

ABSTRACT
Despite the availability of two attenuated vaccines, rotavirus (RV) gastroenteritis remains an important cause of mortality among children in developing countries, causing about 215,000 infant deaths annually. Currently, there are no specific antiviral therapies available. RV is a nonenveloped virus with a segmented double-stranded RNA genome. Viral genome replication and assembly of transcriptionally active double-layered particles (DLPs) take place in cytoplasmic viral structures called viroplasms. In this study, we describe strong impairment of the early stages of RV replication induced by a small molecule known as an RNA polymerase III inhibitor, ML-60218 (ML). This compound was found to disrupt already assembled viroplasms and to hamper the formation of new ones without the need for de novo transcription of cellular RNAs. This phenotype was correlated with a reduction in accumulated viral proteins and newly made viral genome segments, disappearance of the hyperphosphorylated isoforms of the viroplasm-resident protein NSP5, and inhibition of infectious progeny virus production. In in vitro transcription assays with purified DLPs, ML showed dose-dependent inhibitory activity, indicating the viral nature of its target. ML was found to interfere with the formation of higher-order structures of VP6, the protein forming the DLP outer layer, without compromising its ability to trimerize. Electron microscopy of ML-treated DLPs showed dose-dependent structural damage. Our data suggest that interactions between VP6 trimers are essential, not only for DLP stability, but also for the structural integrity of viroplasms in infected cells.
IMPORTANCE Rotavirus gastroenteritis is responsible for a large number of infant deaths in developing countries. Unfortunately, in the countries where effective vaccines are urgently needed, the efficacy of the available vaccines is particularly low. Therefore, the development of antivirals is an important goal, as they might complement the available vaccines or represent an alternative option. Moreover, they may be decisive in fighting the acute phase of infection. This work describes the inhibitory effect on rotavirus replication of a small molecule initially reported as an RNA polymerase III inhibitor. The molecule is the first chemical compound identified that is able to disrupt viroplasms, the viral replication machinery, and to compromise the stability of DLPs by targeting the viral protein VP6. This molecule thus represents a starting point in the development of more potent and less cytotoxic compounds against rotavirus infection.
KEYWORDS: DLP, ML-60218, RNA polymerase III, VP6, antivirals, drug, inhibitor, rotavirus, viroplasms
INTRODUCTION
Rotavirus (RV) is the most common cause of gastroenteritis in young children and infants throughout the world. The impact of RV vaccines on global estimates of RV mortality has been limited, and the rate of deaths due to RV gastroenteritis in developing countries is still approximately between 197,000 and 233,000 per year (1). Currently, there are no specific antivirals available. The virus belongs to the family Reoviridae, is nonenveloped, and contains a genome of 11 segments of double-stranded RNA (dsRNA). During entry into the host cell (enterocytes of the intestinal villi), the virion (triple-layered particle [TLP]) loses the outermost of its three concentric protein layers, formed by the glycoprotein VP7 and the spike protein VP4, and becomes a transcriptionally active double-layered particle (DLP). The viral RNA-dependent RNA polymerase VP1 acts both as a transcriptase (for synthesis of viral mRNAs) and as a replicase (for synthesis of minus strands, resulting in new genome segments) (2). To be transcriptionally active, VP1 must be localized within a DLP. Both DLP layers, the inner one formed by VP2 and the outer one formed by VP6 trimers, are required to guarantee transcriptional activity. Removal of VP6 from DLPs abolishes this activity, which can be recovered by adding native or recombinant VP6 (3, 4). The transcribed plus-strand RNAs act as both messengers for viral protein synthesis and templates for viral genome replication. The viral genome is replicated in viral cytoplasmic structures called viroplasms, where the assembly of progeny DLPs also takes place. These newly synthesized DLPs perform a second round of transcription, called secondary transcription, that produces additional plus-strand RNAs (5, 6). In the final stages of virus morphogenesis, the progeny DLPs bud into the endoplasmic reticulum (ER), acquiring a transient envelope, which is then replaced by the viral proteins (VP7 and VP4) forming the outer layer of progeny TLPs (2).
In addition to the structural proteins forming DLPs (VP1, VP2, the capping enzyme VP3, and VP6), two viral nonstructural proteins are also found in viroplasms: NSP2 and NSP5. Both are essential for viroplasm formation and therefore for viral replication (6–9). During viral infection, NSP5 undergoes a complex hyperphosphorylation process involving different kinases and interactions with other viral proteins (10–12). However, the role of this posttranslational modification in the viral replication cycle is not completely understood. In fact, several lines of evidence show correlation between viroplasm formation and NSP5 phosphorylation (13–15). Also, NSP2 and VP2 were recently found to be phosphorylated, and it has been proposed that viroplasm formation is a phosphorylation-dependent process (11).
The number and size of viroplasms in infected cells are indicative of viral replication efficiency. These values decrease in the presence of compounds with antiviral effects on the early steps of the viral cycle, like inhibitors of proteasome (16), microtubules (MT) (17), Eg5 kinesin (17), and thiazolides (18), that interfere with viroplasm assembly and virus infectivity. Ribavirin has also been reported to affect RV replication (19). In this work, we describe the unexpected antiviral activity of a chemical compound, ML-60218 (referred to here as ML), reported to be an inhibitor of the RNA polymerase III complex (20). The effect on RV shown here is independent of the RNA polymerase III catalytic activity. For the first time, we describe a chemical compound that is able to disrupt viroplasms in virus-infected cells and to compromise the stability of purified DLPs by interfering with interactions between VP6 trimers.
RESULTS
ML impairs viroplasm formation and disrupts already assembled viroplasms.
Treatment of RV-infected cells with ML at 10 μM caused a strong reduction of viroplasms accumulated during viral replication and, in the case of the porcine strain OSU, complete disappearance of those structures, as observed by both immunofluorescence (Fig. 1) and electron microscopy (EM) analyses (Fig. 2). Regardless of whether the compound was added at 1, 3, or 5 h postinfection (hpi), a 4-h treatment had the same effect on viroplasms (Fig. 1A). Viroplasm disruption was correlated with a decrease of accumulated viral proteins, as observed with the porcine OSU and the simian SA11 strains (Fig. 1B). When added before infection, together with the infectious virion, or at 1 hpi (Fig. 1C, top), ML caused a reduction of viral proteins in a dose-dependent manner with concomitant disappearance of the NSP5 hyperphosphorylated isoforms (Fig. 1C, middle). ML added at a concentration of 10 μM together with the infectious virions completely blocked assembly of viroplasms (Fig. 1C, bottom). The same phenotype was observed in two different cell lines (MA104 monkey kidney epithelial cells and U2OS human osteosarcoma cells) upon infection with two different RV strains (OSU and SA11) (Fig. 1D and E).
FIG 1.

ML effect on RV replication. (A to E) Western blot and confocal immunofluorescence analyses with the indicated antibodies of RV-infected cells (MOI, 25 VFU/cell) treated with ML at 10 μM, unless otherwise indicated, or with DMSO (D) for the indicated times. Scale bars, 10 μm. TUB, tubulin. (F) Time course of viral progeny yield of OSU-infected (MOI, 25 VFU/cell) MA104 cells treated with 10 μM ML, added at 2 hpi. The data are presented as averages ± standard deviations of the results of three independent experiments. ***, P < 0.001 (t test). (G) Genome segment analysis of blotted total RNA extracted from noninfected (NI) and OSU-infected (25 VFU/cell) MA104 cells treated with ML (10 μM) or DMSO from 1 to 8 hpi and revealed with an anti-dsRNA antibody. (H) Viability of noninfected or OSU-infected (MOI, 25 VFU/cell) MA104 cells determined by cytofluorometry of propidium iodide-stained cells following treatment at 2 hpi with or without 10 μM ML for up to 12 hpi. The data are presented as averages ± standard deviations of the results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (t test).
FIG 2.

