine to be administered to the populace. However, other endemic pathogens, like Ebola virus Sudan and Marburg, also pose an important health risk to the public and therefore require development of a vaccine prior to the occurrence of an outbreak. The significance of our research was the development of a blended trivalent filovirus vaccine that elicited a balanced immune response when administered as a single dose and provided complete protection against a lethal challenge with all three filovirus pathogens.
KEYWORDS: attenuation, Ebola virus vaccine, Marburg virus vaccine, challenge, glycoprotein, nonhuman primates, protection, rVSV vector, trivalent, vaccine
INTRODUCTION
Ebolavirus, Marburgvirus, and Cuevavirus comprise the Filoviridae (1). The genus Ebolavirus contains five antigenically distinct species: Bundibugyo ebolavirus (BDBV), Reston ebolavirus (RESTV), Tai Forest ebolavirus (TAFV), Sudan ebolavirus (SUDV), and Zaire ebolavirus (EBOV) (2, 3). The genus Marburgvirus (MARV) contains a single species of the same name and a related but genetically distinct subtype named Ravn virus (RAVV) (4). The genus Cuevavirus currently contains a single species, Lloviu cuevavirus (LLOV), which was discovered by the isolation of RNA from dead bats found in southern European caves (5). In sub-Saharan Africa, BDBV, SUDV, EBOV, and MARV have caused sporadic epidemics of lethal hemorrhagic fever in humans, with case fatality rates of 23 to 90% (6, 7). In December 2013, an outbreak of EBOV in West Africa was the largest ever recorded, with case fatality rates of up to 70%. As of 30 March 2016, the World Health Organization (WHO) had reported a total of 28,647 suspected cases and 11,322 deaths (8), though the WHO believes that this substantially understates the magnitude of the epidemic (9) At present, there are no licensed postinfection interventions, although some approaches have shown promise in nonhuman primates (NHPs) (10–12), and there is currently no licensed vaccine available for disease prevention in communities where new outbreaks may occur. To meet this ever-present challenge, an efficacious, single-dose, multivalent vaccine that rapidly provides protection against the entire group of disease-causing viruses would be most desirable.
A number of vaccines expressing filovirus glycoproteins (GPs) have demonstrated protective efficacy in NHP challenge studies, including recombinant human (rAd5, rAd36, and rAd35) and chimpanzee (rChAd3) adenoviruses, plasmid DNA (pDNA), modified vaccinia virus Ankara (MVA), virus-like particles (VLPs), alphavirus replicons, recombinant human parainfluenza virus 3 (rHPIV 3), recombinant rabies virus (rRV), and recombinant vesicular stomatitis virus pseudotyped with a filovirus GP (rVSVΔG-GP; filovirus GP replacing VSV G glycoprotein) (reviewed by Falzarano et al. [13]). Of these vaccine vectors, only rChAd3 and the rVSVΔG-GP pseudotype vaccines have demonstrated single-dose protection in NHP filovirus challenge studies (14, 15), and only rVSV-filovirus GP in a trivalent (trival) formulation has demonstrated single-dose protection against EBOV, SUDV, and MARV in NHPs (16). The rVSVΔG-GP pseudotype vector has also demonstrated protective efficacy in phase II/III clinical trials conducted during the recent EBOV (strain Makona) outbreak in West Africa (17). However, the rVSVΔG-GP vaccine has been associated with a number of adverse reactions, including transient postvaccination arthritis, a relatively high level of vaccine vector RNA in the blood of trial participants, and vesicles on the skin distal to the inoculation site containing infectious vaccine virus (18, 19).
To reduce these undesirable side effects, a more attenuated rVSV vector expressing filovirus GP has been developed. Attenuation was achieved by combining VSV N gene translocation (N4) with truncation of the VSV G protein cytoplasmic tail domain (CT1) (20). These modifications reduced the intracellular abundance of the major structural N protein (21) and diminished the interaction between the VSV G protein CT domain and the viral nucleocapsid core during particle maturation (22). The resulting rVSV-N4CT1 vectors (also known as the VesiculoVax vaccine vector platform technology) are profoundly attenuated in a highly sensitive mouse neurovirulence (NV) model yet retain immunogenicity similar to that of the more virulent wild-type rVSV vectors (23). The attenuated rVSV-N4CT1 vector also demonstrated robust immunogenicity in NHPs following intramuscular (i.m.) inoculation and only a very mild, transient inflammatory response after direct intrathalamic inoculation (24). One of these vectors, HVTN 087, expressing HIV-1 gag protein (rVSV-N4CT1gag1), demonstrated immunogenicity and safety in two completed phase I clinical trials (25). The attenuated rVSV-N4CT1 vector genome was modified to express GP protein from the Mayinga strain of EBOV, and the resulting vector demonstrated complete single-dose protection of NHPs challenged with either the Kikwit or Makona strain of EBOV (26, 27).
Here, we describe the generation of various attenuated rVSV vectors, including rVSV-N4CT1, expressing MARV (Angola), EBOV (Mayinga), or SUDV (Boniface) GP; the immunogenicities of the vectors in mice as monovalent (monoval) and trivalent vaccine formulations; and the immunogenicity and protective efficacy of an optimal trivalent vaccine formulation in NHPs challenged with all three viral pathogens.
RESULTS
Recovery and characterization of rVSV-N4CT1 pan-FiloGP vectors.
Vectors expressing EBOV, SUDV, and MARV GPs were recovered from Vero cells following electroporation with pDNAs encoding the full-length rVSV-filoGP genomic cDNAs and VSV helper proteins (Fig. 1A to D). In each case, the rescued virus was plaque purified and amplified to produce virus seed stocks that were purified and concentrated for the in vivo studies. Before proceeding to immunogenicity studies in mice and NHPs, the vector genomes were completely sequenced to verify the fidelity of the open reading frames (ORFs) of all the genes. Expression of each filovirus GP was also demonstrated by Western blot analysis (Fig. 1E to G). Three different vector designs were generated for each filovirus vaccine in order to identify the design providing the best combination of manufacturing yield and immunogenicity for NHP studies. Vectors of design A contained the GP gene in position 1 of the viral genome immediately adjacent to the 3′ transcription promoter in order to maximize GP expression. Vectors of design B were designed to reduce GP expression ∼50% relative to design A by virtue of increased distance of the GP gene from the 3′ transcription promoter in the event that maximal GP protein expression (design A) proved toxic to rVSV replication. Vector C did not contain the major attenuating N4 gene translocation found in designs A and B and was also designed to reduce expression of the VSV G protein, which is known to be a dominant B cell antigen, in order to potentially enhance immune responses to the filovirus GPs. Vector D expressing HIV-1 gag protein was used as a control vaccine for mouse immunogenicity and NHP challenge studies. Vaccines for all three filoviruses were generated in all three vector design configurations.
FIG 1.

Genetic organization of attenuated rVSV filovirus vaccine vectors and GP expression. (A) Vectors of the rVSV-N4CT1 FiloGP1 design contained a filovirus GP gene(s) in position 1, an N gene translocation (N4), and a truncated G gene (CT1). (B) Vectors of the rVSV-N4CT1 FiloGP3 design contained a filovirus GP gene(s) in position 3, an N gene translocation (N4), and a truncated G gene (CT1). (C) rVSV-N1CT1 FiloGP3 vectors retained the VSV N gene in position 1, a filovirus GP gene(s) at position 3, and a truncated form of the VSV G gene at position 6. (D) The rVSV-N4CT1 HIVgag1 vector contained the HIV-1 gag gene in position1, an N gene translocation (N4), and a truncated G gene (CT1). The numbers designate genome positions. Virus leader (Le), trailer (Tr), and intergenic regions are shown in black. The stippled regions represent deletions in the VSV G gene. (E to G) Expression of the EBOV (E), SUDV (F), and MARV (G) GPs by Western blotting.
Immunogenicity of trivalent rVSV-N4CT1 pan-FiloGP vaccines in mice.
In order to identify an optimal rVSV-N4CT1 pan-FiloGP (EBOV, SUDV, and MARV GP) vaccine vector design eliciting robust GP-specific immune responses when given as a trivalent vaccine, a mouse immunogenicity study was designed (Fig. 2). For this study, groups of 15 BALB/c mice were immunized with two i.m. doses in study weeks 0 and 4. Over the course of the study, 5 mice from each group were bled, and serum was prepared in study weeks 0, 2, 4, 6, and 10 for the determination of filovirus GP-specific IgG titers by enzyme-linked immunosorbent assay (ELISA) (Fig. 3A to C). In addition, 5 mice from each group were sacrificed at study week 1 (1 week post-priming immunization), week 5 (1 week post-booster immunization), and week 10 (6 weeks post-booster immunization) to quantitate filovirus GP-specific gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISpot) assay responses (Fig. 4).
FIG 2.
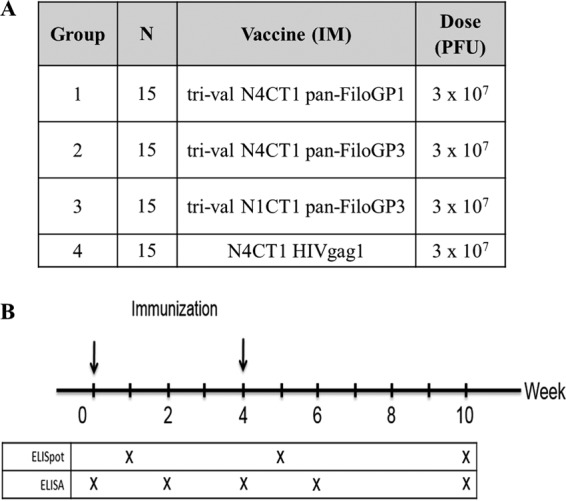
Mouse study design comparing immunogenicities of trivalent vaccine designs. (A) Study design. (B) Study schedule.
FIG 3.
