

Abstract
Microglia, the resident immune cells of the brain, have been implicated in numerous neurodegenerative and neurodevelopmental diseases. Activation of microglia by a variety of stimuli has been shown to induce the release of numerous factors that contribute to neuro-inflammation and oxidative stress, two crucial processes linked to disorders of the central nervous system. The in vitro techniques described here will provide a set of protocols for the isolation and plating of primary cerebellar granule neurons, primary cortical microglia from a mixed glia culture, and methods for co-culturing these two cell types, allowing for mechanistic investigations into how the presence of microglia and the factors they release in this shared environment may mediate the effect of toxicants on neuronal function and survival. The protocols presented here allow for flexibility in experimental design, the study of numerous toxicological endpoints, and the opportunity to explore neuroprotective strategies.
Keywords: Microglia, neurons, co-culture, neuro-inflammation, oxidative stress, glia
Introduction
Microglia, the resident immune cells of the brain, play a defensive and beneficial role in maintaining an environment that supports the functioning and survival of neurons partially through their ability to mediate controlled inflammatory responses (Block et al., 2007). In response to injury or numerous stimuli microglia can become activated and move through a series of morphological and functional changes. They proliferate, retract their ramified extensions, become motile and more amoeboid in shape, and gain the ability to phagocytize debris (Stence et al., 2001). In the activated state, microglia are known to release both protective and cytotoxic factors, through which they can influence neuronal functions and viability (Block et al., 2007; Ransohoff and Perry, 2009; Luo and Chen, 2012). When this process is dysregulated, reactive microglia act as major contributor to oxidative and neuro-inflammatory damage, through the release of various pro-inflammatory cytokines and the generation of reactive oxygen species (Block et al., 2007). Both neuro-inflammation and oxidative stress are believed to play primary roles in neurotoxicity and in neurodevelopmental and neurodegenerative disorders (Kraft and Harry, 2011; Graeber et al., 2011). In fact, microglia have been shown to be actors in the pathology of Alzheimer’s and Parkinson’s diseases, as well as multiple sclerosis and autism (Hong et al., 2016; Block et al., 2007, 2004; Goldmann and Prinz, 2013; Ransohoff and Perry, 2009; Zhan et al., 2014; Takano, 2015; Qian and Flood, 2008).
This unit presents methods that can be used to study the interactions of neurons and microglia, expanding our understanding of the role that microglia play in mediating neurotoxicity after exposure to toxicants. The protocols describe an experimentally flexible, in vitro co-culture system, where mouse primary cerebellar granule neurons (CGNs) and cortical microglia are first grown separately and then combined. Basic Protocol 1 describes the isolation of mouse cerebellar granule neurons and their maintenance and growth. The establishment and maintenance of primary cortical mixed glial cultures are described in Basic Protocol 2, while procedures to isolate and subculture primary cortical microglia from the mixed glia cultures into porous inserts is presented in Basic Protocol 3. Finally, Basic Protocol 4 describes the method used to combine neurons and microglia in a co-culture system (Figure 1), where they will share the environment (e.g. for 24 hours). This system allows comparison of the effect of the toxicant of interest on neurons alone or in the presence of microglia, as well as determination of how treatment impacts microglial. The use of porous inserts also allows for the exchange of secreted factors between these two cell types, while maintaining separation.
Figure 1.
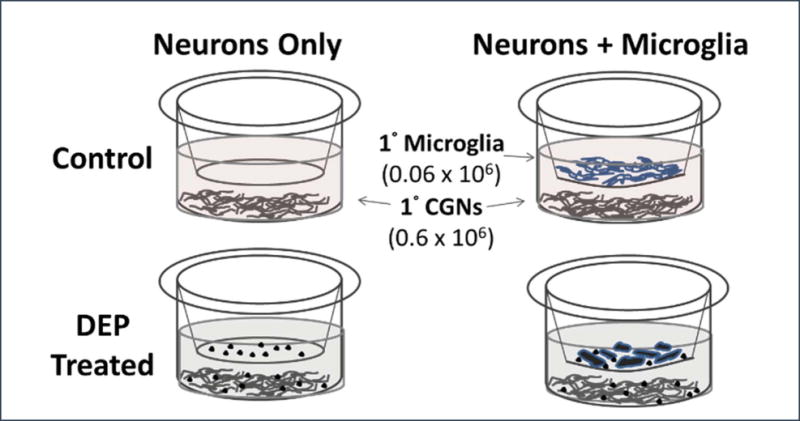
Neuron-Microglia Co-culture treated with diesel particles (DEP) (Roqué et al., 2016).