Electron microscopy of RV-infected cells treated with ML. High-definition electron microscopy of noninfected (NI) and RV-infected (OSU; MOI, 100 VFU/ml) MA104 cells untreated (DMSO) or treated with ML (20 μM) from 1 hpi. At 6 hpi, the cells were fixed with glutaraldehyde and processed for transmission electron microscopy. V, viroplasms; Nu, nucleus, ER, endoplasmic reticulum; Gg, Golgi complex; Vc, vacuoles; Ph, phagosomes; CM, cell membrane; the thin arrows indicate the endoplasmic reticulum membrane surrounding viroplasms; the large arrowheads indicate viral particles.
The absence of viroplasms and the reduction of accumulated viral proteins indicated that ML inhibits RV replication. This result was confirmed by assessing the yield of infectious progeny virus produced at different time points postinfection (Fig. 1F) and the production of newly made dsRNA genome segments (gs) (Fig. 1G). During the time intervals of the analysis, ML was not cytotoxic (Fig. 1H).
Three hours was the minimum time and 10 μM the minimal concentration required for a complete effect on viroplasms (data not shown). Concentrations higher than 20 μM were toxic to the cells, as shown by the decreased levels of actin in Western blots (Fig. 1C). Treatments of up to 12 to 14 h at 10 μM were well tolerated (Fig. 1H).
ML-mediated viroplasm disruption causes NSP5 dephosphorylation.
In order to determine whether the effect of ML on NSP5 phosphorylation was due to activation of phosphatases, 0.5 μM okadaic acid (an inhibitor of serine/threonine phosphatases) was added to OSU-infected cells at 3 hpi (1 h before the addition of ML) and maintained during the following 4-h treatment (Fig. 3, top). Upon inhibition of phosphatases with okadaic acid, the effect of ML on viroplasms remained unaltered (Fig. 3, bottom right), but NSP5 hyperphosphorylated isoforms did not disappear (Fig. 3, bottom left, lane 2). This result suggests that the effect of ML on viroplasms is not mediated by activation of phosphatases and that NSP5 dephosphorylation is the consequence of disruption of viroplasms that, when intact, protect NSP5 from cytosolic phosphatases.
FIG 3.

NSP5 dephosphorylation caused by ML-mediated viroplasm disruption. Shown are Western blot and confocal immunofluorescence analyses with the indicated antibodies of OSU-infected (25 VFU/cell) MA104 cells treated with 10 μM ML and/or 0.5 μM okadaic acid (OA) or DMSO for the indicated times. Scale bars, 5 μm.
ML activity against RV does not require newly synthesized cellular transcripts or proteins and is independent of RNA polymerase III catalytic activity.
To determine whether the phenotype observed under ML treatment was due to de novo synthesis of cellular proteins or RNAs, actinomycin D was added to cells at 3 h postinfection, 1 h before treatment with the drug, and maintained during the following 4-h treatment (Fig. 4A, top). Actinomycin D is a DNA intercalator that inhibits transcription of RNA polymerase I at very low concentrations (>0.01 μg/ml), of RNA polymerase II at concentrations higher than 2 μg/ml, and of RNA polymerase III at concentrations higher than 5 μg/ml (21, 22). Newly synthesized RNAs were labeled with the ribonucleoside homolog 5-ethynyl uridine (EU) and visualized by reacting with an Alexa-488-conjugated azide (Fig. 4A, right, green) to assess the effectiveness of actinomycin D treatment. Actinomycin D at 10 μg/ml did not interfere with the effect of ML, indicating that newly synthesized cellular transcripts are not required for ML antiviral activity. Also, in the absence of ML, treatment with actinomycin D did not compromise viroplasm integrity or affect the accumulation of viral proteins (Fig. 4A, bottom left). On the contrary, it slightly increased the amounts of VP2 and NSP5 compared to the untreated control (Fig. 4A, bottom left, lane 2 versus lane 1). This result explains why actinomycin D was found to compensate for the decrease of VP2 induced by ML (Fig. 4A, bottom left, lane 4 versus lane 3). All these data indicate that RV replication does not require the catalytic activity of RNA polymerase III and that ML activity against RV is not mediated by inhibition of this cellular target. In addition, de novo protein synthesis is not required for ML activity. Addition of ML (at 4 hpi) to cells treated with the protein synthesis inhibitor cycloheximide (CHX) (added at 3 hpi) still caused viroplasm disruption and NSP5 dephosphorylation (Fig. 4B).
FIG 4.

ML antiviral activity is independent of cellular transcription and protein synthesis. (A) Western blot and confocal immunofluorescence analyses with the indicated antibodies of OSU-infected MA104 cells (25 VFU/cell) fed with EU and treated with 10 μM ML and/or 10 μg/ml actinomycin D (Act D) or DMSO for the indicated times. EU-labeled, newly synthesized RNAs were visualized by reaction with an Alexa-488-conjugated azide (green). Scale bars, 5 μm. (B) Western blot and confocal immunofluorescence analyses with the indicated antibodies of OSU-infected (MOI, 25 VFU/cell) MA104 cells treated with 10 μM ML and/or 10 μg/ml CHX or DMSO for the indicated times. Scale bars, 10 μm.
ML compromises DLP stability.
We then investigated whether ML acts on a viral target. RV DLPs were purified from cells infected with strain OSU or SA11, and their transcriptional activity was tested in vitro in the presence of increasing ML concentrations. An irrelevant small molecule (compound 7749832; ChemBridge Corp.) solubilized in the same buffer was used as a control. Upon incubation with ML, the numbers of transcripts produced by DLPs of both strains were significantly decreased in a dose-dependent manner (Fig. 5A) compared to incubation with the vehicle or the irrelevant small molecule. A 50% inhibitory concentration (IC50) of 10 μM was observed for both viruses. In order to be transcriptionally active, DLPs must be intact. The absence of the external layer formed by VP6 trimers alone is sufficient to compromise transcriptional activity (3, 4, 21). Interestingly, EM analyses of purified DLPs showed that 4 h of incubation with ML (in the same buffer used for the in vitro transcription assays) caused structural damage in a dose-dependent manner. DLPs with an irregular shape and partially open were observed (Fig. 5B; quantifications in all samples are plotted in Fig. 5C). Altogether, these results suggest that ML compromises the structural integrity of DLPs, thus impairing their transcriptional activity.
FIG 5.

ML-mediated impairment of DLP stability. (A) Transcriptional activity of purified DLPs. The plot shows the dose-dependent decrease of transcripts produced by SA11 or OSU DLPs incubated in the presence of the indicated concentrations of ML. The small molecule 7749832 (ChemBridge Corp.) at 200 μM was used as an irrelevant compound (irr.). The data represent the means ± standard deviations of the results of at least three independent experiments. **, P < 0.01; ***, P < 0.001; ns, P > 0.01 (t test). (B and C) DLP morphology analyzed by electron microscopy. Purified DLPs were incubated for 4 h with the indicated concentrations of ML or DMSO. The arrows indicate damaged DLPs with an irregular shape and partially open. Quantification of damaged DLPs is shown in panel C. The data are presented as averages ± standard errors of the mean (SEM). ***, P < 0.001 (t test); n > 100.
ML interferes with higher-order VP6 structures.
Although ML clearly altered DLP morphology, the EM images showed the presence of residual structures (Fig. 5B, arrows). These structures could be DLPs partially or totally deprived of the VP6 layer.
We therefore investigated whether ML impaired the interaction of VP6 with VP2 or with itself, as in the formation of trimers or in the interactions between trimers.
The VP6-VP2 interaction was tested by coimmunoprecipitation with an anti-VP6 monoclonal antibody (MAb) (clone RV-138) from cells overexpressing both VP6 and VP2 and treated for 5 h with ML. The inhibitor was also maintained during cell lysis and incubation with the precipitating antibody. The experiment, repeated at various ML concentrations (up to 200 μM, the highest concentration used), showed that ML did not interfere with VP6-VP2 interactions (Fig. 6A).