Filovirus GP-specific IgG ELISA titer responses in BALB/c mice immunized with trivalent vaccine. At study weeks 0 and 4 (arrows), BALB/c mice were immunized i.m. with 3 × 107 PFU of trivalent rVSV/filovirus vaccines. At study weeks 0, 2, 4, 6, and 10, sera were collected and tested for EBOV GP-specific (A), SUDV GP-specific (B), and MARV GP-specific (C) IgG responses by ELISA. The data represent the average log-transformed filovirus GP-specific IgG endpoint titers, with the standard errors of the means indicated by the error bars. *, statistically significant difference (P < 0.05) between trivalent rVSV-N4CT1 pan-FiloGP1 and trivalent rVSV-N4CT1 pan-FiloGP; #, statistically significant difference (P < 0.05) between trivalent rVSV-N4CT1 pan-FiloGP3 and trivalent rVSV-N1CT1 pan-FiloGP3.
FIG 4.
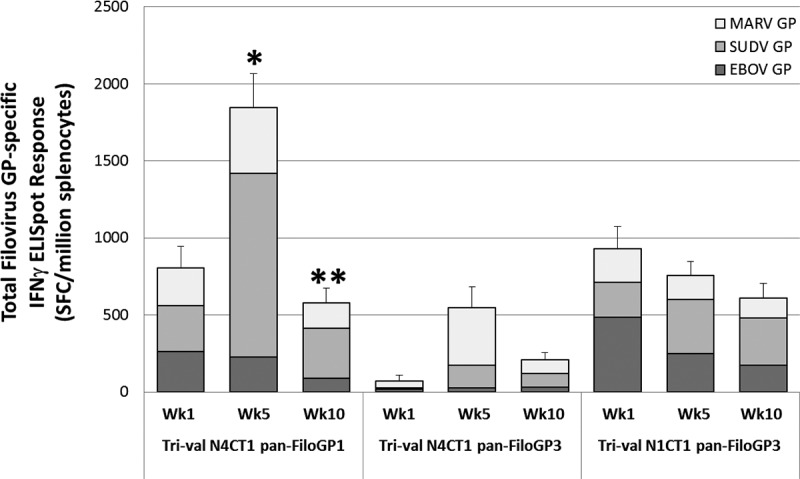
Filovirus GP-specific IFN-γ ELISpot responses in BALB/c mice immunized with trivalent vaccine. At study weeks 1, 5, and 10, splenocytes were collected and tested for EBOV GP, SUDV GP, and MARV GP peptide pool-specific IFN-γ secretion by ELISpot assay. The data represent the average total filovirus-specific IFN-γ ELISpot responses, with the standard errors of the mean indicated by the error bars. *, statistically significant (P < 0.05) difference relative to trivalent rVSV-N4CT1 pan-FiloGP3 and trivalent rVSV-N1CT1 pan-FiloGP3; **, statistically significant (P < 0.05) difference relative to trivalent rVSV-N4CT1 pan-FiloGP3.
As shown in Fig. 3A, for the vaccinated mice in group 1 (trival N4CT1 pan-FiloGP1), group 2 (trival N4CT1 pan-FiloGP3), and group 3 (trival N1CT1 pan-FiloGP3), the EBOV GP-specific IgG ELISA log10 endpoint titers increased in week 2 following vaccination and continued to increase through week 4. At study week 4, EBOV GP-specific log10 endpoint titers in groups 1 and 3 were highest (4.18 ± 0.08 and 4.11 ± 0.11, respectively), with significantly (P < 0.05) lower responses to EBOV GP in group 2 mice vaccinated with trivalent rVSV-N4CT1 pan-FiloGP3 vaccine (3.77 ± 0.22). Following the vaccine boost in week 4, EBOV GP-specific IgG ELISA log10 endpoint titers increased by approximately 1.5 log units and were significantly higher in groups 1 and 3 (trival N4CT1 pan-FiloGP1- and trival N1CT1 pan-FiloGP3-immunized) mice than in the group 2 (trival N4CT1 pan-FiloGP3-immunized) mice at study week 10.
As shown in Fig. 3B, the SUDV GP-specific IgG ELISA responses over time were very similar to the EBOV GP-specific IgG ELISA responses shown in Fig. 3A. Following immunization, the SUDV GP-specific IgG ELISA log10 endpoint titers increased in week 2 and continued to increase through week 4. At study week 4, the SUDV GP-specific log10 endpoint titers in group 1 were highest (4.35 ± 0.09), with lower responses to SUDV GP in group 2 mice vaccinated with trivalent rVSV-N4CT1 pan-FiloGP3 vaccine (3.89 ± 0.12; P < 0.05) and group 3 mice vaccinated with trivalent rVSV-N1CT1 pan-FiloGP3 vaccine (4.01 ± 0.15). Following the vaccine boost in week 4, SUDV GP-specific IgG ELISA log10 endpoint titers increased approximately 1.5 log units and were significantly higher in group 2 (trival N4CT1 pan-FiloGP3-immunized) mice than in the group 1 immunized mice at study week 10.
After a single priming immunization, group 2 and 3 MARV GP-specific log10 endpoint titers at study week 2 were 1 to 1.5 log units lower than the EBOV/SUDV GP-specific IgG ELISA responses (Fig. 3C). As seen for EBOV- and SUDV-specific responses, MARV GP-specific IgG ELISA log10 endpoint titers increased approximately 1.5 log units following the vaccine boost in week 4 but trended approximately 1 log unit lower than the EBOV and SUDV GP-specific responses at study week 10. Notably, the magnitude of the MARV GP-specific response detected in group1 mice vaccinated with trivalent rVSV-N4CT1 pan-FiloGP1 was the highest of all three vector designs at all time points postvaccination.
As shown in Fig. 4, a single immunization with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine candidate elicited an average total filovirus GP-specific IFN-γ ELISpot response of 840 ± 140 spot-forming cells (SFC)/106 splenocytes, a response that was very similar to that seen following a single immunization with the trivalent rVSV-N1CT1 pan-FiloGP3 vaccine candidate (928 ± 147 SFC/106 splenocytes). In contrast, a single immunization with the trivalent rVSV-N4CT1 pan-FiloGP3 vaccine candidate resulted in a substantially reduced average total filovirus GP-specific IFN-γ ELISpot response (70 ± 43 SFC/106 splenocytes).
At 1 week postboost (week 5), the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine candidate elicited an average total filovirus GP-specific IFN-γ ELISpot response of 1,843 ± 224 SFC/106 splenocytes, a response that was significantly increased (P < 0.05) relative to the response seen following two doses of trivalent rVSV-N1CT1 pan-FiloGP3 vaccine (757 ± 91 SFC/106 splenocytes) and the response seen following two doses of trivalent rVSV-N4CT1 pan-FiloGP3 vaccine (547 ± 150 SFC/106 splenocytes).
At 6 weeks postboost (week 10), the IFN-γ ELISpot response elicited by trivalent rVSV-N4CT1 pan-FiloGP1 vaccine decreased to 577 ± 97 SFC/106 splenocytes, a response that was again similar to the response seen following two doses of trivalent rVSV-N1CT1 pan-FiloGP3 vaccine (609 ± 95 SFC/106 splenocytes).
Based on these collective data, the trivalent rVSV-N4CT1 pan-FiloGP1 vector was selected for further studies in mice to investigate any differences in GP-specific immunogenicity between monovalent and trivalent vaccine formulations.
Immunogenicity of monovalent versus trivalent rVSV-N4CT1 pan-FiloGP1 vaccines in mice.
We next sought to determine if immunization with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine formulation resulted in filovirus GP-specific immunocompetition and reduced GP-specific immunity relative to immunization with monovalent rVSV-N4CT1 pan-FiloGP1 vaccines. For this study, mice were immunized i.m. with two doses of vaccine in study weeks 0 and 4. Over the course of the study, 5 mice from each group were bled, and serum was prepared in study weeks 0, 2, 6, and 10 for the determination of filovirus GP-specific IgG titers by ELISA. In addition, 5 mice from each group were sacrificed in study week 1 (1 week post-priming immunization), week 5 (1 week post-booster immunization), and week 10 (6 weeks post-booster immunization) to quantitate filovirus GP-specific IFN-γ ELISpot responses.
As seen in Fig. 5, at 1 week postprime, group 1 mice, which received a single immunization with the monovalent rVSV-N4CT1 EBOVGP1 vaccine, demonstrated an average EBOV GP-specific IFN-γ ELISpot response of 559 ± 99 SFC/106 splenocytes, a response that was significantly higher than the response seen following a single immunization with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine (group 4; 123 ± 33 SFC/106 splenocytes). Similarly, group 2 mice, which received a single immunization with the monovalent rVSV-N4CT1 SUDVGP1 vaccine, demonstrated an average SUDV GP-specific IFN-γ ELISpot response of 452 ± 93 SFC/106 splenocytes, a response that was significantly higher than the response seen following a single immunization with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine (group 4; 144 ± 30 SFC/106 splenocytes). In addition, group 3 mice, which received a single immunization with the monovalent rVSV-N4CT1 MARVGP1 vaccine, demonstrated an average MARV GP-specific IFN-γ ELISpot response of 348 ± 44 SFC/106 splenocytes, a response that was significantly higher than the response seen following a single immunization with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine (group 4; 115 ± 29 SFC/106 splenocytes).
FIG 5.
Filovirus GP-specific IFN-γ ELISpot titer responses in BALB/c mice immunized with monovalent and trivalent rVSV-N4CT1 FiloGP1 vaccines. At study weeks 1, 5, and 10, splenocytes were collected and tested for EBOV GP, SUDV GP, and MARV GP peptide pool-specific IFN-γ secretion by ELISpot assay. The data represent the average total filovirus-specific IFN-γ ELISpot responses, with the standard errors of the mean indicated by the error bars. *, statistically significant difference (P < 0.05) between the monovalent rVSV-N4CT1 FiloGP1- and trivalent rVSV-N4CT1 pan-FiloGP1-immunized mice.
One week postboost, SUDV GP-specific IFN-γ ELISpot responses were similar to those seen in the trivalent-vaccine-immunized mice; however, EBOV and MARV GP-specific IFN-γ ELISpot responses remained significantly higher in the monovalent-vaccine-immunized mice than in trivalent-vaccine-immunized mice. Finally, at 6 weeks postboost, both SUDV and MARV GP-specific IFN-γ ELISpot responses were similar in trivalent- and monovalent-vaccine-immunized mice; however, EBOV GP-specific IFN-γ ELISpot responses remained significantly higher in the monovalent-vaccine-immunized groups than in the trivalent-vaccine-immunized mice.