The co-culture protocol offers flexibility in experimental design and allows for the application of a variety of experimental endpoints. Additionally, mechanistic studies focused on the contribution of each cell type can be performed by designing experiments to investigate the effect of treatment on either neurons or microglia themselves, with methods to study neuro-inflammation being of interest (Monnet-Tschudi et al., 2011). Our laboratory has used this co-culture system to investigate the role of microglia in mediating diesel exhaust particle (DEP) neuronal death (Roqué et al., 2016).
NOTE: All work presented here should be performed in a laminar-flow fume hood to ensure aseptic conditions. All animal procedures should comply with approved Institutional Animal Use and Care Committee (IACUC) protocols. Solutions used for washing or replacing medium should be warmed to 37°C in a water bath prior to use on growing cultures. All incubation steps, unless otherwise mentioned, should be carried out in a humidified, 37.6°C, 5% CO2 environment. Methods to anatomically identify and dissect cerebellar and cortical tissue from mouse brain are not fully described here as they have been well-described elsewhere (Giordano and Costa, 2011; Lee et al., 2009; Spijker, 2011).
Basic Protocol 1
Isolation and culture of mouse primary cerebellar granule neurons
Cerebellar granule neurons (CGNs) have been used extensively in the study of neurotoxicity (Huang et al., 2010; Giordano et al., 2006; Lee et al., 2009; Giordano and Costa, 2011; Oberdoerster, 2001), and the method described here for isolating these primary cells has been modified from previously published protocols (Giordano and Costa, 2011; Oberdoerster, 2001). Because CGNs are among the most numerous neurons in the brain and are relatively easy to obtain, they yield a large number of cells for experimental use (Giordano and Costa, 2011; Llinás et al., 2004; Banker, 1998), making them well-suited as a general neuronal model system.
Materials
Animals: Post-natal day (PND) 6–8 mice
Poly-D-lysine working solution (40 μg/ml) (see Reagents and Solutions)
NB medium (See Reagents and Solutions)
Papain working solution, 1 ml per 2 pups (see Reagents and Solutions)
DNase (4 mg/ml) (see Reagents and Solutions)
Hank’s balanced salt solution, supplemented with 10 mM HEPES (HBSS)
Hank’s balanced salt solution, calcium and magnesium free, supplemented with 10 mM HEPES (HBSS-CMF)
24-well sterile cell culture plates
12 mm, round glass coverslips (optional)
5 and 10 ml serological pipettes
Sterile 35 mm dish for tissue collection (add 1.5 ml HBSS-CMF)
Sterile 100 mm dishes for pup and tissue collection, with filter paper (3)
Ice in a small container to hold 35 mm dish during tissue collection
Sterile filter paper, round, 100 mm
Sterile surgical tools
15 ml conical tubes
50 ml conical tubes
2–50 ml tubes containing 70% ethanol, two-thirds full
100 μm 50 ml tube filter
Centrifuge
10 ml syringe with 0.22 syringe filter for sterilization of papain
Steps and Annotations
Coat 24-well plates or glass coverslips
Depending on needs of the experimental assays planned, CGNs can be coated in 24-well plates for biochemical assays, or on sterile glass coverslips, for imaging assays. The procedures for coating and washing glass coverslips are the same as that for 24-well plates and follow that presented below. Before the start of the coating steps, sterile, 12 mm, glass coverslips should be plated, one per well, in 24-plates.
Plates or glass coverslips should be coated with poly-d-lysine (PDL) overnight, so this task should be performed the day prior to tissue dissection. Plates can be coated earlier, washed and stored for up to one week prior to seeding neurons.
-
1
Prepare PDL working solution (200 μg/ml) to coat 24-well plates, using the PDL stock solution (see Reagents and Solutions). Prepare an appropriate volume to add 1 ml of working solution to each experimental well. Sterile filter.
Larger volumes of working solution can be prepared and stored at 4°C for up to 1 month. Keep sterile.
-
2
Add 1 ml PDL working solution to each well. Incubate overnight in a 37°C incubator.
-
3
Wash each well 2× with sterile water and replace with HBSS until use. Set aside in hood while completing the next steps.
Plates can be pre-coated and stored in an incubator for up to one week. Wells should be coated overnight as above, washed 2× with water after incubation and wells replaced with water and stored in the incubator until use. On the day of use, replace the water with HBSS and set aside at RT until use.
Dissect Cerebellar Tissue
During these steps, to preserve the structural integrity of the brain tissue and maintain a clear distinction between regions, decapitate and obtain tissue from one mouse at a time. Perform this animal work according to approved IACUC protocols. Work quickly to ensure cell survival.
