

Abstract
OBJECTIVE: The purpose of this study was to evaluate the safety and efficacy of ingenol disoxate gel using a once-daily, three-day field treatment regimen in patients with actinic keratosis. DESIGN: This was a Phase II, multicenter, open-label trial (clinicaltrials.gov: NCT02305888). SETTING: The study was conducted in 20 trial sites in the United States. PARTICIPANTS: Participants included patients with 5 to 20 clinically typical actinic keratosis lesions on the full face/chest (250cm2), scalp (25–250cm2), or the trunk/extremities (250cm2). MEASUREMENTS: We measured incidence of dose-limiting events based on local skin responses. Percentage reduction in actinic keratosis lesion count from baseline, complete clearance, and partial clearance (≥75%) of actinic keratosis lesions were assessed at Week 8. RESULTS: Nine of 63 (14.3%) patients in the face/chest group reported dose-limiting events; zero of 63 patients in the scalp group reported dose-limiting events; and 11 of 62 (17.7%) patients in the trunk/extremities group reported dose-limiting events. Mean composite local skin response scores peaked at Day 4, then rapidly declined, reaching or approaching baseline levels by Week 4. Less than five percent of patients reported severe adverse events; the most common treatment-related adverse events were application site pain and pruritus. The reduction in actinic keratosis lesion count was 78.9, 76.3, and 69.1 percent for the face/chest, scalp, and trunk/extremities groups, respectively. Complete clearance was achieved in 36.5, 39.7, and 22.6 percent of patients in the face/chest, scalp, and trunk/extremities groups, respectively. Partial clearance was achieved in 71.4, 65.1, and 50.0 percent of patients in the face/chest, scalp, and trunk/extremities groups, respectively. CONCLUSION: Ingenol disoxate demonstrated adverse events and local skin reaction profiles similar to results seen in trials evaluating shorter two-day regimens and was effective in patients with actinic keratosis. These data support the use of ingenol disoxate gel for actinic keratosis field treatment.
Keywords: Actinic keratosis, ingenol disoxate, safety, efficacy, field treatment
Actinic keratosis (AK) is a common skin condition visibly characterized by thickened, cornified, scaly lesions and histologically characterized by atypical epithelial proliferation.1 AK lesions usually develop on areas of the body that are frequently exposed to the sun (e.g., the face, ears, scalp, neck, forearms, and the back of the hands).2 AK typically arises in people with fair skin (i.e., in those with Fitzpatrick Types I or II skin).3 Reported prevalence rates for AK in Europe and the United States are approximately 11 to 25 percent of the population, while estimates in Australia have been reported to be up to 60 percent.4
Evidence suggests that AK lesions are on a continuum with in situ and invasive squamous cell carcinoma (SCC)5 and, if left untreated, can develop into SCC, resulting in increased morbidity.6 Sun-damaged areas of skin can contain fields of cancerization, which are characterized by multiple subclinical and clinically visible AK lesions, along with the presence of multifocal pre-neoplastic changes and genetic mutations.7 Subclinical AK lesions can progress into clinically visible lesions and/or SCC and new subclinical AK lesions can develop, warranting therapies that treat the entire cancerized field.6 Invasive SCC can develop from lesions that are limited to the epidermal basal layer (AK 1) or in a stepwise progression along the classical pathway.8 This is corroborated by histological data reporting that the severity of dysplasia or expression of tumor protein p53 is not predicted by lesion thickness,9 suggesting that all AKs are equally invasive and should be treated whether subclinical or clinical.
Previous trials with ingenol mebutate gel (Picato®; LEO Pharma, Ballerup, Denmark) have demonstrated that treatment over a small area of 25cm2 provides a high rate of complete clearance as compared with vehicle gel.10 In addition, ingenol mebutate has a favorable safety profile demonstrated by local skin responses (LSR) that return to baseline levels by Week 4 and mild-to-moderate adverse events (AEs) that resolve without sequelae. However, there are patients who require treatment over an area of field cancerization greater than 25cm2.