Interestingly, 6 weeks after two immunizations with the monovalent rVSV-N4CT1 EBOVGP1 vaccine, 5 of 5 (100%) group 1 mice demonstrated a cross-reactive SUDV GP-specific IFN-γ ELISpot response. whereas 0 of 5 (0%) group 2 mice immunized with the monovalent rVSV-N4CT1 SUDVGP1 vaccine demonstrated a detectible cross-reactive EBOV GP-specific IFN-γ ELISpot response. In contrast, 0 of 5 (0%) group 3 mice immunized with the monovalent rVSV-N4CT1 MARVGP1 vaccine demonstrated a detectible cross-reactive EBOV or SUDV GP-specific IFN-γ ELISpot response (data not shown).
As shown in Fig. 6A, for the vaccinated mice in group 1 (monoval N4CT1 EBOVGP1) and group 4 (trival N4CT1 pan-FiloGP1), EBOV GP-specific IgG ELISA log10 endpoint titers increased by week 2 following vaccination and continued to increase after boosting (week 6). At study week 6, 2 weeks after the booster immunization, EBOV GP-specific log10 endpoint titers in group 1 were highest (5.58 ± 0.07) and were significantly (P < 0.05) higher than the EBOV GP-specific log10 endpoint titers in the group 4 mice vaccinated with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine (5.24 ± 0.06). However, by study week 10, the difference in the EBOV GP-specific log10 endpoint titers detected in the monovalent- and the trivalent-vaccine-immunized mice was no longer significant. Interestingly, 5 of 5 mice in group 2 vaccinated with monovalent rVSV-N4CT1 SUDVGP1 demonstrated measurable cross-reactive EBOV GP-specific IgG responses.
FIG 6.
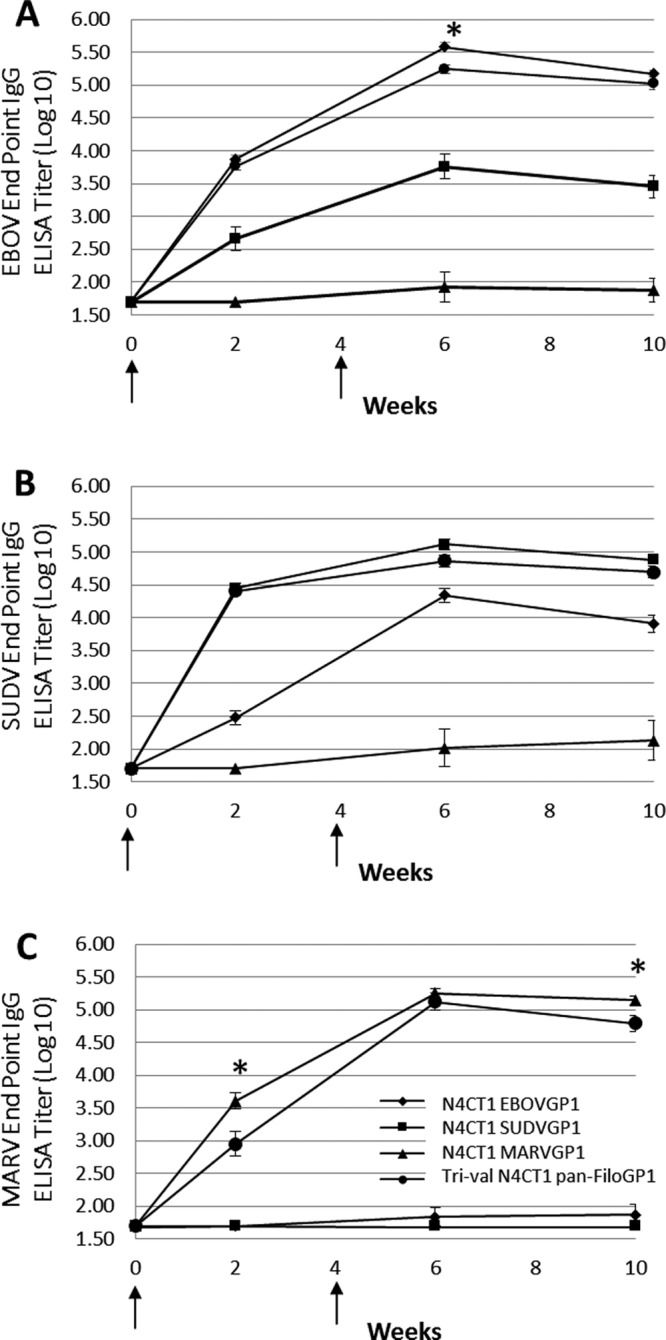
Filovirus GP-specific IgG ELISA titer responses in BALB/c mice immunized with monovalent and trivalent rVSV-N4CT1 FiloGP1 vaccines. At study weeks 0 and 4 (arrows), BALB/c mice were immunized i.m. with 1 × 107 PFU of each monovalent rVSV-N4CT1 FiloGP1 vaccine or with 3 × 107 PFU of the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine. At study weeks 0, 2, 6, and 10, sera were collected and tested for EBOV GP-specific (A), SUDV GP-specific (B), and MARV GP-specific (C) IgG responses by ELISA. The data represent the average log-transformed filovirus GP-specific IgG endpoint titers, with the standard errors of the mean indicated by the error bars. *, statistically significant difference (P < 0.05) between the monovalent rVSV-N4CT1 FiloGP1- and trivalent rVSV-N4CT1 pan-FiloGP1-immunized mice.
As shown in Fig. 6B, for the vaccinated mice in group 2 (monoval N4CT1 SUDVGP1) and group 4 (trival N4CT1 pan-FiloGP1), SUDV GP-specific IgG ELISA log10 endpoint titers increased by week 2 following vaccination and increased only slightly at week 6 following the boost. At study week 6, 2 weeks after the booster immunization, SUDV GP-specific log10 endpoint titers in group 2 were highest (5.12 ± 0.08) but were not significantly different from the SUDV GP-specific log10 endpoint titers in group 4 mice vaccinated with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine (4.86 ± 0.10). Consistent with the IFN-γ ELISpot data, 5 of 5 mice in group 1 vaccinated with monovalent rVSV-N4CT1 EBOVGP1 demonstrated measurable cross-reactive SUDV GP-specific IgG responses.
As shown in Fig. 6C, for the vaccinated mice in group 3 (monoval N4CT1 MARVGP1) and group 4 (trival N4CT1 pan-FiloGP1), MARV GP-specific IgG ELISA log10 endpoint titers increased by week 2 following vaccination and continued to increase following boost at week 6. At study week 6, 2 weeks after the booster immunization, MARV GP-specific log10 endpoint titers in group 3 were highest (5.25 ± 0.08) but were not significantly different from the MARV GP-specific log10 endpoint titers in the group 4 mice vaccinated with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine (5.12 ± 0.13). However, by study week 10, the MARV GP-specific ELISA response in the trivalent rVSV-N4CT1 pan-FiloGP1-vaccinated mice appeared to decline more rapidly, and the 0.35 log10 difference in the MARV GP-specific ELISA response between the monovalent- and the trivalent-vaccine-immunized mice reached statistical significance. Consistent with the IFN-γ ELISpot data, none of the mice in group 1, vaccinated with monovalent rVSV-N4CT1 EBOVGP1 vaccine, and none of the mice in group 2, vaccinated with monovalent rVSV-N4CT1 SUDVGP1 vaccine, demonstrated measurable cross-reactive MARV GP-specific IgG responses.
Immunogenicity of trivalent rVSV-N4CT1 pan-FiloGP1 vaccine in macaques.
A total of 15 cynomolgus macaques were vaccinated i.m. with a trivalent rVSV-N4CT1 pan-FiloGP1 vaccine formulation containing 1 × 107 PFU of each vector, for a total dose of 3 × 107 PFU/macaque (Fig. 7A and B). Six macaques were vaccinated with an equivalent dose of rVSV-N4CT1 HIVgag1 as a study control. Blood samples were drawn 14 days before vaccination, immediately before dosing on the day of vaccination, and 10 or 14 days after vaccination to assess cellular and humoral immune responses elicited against each GP protein. The humoral immune response elicited by each vector was assessed by measuring serum GP-specific IgG binding antibody by ELISA and filovirus GP-specific neutralization activity by a 50% plaque reduction neutralization (PRNT50) test. IgG responses specific for each of the three filovirus GPs were detected by ELISA 10 and 14 days after vaccination and are shown in Fig. 8A.
FIG 7.
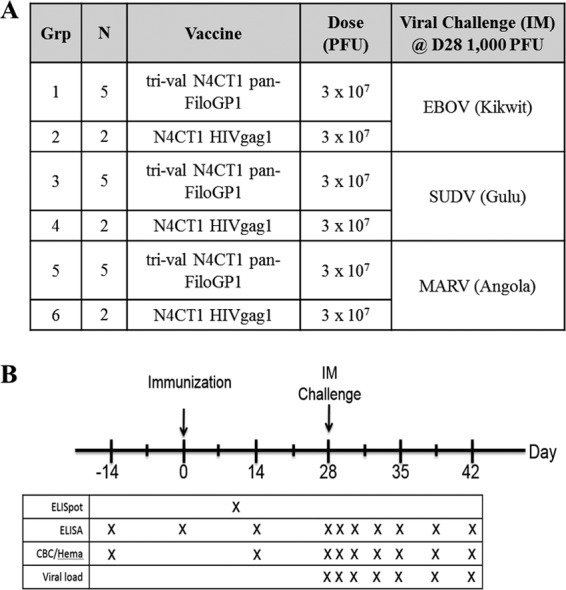
NHP study design to assess immunogenicity and protective efficacy of a trivalent rVSV-N4CT1 pan-FiloGP1 panfilovirus vaccine. (A) Study design. (B) Study schedule of events.
FIG 8.