-
4
Prepare 1 ml of papain solution for every 2 pups (see Reagents and Solutions).
-
5
Warm papain, DNase, Neurobasal medium (NB), and HBSS in a water bath to 36.7°C.
-
6
Decapitate one PND7 pup and sterilize the head by quickly immersing the head in ethanol.
-
7
Carefully remove cerebral hemisphere and place onto the filter paper.
-
8
Dissect the cerebellum.
-
9
Remove the meninges by rolling the tissue over sterile filter paper using a sterile spatula.
Improper removal of the meninges will introduce unwanted cells into the culture.
Complete removal of the meninges is evident by the appearance of red remnants as you roll the tissue. Continue rolling until clear, about 3–4 complete rolls.
-
10
Transfer cerebellar tissue to a 35 mm dish containing 1.5 ml HBSS-CMF, stored on ice.
-
11
Repeat until all tissue is collected.
-
12
Remove excess HBSS from the 35 mm dish using a 5 ml serological pipette and mince the tissue using sterile surgical scissors to ~ 1 mm size.
-
13
Using a syringe and 0.22 μm filter, filter the papain.
Digest and Dissociate the Tissue
-
14
Transfer the minced tissue and the entire volume of papain solution to a sterile 15 ml tube, using the papain to rinse the 35-mm petri dish to maximize tissue collection.
-
15
Gently invert 2–3× to mix and incubate for 30 minutes in a 36.7°C water bath. After 10 minutes, invert tube to ensure proper exposure of settled tissue to papain and DNase.
The tissue pieces should remain distinctly separated after 10 minutes. If the tissue has formed a coagulated mass, add an additional aliquot of DNase (1:50) to the solution and invert to mix. Return to the water bath for the remaining incubation time. (Add DNase in a sterile hood).
-
16
After incubation, centrifuge the solution for 3 minutes at 200 × g (4°C).
-
17
Aspirate the supernatant being careful to avoid the pellet and add 5 ml of HBSS-CMF.
-
18
Triturate the suspension with a 5 ml serological pipette, by taking up and dispensing the solution 12×. Let the tissue settle for 2 minutes.
To aid in dissociation, and to minimize cell death, dispense the solution against the side of the tube. Work quickly, but gently, and avoid creating bubbles.
The dissociation of the cells should be evident by a reduction in visible tissue and the solution becoming less opaque. If the cells are not dissociating from the tissue, add an additional aliquot of DNase (1:50).
-
19
Without disturbing the settled tissue, transfer the cell-containing supernatant to a 50 ml conical tube containing a 100 μm mesh filter.
-
20
Add 5 ml HBSS-CMF to the remaining tissue. Triturate 12× as before to continue the dissociation process. Wait 2 minutes for the tissue to settle and transfer the remaining supernatant to the 50 ml tube. Discard the remaining pellet.
-
21
solution containing the cells to a 15 ml tube and centrifuge for 7 minutes at 200 × g (4°C).
-
22
Aspirate the supernatant and add 5 to 10 ml of warmed NB medium. Thoroughly and evenly resuspend the pelleted cells.
To ensure a concentrated enough dilution for plating, resuspend the cells in 5 ml of medium if the cerebellum from 4 or fewer pups were collected, and 10 ml if greater than 4.
Count and seed cells
-
23
Count the viable cells using a hemocytometer and dilute the cell solution to 0.6 × 106 cells/ml.
-
24
1 ml to each pre-coated well of a 24-well plate.
-
25
Incubate for 1 hour at 37.6°C to allow neurons to adhere.
-
26
After 1 hour, gently wash each well 2× with warmed HBSS and replace with 1 mL warmed NB medium.
When washing or replacing the medium in growing cultures, use the well side wall to dispense the solution so that cells are not directly disturbed by an intense flow of solution.
While most cells will have adhered, it is not unusual for floating, or dead cells to appear at this time point. Dead cells will be removed by washing.
-
27
On DIC4, wash the cells 2× with 1 ml warmed HBSS and replace with 1 ml NB (-AO).
At this point, networks between neurons will have begun to form. It is not unusual at this time point to see cells that have died floating in the medium. The wash will remove the cells.
-
28
On DIC8, remove 500 μl NB (-AO) medium from each well, using a P1000 pipette, and replace it with 500 μl fresh, warmed NB (-AO) medium.
-
29
On day 10, use neurons for experiments (e.g. treatment) or co-culture them with microglia (See Basic Protocol 4).