Ingenol disoxate (LEO 43204) gel is a novel ingenol derivative in development for the field treatment of AK; this compound builds on the established chemical, efficacy, and safety profiles of ingenol mebutate. The biological activity of the mebutate ester is provided by hydroxyl groups in the four-, five-, and 20-positions; however, these groups also reduce chemical thermostability and increase the risk of pH-dependent degradation through acyl migration. The physicochemical characteristics of the disoxate ester, including a reduced migration ability of the ester moieties on the aforementioned hydroxyl groups, improved stability in comparison with ingenol mebutate.11 Furthermore, the disoxate ester has been shown to have increased cytotoxic potency to that of the mebutate derivative.11 Preclinical data suggest that the mode of action of ingenol disoxate involves direct cellular cytotoxicity and the ability to induce proinflammatory mediators.11 As compared with ingenol mebutate 0.05% gel, ingenol disoxate has previously demonstrated a similar safety profile and a dose response relationship for LSRs. In addition, ingenol disoxate 0.075% gel significantly reduced the AK lesion count as compared with ingenol mebutate (p<0.04).12 Previous Phase I/II dose-finding trials of ingenol disoxate on the face, chest, and scalp(25–250cm2) have demonstrated a greater efficacy when compared with vehicle and a well-defined LSR profile, which was associated with high treatment satisfaction.13,14
The aims of this trial were to evaluate the safety, tolerability, and efficacy of ingenol disoxate gel in a once-daily regimen for three consecutive days, following application to the face or chest, scalp, or trunk or extremities. Cosmetic and patient satisfaction outcomes from this trial are presented in the accompanying paper.15
METHODS
Trial deslgn. This was a Phase II, multicenter, open-label, eight-week trial evaluating the safety and efficacy of ingenol disoxate (LEO 43204) applied once daily for three consecutive days on either the face/chest, scalp, or on the trunk/extremities (NCT02305888). The trial consisted of three periods: a 35-day screening period, a three-day treatment period, and an eight-week follow-up period (Figure 1). The protocol was approved by the appropriate independent ethics committees and institutional review boards and conformed to the ethical principles of the Declaration of Helsinki, the International Conference on Harmonisation and Good Clinical Practice, and all applicable regulatory requirements. All patients provided informed written consent.
FIGURE 1.

Trial design—Times shown are representative of the window within which each visit occurred: screening took place within 35 days of Visit 2; Visits 2 and 3 occurred on Days 1 and 4, respectively; Visits 4 and 5 were conducted at Day 8 and Week 2, respectively ±2 days; Visit 6 occurred at Week 4 ±4 days; Visit 7 was at Week 8 ±7 days.
*Represents actual sample size—one patient in the trunk and extremities group was excluded from the full analysis set because of incorrect dose assignment.
AK: actinic keratosis; LSR: local skin response
Inclusion and exclusion criteria. Patients aged 18 years or older with 5 to 20 clinically typical, visible, discrete AKs within a selected treatment area of sun-damaged skin either on the full face or a contiguous area of approximately 250cm2 on their chest, exposed scalp (25–250cm2), or a contiguous area of approximately 250cm2 on their trunk or extremities were included. Patients could also have visible and discrete hyperkeratotic or hypertrophic lesions in this area; these lesions were marked on a transparency and each lesion was followed separately. Patients were excluded from the trial if the selected treatment area was within 5cm of an incompletely healed wound, a suspected basal cell carcinoma, or a suspected SCC. In addition, patients were excluded if they had received prior treatment of the selected area with ingenol mebutate gel within the last 12 months, had atypical nonresponsive lesions (i.e., did not respond to cryotherapy on two occasions), or had a history of skin conditions other than AK (e.g., eczema, unstable psoriasis, or xeroderma pigmentosum).
Treatment. Patients received ingenol disoxate gel at the following concentrations dependent upon the region to be treated: face/chest group at 0.018%; scalp group at 0.037%; and trunk/extremities group at 0.1%. On Day 1, the gel was applied under the supervision of trained research staff and, on Days 2 and 3, the patients self-applied the treatment at home. Patients were assigned to treatment groups via an interactive web response system. If a patient fulfilled the requirement for more than one treatment area, the following order of assignment was used: 1) trunk/extremities; 2) scalp; and 3) face/chest.
Objectives and endpoints. The primary and secondary objectives of the trial were to evaluate the safety and efficacy of ingenol disoxate after a once-daily field treatment for three consecutive days, respectively.
Safety endpoints (i.e., LSRs and AEs) were assessed on Days 1, 4, and 8, and Weeks 2, 4, and 8. The individual LSR components consisted of erythema, flaking/scaling, crusting, swelling, vesiculation/pustulation, and erosion/ulceration. Each was graded on a scale of 0 to 4 (with higher numbers indicating greater severity), giving a maximum possible composite score of 24.16
The primary endpoint of the trial was the number of patients who had dose-limiting events (DLEs) based on LSRs up to and including Day 8. DLEs were recorded according to predefined definitions (Table 1).
TABLE 1.
Defnition of dose-limiting events based on local skin responses
| TREATMENT AREA | LOCAL SKIN RESPONSES |
|---|---|
| Face/chest | At least one of the following LSRs: crusting Grade 4, erosion/ulceration Grade 4, vesiculation/postulation Grade 4; or at least two of the following LSRs: erythema Grade 4, crusting Grade 3, swelling Grade 4, erosion/ulceration Grade 3, vesiculation/postulation Grade 3 |
| Scalp | Erosion/ulceration Grade 4 |
| Trunk/extremities | At least one of the following LSRs: crusting Grade 4, erosion/ulceration Grade 4, vesiculation/postulation Grade 4; or at least two of the following LSRs: crusting Grade 3, swelling Grade 4, erosion/ulceration Grade 3, vesiculation/postulation Grade 3 |
LSR: local skin response
The secondary endpoints of the trial were reduction in AK lesion count, analyzed separately for nonhyperkeratotic/hypertrophic lesions and for hyperkeratotic/hypertrophic lesions, and complete clearance of AK lesions (AKCLEAR 100), excluding hyperkeratotic/hypertrophic lesions. An additional endpoint was partial clearance of AKs (AKCLEAR 75), defined as 75-percent reduction or more in baseline AK lesion count, excluding hyperkeratotic/hypertrophic lesions. AKCLEAR 100 and AKCLEAR 75 have been introduced as more descriptive terminology for the frequently used clinical endpoints of complete and partial clearance of AKs in a field.13,14 AKCLEAR 100 was also analyzed in hyperkeratotic/hypertrophic lesions. Efficacy endpoints were assessed by AK lesion count at Week 4 and Week 8.