Filovirus GP-specific IgG ELISA titers and IFN-γ ELISpot responses in macaques immunized with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine. At study day 0, macaques were immunized i.m. with 3 × 107 PFU of the trivalent rVSVN4CT1 pan-FiloGP1 vaccine. As a control, macaques were immunized with 3 × 107 PFU of rVSV N4CT1gag1. (A) At study days 0, 10, and 14, sera were collected and tested for EBOV GP-, SUDV GP-, and MARV GP-specific IgG responses by ELISA. The data represent the average log-transformed filovirus GP-specific IgG concentrations, with the standard errors of the mean indicated by the error bars. (B) At study day 10, PBMCs were collected and tested for EBOV GP, SUDV GP, and MARV GP peptide pool-specific IFN-γ secretion by ELISpot assay. The data represent the individual NHP total filovirus-specific IFN-γ ELISpot responses. *, antigen-specific IFN-γ ELISpot response of ≥25 SFC/106 PBMCs.
Cell-mediated immune (CMI) responses in the peripheral blood mononuclear cells (PBMCs) of macaques were assessed by IFN-γ ELISpot assay 10 days postvaccination (Fig. 8B). The magnitudes of individual filovirus GP-specific CMI responses were probably not peak values, which typically occur 7 days postvaccination with rVSV vectors (23, 24), but the timing of the assay was chosen to coincide with blood collection for the IgG assay in order to minimize the number of bleeds and the resulting discomfort for the macaques. Above-background CMI responses were detected in 6 of the 15 macaques receiving the trivalent vaccine. Of those positive responders, 4 of 6 demonstrated balanced CMI responses to all three filovirus GP proteins. Collectively, the antibody and CMI data indicated that a single i.m. dose of trivalent rVSV-N4CT1 pan-FiloGP1 vaccine elicited a rapid and balanced GP-specific immune response in macaques.
Efficacy of trivalent rVSV-N4CT1 pan-FiloGP1 vaccine in macaques.
At day 28 postvaccination, 3 groups of 7 macaques, each group containing 5 vaccinees and 2 controls, were challenged i.m. with 1,000 PFU of low-passage-number EBOV (strain Kikwit), SUDV (strain Gulu), or MARV (strain Angola). By days 6 to 9 postchallenge, control macaques in all three groups had succumbed, while all 15 macaques receiving trivalent rVSV-N4CT1 pan-FiloGP1 vaccine remained generally healthy and survived the lethal filovirus challenge (Fig. 9). Following EBOV challenge, one macaque vaccinated with the trivalent vaccine had a pattern of altered serum chemistry and hematology similar to that of the 2 sham-vaccinated control macaques and also showed signs of mild depression and anorexia; however, this macaque did not display any other signs of illness and returned to normal health by day 14 postchallenge. Similar observations were made for 2 macaques receiving the trivalent vaccine and challenged with SUDV and 1 macaque receiving trivalent vaccine and challenged with MARV.
FIG 9.
Postchallenge survival of cynomolgus macaques immunized with the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine. At study day 0, macaques were immunized i.m. with 3 × 107 PFU of the trivalent rVSV-N4CT1 pan-FiloGP1 vaccine. As a control, macaques were immunized with 3 × 107 PFU of an rVSV-N4CT1gag1 vector. At study day 28, the macaques were challenged i.m. with 1,000 PFU 7U EBOV Kikwit (A), SUDV Gulu (B), or MARV Angola (C).
Postchallenge filovirus level in plasma.
The levels of postchallenge filovirus genomic RNA and infectious virus present in plasmas from macaques were assessed by real-time quantitative PCR (RT-qPCR) (Fig. 10A, C, and E) and plaque assay (Fig. 10B, D, and F), respectively. There was no detectible infectious virus present in plasma from any of the 15 macaques receiving trivalent vaccine at any time postchallenge, contrasting with peak levels of infectious virus (7 to 8 log10 PFU/ml) detected in plasma from sham-vaccinated macaques by day 6 postchallenge (Fig. 10B, D, and F). Peak levels of EBOV genomic RNA (∼2 × 106 to 1 × 109/ml) were detected in plasma at day 6 postchallenge in 4 of the 5 vaccinated macaques, remaining detectable at reduced levels until day 21 postchallenge in one of these macaques. However, it is notable that the peak levels of EBOV genomic RNA detected in the 2 sham-vaccinated macaques at day 6 postchallenge was at least 3 orders of magnitude greater than in vaccinated macaques. Similarly, very high levels of filovirus genomic RNA were detected in sham-vaccinated macaques at day 6 postchallenge with either SUDV or MARV, while viral genomic RNA was detected at a relatively low level in only 1 of the 5 vaccinated macaques at day 6 post-SUDV challenge and not in any macaques at any time post-MARV challenge.
FIG 10.

Filovirus viremia in trivalent rVSV-N4CT1 pan-FiloGP1-vaccinated cynomolgus macaques challenged i.m. with EBOV, SUDV, or MARV. Trivalent rVSV-N4CT1 pan-FiloGP1- or control rVSV-vaccinated macaques were challenged i.m. with 1,000 PFU 7U EBOV Kikwit (A and B), SUDV Gulu (C and D), or MARV Angola (E and F). Postchallenge viremia was monitored by RT-qPCR (A, C, and E) or by infectious plaque assay (B, D, and F).
DISCUSSION
Previous work demonstrated protective efficacy of a single i.m. dose of an attenuated rVSV-N4CT1 EBOVGP1 vaccine against lethal EBOV challenge in NHPs (26, 27). As an extension of that work, we evaluated the immunogenicities of various similarly attenuated vectors expressing EBOV (Mayinga), SUDV (Boniface), and MARV (Angola) GP proteins in mice. The studies identified an optimal vector design for further evaluation of the immunogenicity and protective efficacy of a single-dose trivalent vaccine formulation following lethal challenge with EBOV, SUDV, and MARV in NHPs.
The recent epidemic of the Makona strain of EBOV in West Africa has highlighted the urgent need for an efficacious vaccine to combat future filovirus outbreaks and to save lives and suffering in sub-Saharan Africa. One such candidate vaccine developed by Merck has recently demonstrated efficacy in a phase III clinical trial using a ring vaccination strategy at sites of disease in West Africa (17). This vaccine is also an rVSV-based vector, modified by exchanging the VSV G gene with the GP gene from the Mayinga strain of EBOV (14, 28). While the vaccine appears to be efficacious, there are some undesirable vaccine-associated side effects, including postvaccination arthritis, vesicle formation distal to the inoculation site from which viral RNA can be isolated, moderate to severe postvaccination fever persisting for 2 to 3 days, and relatively robust viremia indicated by the presence of viral RNA in blood 2 to 3 days after vaccination (18, 19). While robust vaccine virus replication likely contributes to vaccine efficacy, circulation of high levels of virus throughout the body raises concerns about potential long-term vaccine side effects, in addition to the undesirable short-term adverse events. In an effort to address the short- and potential long-term side effects associated with the rVSVΔG FiloGP pseudotype vaccine, we developed a more attenuated rVSV-N4CT1 vector expressing GP protein that can protect NHPs from lethal EBOV challenge (26, 27) and that is currently under phase I clinical evaluation. However, a monovalent EBOV vaccine is unlikely to provide protection from antigenically distinct filoviruses, such as SUDV and MARV, both of which have been responsible for sporadic deadly outbreaks of disease in sub-Saharan Africa (6, 7). We address this need with the development of the trivalent vaccine described here.
One critical consideration for multivalent, replication-competent viral vaccines is the potential for interference between vaccine components, either through replicative competition among constituent vectors or through immunological competition among target antigens, creating an imbalance in the resulting immune response to each vaccine component. Such an interference phenomenon has been observed for multivalent, live attenuated influenza vaccine (29, 30). Here, all three vaccine vectors were based on the same attenuated rVSV-N4CT1 vector; differing only in filovirus GP protein amino acid composition and structure. Findings from mouse immunogenicity studies indicated that the magnitudes of the filovirus GP-specific immune responses were significantly different for different vector designs, particularly after a single vaccine dose, and a comparison of filovirus GP-specific cell-mediated and humoral immune responses elicited by the various vaccine designs enabled identification of the rVSV-N4CT1 pan-FiloGP1 vector as an optimal design for further immunogenicity studies. Additional mouse studies also demonstrated that there were negligible differences between the SUDV and EBOV GP-specific IgG responses elicited by either of the corresponding monovalent formulations or the trivalent vaccine formulation following a single vaccine dose. However, the MARV GP-specific immune response was significantly lower following single-dose vaccination with the trivalent formulation than after single-dose vaccination with the corresponding monovalent formulation, possibly indicating some form of vector interference or immunological competition between the different filovirus GP proteins when given as a multivalent vaccine. Nevertheless, IgG responses to all three filovirus GP proteins were relatively robust at all times after one and two doses of vaccine given either as monovalent or in a trivalent formulation, indicating that the efficacy of a trivalent vaccine might be similar to that of a monovalent vaccine. Notably, and consistent with known antigenic relatedness, there was substantial and significant filovirus GP-specific IgG cross-reactivity between the EBOV and SUDV GP proteins that was absent in the MARV GP-specific response in the mouse studies (3, 31). However, this cross-reactivity between EBOV and SUDV GPs does not confer protection on monovalent-vaccine-vaccinated mice and NHP's when challenged with a heterologous filovirus.
The relatively robust filovirus GP-specific IgG responses and modest but balanced cellular immune responses to the rVSV-N4CT1 pan-FiloGP1 vectors in mice provided sufficient encouragement to test the efficacy of a trivalent vaccine formulation in NHPs. Consistent with the GP-specific IgG responses detected in mice, at 2 weeks postvaccination with trivalent vaccine, significant IgG responses to the EBOV, SUDV, and MARV GPs were observed. Also notable was the absence of detectable neutralizing antibody responses against EBOV and MARV relative to control macaques 14 days after vaccination and a SUDV neutralizing response that was only marginally above control levels (data not shown). Nevertheless, all the macaques that received a single dose of trivalent vaccine were protected from serious disease and death after i.m. challenge with 1,000 PFU of EBOV, SUDV, or MARV on study day 28.