Basic Protocol 2
Preparation of cortical mixed glial cultures
In order to obtain primary microglia, cultures of mixed glia are first established from cerebral cortices of PND3 mouse pups. The methods described here and in Basic Protocol 3 are largely based, with modifications, on previously published protocols (Ni and Aschner, 2010; Witting and Möller, 2011). This protocol describes the first of two steps necessary to obtain primary microglia, i.e. the preparation and maintenance of cortical mixed glia cultures, while Basic Protocol 3 with present techniques for sub-culturing microglia for experimental use.
Materials
Animals: post-natal day 3 (PND3) mice
Centrifuge
Laminar-flow cell culture hood
PDL working solution (40 μg/ml) (See Reagents and Solutions)
Mixed Glia and Microglia Medium (DMEM+) (See Reagents and Solutions)
Trypsin (0.25% in PBS), 1 ml per pup cortex
DNase: 4 mg/ml prepared in HBSS-CMF, sterile filtered
Phosphate-buffered saline
Hank’s buffered salt solution (HBSS)
HBSS without calcium or magnesium (HBSS-CMF)
T175 flasks pre-coated with PDL, 1 flask per 4–6 pup cortices
Sterile 35-mm petri dish for tissue collection (add 1.5 ml HBSS-CMF)
Sterile 100 mm dishes for pup and tissue collection
Sterile filter paper, round, 100 mm
Sterile surgical tools
2–50 ml tubes containing 70% ethanol
100 μm 50 ml tube filter
5, 10 and 25 ml serological pipettes
Steps and Annotations
Coat flasks
-
1
Prepare PDL working solution (40 μg/ml) to coat T175 flasks. Prepare an appropriate volume to coat 1 flask per 4–6 pups and with 25 mls of PDL per flask. Sterile filter.
Larger volumes can be prepared and stored at 4°C for up to 1 month. Keep sterile.
Because large amounts of this concentration of PDL is used each time (25 ml per flask and for coating inserts), you may wish to prepare a 250 or 500 ml working solution.
-
2
Add 25 ml PDL working solution to each flask. Incubate for 1.5 hours at RT in laminar-flow hood.
-
3
Wash flasks 1× with sterile water and replace with PBS until use.
In our experience, the use of 25 ml serological pipettes for removing and replacing medium, as opposed to a glass pipette-fitted aspirator, is beneficial to help reduce potential contamination. The T175 flasks are quite large and the longer, 9” Pasteur pipettes do not adequately allow for solution removal without portions of the aspirator entering the neck of the flask. Use 25 ml pipettes for all wash steps involving T175 flasks in this protocol.
Flasks can be pre-coated and stored containing water in an incubator for 1–2 weeks.
Remove water and replace with PBS prior to use.
Dissect cortices
During these steps, to preserve the structural integrity of the brain tissue and maintain a clear distinction between regions, decapitate and obtain tissue from one mouse at a time. Perform this animal work according to approved IACUC protocols. Work quickly to ensure cell survival.
-
4
Warm DMEM, trypsin and DNase to 36.7°C in a water bath.
-
5
Decapitate one PND 3 pup and sterilize the head by quickly immersing the head in ethanol.
-
6
Carefully remove the entire cerebral hemisphere with a sterile spatula and place onto filter paper.
-
7
Using a scalpel, dissect away the olfactory bulbs, cerebellum, and hind brain. Lift the cortical tissue off the mid brain and move the cortices to a clean section of filter paper.
-
8
Remove the meninges by rolling the tissue over sterile filter paper using a sterile spatula.
Improper removal of the meninges will introduce unwanted cells into the culture.
Complete removal of the meninges is evident by the appearance of red remnants as you roll the tissue. Continue rolling until clear, about 3–4 complete rolls.
-
9
Transfer cortical tissue to 35-mm petri dish containing 1.5 ml HBSS-CMF, stored on ice.
-
10
Repeat until all cortical tissue from litter has been collected.
-
11
Remove excess HBSS-CMF with a 5 ml pipette and mince tissue into approximately 1 mm pieces using sterile surgical scissors.
Digest tissue and prepare mixed glia cultures
-
12
Transfer the minced tissue and trypsin (1 ml/pup) into a sterile 15 mL tube, using trypsin to rinse the 35-mm petri dish to maximize tissue collection.
-
13
Add 1:200 DNase. Cover and invert the tube 3–4 times gently to mix.
-
14
Incubate for 30 minutes in a 37°C water bath.
-
15
Aspirate trypsin without disturbing the settled tissue.
Prior to aspiration, check to ensure that the tissue remains as distinct pieces. At times, the negatively charged portion of disrupted DNA can cause the tissue to attract as a mass. If this occurs, do not aspirate, but remove the trypsin using a 10 ml serological tube, otherwise the force of the aspiration can cause a loss of a significant amount, if not all of the tissue. This should not affect the tissue dissociation process.