FIG 6.

Effect of ML on RV VP6. (A) VP6-VP2 interaction. Shown is Western blot analysis with anti-VP2 and anti-VP6 antibodies of immunoprecipitates (IP) obtained with anti-VP6 MAb RV138 from extracts of MA104 cells transfected with VP6 and VP2 and treated for 5 h with 10 μM ML or DMSO. The inhibitor (200 μM) was maintained during cell lysis and incubation with the precipitating antibody. The numbers on the left are kilodaltons. (B) VP6 trimer stability. (Left) Western blot analysis with anti-VP6 antibody of nonboiled extracts from MA104 cells infected with a recombinant vaccinia virus expressing VP6 (VVVP6) and treated with 10 μM ML or DMSO from 1 to 7 hpi. (Right) Western blot analysis of nonboiled extracts from cells infected with OSU (MOI, 25 VFU/cell) and treated with 10 μM ML or DMSO from 1 to 5 hpi. The numbers on the right are kilodaltons. (C) Confocal immunofluorescence (IF) analysis with the anti-VP6 MAb 4B2D2 of MA104 cells overexpressing VP6 (infected with VVVP6) and treated with 10 μM ML or DMSO from 1 to 7 hpi. The arrow indicates a VP6 higher-order structure observed in the absence of other RV proteins. Scale bars, 5 μm. (D) Representative images of VP6 tubes and spheres visualized by negative-staining electron microscopy after treatment with 25 μM ML for 4 h at 37°C. (E) Interaction of VP6 with ML evaluated by nanoscale thermophoresis. The fraction of Cys- or Lys-labeled VP6 bound to ML was plotted against increasing concentrations of the inhibitor. The data were fitted with two state equations, and an EC50 of 294 ± 62 μM was calculated as the average of the results of three independent measurements. The error bars indicate standard deviations.
VP6 trimer formation was assessed by Western blotting of nonboiled lysates from cells overexpressing VP6 and treated with ML. In fact, VP6 trimers (mass = 135 kDa) resist SDS denaturing and reducing conditions, but not high temperatures (23). Cells infected with a recombinant vaccinia virus expressing VP6 (VVVP6) were treated with ML at 1 hpi for 7 h. VP6 trimers formed in vivo were not affected by the presence of ML (Fig. 6B, left), which did not compromise the total levels of VP6 expression. Intact VP6 trimers were also found in RV-infected cells treated with ML, although in decreased amounts because of the reduced viral replication (Fig. 6B, right). ML, however, appeared to interfere with the formation of the VP6 higher-order structures typically observed in cells overexpressing VP6 in the absence of other RV proteins (24) (Fig. 6C). In immunofluorescence assays, VP6 staining showed tubular structures or aggregates, while in the presence of ML it showed homogeneous distribution (Fig. 6C). In contrast, other structures, such as viroplasm-like structures (VLS), generated by overexpression of NSP5 with NSP2 (VLS-NSP2i) or with VP2 (VLS-VP2i) (24, 25) or with both, were not affected by ML treatment (Fig. 7A and C). Under these conditions, NSP5 remained hyperphosphorylated and VP6 recruitment into VLS was not compromised (Fig. 7B and C).
FIG 7.
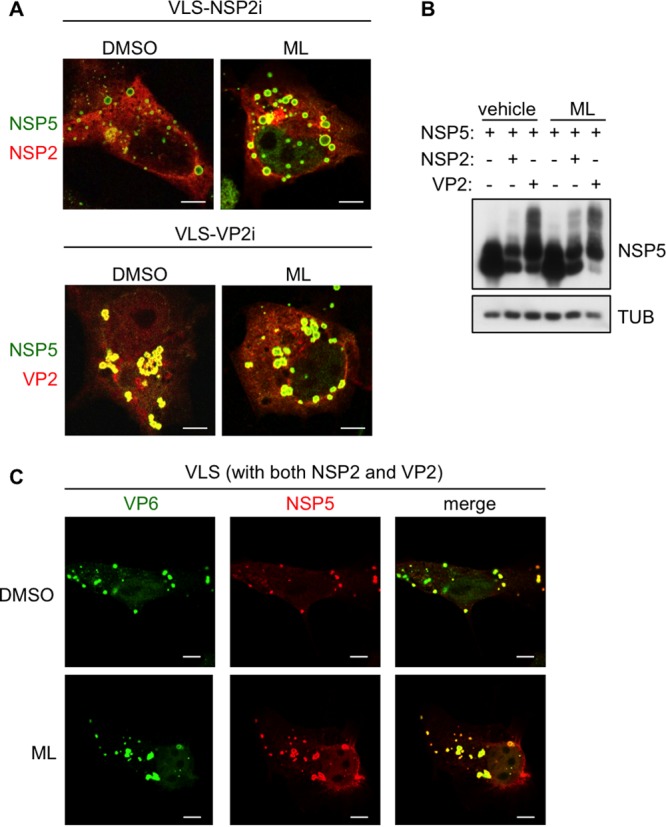
ML effect on VLS. Confocal immunofluorescence assay of VLS (A and C) and Western blot analysis (B) with the indicated antibodies of MA104 cells transfected with NSP5, NSP2, VP2, and VP6, as indicated. (A) NSP5 is shown in green and NSP2 or VP2 in red. (C) NSP5 is shown in red and VP6 in green. Cells were treated for 5 h with 10 μM ML or DMSO at 18 h posttransfection. Bars, 5 μm.
Depending on the pH and ionic strength, purified VP6 self-assembles into helical tubes or spherical particles (26) (Fig. 6D, left). Two different types of tubes can be reconstituted in vitro, characterized by a constant diameter of either 45 or 75 nm with a nonfixed length of several micrometers. On the other hand, VP6 spheres are heterogeneous in size, with diameters varying from 75 to 100 nm (26, 27). In order to evaluate whether ML had an effect on the assembly of these VP6 structures, we performed a negative-staining electron microscopy analysis of purified VP6 in the presence or absence of ML. Upon incubation with 25 μM ML, both VP6 tubes (pH 6.0) and spherical particles (pH 4.0) were severely damaged (Fig. 6D). In fact, a thorough visual inspection of the EM grids failed to reveal any intact tube or sphere in the presence of ML (Fig. 6D, right). Thus, ML has a direct effect on the higher-order interactions of VP6 trimers.
To further study the effect of ML on VP6, the interaction between the protein and the drug was evaluated in nanoscale thermophoresis assays. Upon addition of the inhibitor, concentration-dependent quenching of the labeled protein was observed, which allowed us to calculate a 50% effective concentration (EC50) of 294 ± 62 μM (Fig. 6E). Since millimolar concentrations of divalent cations are known to destabilize the VP6 higher-order structures, shifting the equilibrium toward isolated trimers (26), thermophoresis mobility was also tested in the presence of CaCl2 (up to 500 mM). No significant changes were observed (data not shown). Thus, under the thermophoresis experimental conditions, the signal observed was due to direct binding of ML to VP6 trimers rather than to destabilization of its oligomerization state. Altogether, these results suggest that, although ML binds VP6 trimers with moderate affinity, it likely prevents the optimal packing of its higher-order oligomeric structures on the DLP outer layer. Whether this is due to a conformational change induced by the molecule or to a steric hindrance effect is not known and awaits structural confirmation.
The effects of ML on viroplasms and VP6 strongly indicate that VP6 plays an essential role in the structural integrity of viroplasms. In fact, as reported previously (5), silencing VP6 expression with a specific small interfering RNA (siRNA) led to a significant reduction of viroplasm numbers in RV-infected cells (Fig. 8A), strengthening the hypothesis that VP6 is essential for maintaining the structure of viroplasms. Of note, in ML-treated virus-infected cells, VP6 was found to be diffused throughout the cytosol (Fig. 8B).