Cosmetic and patient-reported outcomes including the Treatment Satisfaction Questionnaire for Medication (TSQM),17 a cosmetic outcome questionnaire, and a global photo damage outcome assessment were also assessed at Week 8 (detailed analysis reported separately in Berman et al15 ).
Statistical methods. A formal sample size calculation was not performed for the initial part of the trial (for the initial 18 patients); however, a sample size was selected to determine an adequate precision rate of the DLEs. The numbers of subjects experiencing DLEs up to and including Day 8 were tabulated for the first 18 patients recruited in each treatment group and the results were presented for the Early Data Review Committee (Interim analysis). The sample size (n=62, including the initial 18 patients) for the latter part of the trial was selected to obtain a precision rate with respect to the secondary endpoints (i.e., reduction in AK lesion count and AKCLEAR 100) and as per previous trials.13 The ratio of number of AK lesions at Week 8 relative to baseline was analyzed separately for each treatment group, using a negative binomial regression on the AK lesion count at Week 8 with the log baseline value as an offset variable. The estimated percent reduction in AK lesion count and a corresponding 95-percent confidence interval (CI) are presented. For the analysis of AK lesion count at Week 8 and Week 4, missing values were imputed using last observation carried forward.
RESULTS
Trial population. A total of 253 patients from 20 trial sites in the United States were enrolled in the trial. Of those, 189 were assigned to treatment groups. In total, 188 patients were included in the full analysis set and safety analysis set; one patient was excluded due to an incorrect dose assignment (face/chest, n=63, scalp; n=63; and trunk/extremities, n=62; Table 2, Figure 2). The majority of patients in the trial were male (face/chest, 63.5%; scalp, 98.4%; trunk/extremities, 62.9%) and had a mean (standard deviation [SD]) age of 64.0 (8.8) years in the face/chest group, 67.7 (9.6) years in the scalp group, and 66.7 (9.6) years in the trunk/extremities group. The median duration of AK was seven years for the face/chest group, 10 years for the scalp group, and seven years for the trunk/extremities group (Table 2). Nine patients in the face/chest group, 10 patients in the scalp group, and 16 patients in the trunk/extremities group had hypertrophic/hyperkeratotic lesions at baseline. The majority of patients completed the eight-week follow-up, except one patient in the scalp group who withdrew due to an adverse event (AE) and one patient in the trunk/extremities group who was lost to follow-up.
TABLE 2.
Baseline patient characteristics (full analysis set)
| CHARACTERISTIC | INGENOL DISOXATE GEL | ||
|---|---|---|---|
| FACE/CHEST 0.018% (n=63) |
SCALP 0.037% (n=63) |
TRUNK/EXTREMITIES 0.1% (n=62) |
|
| Sex, n (%) | |||
| Male | 40 (63.5%) | 62 (98.4%) | 39 (62.9%) |
| Female | 23 (36.5%) | 1 (1.6%) | 23 (37.1%) |
| Age, mean (SD) | 64.0 years (8.8 years) | 67.7 years (9.6 years) | 66.7 years (9.6 years) |
| Race, n (%) | |||
| White | 62 (98.4%) | 62 (98.4%) | 62 (100.0%) |
| American Indian/Alaska native | 1 (1.6%) | 0 | 0 |
| Other | 0 | 1 (1.6) | 0 |
| Fitzpatrick skin type, n (%) | |||
| I (burns easily, never tans) | 8 (12.7%) | 5 (7.9%) | 12 (19.4%) |
| II (burns easily, tans minimally) | 30 (47.6%) | 25 (39.7%) | 36 (58.1%) |
| III (burns moderately, tans gradually) | 22 (34.9%) | 30 (47.6%) | 12 (19.4%) |
| IV (burns minimally, always tans well) | 3 (4.8%) | 3 (4.8%) | 2 (3.2%) |
| Median duration of AK (range) | 7 years (0-40 years) | 10 years (0-36 years) | 7 years (0-32 years) |
| AK lesion count,* median (range) | 10 years (5-20 years) | 11 years (6-20 years) | 11 years (5-20 years) |
| Hyperkeratotic/hypertrophic lesions, n (%) | |||
| 0 lesions | 54 (85.7%) | 53 (84.1%) | 46 (74.2%) |
| 1-2 lesions | 7 (11.1%) | 7 (11.1%) | 7 (11.3%) |
| ≥3 lesions | 2 (3.2%) | 3 (4.8%) | 9 (14.5%) |
| Previously treated for AK, n (%) | 48 (76.2%) | 52 (82.5%) | 56 (90.3%) |
| Non-melanoma skin cancer history, n (%) | 24 (38.1%) | 22 (34.9%) | 20 (32.3%) |
excluding hyperkeratotic or hypertrophic lesions
AK: actinic keratosis; SD: standard deviation
FIGURE 2.