-
16
Add 5 ml of HBSS-CMF to wash the tissue. Cover and invert the mixture 3–4 times. Let tissue settle and remove the HBSS-CMF.
-
17
Add an additional 5 ml HBSS-CMF. Cover and invert the mixture 3–4 times. Let tissue settle and remove the HBSS-CMF and repeat step 43.
-
18
Add 10 ml of warmed DMEM medium and triturate 12–15 times using a 10 ml pipette to dissociate tissue.
Optional: Add DNase (1:500) if tissue does not easily dissociate.
-
19
Filter cell solution through a 100 μm mesh filter into a 50 ml conical tube and transfer the filtrate to a 15 ml sterile tube.
-
20
Centrifuge for 10 minutes at 200 × g (4°C).
-
21
Aspirate supernatant and add 10 mL warmed DMEM medium.
-
22
Resuspend the cells by triturating with a 10 ml pipette, 10–12× or until the cells are evenly suspended.
-
23
Seed flasks with a volume of cell suspension equivalent to 4–6 pups per flask in a total of 25 ml of medium per flask.
-
24
Incubate the seeded flasks overnight at 37.6°C.
-
25
On the next day, wash flasks 1× with PBS and replace with 25 ml warmed DMEM medium (25 ml).
Visualize the cells before and after wash. A large amount of debris will be present before the wash step, which is typical of this mixed glia protocol. Some cells will have adhered and will be more clearly visible post-wash. After the wash, some debris may still remain after washing, which will not harm the culture. Adhered cells will be sparse and seemingly diverse at this point, which is to be expected. These cells will proliferate to confluency within the first week in culture.
-
26
Change the medium on DIC 8, or 7 days after the last medium change.
-
27
Monitor the proliferation of the microglia and subculture microglia on DIC15 (see Basic Protocol 3). If microglia are not used at this time point, change the medium to ensure the health of the mixed glia monolayer.
Basic Protocol 3
Isolate primary microglia from mixed glial cultures
This protocol describes the methods to isolate primary microglia from the previously established mixed glia cultures (see Basic Protocol 2). Microglia begin to appear in culture soon after the mixed glial culture reaches confluency, or near in time to the DIC8 medium change. They will appear as small, bright, round cells attached to the surface of the glial monolayer, although some can be seen suspended within the culture medium. This population of cells will continue to proliferate and increase in number over the next week in culture. In the system described here, microglia are isolated on or near DIC15, which, in our experience, has been the time point when sufficient numbers of microglia are available for experimental use. Cultures of mixed glia will continue to produce microglia for subsequent passages, though as the cultures age, the proliferative ability decreases. Because of this and to maintain consistency between distinct cultures, only two populations of primary microglia are obtained from the same flask and the flasks are retired after 1 – 1.5 months.
Isolated microglia are plated into pre-coated inserts, which fit into wells of 24-well plates. The plating surface of the insert consists of a porous nylon mesh, which allows soluble factors secreted by microglia or neurons to become a part of the shared neuron-microglia environment. The percentage of microglia in the brain varies depending on brain region, and ranges from 6–17% (Block et al., 2007). In an attempt to more accurately reflect in vivo cell distributions, microglia are plated at a density of 60,000 cells/insert, which reflects 10% of the plating density of neurons in this co-culture system. The isolation of microglia for experimental use takes place 2 days prior to their co-culturing with neurons, or on neuronal DIC8.
Materials
Established mixed glia cultures, DIC12-15 (see Basic Protocol 2)
DMEM medium (see Reagents and Solutions)
PDL (40 μg/ml) working stock, prepared from stock solution (see Reagents and Solutions)
PBS
24-well plates
24-well inserts (3.0 μm pores)
Tweezers to move inserts
Laminar-flow hood
Steps and Annotations
Microglia are cultured two days prior to co-culturing with neurons or on neuronal DIC8.
Coat inserts
-
1
Prepare PDL (40 μg/ml) from stock solution, making enough for 1.3 mls per insert.
Because this concentration of PDL is also used in large volumes for the coating of flasks for mixed glia cultures, it may be helpful to prepare 250 or 500 ml of working solution. Larger volumes can be stored at 4°C for up to 1 month. Keep sterile.
-
2
Using sterile tweezers, hold the tabs on top of the insert, and transfer the insert to wells of a sterile 24-well plate.
Prepare one insert for each neuronal well of the experiment. If the experimental plan includes comparing effects of treatment on neurons in the presence of microglia to treatment of neurons alone, inserts should also be prepared for those neuronal wells to control for the effect of the insert on the culture.