Our data indicated that the observed level of protection in the vaccinated macaques was due primarily to the elevated level of circulating virus-specific antibody at the time of challenge. Therefore, the following question remained: if EBOV-specific antibody was permitted to return to a baseline steady state, would the same level of protection be achieved? A pilot study to address this question showed that cynomolgus macaques vaccinated with a monovalent N4CT1-EBOV vaccine provided slightly reduced protection 6 months postvaccination following challenge with a lethal dose of EBOV (unpublished results). Even though the circulating virus-specific antibody titers declined through time, the vaccinated macaques mounted a sufficient immune response against EBOV challenge, providing significant protection from death (and possibly disease). Further studies are needed to determine the level of protection when the time interval between vaccination and challenge is increased beyond 6 months and when vaccinated macaques are challenged with a heterologous filovirus.
Significant levels of postchallenge EBOV and SUDV genomic RNA in blood from 5 of 15 vaccinated macaques indicated that sterilizing immunity was not achieved by a single vaccine dose and is not necessary for protection from disease. However, the extent of the infection was blunted, since no replicating postchallenge EBOV or SUDV was found in the vaccinated NHPs. Collectively, these observations suggest that filoviruses can replicate and spread throughout the bodies of vaccinated macaques in the days following challenge; however, it is likely that the vaccine-induced GP-specific humoral and CMI responses impede challenge virus growth despite the absence of detectable neutralizing antibodies early in the infection, allowing time for expansion of the anamnestic GP-specific response, which then shuts down the virus infection before it reaches a critical disease threshold.
Supporting this contention is the relatively high-level and persistent postchallenge EBOV genomic RNA detected in plasma from macaque number 68453, correlating with a relatively low corresponding GP-specific IgG ELISA titer at day 14 postvaccination. This macaque also showed very mild signs of clinical disease, which resolved without complications. However, the relatively small number of macaques in each challenge group make any clear correlation between the level of immune response, the level of challenge virus replication, and the relative level of disease symptoms very difficult.
The results of these studies clearly demonstrate that a single i.m. dose of a trivalent formulation composed of attenuated rVSV-N4CT1GP1 vectors expressing the GP protein(s) of EBOV, SUDV, and MARV protects macaques from significant disease and death following i.m. challenge with a lethal dose of any of the three filoviruses. These findings, together with very promising preliminary safety data from an ongoing phase I clinical study with an analogous monovalent rVSV-N4CT1GP1 EBOV vaccine (Profectus BioSciences; https://clinicaltrials.gov/ct2/show/NCT02718469), support testing of a trivalent filovirus vaccine formulation in phase I clinical trials as the next step in identifying a vaccine that is safe and has broad-spectrum protective efficacy against this group of viral pathogens. In addition, new filovirus vaccine targets, such as the Bundibugyo GP, could be incorporated into the attenuated rVSV backbone to combat new emerging filovirus strains. In the future, blanket immunization with such a vaccine in sub-Saharan Africa may either prevent or greatly limit any future outbreak(s) of disease caused by members of the genera Ebolavirus and Marburgvirus.
MATERIALS AND METHODS
Generation and characterization of filovirus vectors.
As previously described (26), an attenuated rVSV-N4CT1 EBOVGP1 vector (serotype Indiana) expressing the EBOV GP (1976 Mayinga isolate) was generated and used as the backbone for generating two additional rVSV-N4CT1 vectors encoding the SUDV and MARV GPs in the 1st position of the genome (Fig. 1A). The cDNA for Sudan GP (1976 Boniface isolate) was kindly provided by Heinz Feldmann, Rocky Mountain Laboratories, NIH, and the gene encoding Marburg GP (2005 Angola isolate; GenBank accession no. DQ447653.1) was de novo synthesized (Genewiz, South Plainfield, NJ). All full-length EBOV, SUDV, and MARV GPs were generated by PCR using primers to create flanking XhoI/NotI restriction sites for cloning into the rVSV genome cDNA. A vector expressing filovirus GP from the 3rd position in the genome was generated by cloning the GP genes into XhoI/NotI restriction sites contained within a multiple cloning site (MCS) in a transcriptional cassette located between VSV M and N genes, where the N gene was in position 4 of the genome. This manipulation was followed by the insertion of a PCR fragment containing a portion of the VSV L gene, a modified VSV G CT1 gene, and trailer sequence into the resulting N4 genome at position 6 via unique HindIII/RsrII sites, generating the rVSV-N4(G*CT1)6 FiloGP3 genomic cDNA (Fig. 1B). The modified G*CT1 component was generated by deleting nucleotides 52 to 1209 in the G CT1 cDNA and inserting a cytoplasmic epitope of VSV G (QIYTDIEMNRLGK) into the extracellular- membrane-proximal region of the G stem close to the G trimerization signal. The less attenuated N1 vectors were generated by first cloning EBOV, SUDV, or MARV GP genes into a VSV-N1ΔG backbone via XhoI/NotI restriction sites contained in an MCS within a transcriptional cassette located at position 3 of the genome. Then, PCR primers AATAACATTAGTCGACGGTATAAGCTTTTGG and AATAACATTAGCTAGCCCGCACTAGTATCGAG were used to generate a DNA fragment containing a portion of VSV L, a modified G*CT1 gene, and trailer sequence at position 6 of the genome via unique HindIII/RsrII sites to generate the rVSV-N1(G*CT1)6 FiloGP3 cDNA (Fig. 1C). For control vaccine, an attenuated rVSV-N4CT1 HIVgag1 design (20) expressing HIV-1 gag in the first position of the genome was used in animal studies (Fig. 1D).
The rVSV-filovirus vectors were rescued from genomic cDNA as previously described (32). The rescued virus was plaque purified and amplified on Vero cell monolayers (ATCC CCL-81). For animal studies, virus vectors were purified from infected BHK-21 (ATCC CCL-10) cell supernatants by centrifugation through a 10% sucrose cushion. The purified virus was resuspended in phosphate-buffered saline (PBS), pH 7.0, mixed with a sucrose phosphate (SP) stabilizer (7 mM K2HPO4, 4 mM KH2PO4, 218 mM sucrose), snap-frozen in ethanol-dry ice, and stored at −80°C until it was ready for use. Virus titers were determined by plaque assay on Vero-E6 cells and stained with a 1% crystal violet solution for fixing and visualization.
All rVSV-filovirus vaccine vectors were characterized by full genomic sequencing to ensure that attenuating mutations were intact and that additional deleterious mutations were not introduced into GP genes; expression of each filovirus GP was also confirmed by Western blotting and plaque immunostaining with GP-specific monoclonal antibodies (detailed below). For sequence analysis, extracted viral RNA (vRNA) was used to generate six overlapping cDNA fragments spanning the viral genome using VSV-specific primers in the Superscript III one-step RT-PCR system with Platinum Taq High Fidelity (Invitrogen). The PCR products were purified, and the sequences of both DNA strands were determined by dye terminator cycle sequencing. The resulting viral genome consensus sequence(s) was aligned with the plasmid DNA encoding the full-length viral genome used for virus rescue in the SeqMan program (DNAStar).
GP expression was analyzed by Western blot analysis of rVSV-filovirus-infected cell lysates run on a 4 to 12% PAGE gel and probed with either monoclonal mouse anti-EBOV GP (clone 4F3; IBT Services; catalog no. 0201-020), anti-SUDVGP (clone 2H5; IBT Services; catalog no. 0202-029), or polyclonal rabbit anti-MARVGP (IBT Services; catalog no. 0303-007) antibodies at 1:1,000 dilution. The GP bands were then detected with an anti-mouse or anti-rabbit-alkaline phosphatase (AP) conjugate IgG(H+L) (Promega) and developed with a chromogenic substrate (Western Blue; Promega). Bands were seen at approximately 160, 140, and 110 kDa.
For plaque staining of each filovirus vector, virus samples were serially diluted with Dulbecco's minimal essential medium (DMEM) and adsorbed to confluent monolayers of Vero-E6 cells in six-well plates for 15 min at room temperature, followed by 30 min at 37°C. The plates were then overlaid with 0.8% SeaPlaque agarose (Lonza; catalog no. 50100) prepared in DMEM supplemented with 10% fetal bovine serum (FBS) and incubated at 32°C in a 5% CO2 humidified incubator for 4 to 6 days. The agar overlay was removed, and the cell monolayer was fixed with acetone-methanol (1:1) fixative solution for 30 min and then rinsed three times with PBS. The plates were initially blocked with 5% nonfat milk in PBS and 1% Tween 20 (PBST), followed by incubation with either monoclonal mouse anti-EBOV GP (clone 4F3; IBT Services; catalog no. 0201-020), anti-SUDVGP (clone 2H5; IBT Services; catalog no. 0202-029), or polyclonal rabbit anti-MARVGP (IBT Services; catalog no. 0303-007) antibody at 1:1,000 dilution for 1 h at room temperature. The plates were washed three times with PBST and incubated with either goat anti-mouse IgG horseradish peroxidase (HRP) conjugate or goat anti-rabbit IgG HRP conjugate at 1:2,000 dilution for 1 h at room temperature, followed by three PBST washes. Protein expression within the plaques was then visualized with a chromogenic substrate (3,3′-diaminobenzidine [DAB] tablets; Sigma; catalog no. D5905) until a strong color reaction occurred.
Murine studies.
For murine studies, 8- to 10-week-old female BALB/c mice were used. The mice were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (33). In addition, procedures for the use and care of the mice were approved by Profectus Bioscience's and New York Medical College's Institutional Animal Care and Use Committees.
Murine IFN-γ ELISpot assay.
Vaccine-elicited IFN-γ ELISpot responses were determined using a mouse IFN-γ ELISpot kit (material number 551881; BD Biosciences, San Diego, CA). The input cell numbers were 4 × 105 splenocytes/well (4 × 106 splenocytes/ml) assayed in duplicate wells. Briefly, the splenocytes were incubated for 18 to 20 h at 37°C in complete R10 culture medium (RPMI 1640 medium supplemented with 10% FBS, 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin sulfate, 1 mM sodium pyruvate, 1 mM HEPES, 100 mM nonessential amino acids) containing either 2 μg/ml concanavalin A (ConA) (Sigma) or peptide pools (15-mers overlapping by 11 amino acids; final peptide concentration, 1 μM [each]) spanning EBOV GP, SUDV GP, or MARV GP. Following development and drying, the plates were then scanned and counted with a CTL ImmunoSpot analyzer using the SmartCount data analysis module. The results are reported as IFN-γ SFC per 106 cells for each individual peptide pool. The peptide-specific IFN-γ ELISpot responses were considered positive if the response (minus medium background) was ≥3-fold above the medium response and ≥50 SFC/106 cells.