Patient disposition
Safety. A total of nine patients (14.3%) reported DLEs in the face/chest group, no DLEs were reported in the scalp treatment group, and 11 patients (17.7%) reported DLEs in the trunk/extremities group. Mean composite LSR scores peaked at Day 4 with values of 10.5 for the face/chest group, 10.4 for the scalp treatment group, and 9.1 for the trunk/extremities treatment group (Figure 3). In the face/chest and scalp groups, these scores declined rapidly after peaking, reaching mild levels by Week 2 and baseline levels by Week 4, respectively. In the trunk/extremities group, these scores remained high until Day 8, then rapidly declined, reaching mild levels by Week 2 and approaching baseline levels by Week 4 (Figure 3).
FIGURE 3.

Composite local skin response (LSR) score: thin lines denote individual scores; bold lines denote mean scores
Throughout the trial, erythema was the most common LSR component in each treatment group (Figure 4). Vesiculation/pustulation and swelling were relatively high at Day 4, but quickly approached baseline levels thereafter. The least common LSR component was erosion/ulceration in each treatment group throughout the trial period (Figure 4). Of the Grade 4 LSRs, erythema was the most common (face/chest, n=7; scalp, n=13; trunk/extremities, n=13). Flaking/scaling was the next most common Grade 4 LSR reported by patients (face/chest, n=3; scalp, n=4; trunk/extremities, n=7); similar proportions were reported for crusting, swelling, and vesiculation/pustulation. No patients had Grade 4 erosion/ulceration (Figure 4). Overall, LSR components resolved by Week 8 (Figure 4); vesiculation/pustulation and erosion/ulceration resolved by Week 4 in the majority of patients. This is reflected in the composite LSR scores (Figure 3).
FIGURE 4.

Individual local skin response (LSR) components by visit
Adverse events were reported by 35 patients (55.6%) in the face/chest group, 47 patients (74.6%) in the scalp group, and 43 (69.4%) in the trunk/extremities group. Nine patients had 12 AEs of severe intensity (3 [4.8%] patients in each treatment group). Treatment-related AEs were reported by 32 patients (50.8%) in the face/chest group, 44 patients (69.8%) in the scalp group, and 41 patients (66.1%) in the trunk/extremities group. Treatment-related AEs of severe intensity were application site pain (n=1 in each treatment group), application site discomfort (scalp, n=1; trunk/extremities, n=2), application site pruritus (scalp, n=1; trunk/extremities, n=1), application site dermatitis (face/chest, n=1), and eyelid irritation (scalp, n=1). One patient in the face/chest group reported a serious AE (SAE) of inguinal hernia, which was considered as not related to treatment by the investigator. One patient in the scalp group had an AE of migraine that led to withdrawal from the trial. Two patients in the trunk/extremities group reported SCC in the treatment area; one patient had one SCC considered possibly related to the treatment by the investigator and one patient had two SCCs considered probably related to treatment by the investigator. The most common treatment-related AE was application site pain, reported by 28 patients (44.4%) in the face/chest group, 34 patients (54.0%) in the scalp group, and 26 patients (41.9%) in the trunk/extremities group (Table 3). No deaths were reported during the trial.
TABLE 3.
Treatment-related AEs reported in ≥3% of patients in any treatment group
| TYPE | FACE/CHEST 0.018% (n=63) |
SCALP 0.037% (n=63) |
TRUNK/EXTREMITIES 0.1% (n=62) |
|---|---|---|---|
| Application site pain | 28 (44.4%) | 34 (54.0%) | 26 (41.9%) |
| Application site pruritus | 18 (28.6%) | 23 (36.5%) | 21 (33.9%) |
| Application site discomfort | 1 (1.6%) | 1 (1.6%) | 2 (3.2%) |
| Periorbital edema | 2 (3.2%) | 3 (4.8%) | 2 (3.2%) |
| Eyelid edema | 0 | 2 (3.2%) | 0 |
| Headache | 1 (1.6%) | 2 (3.2%) | 0 |
| Erythema | 0 | 0 | 2 (3.2%) |
| SCC of skin | 0 | 0 | 2 (3.2%) |
AEs: adverse events; SCC: squamous cell carcinoma
Efficacy. For nonhyperkeratotic/hypertrophic lesions, reduction in AK lesion count from baseline to Week 8 was 78.9 percent (95% CI 70.9%, 84.7%; n=63) for the face/chest group, 76.3 percent (95% CI 66.7%, 83.1%; n=63) for the scalp group, and 69.1 percent (95% CI 60.3%, 76.0%; n=62) for the trunk/extremities group (Figure 5). Similar results were seen for the reduction from baseline to Week 4 for the face/chest group (81.2% [95% CI 74.7%, 86.1%; n=63]), the scalp group (78.8% [95% CI 71.6%, 84.1%; n=63]), and the trunk/extremities group (59.0% [95% CI 50%, 66.3%; n=62]).