-
3
Coat both sides of the insert by adding 1 mL PDL working solution to the well portion of the system and 200 μL of the same solution to the insert itself.
A 5 ml serological pipette can be used to add 1 ml of medium to the well, using the pipette tip to push the insert slightly to the side of the well. For adding solutions or cells to the insert, a P200 pipette works best.
-
4
Incubate for 1.5 hours at RT in the laminar-flow hood.
-
5
At incubation end, aspirate the PDL from both the insert and the well, wash both 2× with sterile water and replace with sterile PBS. Set aside until use.
To effectively remove solution from the well and insert, it helps to tilt the plate toward you so that the solution pools at the bottom of the each. Use the glass pipette on the aspirator to move the insert to the side to aid in complete removal of the solution in the well. To avoid the suction of the aspirator causing the insert to attach to the pipette, avoid the mesh by inserting the aspirator pipette along the plastic wall.
Sub culturing microglia from established cultures is a relatively fast process. Inserts can be pre-coated and stored in water an incubator until use, or coated just before the passage. If you are coating the inserts on the same day as passing the cells, start the coating process at least one-half hour to an hour before you begin the process of isolating microglia. By the end of the cell collection and counting, the inserts should be ready to use, but note because of the presence of the inserts, the washing steps take a bit of time.
Isolate microglia
-
6
Warm DMEM medium to 36° in a water bath.
-
7
Observe the cultures under the microscope to visualize the quantity of cells adhered to the monolayer.
-
8
Working in the hood, manually dislodge the microglia from the monolayer by firmly tapping the flasks by hand, 5 times on each side.
The goal here is to tap hard enough to dislodge the microglia but not so hard that the lower monolayer is disturbed, bubbles are produced or the medium splashes in the flask.
Providing resistance by firmly holding the flasks in one hand while tapping with the other is helpful. Microglia from two flasks can be dislodged at one time by stacking the flasks.
-
9
Rock the flasks back and forth 10× so that the medium washes over the surface.
-
10
Repeat steps 8 and 9 for a total of 3 times.
The number of taps and cycles can be adjusted depending on the force of your tap and the state of the cells in culture when visualized.
-
11
Observe the cells under the microscope. If more than 10–15% of the cells remain adhered to the monolayer, repeat the dislodging steps. If 85–90% of the cells have dislodged, then continue to the next step.
While the desire is to obtain the highest number of cells for passage, from these microglia cultures, it is thought that microglia remaining within the wells will act as seeds for further proliferation (Witten and Möller, 2011). Attempt to isolate about 85–90% of the starting population of microglia.
-
12
Transfer the microglia-containing medium from flasks to 50 ml tubes, using a 25 ml pipette.
-
13
Replace the flask medium by adding 25 ml warmed DMEM to each flask and return them to the incubator.
Mixed glia flasks are used for two passages of microglia, so after the first passage, the medium is replaced and the flasks returned to the incubator. The proliferative ability of the mixed glia cultures decreases with age and so each flask is retired after the second passage or after 1 to 1.5 months.
-
14
If microglia are being isolated from additional flasks, repeat steps 7–13.
-
15
When all microglia-medium has been collected, centrifuge for 7 minutes at 200 × g, (4°C).
-
16
Aspirate the supernatant leaving approximately 1 to 1.5 ml of medium.
A pellet will not be visible after centrifugation, so use care with aspiration.
-
17
Using a 5 ml serological pipette, gently resuspend the cells in the remaining solution, taking care to minimize the formation of foam or bubbles. If more than one tube of cells were centrifuged, transfer the cells to the next tube and resuspend the mixture. Continue until all cells have been resuspended.
-
18
Count the cells using a hemocytometer.
-
19
Dilute the solution in DMEM to 0.120 × 106 per ml.
Cells will be seeded at a density of 60,000 cells per insert, in a volume of 200 μl, so 120,000 cells per ml.
-
20
Aspirate the PBS from the inserts and wells. Add 1 mL of warmed DMEM to each well. Remix the cell solution and add 200 μl to each insert.
-
21
Incubate at 36.7°C.
-
22
After two days, co-culture with DIC10 CGNs (see Basic Protocol 4).
Basic Protocol 4
Primary Neuron and Microglia Co-cultures
In this protocol, inserts containing microglia (see Basic Protocol 3) are combined with neurons and co-treated for 24-hours with a toxicant of interest. As previously discussed, this co-culture system allows for the investigation of the role of microglia in mediating neuronal toxicity and allows for the measure of numerous endpoints. Additionally, various aspects of the experimental design can be modified to address particular experimental questions. For example, the length of treatment and co-culture can be adjusted to better assess the time-course of an observed effect.