Murine filovirus GP-specific ELISA.
Immulon 2HB flat-bottom ELISA plates (Thermo Labsystem; catalog no. 3455) were coated for 18 h at 4°C with 100 ng/well recombinant proteins (recombinant EBOV GP minus the transmembrane region [EBOV rGPdTM; catalog no. 0501-015], recombinant SUDV GP minus the transmembrane region [SUDV rGPdTM; catalog no. 0502-015], and recombinant MARV strain Angola GP minus the transmembrane region [MARV-Angola rGPdTM; catalog no. 0506-015]). All the recombinant proteins were purchased from IBT Bioservices, Gaithersburg, MD, and diluted in 0.1 ml carbonate/bicarbonate buffer (carbonate/bicarbonate buffer with azide tablets from Sigma; catalog no. 08058-50TAB-F). The plates were then washed five times with 300 μl of 1× PBS containing 0.1% Tween 20 and blocked for 2 h at room temperature with 1× PBS containing 0.1% Tween 20 and 3% bovine serum albumin (BSA). Murine serum samples, diluted with 1× PBS containing 1% BSA and 0.1% Tween 20, were added to the ELISA plates at a starting dilution of 1:100 and further diluted 3-fold across the plates. The plates were kept at 4°C overnight, after which they were washed five times with 300 μl of 1× PBS containing 0.1% Tween 20, a biotin-SP-AffiniPure goat anti-mouse IgG (subclasses 1, 2a, 2b, and 3), Fcγ fragment specific (min X Hu, Bov, Rb Sr Prot; catalog no. 115-065-164; Jackson Laboratory), diluted 1:20,000 with 1× PBS supplemented with 0.1% Tween 20, and 1% BSA was then added (100 μl/well) for 1 h at room temperature. After five washes with 300 μl of 1× PBS containing 0.1% Tween 20, the plates were incubated for 1 h at room temperature with 100 μl/well of a streptavidin-horseradish peroxidase-conjugated anti-biotin antibody (500 units/ml stock; Roche Immunochemical, Indianapolis, IN) diluted 1:10,000 with 1× PBS supplemented with 0.1% Tween 20 and 1% BSA for antigen-specific immunoglobulin detection. Then, the plates were washed five times with 300 μl of 1× PBS containing 0.1% Tween 20. Finally, the plates were developed by the addition of 100 μl/well of TMB (3,3′,5,5′-tetramethyl benzidine) (Sigma; catalog no. T-8665). After 3 min, the reaction was stopped by adding 100 μl/well of 1 N sulfuric acid (VWR; catalog no. VW3232-1). Absorbance was measured at 450 nm in a VersaMax plate reader (Molecular Devices, Sunnyvale, CA). Antigen-specific serum IgG endpoint titers were defined as the reciprocal of the last serum dilution giving an optical density at 450 nm (OD450) greater than 0.1.
Challenge viruses.
Ebola virus Zaire isolate 199510621 (strain Kikwit) originated from a 65-year-old female patient who had died on 5 May 1995. The study challenge material was from the second Vero E6 passage of Zaire isolate 199510621. Briefly, the first passage at the University of Texas Medical Branch at Galveston (UTMB) consisted of inoculating CDC 807223 (passage 1 of Ebola virus isolate 199510621) at a multiplicity of infection (MOI) of 0.001 onto Vero E6 cells. The cell supernatants were subsequently harvested at 10 days postinfection and put in vials in 1-ml aliquots. Deep sequencing indicated the Zaire virus was greater than 98% 7U (consecutive stretch of 7 uridines). No detectable mycoplasma or endotoxin levels were measured at <0.5 endotoxin units (EU)/ml. Ebola virus Sudan isolate 200011676 (strain Gulu) originated from a 35-year-old male patient who had died on 16 October 2000. The study challenge material was from the second Vero E6 cell passage of Sudan isolate 200011676. Briefly, the first passage at UTMB consisted of inoculating CDC 808892 (CDC passage 1 of Sudan isolate 200011676) at an MOI of 0.001 onto Vero E6 cells. The cell supernatants were subsequently harvested at 7 days postinfection and put into vials in 1-ml aliquots. No detectable mycoplasma or endotoxin levels were measured at <0.5 EU/ml. Marburg virus Angola isolate 200501379 originated from human serum from an 8-month-old female patient in Uige, Angola. The serum was collected 17 days postonset, 1 day before death. The study challenge material was from the second Vero E6 cell passage of Marburg virus Angola isolate 200501379. Briefly, the first passage at UTMB consisted of inoculating CDC 810820 at an MOI of 0.001 onto Vero E6 cells. The cell supernatants were subsequently harvested at 6 days postinfection and put into vials in 1-ml aliquots. No detectable mycoplasma or endotoxin levels were measured at <0.5 EU/ml.
Nonhuman primate vaccination and challenge.
Twenty-one healthy, filovirus-naive, adult (∼3 to 9.5 kg) cynomolgus macaques (Macaca fascicularis; Covance Research Products) were randomized into three groups of five experimental animals and two control animals each. At Battelle Memorial Institute, the experimental animals were vaccinated by intramuscular injection of 3 × 107 PFU of the trival rVSV-N4CT1 pan-FiloGP1 vaccine, and the control animals were vaccinated by intramuscular injection of 3 × 107 PFU of an irrelevant rVSV-N4CT1-HIVgag1 vaccine. The macaques were transferred to UTMB and challenged 4 weeks after the single vaccination by intramuscular injection with 1,000 PFU of EBOV strain Kikwit for group 1, 1,000 PFU of SUDV strain Gulu for group 2, or 1,000 PFU of MARV strain Angola for group 3. All the macaques were given physical examinations, and blood was collected before vaccination; at day 10 after vaccination; at the time of challenge; and on days 3, 6, 10, 14, 22, and 28 after challenge. The macaques were monitored daily and scored for disease progression with an internal filovirus scoring protocol approved by the UTMB Institutional Animal Care and Use Committee (IACUC) in accordance with state and federal statutes and regulations relating to experiments involving animals and by the UTMB Institutional Biosafety Committee. The scoring changes measured from baseline included posture/activity level; attitude/behavior; food and water intake; weight; respiration; and disease manifestations, such as visible rash, hemorrhage, ecchymosis, or flushed skin, with increased scores resulting in euthanasia.
Clinical observations.
The macaques were observed daily and scored for general distress, anorexia (food consumption), weakness, recumbency, hunched posture, unresponsiveness, dyspnea, nasal exudate, edema (facial, cervical, or other), rash (petechial or ecchymotic), bleeding (oral, nasal, rectal, or other), emesis/hematemesis, dehydration, and diarrhea. In addition, the macaques were given physical examinations on days 0, 3, 6, 10, 14, and 28 after filovirus challenge. The physical examinations consisted of assessing body weight, rectal temperature, dyspnea, nasal exudate, edema (facial, cervical, or other), rash (petechial or ecchymotic), bleeding (oral, nasal, rectal, or other), emesis/hematemesis, dehydration, and diarrhea. A humane endpoint scoring sheet was approved by the UTMB IACUC. The scoring criteria included four parameters: (i) respiration (increased or decreased rate, abdominal breathing, or agonal breathing), (ii) appetite (eating biscuits and/or enrichment), (iii) activity and appearance (hunched or prostate), and (iv) bleeding/hemorrhage (petechial rash, ecchymotic rash, controlled bleeding, or uncontrolled bleeding). The scores in each parameter ranged from 0 for normal to 9 for increased disease. The scores from all the parameters were totaled, and any animal that reached a score of greater than or equal to 9 was euthanized.
Macaque IFN-γ ELISpot assay.
Vaccine-elicited IFN-γ ELISpot responses were determined using a T-cell human IFN-γ ELISpot kit (material number CTL HIFNG-10; Cellular Technology Ltd., Shaker Heights, OH). Briefly, heparinized whole blood was collected 10 days after immunization of the macaques, and PBMCs were isolated from the whole blood by Ficoll-Hypaque density gradient centrifugation and cryopreserved. The cryopreserved macaque PBMCs were then thawed and first cultured overnight with complete R10 culture medium. The cells were then collected, counted, and resuspended in complete R05 culture medium (RPMI 1640 medium supplemented with 5% FBS, 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin sulfate, 1 mM sodium pyruvate, 1 mM HEPES, 100 mM nonessential amino acids) containing either 5 μl/ml phytohemagglutinin mucoprotein (Sigma); peptide pools (15-mers overlapping by 11 amino acids; final peptide concentration, 1 μM [each]) spanning the EBOV GP, SUDV GP, or MARV GP; or medium alone. The input cell number was 2 × 105 PBMCs per well (2 × 106 PBMCs/ml), and the cells were assayed in duplicate wells. The cells were incubated for 20 to 24 h at 37°C. Following development and drying, the plates were scanned and cells were counted with a CTL ImmunoSpot analyzer using the SmartCount data analysis module. Peptide-specific IFN-γ ELISpot responses were considered positive if the responses (minus the medium background) were 3-fold above the medium response and ≥25 SFC/106 PBMCs. Unpaired t test analysis of IFN-γ ELISpot data was performed with GraphPad Prism version 5.02 software. Two-tailed P values of less than 0.05 indicated that the test results were statistically significant.
NHP filovirus anti-GP IgG ELISA.