FIGURE 5.
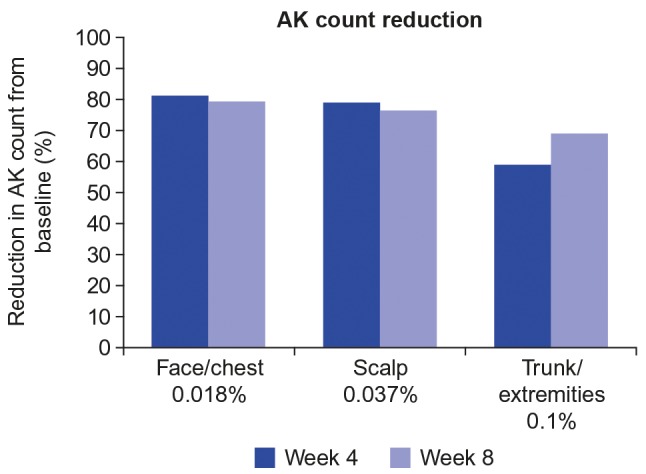
Reduction in actinic keratosis (AK) lesion count at Weeks 4 and 8. Excludes hyperkeratotic/hypertrophic lesions.
The reduction in AK lesion count from baseline to Week 8 for hypertrophic/hyperkeratotic lesions was 68.5 percent (95% CI 4.5%, 89.6%; n=9) for the face/chest group, 62.5 percent (95% CI 13.2%, 87.5%; n=10) for the scalp group, and slightly lower for the trunk/extremities group at 39.2 percent (95% CI 7.9%, 59.9%; n=16). Similar results were observed for the mean percent reduction in AK lesion count of hypertrophic/hyperkeratotic lesions from baseline to Week 4 (face/chest, 78.7% [95% CI 38.7%, 92.6%; n=9]; scalp, 58.1% [95% CI 6.8%, 81.2%; n=10]; and trunk/extremities, 36.0% [95% CI 1.3%, 58.5%; n=16]).
For nonhyperkeratotic/hypertrophic lesions, AKCLEAR 100 at Week 8 was achieved in 36.5 percent of patients in the face/chest group, 39.7 percent of patients in the scalp group, and 22.6 percent of patients in the trunk/extremities group (Figure 6). Comparable values were seen at Week 4 for the face/chest group (36.5%), scalp group (31.7%), and trunk/extremities group (14.5%).
FIGURE 6.
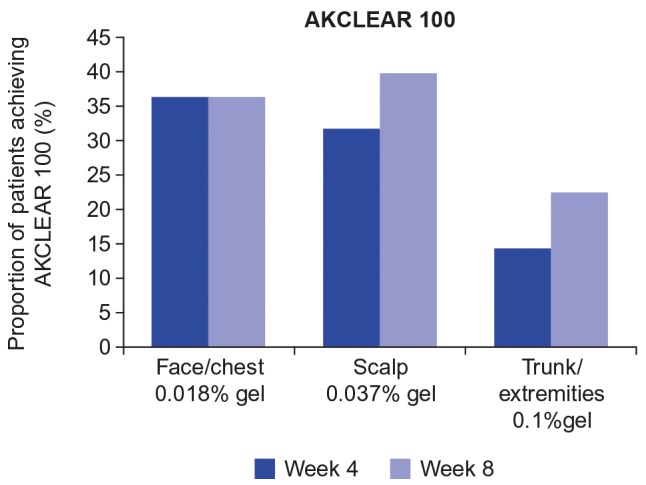
Percentage of patients with complete clearance of actinic keratoses (AKCLEAR 100) at Weeks 4 and 8; excludes hyperkeratotic/hypertrophic lesions
AKCLEAR 100 at Week 8 for the hyperkeratotic/hypertrophic lesions was achieved in five of nine (55.6%) patients in the face/chest group, six of 10 (60.0%) patients in the scalp group, and four of 16 (25.0%) patients in the trunk/extremities group.
For nonhyperkeratotic/hypertrophic lesions, AKCLEAR 75 at Week 8 was achieved in 71.4 percent of patients in the face/chest group, 65.1 percent of patients in the scalp group, and 50.0 percent of patients in the trunk/extremities group (Figure 7). Comparable levels were observed at Week 4 for the face/chest (76.2%) and scalp (74.6%) groups, but were slightly lower in the trunk/extremities group (37.1%).
FIGURE 7.