FIG 8.

VP6 in RV-infected cells. Shown is confocal immunofluorescence of MA104 cells infected with either OSU or SA11 (MOI, 25 VFU/cell) and transfected with siRNAs specific for SA11 VP6 or OSU VP6 or with a nontargeting siRNA (siNT) (A) or treated with 10 μM ML or DMSO (B). At the indicated times postinfection, viroplasms were visualized with anti-NSP5 antibody (red) and VP6 with MAb 4B2D2 (green). Scale bars, 5 μm.
DISCUSSION
We describe for the first time a compound able to disrupt already formed RV viroplasms in infected cells, thus impairing RV replication, as demonstrated by decreases in viral protein translation, de novo synthesis of viral genomic dsRNA, and yield of progeny virus. This effect of ML on RV replication was initially surprising, because the drug, a cell-permeating indazole-sulfonamide small molecule, has been reported to favor replication of some DNA viruses and bacteria by inhibiting the RNA polymerase III-mediated mechanism that activates the RIG-I pathway (28–30). RV, however, is an RNA virus. We ruled out the possibility that ML inhibition was mediated by RNA polymerase III activity, as actinomycin D, used at a concentration that inhibits RNA polymerase III transcription, still allowed RV replication. In addition, actinomycin D was unable to counteract the ML antiviral effect, indicating that ML antiviral activity does not require synthesis of newly made cellular transcripts, including those of RNA polymerase III. This is consistent with the finding that the ML target is of viral nature. We showed that ML impaired DLP stability in vitro by inhibiting the interactions between VP6 trimers. In the absence of a correctly assembled VP6 layer, DLPs lose the capacity to transcribe plus-strand RNAs (3, 31). These results explain the decreased number of transcripts obtained in vitro from purified DLPs and provide a basis for the reduced amounts of viral proteins and dsRNAs observed in infected cells treated with ML. Altogether, these data indicate that the inhibitor binds VP6, compromising both the integrity of DLPs and the structure of viroplasms.
The mechanism of viroplasm assembly is still not well understood. Viroplasms contain six viral proteins: two nonstructural proteins, NSP5 and NSP2 (both shown to be essential for viroplasm formation [6, 7, 32]), and four structural proteins, VP1, VP2, VP3, and VP6 (33). Electron microscopy studies have suggested that newly made DLPs assemble at the peripheries of viroplasms (34). Immunogold labeling of VP6 indicated that the protein localizes exclusively at the peripheries of viroplasms, while NSP5, NSP2, and VP2 were also detected in the viroplasm interior (F. Arnoldi, C. Gagliani, R. Contin, C. Tacchetti, and O. R. Burrone, unpublished results). Silencing VP6 expression leads to fewer and smaller viroplasms and reduced amounts of viral mRNAs, viral dsRNAs, and viral proteins (5), indicating that the protein is also required for the structural and functional integrity of viroplasms. We confirmed that knocking down VP6 expression produced a sharp reduction in the number of viroplasms (Fig. 8) (5), consistent with the capacity of ML to hamper viroplasms by interaction with VP6. However, it is not clear whether the destabilizing effect on viroplasm structure is the consequence of damaging the VP6 layer in newly assembled DLPs or of impairing VP6 interactions outside the context of DLPs.
Disruption of already assembled viroplasms was particularly evident with OSU and less pronounced with SA11, suggesting that viroplasms may have different stabilities depending on the strain. The decreases of accumulated viral proteins were nevertheless comparable in the two strains and suggest that ML targets a structural region of VP6 that is conserved among different viruses.
Notably, ML did not interfere with the formation of VLS or with recruitment of VP6 into VLS, suggesting that (i) ML does not target the interactions required for VLS assembly (NSP5 with either NSP2 or VP2) or for recruitment of VP6 into VLS, which depends on its interaction with VP2 (24), and (ii) VLS are clearly different from viroplasms, as previously reported (16). The lack of an ML effect on VP2-VP6 interaction was further confirmed by the coimmunoprecipitation experiments.
Interestingly, a strong reduction of NSP5 hyperphosphorylation was observed upon treatment with ML. NSP5 hyperphosphorylation has been associated with RV replication because of its link with viroplasm formation: (i) the two events have been found to be correlated during the course of viral infection (15); (ii) overexpression of NSP5 with either NSP2 or VP2 in uninfected cells induces both NSP5 hyperphosphorylation and VLS formation (13, 24, 25, 35); and (iii) silencing of casein kinase 1-alpha, which affects NSP5 phosphorylation (but does not abolish it entirely), produces viroplasms with irregular shapes (14). Recently, it was shown that at the beginning of infection hypophosphorylated NSP5 interacts with a cytoplasmically dispersed form of NSP2, and it was then proposed that during infection phosphorylation of not only NSP5, but also NSP2 and possibly VP2, leads to viroplasm maturation (11). Our data for cells treated with both ML and the phosphatase inhibitor okadaic acid show for the first time that NSP5 can remain hyperphosphorylated in infected cells in the absence of viroplasms. Such a finding indicates that viroplasms protect NSP5 from dephosphorylation. This result in part supports the model suggested by Criglar et al. (11), which proposes an initial interaction among non- or hypophosphorylated viral proteins. However, although a mechanism of concerted phosphorylation involving several proteins of viroplasms might be necessary for subsequent viroplasm maturation and functioning, our data suggest that NSP5 hyperphosphorylation is necessary, but not sufficient, for viroplasm formation. Thus, treatment of uninfected cells expressing recombinant NSP5 with inhibitors of cellular phosphatases leads to NSP5 hyperphosphorylation, but not to VLS formation (36).
So far, a few types of molecules, such as proteasome inhibitors (MG132 and bortezomib) (16), MT-depolymerizing drugs (nocodazole and vinblastine) (17), an allosteric inhibitor of Eg5 kinesin (monastrol) (17), and thiazolides (18), have been shown to affect RV viroplasms. Proteasome inhibitors impair the formation of new viroplasms without affecting the stability of those already assembled, do not show any inhibitory activity on RV particles in vitro, and do not interfere with the formation of VLS in transfected cells. Although the inhibition mechanism remains obscure, it was hypothesized that the effect was the consequence of blocking proteasome-mediated degradation of an unknown host factor capable of impairing formation of viroplasms (16, 37). Both MT-depolymerizing drugs and Eg5 kinesin inhibitor destabilize the viroplasm structure and coalescence but do not reduce viral protein expression (17). Finally, treatment with thiazolides causes a reduction in viroplasm size, resulting in inhibition of dsRNA genome segment synthesis, but without impairing viral protein expression or affecting the stability of RV particles (18). Importantly, the compound described in this work is the only RV inhibitor so far identified that can disrupt viroplasms and act on a viral target. This makes the chemical structure of ML particularly appealing for further studies in the field of antivirals. As such, ML could not be used as an antiviral drug because of its cytotoxicity. However, future research and structural analyses might lead to the development of more potent and selective inhibitors as potential drugs against RV infections.
MATERIALS AND METHODS
Cells and viruses.
MA104 cells (embryonic African green monkey kidney cells; ATCC CRL-2378) were grown in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies) containing 10% fetal bovine serum (Life Technologies) and 50 μg/ml gentamicin (Biochrom AG). BSC-40 cells (African green monkey kidney epithelial cells; ATCC CRL-2761) were cultured in DMEM supplemented with 10% fetal calf serum (Amimed, Switzerland) and penicillin (100 units/ml)-streptomycin (0.1 mg/ml) (Sigma). Sf9 cells (Spodoptera frugiperda ovary cells; ATCC CRL-1711) were grown in suspension in Sf-900 II SFM medium (Thermo Fisher Scientific) at 27°C.