Materials
Primary CGNs, DIC10 (see Basic Protocol 1)
Primary microglia in inserts, DIC2 (See Basic Protocol 2)
Hank’s balanced salt solution (HBSS) supplemented with 10 mM HEPES
PBS
NB minus AO medium
24-well inserts (3.0 μm pores)
Tweezers to transfer inserts
Steps and Annotations
-
1
Prepare treatment medium in warmed NB minus AO.
Prepare enough treatment medium for 1 ml per well and 200 μl per insert.
-
2
Wash neurons 2× with warmed HBSS.
-
3
Add 1 ml of treatment medium to each neuronal well, following the experimental plan.
-
4
Aspirate medium from the wells and inserts of microglia cultures and wash 1× with PBS. Transfer one insert to each neuronal well and add 200 μl of the appropriate treatment medium.
Cells in culture become stressed or may begin to die if left too long out of solution between wash steps. Due to the increased time it takes to aspirate, wash, empty and transfer inserts to neuronal wells, do not attempt to complete the steps above for the entire group at one time if you have more than 12 inserts, or if the cells will be outside of solution for greater than one minute. Instead, complete these steps in sets. Wash, transfer and treat one set of inserts and then proceed to the next set.
-
5
Incubate the treated neurons and microglia for 24 hours.
Depending on your experimental question, the timing of treatment can be varied. Our laboratory has not performed the co-culture for longer than 24 hours, so optimization of the system may be necessary if significantly longer time points are of interest.
-
6
After 24 hours, stop the treatment and perform the experimental assay of choice.
REAGENTS AND SOLUTIONS
Deoxyribonuclease- I (DNAse) stock solution (4 mgs/ml)
Dissolve 8 mgs DNAse from bovine pancreas in 2 ml HBSS-CMF.
Store at −20°C and avoid excessive freeze/thaw cycles to persevere enzymatic activity.
At first preparation, warm to 36°C in a water bath for 20 minutes to solubilize then sterile filter.
Hank’s balanced salt solution (HBSS)
10 mM 4-(2-hydroxyethyl)-1-piperazineethane sulfonic acid (HEPES) buffer solution
Hank’s balanced salt solution, calcium and magnesium free (HBSS-CMF)
10 mM HEPES buffer solution.
Mixed Glia and Microglial Medium (DMEM)
Dulbecco’s Modified Eagle’s Medium (DMEM) with low glucose
10% (v/v) fetal bovine serum (FBS)
100 U/mL penicillin
100 μg/mL streptomycin (P/S)
Keep sterile and store up to 1 month at 4°C.
Neuronal Medium plus antioxidant (NB)
(All listed concentrations represent final concentrations).
Neurobasal-A medium (Gibco)
2% B-27 supplement (v/v) (Gibco)
25 mM potassium chloride (KCl)
1.5 mM GlutaMax
100 μg/mL gentamicin
1 μg/mL fungizone
Neuronal Medium minus antioxidant (NB minus AO)
Follow NB medium recipe but replace B-27 supplement with 2% B-27 supplement minus antioxidant (AO) (v/v) (Gibco).
Papain
Papain from papaya latex (1.5 mg/ml)
HBSS-CMF
DNase (1:500)
5 mM MgCl2
Prepare 1 ml solution for every 2 pups, fresh at each use.
Invert tube to mix. Do not vortex.
Solution will appear cloudy and particles may appear on the side of the tube.
Place solution in water bath at 36°C to solubilize until use (minimum 30 minutes).
Just prior to use, invert tube to mix well and sterile filter.
Store papain powder at −20°C.
Poly-D-Lysine hydrobromide (PDL) stock solution (10 mg/ml)
Dissolve PDL in sterile water for a final concentration of 10 mg/ml.
To minimize loss of substrate, aliquot PDL volumes that allow minimal freeze/thaw cycles.
Store at −20°C until use.
Trypsin stock
0.25% trypsin in phosphate buffered saline (PBS).
Keep sterile during preparation.
Store in 20-ml aliquots at −20°C.
Limit aliquot re-freezing to 1× to minimize enzymatic inactivation.