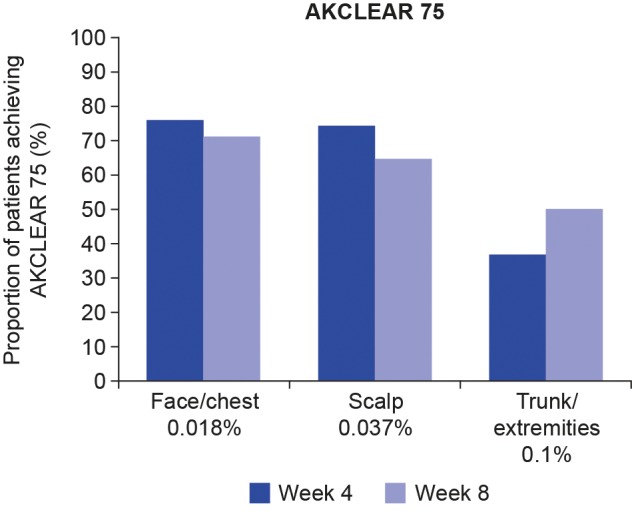
Percentage of patients with AKCLEAR 75 at Weeks 4 and 8; excludes hyperkeratotic/hypertrophic lesions AKCLEAR 75: partial clearance of actinic keratoses defined as 75 percent reduction or more in baseline AK lesion count
A detailed analysis of cosmetic outcomes, patient treatment satisfaction, and treatment adherence from this trial can be found in the study by Berman et al.15
DISCUSSION
AK presents itself not only in visibly discrete AK lesions, but also subclinically in areas of field cancerization. Ingenol disoxate is a new ingenol derivative with improved stability characteristics and preserved or improved anti-tumor characteristics as compared with ingenol mebutate, including cell death mechanisms, activation of protein kinase C, and induction of local immune response.11 This Phase II trial examined the safety and efficacy of ingenol disoxate following daily application of 0.018% gel on the full face/chest (approximately 250cm2), 0.037% gel on the scalp (25–250cm2), or 0.1% gel on the trunk/extremities (approximately 250cm2) over three consecutive days.
Safety. The overall number of DLEs per treatment group was comparable with the incidence observed in previous dose-finding studies with once-daily dosing for two consecutive days.13,14
The LSR profile of the ingenol disoxate regimen in this trial was comparable with those of previous dose-finding studies that evaluated a shorter two-day regimen; Weiss et al13 and Bourcier et al14 reported that, following ingenol disoxate application, LSR scores peaked on Day 3, rapidly declined, and reached or approached baseline levels by Week 4. Similarly, in the present trial, the mean composite LSR score peaked the day after the final application of ingenol disoxate (Day 4) and rapidly declined, reaching or approaching baseline levels by Week 4.
The peak composite LSR scores in the present trial were slightly higher than those reported in the previous trials, in line with a longer duration of treatment; Weiss et al13 observed mean LSR scores on Day 3 of 8.6 and 8.7 for ingenol disoxate gels at 0.037% and 0.05%, respectively; Bourcier et al14 reported a mean composite LSR score on Day 3 for ingenol disoxate gel at 0.018%, 0.012%, and 0.006% of 8.6, 8.0, and 6.0, respectively. However, higher composite LSRs in this trial resolved over a similarly short time period to the previous trials, and did not affect adherence to treatment, as 97 percent of patients overall completed the three-day treatment regimen.15
The most common LSRs across all treatment groups, in the previous trials evaluating ingenol disoxate, were erythema and flaking/scaling.13,14 In the present trial, erythema was the most common LSR reported by patients. In the majority of patients, the LSRs of swelling and vesiculation/pustulation resolved by Week 4 despite being relatively high on Day 4, suggesting these individual LSR components resolved quicker than the others. Erosion/ulceration was the least dominant LSR and resolved in the majority of patients by Week 4.
Three-day dosing with ingenol mebutate has also demonstrated similar safety findings.10 When ingenol mebutate 0.015% gel was applied to the face and scalp, local skin reactions peaked on Day 4, with a mean maximum composite LSR score of 9.1. They then rapidly decreased, approaching baseline scores by approximately Week 4.
It is always important that proper patient education is provided regarding expected skin reactions and their management when treating patients with AK—particularly those without prior experience of topical treatment. Approximately 80 to 90 percent of patients on topical therapy will experience LSRs such as erythema, dryness, burning, and pruritus.18
Thus, providing clear information (including photographs to support communication) can help to alleviate fear, and patients can also be provided with techniques to use at home to help reduce any pain or itching.
Less than 5 percent of patients in each treatment group had severe AEs. Treatment-related AEs were reported in 50.8, 69.8, and 66.1 percent of patients in the face/chest, scalp, and trunk/extremities groups, respectively; approximately half of the patients had application site pain and approximately one-third had application site pruritus. Based upon these findings, the safety of the dosing regimen with once-daily dosing for three consecutive days was in line with previous dose finding trials evaluating ingenol disoxate two-day regimens.13,14
Efficacy. The three-day treatment regimen evaluated in this trial demonstrated clinically relevant efficacy. This was demonstrated both by the high percentage reduction in AK lesion count from baseline and also the percentage of patients who achieved complete clearance (AKCLEAR 100) by the end of the trial at Week 8. It should also be noted that approximately two-thirds of patients in the face/chest and scalp groups and half of the patients in the trunk/extremities group achieved partial clearance (AKCLEAR 75) by Week 8. Comparable levels were observed for reduction in AK lesion count, as well as partial and complete clearance at Week 4, indicating that the majority of the effect on AK lesions was already achieved by this time point.