For RV infection experiments, the porcine OSU (G5; P9[7]) and simian SA11 4F (G3; P6[1]) strains of RV were used; they were propagated in MA104 cells as described previously (38, 39). Virus titers were determined as described by Eichwald et al. (17) and expressed as viroplasm-forming units (VFU) per milliliter. Purified DLPs were obtained by CsCl gradient purification from infected MA104 cells, essentially as described by Patton et al. (40).
For experiments with VLS production and VP6 overexpression in uninfected cells, MA104 cells were infected, respectively, with a T7 RNA polymerase recombinant vaccinia virus (strain VVTF7.3) (41) and with VVT7/LacOI/VP6 virus (VVVP6), an IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible recombinant vaccinia virus driving expression of both the T7 RNA polymerase and VP6 under induction with 1 mM IPTG. For generation of VVVP6, BSC-40 cells were infected with recombinant vaccinia virus VVT7/LacOI (42) and transfected with pVOTE.1-VP6. Selection and amplification were carried out as described by Earl et al. (43). The plasmid pVOTE.1 and VVT7/LacOI were kindly provided by B. Moss.
For the heterologous production of VP6 protein, the recombinant baculovirus BacVP6C (44) was kindly provided by D. Poncet.
Chemicals.
Cells were treated with ML (from 5 to 20 μM; Merck Millipore) following the kinetics indicated in Results above. Actinomycin D (0.1 μg/ml; Sigma), okadaic acid (0.5 μM; Sigma), and cycloheximide (0.1 μg/ml; Sigma) were added to the cells from 3 to 8 hpi. Purified DLPs were treated with ML (from 2.5 to 200 μM, as indicated in Results) or with 200 μM irrelevant compound (number 7749832; ChemBridge Corp.).
Plasmid construction.
The plasmids pcDNA3-NSP5, -NSP2, -VP6, and -VP2 used to overexpress RV proteins in uninfected cells have been described previously (13, 35, 45). The plasmid pVOTE.1-VP6 was obtained by PCR amplification of the VP6 mouse strain EC, using specific primers (5′-ATGCCCATGGATGTGCTGTACTCCATC-3′ and 5′-GATCGGATCCTCACTTTACCAGCATGCTTCT-3′) to incorporate NcoI and BamHI restriction sites at the 5′ and 3′ ends, respectively. The amplified fragment was ligated between NcoI and BamHI restriction sites in pVOTE.1 (42). The sequences of all the primers used in this study for PCR and sequencing are available upon request.
Infections and transient transfections.
Infection experiments with RV were carried out at an MOI of 25 VFU/cell (17). For overexpression of RV proteins in uninfected cells, confluent monolayers of MA104 cells in 12-well plates were infected with VVTF7.3 at an MOI of 10 PFU/cell (41, 46). At 1 hpi, the cells were transfected with a total of 2 μg of DNA plasmid using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer's instructions. At 16 hpi, the cells were washed once to remove serum and then treated with 10 μM ML in serum-free DMEM for 5 h. The cells were then collected for immunofluorescence or Western blot analysis. For VP6 overexpression, confluent monolayers of MA104 cells in 12-well plates were infected with VVVP6 (multiplicity of infection [MOI], 10 PFU/cell). At 1 hpi, both 1 mM IPTG and 10 μM ML were added, and 7 h later, the cells were collected for immunofluorescence or Western blot analysis.
For experiments with siRNAs against VP6 (siVP6-OSU, UGGAACCAUCAUAGCUAGAAA; siVP6-SA11, UGGAACUAUCGUAGCUAGAAA), 5 × 104 MA104 cells per well were seeded into 12-well plates and transfected the next day with 0.1 nmol of annealed duplex siRNA (Sigma) using 5 μl of RNAiMax Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions. The control siRNAs were siNSP5-SA11 and siNSP5-OSU, here referred as siNT and described by Campagna et al. (7). At 48 h posttransfection (hpt), the cells were infected at the same MOI and collected at 6 hpi (for viroplasm counting by immunofluorescence or Western blot analysis).
Cellular extracts (about 3 × 105 cells) were prepared with 50 μl of reducing SDS buffer (125 mM Tris-HCl, pH 6.8, 6% SDS, 40% glycerol, 5% β-mercaptoethanol, 0.04% bromophenol blue) and subsequently sonicated with a VialTweeter (Hielscher Ultrasonics GmbH) for 1 min (10 W; 0.5-s pulse) to disrupt DNA. Typically, 10 μl of cellular extracts was loaded onto an SDS-polyacrylamide gel for Western blot analyses. For VP6 trimer analyses, cellular extracts were prepared in TNN lysis buffer (100 mM Tris-HCl, pH 8, 250 mM NaCl, 0.5% NP-40, and 30 mM N-ethylmaleimide) and centrifuged at 5,000 × g for 5 min at 4°C. For immunoprecipitation assays, cellular extracts were prepared in radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl, 1% Triton X-100, 0.5% sodium deoxycholate, 1 mM EDTA, 150 mM NaCl [pH 7.4]), incubated on ice for 15 min, and then centrifuged at 13,000 × g for 5 min at 4°C.
Immunoprecipitation and protein analyses.
For immunoprecipitation assays, usually 4/5 of the total extract, i.e., approximately 80 μl, was immunoprecipitated for 2 h at 4°C after addition of 100 μl of an undiluted mouse anti-VP6 (clone RV138) monoclonal antibody supernatant (47) (kindly provided by D. Agnello and P. Pothier), 1 μl of a protease inhibitor cocktail (Sigma), and 50 μl of 50% immobilized rProtein A beads (Repligen Bioprocessing) in RIPA buffer. The beads were washed four times with RIPA buffer, followed by one wash with PBS, and resuspended in 20 μl of loading buffer. Proteins were separated on an SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore; IPVH00010) (48). For protein analysis of either cellular extracts or immunoprecipitated proteins, membranes were incubated with the following primary antibodies: anti-NSP5 (1:10,000) (46), anti-VP2 (1:5,000) (35), anti-RV (1:2,500) (17), anti-VP1 (1:5,000) (35), guinea pig sera, anti-NSP3 rabbit serum (1:1,500) (kindly provided by S. López), anti-NSP4 rabbit serum (1:1,1000) (kindly provided by D. Luque), anti-VP5 clone 2G4 mouse monoclonal antibody (1:2,000) (kindly provided by H. Greenberg), anti-alpha-tubulin mouse monoclonal antibody (1:3,000; Calbiochem), and anti-actin rabbit polyclonal antibody (1:1,000; Sigma). The membranes were then incubated with the corresponding horseradish peroxidase (HRP)-conjugated goat anti-guinea pig (1:10,000; Jackson ImmunoResearch), goat anti-mouse (1:5,000; Jackson ImmunoResearch), and goat anti-rabbit (1:5,000; Thermo Scientific Pierce) secondary antibodies. Signals were detected by using the enhanced chemiluminescence system (Pierce ECL Western blotting substrate; Thermo Scientific).
Immunofluorescence microscopy.
Immunofluorescence experiments were performed as described previously (49) using the following secondary-antibody dilutions: anti-NSP5 guinea pig serum, 1:1,000; anti-VP6 mouse monoclonal antibody (clone 4B2D2), 1:1,000 (kindly provided by J. L. Zambrano and F. Liprandi); Alexa Fluor 488-conjugated anti-mouse, 1:500 (Life Technologies), and Alexa Fluor 546-conjugated anti-rabbit, 1:500 (Life Technologies). Newly synthesized RNAs were labeled using 2 mM EU ribonucleotide homolog containing an alkyne-reactive group, and the modified incorporated nucleotide was revealed with an azide-containing fluorophore (Alexa-488; green) as described for the Click-iT RNA Alexa Fluor 488 imaging kit (Thermo Fisher Scientific). Cell nuclei were stained with 2 μg/ml Hoechst 33342 (Molecular Probes, Life Technologies). Samples were analyzed by confocal microscopy (Zeiss LSM510 equipped with a 100×, numerical aperture [NA] 1.3 objective), and the images were processed using LSM Image Examiner 4.0 software.