Immulon 2HB flat-bottom plates (Fisher Scientific; catalog no. 14-245-61; lot no. NT520833) were coated with 50 ng/well of purified recombinant GP (rGP) (Advanced Bioscience Laboratories; EBOV Kikwit lot 17OCT13; SUDV Gulu lot 22JUL13; MARV Ci67 lot 12DEC2013). The plates were incubated overnight at 2 to 8°C. After incubation, the unbound rGP was removed by washing three times with 300 μl/well ELISA wash buffer (1× PBS, 0.1% Tween 20). The reference standard (RS) (Battelle Memorial Institute; EBOV RS, lot BMIZAIRE007; SUDV RS, lot BMISUDAN001; MARV RS, lot BMIMARV001) was diluted in ELISA diluent (1× PBS, 5% skim milk, 0.1% Tween 20) and added to rows A and B, columns 1 to 11, of each plate, creating an 11-point 2-fold dilution series (100 μl/well). The negative control (NC) (BMI, lot BMI298) was diluted 1:50 in ELISA diluent, and 100 μl was added to wells A12 and B12. The test serum samples and quality controls were added to row C and diluted down the remainder of the plate to create a six-point, 2-fold dilution series (rows C through H). The prepared ELISA plates were incubated for 60 ± 5 min at 37 ± 2°C. During this incubation period, the goat anti-NHP IgG HRP-conjugated secondary antibody (conjugate; Fitzgerald; catalog no. 43R-IG020HRP) was diluted in ELISA diluent (EBOV, 1:26,000; SUDV, 1:38,000; MARV, 1:26,000). The plates were washed again three times with 300 μl/well ELISA wash buffer, and 100 μl of the appropriate diluted conjugate was added to each plate. The plates were incubated for 60 ± 5 min at 37 ± 2°C. Following incubation, the plates were washed three times, and 100 μl of TMB (Fisher Scientific; catalog no. N301) substrate was added to each well of the plate and incubated for 10 ± 2 min at room temperature to allow the colorimetric reaction to take place. Following incubation, 100 μl of TMB stop solution (Fisher Scientific; catalog no. N600) was added. Absorbance was measured at 450 nm (reference wavelength, 650 nm) using a BioTek (Winooski, VT) ELx405 microplate reader accompanied by Gen5 software version 2.01.12. Quality control and test sample concentrations (in ELISA units per milliliter) were calculated using SoftMax Pro version 4.7 software (Molecular Devices, Sunnyvale, CA) based on the reference curve.
Detection of viremia by plaque assay.
Titration of the rVSV-N4CT1 vaccine vectors; confirmation of the challenge dose; and titration of EBOV, SUDV, or MARV in plasma after challenge were performed by standard plaque assay on Vero E6 cell monolayers as previously described (16). Briefly, serial 10-fold dilutions of the samples were adsorbed to Vero E6 monolayers in duplicate wells of six-well plates (200 μl/well). Following adsorption, the inoculum was removed by aspiration, and the monolayers were overlaid with 3 ml of 0.8% SeaPlaque agarose in DMEM. The agarose was permitted to set at room temperature for 15 min, and the plates were then incubated at 32°C under 5% CO2 for 3 to 4 days (for rVSV-N4CT1 vectors) or 7 to 10 days (for filoviruses) to allow plaque development. The limit of detection was 25 PFU/ml.
Detection of viremia by RT-qPCR.
RNA was isolated from whole blood utilizing the Viral RNA mini-kit (Qiagen) using 100 μl of blood added to 600 μl of the viral lysis buffer supplied in the kit. Primers and probe targeting the VP30 gene of EBOV, the L gene of SUDV, and the NP gene of MARV were used for real-time quantitative PCR (RT-qPCR) with the following probes: EBOV, 6-carboxyfluorescein (FAM)-5′ AGG CTT CCC TCG CTG CCG TTA TG 3′-6 carboxytetramethylrhodamine (TAMRA); SUDV, FAM-5′ CAT CCA ATC AAA GAC ATT GCG A 3′-TAMRA; and MARV, FAM-5′ CCC ATA AGG TCA CCC TCT TC 3′-TAMRA (Life Technologies). Viral RNA was detected using the CFX96 detection system (Bio-Rad Laboratories, Hercules, CA) in one-step probe RT-qPCR kits (Qiagen) with the following cycle conditions: EBOV, 50°C for 10 min, 95°C for 10 s, and 40 cycles of 95°C for 10 s and 57°C for 30 s; SUDV and MARV, 50°C for 10 min, 95°C for 10 s, and 40 cycles of 95°C for 10 s and 59°C for 30 s. Threshold cycle (CT) values representing viral genomes were analyzed with CFX Manager software, and the data are shown as genome equivalents (GEq) per milliliter. To create the GEq standard, RNA from viral stocks was extracted, and the number of strain-specific genomes was calculated using Avogadro's number and the molecular weight of each viral genome.
Hematology and serum biochemistry.
Total white blood cell counts, white blood cell differentials, red blood cell counts, platelet counts, hematocrit values, total hemoglobin concentrations, mean cell volumes, mean corpuscular volumes, and mean corpuscular hemoglobin concentrations were analyzed from blood collected in tubes containing EDTA using a laser-based hematologic analyzer (Beckman Coulter). Serum samples were tested for concentrations of albumin, amylase, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), gammaglutamyltransferase (GGT), glucose, cholesterol, total protein, total bilirubin (TBIL), blood urea nitrogen (BUN), creatinine (CRE), and C-reactive protein (CRP) by using a Piccolo point-of-care analyzer and Biochemistry Panel Plus analyzer discs (Abaxis).
Statistical analysis.
Unpaired t test analysis of murine and NHP IFN-γ ELISpot data was performed on GraphPad Prism version 5.02 software. Two-tailed P values of less than 0.05 indicated that the tests were statistically significant.
ACKNOWLEDGMENTS
We thank the Daniel J. Deer and Animal Resource Center staff and the Research Histopathology Core for assistance at the University of Texas Medical Branch (UTMB) and the Galveston National Laboratory (GNL).
These studies were funded under contract W911QY-14-1-0001 to Profectus BioSciences from the Department of Defense (DOD) Medical Countermeasure Systems' Joint Vaccine Acquisition Program (MCS-JVAP).
D.M., D.K.C., and T.L. conceived and designed the vaccine vectors and did preparative work. J.H.E., M.A.E., A.O.-S., and R.X. designed, conducted, and analyzed the vaccine characterization studies in mice. D.M., M.A.E., D.K.C., J.H.E., D.J.K., A.B., C.L.S., and T.W.G. conceived and designed the NHP study. D.J.K. performed the NHP vaccination experiments and conducted clinical observations of the animals. A.O.-S., R.X., M.Q.S.W., T.L.R., and C.L.S. performed NHP immunological assays. C.E.M., J.B.G., and T.W.G. performed the NHP challenge experiments and conducted clinical observations of the animals. J.B.G. and K.N.A. performed the clinical pathology assays. J.B.G. performed the infectivity assays. D.M., D.K.C, M.A.E., J.H.E., R.X., C.E.M., T.W.G., A.B., and C.L.S., analyzed the data. D.M., D.K.C., M.A.E., C.E.M., T.W.G., D.J.K., A.B., and C.L.S. wrote the paper.
D.M., T.L., R.X., A.O.-S., M.A.E., J.H.E., and D.K.C. are paid employees of Profectus BioSciences, Inc. D.K.C. is a coinventor of the attenuated VSV vector (U.S. patent 8,287,878) and does not receive royalties or other payments. T.W.G. has patents (U.S. patent 61/014,626, U.S. patent 61/014,669, and U.S. patent 61/070,748) pending to Boston University. We report no other potential conflicts.
The opinions, interpretations, conclusions, and recommendations contained here are ours and are not necessarily endorsed by UTMB at Galveston or the Department of Defense.
The following materials may be provided upon request and execution of a Material Transfer Agreement: rVSV-N4CT1-EBOVGP3, rVSV-N4CT1-SUDVGP3, rVSV-N4CT1-MARVGP3, rVSV-N1CT1-EBOVGP3, rVSV-N1CT1-SUDVGP3, and rVSV-N1CT1-MARVGP3. The following materials are included in ongoing or projected human clinical trials and will not be provided: rVSV-N4CT1-EBOVGP1, rVSV-N4CT1-SUDVGP1, rVSV-N4CT1-MARVGP1, and rVSV-N4CT1-HIVgag1.