Similar findings have been reported using ingenol disoxate daily dosing for two consecutive days. Weiss et al13 observed a 72.7-percent reduction in AK lesion count from baseline to Week 8 when ingenol disoxate was administered at 0.037% on the scalp; the majority of this effect was reached by Week 4. Similarly, Bourcier et al14 reported a 79.0-percent reduction in AK lesion count from baseline at Week 8 when ingenol disoxate 0.018% gel was administered on the face/chest. Weiss et al13 reported that when patients were treated with ingenol disoxate 0.037% gel on the scalp, 21.9 percent of patients achieved complete clearance, while 54.7 percent achieved partial clearance at Week 8. In the Bourcier et al trial,14 when treated with ingenol disoxate 0.018% gel on the face/chest, 24.2 percent of patients achieved complete clearance and 62.9 percent achieved partial clearance at Week 8. The rate of complete and partial clearance, at the same concentrations of ingenol disoxate as used in previous trials, was increased in the present trial, which is likely due to the additional treatment day.
The efficacy of ingenol disoxate in hyperkeratotic/hypertrophic lesions was also demonstrated by a percentage reduction in AK lesion count, although this was slightly less than that observed in the nonhyperkeratotic/hypertrophic AK lesions. However, due to the small number of patients with this subtype of AK lesions and also the low number of lesions per patient, the CIs for the estimated mean percent reduction were wide. Approximately half of patients in the face/chest and scalp groups and a quarter of patients in the trunk/extremities group demonstrated complete clearance of hyperkeratotic/hypertrophic lesions. These data suggest that ingenol disoxate might be effective on these lesions, which are notoriously difficult to treat.
It is important to note that a field therapy that is highly effective at eliminating clinically visible lesions, as demonstrated in the reduction of AK lesion count and clearance rates, is also likely to eliminate subclinical lesions that are not visible.19
Treatment regimen and dose concentration. This trial evaluated the safety and efficacy of ingenol disoxate gel at different concentrations on different anatomical locations when applied once daily for three consecutive days. The same treatment regimen (once daily for three consecutive days) has proved effective with ingenol mebutate when 0.015% of such was applied to the face or scalp (25cm2).10 Although complete clearance was slightly lower in the present trial, it is important to note that the treatment area was up to 10 times larger with a greater number of baseline AK lesions, making complete clearance more difficult to achieve. A more appropriate comparison for trials in which treatment area sizes are different is percentage reduction in AK lesion count; in two trials evaluating the application of ingenol mebutate gel on the face and scalp, Lebwohl et al10 reported a mean percent reduction of 77.7 percent for the face (both trials) and 55 percent and 50 percent for the scalp, which compares favorably with results in the present trial (face/chest, 78.9%; scalp, 76.3%).
The different doses selected in this trial were determined in Phase I/II dose-finding trials,13,14 and were partly based upon findings of the pivotal ingenol mebutate trials.10 Lebwohl et al10 evaluated one dose concentration to treat both the face and scalp and reported lower efficacy on the scalp. Similar findings for reduced efficacy on the scalp have also been reported in trials evaluating imiquimod.20 Accordingly, the dose evaluated in the present trial has been optimized specifically for treatment on the scalp, which is more difficult to treat. Disparities in efficacy between different regions of skin may relate to differences in absorption and thus warrant optimized dosing concentrations.
Limitations. It should be noted that the safety and efficacy data from this trial should be interpreted with caution, as the trial was open-label, potentially introducing patient and/or investigator bias. An additional consideration is that LSRs following ingenol disoxate application on the first day of treatment could have affected how patients self-applied treatment on the two subsequent days, potentially avoiding certain parts of the treatment area, or applying smaller amounts of gel.
CONCLUSION
In conclusion, ingenol disoxate, when administered once daily over three consecutive days in patients with AK, demonstrated similar findings as previous trials with respect to AE and LSR profiles. It might be an effective field treatment as evidenced by the reduction in AK count and the complete and partial clearance (AKCLEAR 100 and AKCLEAR 75) results. These data support the use of ingenol disoxate for the field treatment of AK. Phase III assessments of ingenol disoxate at the concentrations reported in this trial are being pursued.
ACKNOWLEDGMENTS
The study was funded by LEO Pharma. Medical writing services were provided by Louise Prince, PhD, and Patrick Griffin, MSc, of iMed Comms, an Ashfield Company, part of UDG Healthcare plc, and funded by LEO Pharma in accordance with Good Publication Practice guidelines. The authors thank the investigators who participated in this study.