Analysis of RV genome segments.
Total RNA was purified from MA104 cells infected with OSU (MOI, 25 VFU/cell) after treatment with 10 μM ML or a control vehicle (dimethyl sulfoxide [DMSO]) from 1 to 6 hpi. Crude viral preparations were digested with 10 μg/ml of proteinase K (Thermo Fisher Scientific) in the presence of 5 mM EDTA and 0.5% SDS for 30 min at 60°C. RNA was then purified by phenol-chloroform extraction and precipitation with 5 M ammonium acetate and quantified using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). Total RNA (3.5 μg) was separated using Tris-glycine nondenaturing polyacrylamide (4% stacking gel; 10% resolving gel) and transferred to a charged nylon membrane (GeneScreen Plus hybridization transfer membrane; PerkinElmer). The membrane was blocked with 5% milk and 50 μg/ml DNA fish sperm (Affymetrix) in phosphate-buffered saline (PBS) and incubated overnight with mouse anti-dsRNA (clone J2) monoclonal antibody (1:1,000; English and Scientific Consulting Bt., Hungary) (50), followed by reaction with HRP-conjugated goat anti-mouse antibody (1:5,000; Jackson ImmunoResearch).
Cell viability assay.
MA104 cells (2 × 105) were seeded in 12-well plates. After infection and drug treatment for the indicated times, the supernatants were removed and stored and the cells were trypsinized (0.5% trypsin-EDTA; Gibco, Thermo Fisher Scientific) and then mixed with the supernatants in order not to lose distressed cells that were possibly detached. After centrifugation at 900 × g for 2 min at room temperature (RT), the pellets were resuspended in 200 μl of 0.5-ng/μl propidium iodide in PBS and incubated in the dark for 15 min at RT. Samples were diluted to 1 ml with PBS, filtered using a cell strainer snap-cap tube (BD Falcon), and immediately acquired in a Gallios flow cytometer (Beckman Coulter, Inc.). For this purpose, 10,000 events were acquired, with excitation at 488 nm with an argon laser and a band pass filter of 675/20 nm. The data were analyzed by gating on live cells using Kaluza flow analysis software. Statistical analysis and plotting were performed using Microsoft Excel for Mac 2011 version 14.7.0.
Determination of progeny virus yield.
MA104 cells (2 × 105) seeded in 12-well plates were infected with RV at an MOI of 25 VFU/cell. The virus was allowed to adsorb for 1 h at 4°C, followed by incubation at 37°C. At 2 hpi, the cells were washed twice with PBS, and 10 μM ML or vehicle (2% DMSO) diluted in 500 μl DMEM was added. At the indicated time points, the plates were frozen at −80°C. The cells were then treated with three freeze-thaw cycles, harvested, and centrifuged at 17,000 × g for 5 min at 4°C. The supernatant was recovered and activated with 80 μg/ml of trypsin for 30 min at 37°C. Serial dilutions were prepared and used to determine the viral titers as described by Eichwald et al. (17).
In vitro transcription assays with purified DLPs.
One microgram of purified DLPs was incubated for 4 h at 42°C in a total volume of 100 μl of 1× T7 transcription buffer (Promega), 2 mM each nucleoside triphosphate (NTP), 0.5 mM S-adenosylmethionine (SAM), 0.1 mM MnCl2, 1 mM dithiothreitol (DTT), 0.4 U RNAsin (Promega). RNA was isolated using the GeneJet RNA cleanup and concentration microkit (Thermo Fisher Scientific) and quantified using Qubit RNA assay kits associated with a Qubit 2.0 fluorometer (Thermo Fisher Scientific). The statistical analyses were performed using Student's t test by comparing vehicle- and ML-treated particles of each viral strain.
Production of recombinant baculovirus VP6.
An Sf9 suspension culture (1 × 106 cells/ml) was infected with the recombinant baculovirus BacVP6C (27). At 3 days postinfection, cells were harvested by centrifugation at 180 × g for 20 min at 4°C. The cellular pellet was resuspended in 10 ml of 20 mM MOPS (morpholinepropanesulfonic acid) buffer (pH 6.8), followed by dilution in 1 volume of trichlorofluoromethane (Sigma). The sample was vortexed for 1 min and centrifuged at 4,500 × g for 5 min at 4°C. The aqueous phase was transferred to a new tube, and this step was repeated twice. The sample was then ultracentrifuged at 100,000 × g with a Beckman SW70.2 rotor for 3 h at 4°C. The pellet was resuspended in 3 ml of 50 mM Tris-HCl, pH 8.0, 150 mM CaCl2, 200 mM NaCl to promote formation of isolated VP6 trimers. The trimeric VP6 was further purified by size exclusion chromatography (Superdex200 16/60 equipped with an AKTA system) and stored at 4°C.
Transmission electron microscopy.
For detection of viroplasms, MA104 cells were seeded at 1 × 105 cells in a 2-cm2 well onto sapphire discs and infected with OSU (MOI, 100 VFU/cell) (17). At 1 hpi, 20 μM ML was added. At 6 hpi, the cells were fixed with 2.5% glutaraldehyde in 100 mM Na/K phosphate buffer, pH 7.4, for 1 h at 4°C and kept in 100 mM Na/K phosphate buffer overnight at 4°C. Afterward, samples were postfixed in 1% osmium tetroxide in 100 mM Na/K phosphate buffer for 1 h at 4°C and dehydrated in a graded ethanol series starting at 70%, followed by two changes in acetone, and embedded in Epon. Ultrathin sections (60 to 80 nm) were cut and stained with uranyl acetate and lead citrate. For EM analyses of purified DLPs incubated with ML for 4 h at 42°C (in the same buffer used in in vitro transcription assays), the DLPs were adsorbed for 10 min on glow-discharged carbon-coated Parlodion films mounted on 300 mesh per inch copper grids (Electron Microscopy Science, Hatfield, PA, USA). Samples were washed once with distilled water and stained with saturated uranyl acetate (Fluka) for 1 min at RT. The samples were analyzed in a transmission electron microscope (CM12; Philips, Eindhoven, The Netherlands) equipped with a charge-coupled-device (CCD) camera (Ultrascan 1000; Gatan, Pleasanton, CA, USA) at an acceleration of 100 kV. For negative-staining EM experiments with purified VP6, aliquots of trimeric VP6 (0.5 mg/ml) were dialyzed overnight at 4°C against either 50 mM MOPS, pH 6.0, 50 mM NaCl, or 50 mM sodium acetate, pH 4.0, to reconstitute VP6 nanotubes and spheres. After dialysis, VP6 samples were recovered and incubated in the presence or absence of 25 μM ML for 4 h at 37°C (1% DMSO under both conditions). Prior to sample application, the 400-mesh copper carbon-coated grids (Agar Scientific) were glow discharged for 30 s at 30 mA using a GloQube system (Quorum Technologies). A 4-μl drop of the incubated VP6 samples at a final concentration of 0.25 mg/ml was applied to the glow-discharged grid and incubated for 1 min. The grid was immediately stained by gentle stirring for 1 min using 2% (wt/vol) uranyl acetate solution. After staining, the grid was blotted dry and imaged on a Tecnai G2 T20 LaB6 transmission electron microscope (FEI) operating at 200 keV. Images were manually acquired on an Eagle 2K CCD camera (FEI) at a nominal magnification of ×50,000 (corresponding to a pixel size of 4.55 Å at the specimen level) and defocus values in the range of 1.5 μm to 2.5 μm.
Microscale thermophoresis.