REFERENCES
- 1.Feldmann H, Sanchez A, Geisbert T. 2013. Filoviridae: Marburg and Ebola viruses, p 923–956. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed (electronic) Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Kuhn JH, Becker S, Ebihara H, Geisbert TW, Johnson KM, Kawaoka Y, Lipkin WI, Negredo AI, Netesov SV, Nichol ST, Palacios G, Peters CJ, Tenorio A, Volchkov VE, Jahrling PB. 2010. Proposal for a revised taxonomy of the family Filoviridae: classification, names of taxa and viruses, and virus abbreviations. Arch Virol 155:2083–2103. doi: 10.1007/s00705-010-0814-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macneil A, Reed Z, Rollin PE. 2011. Serologic cross-reactivity of human IgM and IgG antibodies to five species of Ebola virus. PLoS Negl Trop Dis 5:e1175. doi: 10.1371/journal.pntd.0001175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson ED, Johnson BK, Silverstein D, Tukei P, Geisbert TW, Sanchez AN, Jahrling PB. 1996. Characterization of a new Marburg virus isolated from a 1987 fatal case in Kenya. Arch Virol Suppl 11:101–114. [DOI] [PubMed] [Google Scholar]
- 5.Negredo A, Palacios G, Vazquez-Moron S, Gonzalez F, Dopazo H, Molero F, Juste J, Quetglas J, Savji N, de la Cruz Martinez M, Herrera JE, Pizarro M, Hutchison SK, Echevarria JE, Lipkin WI, Tenorio A. 2011. Discovery of an ebolavirus-like filovirus in Europe. PLoS Pathog 7:e1002304. doi: 10.1371/journal.ppat.1002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feldmann H. 2014. Ebola—a growing threat? N Engl J Med 371:1375–1378. doi: 10.1056/NEJMp1405314. [DOI] [PubMed] [Google Scholar]
- 7.Kucharski AJ, Edmunds WJ. 2014. Case fatality rate for Ebola virus disease in west Africa. Lancet 384:1260. doi: 10.1016/S0140-6736(14)61706-2. [DOI] [PubMed] [Google Scholar]
- 8.Gaidamovich S, Altukhova LM, Obukhova VR, Ponirovskii EN, Sadykov VG. 1980. Isolation of the Isfahan virus in Turkmenia. Vopr Virusol 1980:618–620. (In Russian.) [PubMed] [Google Scholar]
- 9.Meltzer MI, Atkins CY, Santibanez S, Knust B, Petersen BW, Ervin ED, Nichol ST, Damon IK, Washington ML, Centers for Disease Control and Prevention . 2014. Estimating the future number of cases in the Ebola epidemic—Liberia and Sierra Leone, 2014-2015. MMWR Suppl 63:1–14. [PubMed] [Google Scholar]
- 10.Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, Young HA, Fredeking TM, Rote WE, Vlasuk GP. 2003. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 362:1953–1958. doi: 10.1016/S0140-6736(03)15012-X. [DOI] [PubMed] [Google Scholar]
- 11.Pettitt J, Zeitlin L, Kim DH, Working C, Johnson JC, Bohorov O, Bratcher B, Hiatt E, Hume SD, Johnson AK, Morton J, Pauly MH, Whaley KJ, Ingram MF, Zovanyi A, Heinrich M, Piper A, Zelko J, Olinger GG. 2013. Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci Transl Med 5:199ra113. doi: 10.1126/scitranslmed.3006608. [DOI] [PubMed] [Google Scholar]
- 12.Takada A, Ebihara H, Jones S, Feldmann H, Kawaoka Y. 2007. Protective efficacy of neutralizing antibodies against Ebola virus infection. Vaccine 25:993–999. doi: 10.1016/j.vaccine.2006.09.076. [DOI] [PubMed] [Google Scholar]
- 13.Falzarano D, Geisbert TW, Feldmann H. 2011. Progress in filovirus vaccine development: evaluating the potential for clinical use. Expert Rev Vaccines 10:63–77. doi: 10.1586/erv.10.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones SM, Feldmann H, Stroher U, Geisbert JB, Fernando L, Grolla A, Klenk HD, Sullivan NJ, Volchkov VE, Fritz EA, Daddario KM, Hensley LE, Jahrling PB, Geisbert TW. 2005. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med 11:786–790. doi: 10.1038/nm1258. [DOI] [PubMed] [Google Scholar]
- 15.Stanley DA, Honko AN, Asiedu C, Trefry JC, Lau-Kilby AW, Johnson JC, Hensley L, Ammendola V, Abbate A, Grazioli F, Foulds KE, Cheng C, Wang L, Donaldson MM, Colloca S, Folgori A, Roederer M, Nabel GJ, Mascola J, Nicosia A, Cortese R, Koup RA, Sullivan NJ. 2014. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat Med 20:1126–1129. doi: 10.1038/nm.3702. [DOI] [PubMed] [Google Scholar]
- 16.Geisbert TW, Geisbert JB, Leung A, Daddario-DiCaprio KM, Hensley LE, Grolla A, Feldmann H. 2009. Single-injection vaccine protects nonhuman primates against infection with Marburg virus and three species of Ebola virus. J Virol 83:7296–7304. doi: 10.1128/JVI.00561-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henao-Restrepo AM, Longini IM, Egger M, Dean NE, Edmunds WJ, Camacho A, Carroll MW, Doumbia M, Draguez B, Duraffour S, Enwere G, Grais R, Gunther S, Hossmann S, Konde MK, Kone S, Kuisma E, Levine MM, Mandal S, Norheim G, Riveros X, Soumah A, Trelle S, Vicari AS, Watson CH, Keita S, Kieny MP, Rottingen JA. 2015. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet 386:857–866. doi: 10.1016/S0140-6736(15)61117-5. [DOI] [PubMed] [Google Scholar]
- 18.Agnandji ST, Huttner A, Zinser ME, Njuguna P, Dahlke C, Fernandes JF, Yerly S, Dayer JA, Kraehling V, Kasonta R, Adegnika AA, Altfeld M, Auderset F, Bache EB, Biedenkopf N, Borregaard S, Brosnahan JS, Burrow R, Combescure C, Desmeules J, Eickmann M, Fehling SK, Finckh A, Goncalves AR, Grobusch MP, Hooper J, Jambrecina A, Kabwende AL, Kaya G, Kimani D, Lell B, Lemaitre B, Lohse AW, Massinga-Loembe M, Matthey A, Mordmuller B, Nolting A, Ogwang C, Ramharter M, Schmidt-Chanasit J, Schmiedel S, Silvera P, Stahl FR, Staines HM, Strecker T, Stubbe HC, Tsofa B, Zaki S, Fast P, Moorthy V, Kaiser L, Krishna S, Becker S, Kieny MP, Bejon P, Kremsner PG, Addo MM, Siegrist CA. 2016. Phase 1 trials of rVSV Ebola vaccine in Africa and Europe: preliminary report. N Engl J Med 374:1647–1660. doi: 10.1056/NEJMoa1502924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huttner A, Dayer JA, Yerly S, Combescure C, Auderset F, Desmeules J, Eickmann M, Finckh A, Goncalves AR, Hooper JW, Kaya G, Krahling V, Kwilas S, Lemaitre B, Matthey A, Silvera P, Becker S, Fast PE, Moorthy V, Kieny MP, Kaiser L, Siegrist CA, VSV-Ebola Consortium . 2015. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: a randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect Dis 15:1156–1166. doi: 10.1016/S1473-3099(15)00154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke DK, Nasar F, Lee M, Johnson JE, Wright K, Calderon P, Guo M, Natuk R, Cooper D, Hendry RM, Udem SA. 2007. Synergistic attenuation of vesicular stomatitis virus by combination of specific G gene truncations and N gene translocations. J Virol 81:2056–2064. doi: 10.1128/JVI.01911-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wertz GW, Perepelitsa VP, Ball LA. 1998. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc Natl Acad Sci U S A 95:3501–3506. doi: 10.1073/pnas.95.7.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts A, Buonocore L, Price R, Forman J, Rose JK. 1999. Attenuated vesicular stomatitis viruses as vaccine vectors. J Virol 73:3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cooper D, Wright KJ, Calderon PC, Guo M, Nasar F, Johnson JE, Coleman JW, Lee M, Kotash C, Yurgelonis I, Natuk RJ, Hendry RM, Udem SA, Clarke DK. 2008. Attenuation of recombinant vesicular stomatitis virus-human immunodeficiency virus type 1 vaccine vectors by gene translocations and G gene truncation reduces neurovirulence and enhances immunogenicity in mice. J Virol 82:207–219. doi: 10.1128/JVI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clarke DK, Nasar F, Chong S, Johnson JE, Coleman JW, Lee M, Witko SE, Kotash CS, Abdullah R, Megati S, Luckay A, Nowak B, Lackner A, Price RE, Little P, Kalyan N, Randolf V, Javadian A, Zamb TJ, Parks CL, Egan MA, Eldridge J, Hendry M, Udem SA. 2014. Neurovirulence and immunogenicity of attenuated recombinant vesicular stomatitis viruses in nonhuman primates. J Virol 88:6690–6701. doi: 10.1128/JVI.03441-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchs JD, Frank I, Elizaga ML, Allen M, Frahm N, Kochar N, Li S, Edupuganti S, Kalams SA, Tomaras GD, Sheets R, Pensiero M, Tremblay MA, Higgins TJ, Latham T, Egan MA, Clarke DK, Eldridge JH, HVTN 090 Study Group and the National Institutes of Allergy and Infectious Diseases HIV Vaccine Trials Network, Mulligan M, Rouphael N, Estep S, Rybczyk K, Dunbar D, Buchbinder S, Wagner T, Isbell R, Chinnell V, Bae J, Escamilla G, Tseng J, Fair R, Ramirez S, Broder G, Briesemeister L, Ferrara A. 2015. First-in-human evaluation of the safety and immunogenicity of a recombinant vesicular stomatitis virus human immunodeficiency virus-1 gag vaccine (HVTN 090). Open Forum Infect Dis 2:ofv082. doi: 10.1093/ofid/ofv082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matassov D, Marzi A, Latham T, Xu R, Ota-Setlik A, Feldmann F, Geisbert JB, Mire CE, Hamm S, Nowak B, Egan MA, Geisbert TW, Eldridge JH, Feldmann H, Clarke DK. 2015. Vaccination with a highly attenuated recombinant vesicular stomatitis virus vector protects against challenge with a lethal dose of Ebola virus. J Infect Dis 212(Suppl 2):S443–S451. doi: 10.1093/infdis/jiv316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mire CE, Matassov D, Geisbert JB, Latham TE, Agans KN, Xu R, Ota-Setlik A, Egan MA, Fenton KA, Clarke DK, Eldridge JH, Geisbert TW. 2015. Single-dose attenuated Vesiculovax vaccines protect primates against Ebola Makona virus. Nature 520:688–691. doi: 10.1038/nature14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marzi A, Feldmann H, Geisbert TW, Falzarano D. 2011. Vesicular stomatitis virus-based vaccines for prophylaxis and treatment of filovirus infections. J Bioterror Biodef S1:2157–2526-S1-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potter CW, Jennings R, Clark A, Ali M. 1983. Interference following dual inoculation with influenza A (H3N2) and (H1N1) viruses in ferrets and volunteers. J Med Virol 11:77–86. doi: 10.1002/jmv.1890110110. [DOI] [PubMed] [Google Scholar]
- 30.Wareing MD, Tannock GA. 2001. Live attenuated vaccines against influenza; an historical review. Vaccine 19:3320–3330. doi: 10.1016/S0264-410X(01)00045-7. [DOI] [PubMed] [Google Scholar]
- 31.Pourrut X, Souris M, Towner JS, Rollin PE, Nichol ST, Gonzalez JP, Leroy E. 2009. Large serological survey showing cocirculation of Ebola and Marburg viruses in Gabonese bat populations, and a high seroprevalence of both viruses in Rousettus aegyptiacus. BMC Infect Dis 9:159. doi: 10.1186/1471-2334-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Witko SE, Kotash CS, Nowak RM, Johnson JE, Boutilier LA, Melville KJ, Heron SG, Clarke DK, Abramovitz AS, Hendry RM, Sidhu MS, Udem SA, Parks CL. 2006. An efficient helper-virus-free method for rescue of recombinant paramyxoviruses and rhadoviruses from a cell line suitable for vaccine development. J Virol Methods 135:91–101. doi: 10.1016/j.jviromet.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Anonymous. 1996. Guide for the care and use of laboratory animals. New York Medical College’s Institutional Animal Care and Use Committees, Tarrytown, NY. [Google Scholar]