REFERENCES
- 1.Einspahr JG, Stratton SP, Bowden GT, Alberts DS. Chemoprevention of human skin cancer. Crit Rev Oncol Hematol. 2002;41(3):269–285. doi: 10.1016/s1040-8428(01)00185-8. [DOI] [PubMed] [Google Scholar]
- 2.Berman B, Amini S. Pharmacotherapy of actinic keratosis: an update. Expert Opin Pharmacother. 2012;13(13):1847–1871. doi: 10.1517/14656566.2012.716039. [DOI] [PubMed] [Google Scholar]
- 3.Alam M. Actinic keratoses: prevalence, pathogenesis, presentation, and prevention. Adv Stud Med. 2006;6(8A):785–790. [Google Scholar]
- 4.Frost CA, Green AC. Epidemiology of solar keratoses. Br J Dermatol. 1994;131(4):455–464. doi: 10.1111/j.1365-2133.1994.tb08544.x. [DOI] [PubMed] [Google Scholar]
- 5.Malvehy J. A new vision of actinic keratosis beyond visible clinical lesions. J Eur Acad Dermatol Venereol. 2015;29(Suppl 1):3–8. doi: 10.1111/jdv.12833. [DOI] [PubMed] [Google Scholar]
- 6.Fu W, Cockerell CJ. The actinic (solar) keratosis: a 21st-century perspective. Arch Dermatol. 2003;139(1):66–70. doi: 10.1001/archderm.139.1.66. [DOI] [PubMed] [Google Scholar]
- 7.Vatve M, Ortonne JP, Birch-Machin MA, Gupta G. Management of field change in actinic keratosis. Br J Dermatol. 2007;157(Suppl 2):21–24. doi: 10.1111/j.1365-2133.2007.08268.x. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Figueras MT, Carrato C, Saenz X, et al. Actinic keratosis with atypical basal cells (AK I) is the most common lesion associated with invasive squamous cell carcinoma of the skin. J Eur Acad Dermatol Venereol. 2015;29(5):991–997. doi: 10.1111/jdv.12848. [DOI] [PubMed] [Google Scholar]
- 9.Heerfordt IM, Nissen CV, Poulsen T, et al. Thickness of actinic keratosis does not predict dysplasia severity or p53 expression. Sci Rep. 2016;6:33952. doi: 10.1038/srep33952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lebwohl M, Swanson N, Anderson LL, et al. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366(11):1010–1019. doi: 10.1056/NEJMoa1111170. [DOI] [PubMed] [Google Scholar]
- 11.Bertelsen M, Stahlhut M, Grue-Sorensen G, et al. Ingenol disoxate: a novel 4-isoxazolecarboxylate ester of ingenol with improved properties for treatment of actinic keratosis and other non-melanoma skin cancers. Dermatol Ther (Heidelb). 2016;6(4):599–626. doi: 10.1007/s13555-016-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sinnya S, Tan JM, Prow TW, et al. A randomised, Phase IIa exploratory trial to assess the safety and preliminary efficacy of LEO 43204 in patients with actinic keratosis. Br J Dermatol. 2015;174(2):305–311. doi: 10.1111/bjd.14245. [DOI] [PubMed] [Google Scholar]
- 13.Weiss J, Ulrich M, Bukhalo M, et al. A seamless Phase I/II dose-finding trial assessing ingenol disoxate (LEO 43204) for field treatment of actinic keratosis on the scalp. Br J Dermatol. 2017;176(6):1456–1464. doi: 10.1111/bjd.15304. [DOI] [PubMed] [Google Scholar]
- 14.Bourcier M, Stein Gold L, Guenther L, et al. A dose finding trial with a novel ingenol derivative (ingenol disoxate; LEO 43204) for field treatment of actinic keratosis on full face or 250cm2 on the chest. J Dermatolog Treat. 2017;28(7):652–658. doi: 10.1080/09546634.2017.1303568. [DOI] [PubMed] [Google Scholar]
- 15.Berman B, Tyring S, Nahm WK, et al. Three-day field treatment with ingenol disoxate (LEO 43204) for actinic keratosis: cosmetic outcomes and patient satisfaction from a phase 2 trial. J Clin Aesthet Dermatol. 2017;10(11):26–32. [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen R, Marmur E, Anderson L, et al. A new, objective, quantitative scale for measuring local skin responses following topical actinic keratosis therapy with ingenol mebutate. Dermatol Ther (Heidelb). 2014;4(2):207–219. doi: 10.1007/s13555-014-0059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atkinson MJ, Sinha A, Hass SL, et al. Validation of a general measure of treatment satisfaction, the treatment satisfaction questionnaire for medication (TSQM), using a national panel study of chronic disease. Health Qual Life Outcomes. 2004;2:12. doi: 10.1186/1477-7525-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ceilley Rl, Jorizzo JL. Current issues in the management of actinic keratosis. J Am Acad Dermatol. 2013;68(1 Suppl 1):S28–S38. doi: 10.1016/j.jaad.2012.09.051. [DOI] [PubMed] [Google Scholar]
- 19.Ulrich M, Lange-Asschenfeldt S, Skak K, et al. Biological efects of ingenol mebutate gel in moderate to severe actinic fields assessed by reflectance confocal microscopy: a phase I study. J Drugs Dermatol. 2016;15(10):1181–1189. [PubMed] [Google Scholar]
- 20.Swanson N, Smith CC, Kaur M, Goldenberg G. Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: two phase 3, multicenter, randomized, double-blind, placebo-controlled studies. J Drugs Dermatol. 2014;13(2):166–169. [PubMed] [Google Scholar]