Thermophoresis was used to determine the binding affinities between recombinant purified VP6 and ML. Experiments were performed in standard capillaries using a Monolith NT.115 instrument (NanoTemper Technologies). Either cysteines or lysines of VP6 were labeled with maleimide or N-hydroxysuccinimide (NHS) dye, respectively, following the manufacturer's protocol. The labeled VP6 (100 nM) was incubated at 37°C for 4 h with increasing concentrations of ML (0 to 106 nM) in MST buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 0.05% Tween 20) supplemented with 10% DMSO. Thereafter, measurements were performed at 24°C using 20% LED power (intensity of the incident light) and 20% MST power (intensity of the infrared laser). The addition of ML causes concentration-dependent quenching of the fluorescent protein. Titration of boiled VP6 in the presence of 4% sodium dodecyl sulfate (SD test) confirmed specificity of the binding. The EC50 was obtained using the Hill equation, which describes cooperative binding, and MO. Affinity Analysis software (NanoTemper Technologies). The reported affinity value is the average of the results of three independent experiments.
ACKNOWLEDGMENTS
We are grateful to Didier Poncet (Institut de Biologie Intégrative de la Cellule, UMR CNRS, CEA, University of Paris XI, Paris, France) for providing us with BacVP6C, to Bernard Moss (National Institutes of Health, Bethesda, MD, USA) for the pVOTE.1 plasmid and the VVT7/LacOI virus, to Davide Agnello and Pierre Pothier (CNR des Virus des Gastroentérites, CHU de Dijon, Dijon, France) for the mouse anti-VP6 (clone RV138) monoclonal antibody, to Jose Luis Zambrano and Ferdinando Liprandi (Instituto Venezolano de Investigaciones Científicas, Caracas, Venezuela) for providing us with the mouse anti-VP6 (clone 4B2D2) monoclonal antibody, to Susana López (Instituto de Biotecnología, UNAM, Cuernavaca, Mexico) for the rabbit anti-NSP3 antibody, to Daniel Luque (Centro Nacional de Microbiología, ISCIII, Madrid, Spain) for the rabbit anti-NSP4 antibody, and to Harry Greenberg (Stanford University, Stanford, CA, USA) for the mouse anti-VP5 monoclonal antibody (clone 2G4). We thank Peter Wild for his advice and critical reading of the manuscript.
Most of the experiments in this study and the work of F.A. and M.B. were supported by a FIRB-Futuro in Ricerca grant (RBFR13209E) funded by the Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR), Italy. Also, this work was supported by a private donation from the late Robert Wyler to M.A. (F-52601-10-01), as well as by general funds allocated to M.A. by the University of Zurich. G.D.L. and G.P. were supported by an ICGEB predoctoral fellowship. The work of M.D.R., E.M., and M.M. was supported by PRIN 2012 NOXSS (X-ray Single Shots of Nano-Objects), MIUR prot. 2012Z3N9R9.
REFERENCES
- 1.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, World Health Organization-Coordinated Global Rotavirus Surveillance Network . 2016. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis 62(Suppl 2):S96–S105. doi: 10.1093/cid/civ1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desselberger U. 2014. Rotaviruses. Virus Res 190:75–96. doi: 10.1016/j.virusres.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 3.Bican P, Cohen J, Charpilienne A, Scherrer R. 1982. Purification and characterization of bovine rotavirus cores. J Virol 43:1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohli E, Pothier P, Tosser G, Cohen J, Sandino AM, Spencer E. 1993. In vitro reconstitution of rotavirus transcriptional activity using viral cores and recombinant baculovirus expressed VP6. Arch Virol 133:451–458. doi: 10.1007/BF01313782. [DOI] [PubMed] [Google Scholar]
- 5.Ayala-Breton C, Arias M, Espinosa R, Romero P, Arias CF, Lopez S. 2009. Analysis of the kinetics of transcription and replication of the rotavirus genome by RNA interference. J Virol 83:8819–8831. doi: 10.1128/JVI.02308-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez T, Rojas M, Ayala-Breton C, Lopez S, Arias CF. 2005. Reduced expression of the rotavirus NSP5 gene has a pleiotropic effect on virus replication. J Gen Virol 86:1609–1617. doi: 10.1099/vir.0.80827-0. [DOI] [PubMed] [Google Scholar]
- 7.Campagna M, Eichwald C, Vascotto F, Burrone OR. 2005. RNA interference of rotavirus segment 11 mRNA reveals the essential role of NSP5 in the virus replicative cycle. J Gen Virol 86:1481–1487. doi: 10.1099/vir.0.80598-0. [DOI] [PubMed] [Google Scholar]
- 8.Patton JT, Silvestri LS, Tortorici MA, Vasquez-Del Carpio R, Taraporewala ZF. 2006. Rotavirus genome replication and morphogenesis: role of the viroplasm. Curr Top Microbiol Immunol 309:169–187. [DOI] [PubMed] [Google Scholar]
- 9.Vascotto F, Campagna M, Visintin M, Cattaneo A, Burrone OR. 2004. Effects of intrabodies specific for rotavirus NSP5 during the virus replicative cycle. J Gen Virol 85:3285–3290. doi: 10.1099/vir.0.80075-0. [DOI] [PubMed] [Google Scholar]
- 10.Afrikanova I, Miozzo MC, Giambiagi S, Burrone O. 1996. Phosphorylation generates different forms of rotavirus NSP5. J Gen Virol 77:2059–2065. doi: 10.1099/0022-1317-77-9-2059. [DOI] [PubMed] [Google Scholar]
- 11.Criglar JM, Hu L, Crawford SE, Hyser JM, Broughman JR, Prasad BV, Estes MK. 2014. A novel form of rotavirus NSP2 and phosphorylation-dependent NSP2-NSP5 interactions are associated with viroplasm assembly. J Virol 88:786–798. doi: 10.1128/JVI.03022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poncet D, Aponte C, Cohen J. 1996. Structure and function of rotavirus nonstructural protein NSP3. Arch Virol Suppl 12:29–35. [DOI] [PubMed] [Google Scholar]
- 13.Afrikanova I, Fabbretti E, Miozzo MC, Burrone OR. 1998. Rotavirus NSP5 phosphorylation is up-regulated by interaction with NSP2. J Gen Virol 79:2679–2686. doi: 10.1099/0022-1317-79-11-2679. [DOI] [PubMed] [Google Scholar]
- 14.Campagna M, Budini M, Arnoldi F, Desselberger U, Allende JE, Burrone OR. 2007. Impaired hyperphosphorylation of rotavirus NSP5 in cells depleted of casein kinase 1alpha is associated with the formation of viroplasms with altered morphology and a moderate decrease in virus replication. J Gen Virol 88:2800–2810. doi: 10.1099/vir.0.82922-0. [DOI] [PubMed] [Google Scholar]
- 15.Poncet D, Lindenbaum P, L'Haridon R, Cohen J. 1997. In vivo and in vitro phosphorylation of rotavirus NSP5 correlates with its localization in viroplasms. J Virol 71:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contin R, Arnoldi F, Mano M, Burrone OR. 2011. Rotavirus replication requires a functional proteasome for effective assembly of viroplasms. J Virol 85:2781–2792. doi: 10.1128/JVI.01631-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eichwald C, Arnoldi F, Laimbacher AS, Schraner EM, Fraefel C, Wild P, Burrone OR, Ackermann M. 2012. Rotavirus viroplasm fusion and perinuclear localization are dynamic processes requiring stabilized microtubules. PLoS One 7:e47947. doi: 10.1371/journal.pone.0047947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.La Frazia S, Ciucci A, Arnoldi F, Coira M, Gianferretti P, Angelini M, Belardo G, Burrone OR, Rossignol JF, Santoro MG. 2013. Thiazolides, a new class of antiviral agents effective against rotavirus infection, target viral morphogenesis, inhibiting viroplasm formation. J Virol 87:11096–11106. doi: 10.1128/JVI.01213-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smee DF, Sidwell RW, Clark SM, Barnett BB, Spendlove RS. 1982. Inhibition of rotaviruses by selected antiviral substances: mechanisms of viral inhibition and in vivo activity. Antimicrob Agents Chemother 21:66–73. doi: 10.1128/AAC.21.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Pan J, Thoroddsen V, Wysong DR, Blackman RK, Bulawa CE, Gould AE, Ocain TD, Dick LR, Errada P, Dorr PK, Parkinson T, Wood T, Kornitzer D, Weissman Z, Willis IM, McGovern K. 2003. Novel small-molecule inhibitors of RNA polymerase III. Eukaryot Cell 2:256–264. doi: 10.1128/EC.2.2.256-264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perry RP, Kelley DE. 1970. Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species. J Cell Physiol 76:127–139. doi: 10.1002/jcp.1040760202. [DOI] [PubMed] [Google Scholar]
- 22.Song W, Filonov GS, Kim H, Hirsch M, Li X, Moon JD, Jaffrey SR. 2017. Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex. Nat Chem Biol 13:1187–1194. doi: 10.1038/nchembio.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sabara M, Ready KF, Frenchick PJ, Babiuk LA. 1987. Biochemical evidence for the oligomeric arrangement of bovine rotavirus nucleocapsid protein and its possible significance in the immunogenicity of this protein. J Gen Virol 68:123–133. doi: 10.1099/0022-1317-68-1-123. [DOI] [PubMed] [Google Scholar]
- 24.Contin R, Arnoldi F, Campagna M, Burrone OR. 2010. Rotavirus NSP5 orchestrates recruitment of viroplasmic proteins. J Gen Virol 91:1782–1793. doi: 10.1099/vir.0.019133-0. [DOI] [PubMed] [Google Scholar]
- 25.Fabbretti E, Afrikanova I, Vascotto F, Burrone OR. 1999. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J Gen Virol 80:333–339. doi: 10.1099/0022-1317-80-2-333. [DOI] [PubMed] [Google Scholar]
- 26.Lepault J, Petitpas I, Erk I, Navaza J, Bigot D, Dona M, Vachette P, Cohen J, Rey FA. 2001. Structural polymorphism of the major capsid protein of rotavirus. EMBO J 20:1498–1507. doi: 10.1093/emboj/20.7.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petitpas I, Lepault J, Vachette P, Charpilienne A, Mathieu M, Kohli E, Pothier P, Cohen J, Rey FA. 1998. Crystallization and preliminary X-Ray analysis of rotavirus protein VP6. J Virol 72:7615–7619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. 2009. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol 10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiu YH, Macmillan JB, Chen ZJ. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karijolich J, Abernathy E, Glaunsinger BA. 2015. Infection-induced retrotransposon-derived noncoding RNAs enhance herpesviral gene expression via the NF-kappaB pathway. PLoS Pathog 11:e1005260. doi: 10.1371/journal.ppat.1005260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charpilienne A, Lepault J, Rey F, Cohen J. 2002. Identification of rotavirus VP6 residues located at the interface with VP2 that are essential for capsid assembly and transcriptase activity. J Virol 76:7822–7831. doi: 10.1128/JVI.76.15.7822-7831.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silvestri LS, Taraporewala ZF, Patton JT. 2004. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J Virol 78:7763–7774. doi: 10.1128/JVI.78.14.7763-7774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrie BL, Greenberg HB, Graham DY, Estes MK. 1984. Ultrastructural localization of rotavirus antigens using colloidal gold. Virus Res 1:133–152. doi: 10.1016/0168-1702(84)90069-8. [DOI] [PubMed] [Google Scholar]
- 34.Cuadras MA, Bordier BB, Zambrano JL, Ludert JE, Greenberg HB. 2006. Dissecting rotavirus particle-raft interaction with small interfering RNAs: insights into rotavirus transit through the secretory pathway. J Virol 80:3935–3946. doi: 10.1128/JVI.80.8.3935-3946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnoldi F, Campagna M, Eichwald C, Desselberger U, Burrone OR. 2007. Interaction of rotavirus polymerase VP1 with nonstructural protein NSP5 is stronger than that with NSP2. J Virol 81:2128–2137. doi: 10.1128/JVI.01494-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blackhall J, Munoz M, Fuentes A, Magnusson G. 1998. Analysis of rotavirus nonstructural protein NSP5 phosphorylation. J Virol 72:6398–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez T, Silva-Ayala D, Lopez S, Arias CF. 2011. Replication of the rotavirus genome requires an active ubiquitin-proteasome system. J Virol 85:11964–11971. doi: 10.1128/JVI.05286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Estes MK, Graham DY, Gerba CP, Smith EM. 1979. Simian rotavirus SA11 replication in cell cultures. J Virol 31:810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graham A, Kudesia G, Allen AM, Desselberger U. 1987. Reassortment of human rotavirus possessing genome rearrangements with bovine rotavirus: evidence for host cell selection. J Gen Virol 68:115–122. doi: 10.1099/0022-1317-68-1-115. [DOI] [PubMed] [Google Scholar]
- 40.Patton J, Chizhikov V, Taraporewala Z, Chen DY. 2000. Virus replication, p 33–66. In Gray J, Desselberger U (ed), Rotaviruses methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 41.Fuerst TR, Moss B. 1989. Structure and stability of mRNA synthesized by vaccinia virus-encoded bacteriophage T7 RNA polymerase in mammalian cells. Importance of the 5′ untranslated leader. J Mol Biol 206:333–348. [DOI] [PubMed] [Google Scholar]
- 42.Ward GA, Stover CK, Moss B, Fuerst TR. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc Natl Acad Sci U S A 92:6773–6777. doi: 10.1073/pnas.92.15.6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Earl PL, Moss B, Wyatt LS, Carroll MW. 2001. Generation of recombinant vaccinia viruses. Curr Protoc Mol Biol Chapter 16:Units 16 17. [DOI] [PubMed] [Google Scholar]
- 44.Tosser G, Labbe M, Bremont M, Cohen J. 1992. Expression of the major capsid protein VP6 of group C rotavirus and synthesis of chimeric single-shelled particles by using recombinant baculoviruses. J Virol 66:5825–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Lorenzo G, Eichwald C, Schraner EM, Nicolin V, Bortul R, Mano M, Burrone OR, Arnoldi F. 2012. Production of in vivo-biotinylated rotavirus particles. J Gen Virol 93:1474–1482. doi: 10.1099/vir.0.040089-0. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez SA, Burrone OR. 1991. Rotavirus NS26 is modified by addition of single O-linked residues of N-acetylglucosamine. Virology 182:8–16. doi: 10.1016/0042-6822(91)90642-O. [DOI] [PubMed] [Google Scholar]
- 47.Kohli E, Maurice L, Vautherot JF, Bourgeois C, Bour JB, Cohen J, Pothier P. 1992. Localization of group-specific epitopes on the major capsid protein of group A rotavirus. J Gen Virol 73:907–914. doi: 10.1099/0022-1317-73-4-907. [DOI] [PubMed] [Google Scholar]
- 48.Eichwald C, Jacob G, Muszynski B, Allende JE, Burrone OR. 2004. Uncoupling substrate and activation functions of rotavirus NSP5: phosphorylation of Ser-67 by casein kinase 1 is essential for hyperphosphorylation. Proc Natl Acad Sci U S A 101:16304–16309. doi: 10.1073/pnas.0406691101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eichwald C, Vascotto F, Fabbretti E, Burrone OR. 2002. Rotavirus NSP5: mapping phosphorylation sites and kinase activation and viroplasm localization domains. J Virol 76:3461–3470. doi: 10.1128/JVI.76.7.3461-3470.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeWitte-Orr SJ, Mehta DR, Collins SE, Suthar MS, Gale M Jr, Mossman KL. 2009. Long double-stranded RNA induces an antiviral response independent of IFN regulatory factor 3, IFN-beta promoter stimulator 1, and IFN. J Immunol 183:6545–6553. doi: 10.4049/jimmunol.0900867. [DOI] [PMC free article] [PubMed] [Google Scholar]