Commentary
General considerations
Knowledge of the interactions between differing cell types in the central nervous system (CNS) is important for understanding the effect of toxicants on the CNS. Because each cell type may possess unique functions, disregarding these interactions in studies of effects may lead to missed opportunities to completely understand and treat disease. To study these interactions, this unit describes a co-culture system of primary CGNs and cortical microglia. Co-culture systems have been used extensively to study neuron-glia interactions (Ullian et al., 2001; Christopherson et al., 2005; Block et al., 2004; Roqué et al., 2016; Viviani, 2006, 2003). While in vivo studies reflect the systemic complexity of the CNS, the benefit of a co-culture system is that it provides a controlled, in vitro environment where each cell type can be experimentally manipulated to better understand their unique mechanistic contributions.
Studies using co-culture systems to investigate interactions between cell types span a range of experimental designs. Some studies of neuron-glia interactions use mixed cultures, where two distinct cell types are grown together, in direct contact, reflecting an important aspect of the in vivo environment (Ullian et al., 2001; Christopherson et al., 2005; Block et al., 2004). In other systems, like the sandwich co-culture system (Viviani, 2006; Roqué et al., 2011, 2014) and the one described in this unit and elsewhere (Roqué et al., 2016), neurons and glia are physically separated, but secreted factors can be exchanged. Because microglia are known to release both pro-inflammatory and oxidative factors in response to various stimuli and toxicants (Block et al., 2007; Ransohoff and Perry, 2009; Luo and Chen, 2012), the use of a co-culture system that focuses on the exchange of factors is of benefit when studying the possible role of microglia in neurotoxicity.
The protocols described here for obtaining primary microglia were modified from those of others (Witting and Möller, 2011; Ni and Aschner, 2010). A particularly important aspect of any protocol is consistency in treatment, handling and timing of cultures (Witting and Möller, 2011). Because microglia are inherently reactive cells, within and between experimental inconsistencies may induce differential effects and could result in subcultures of microglia that are either more or less activated than previous cultures, which can influence results by introducing variability. While some protocols include the addition of growth factors to mixed glia cultures to stimulate microglia proliferation and yield, in an effort to reduce baseline microglial stimulation, the method described in the present unit does not include additional factors.
Microglia are ideally isolated from PND1 to PND5 mice (Tamashiro et al., 2012; Witting and Möller, 2011), and our protocol uses PND3 pups. It may be possible to increase the yield microglia by preparing cultures from younger pups; certainly, cultures derived from older mice (e.g. PND7) result in a lower yield of microglial cells. Additionally, while others have shown that microglia from a mixed glia culture, when successively isolated three times from the same culture flask retain the phenotype of the first passage (Flode and Combs, 2007), it has been our experience that the proliferative ability of microglia decreases with the age of the culture. For this reason, we limit microglia passage from each flask to two times, or to 1 to 1.5 months after initial establishment.
Cerebellar granule neurons were chosen as a model neuronal system. Primary CGNs have the benefit of yielding high numbers of neurons for experimental use and as such, have been extensively used to study neurotoxicity (Giordano and Costa, 2011; Huang et al., 2010; Lee et al., 2009; Giordano et al., 2006; Oberdoerster, 2001). Unlike neurons from other brain regions, CGNs are the last neurons to proliferate in the brain, reaching the height of proliferation in mice between PND6 and PND8 (Lee et al., 2009; Llinás et al., 2004; Selvakumar and Kilpatrick, 2013). The ability to isolate neurons from mice after birth makes mouse colony maintenance and planning practical. Although, this unit describes methods using mice from PND7, successful cultures can also be obtained at the PND6 and PND8 time points. However, to ensure biological reproducibility between experiments, choosing one age for all experiments is recommended. Depending on the experimental focus, neurons from other brain regions may be substituted in the co-culture system to more adequately investigate a particular experimental question or region specific effects. Numerous protocols for isolating specific neuronal types from distinct areas of the brain are readily available (Roqué et al., 2011; Viviani, 2006; Giordano and Costa, 2011; Hilgenberg and Smith, 2007; Kaech and Banker, 2006; Roqué et al., 2014).
The co-culture system described in this Unit makes use of CGNs from PND7 mice and cerebral cortex microglia from PND3 mice; hence, some comments on the relevance of animal age and on brain regions are of relevance. Whereas microglia are best cultured early post-natally (Ransohoff and Cardona, 2010), except in the case of CGNs, which are the last neurons to proliferate in the brain, the most opportune time to successfully culture primary neurons from mice is from the late-prenatal period. However, a controlled co-culture system in which mice of different ages can be used to generate two distinct cultures is advantageous to provide maximal cell yield. In addition to age of mice, the fact that cells of the same type developing and residing in different brain regions have been found to be heterogeneous, should also be considered (Grabert et al., 2016; Oberheim et al., 2006; Schitine et al., 2015; Block et al., 2007). Ideally, to more accurately reflect the in vivo environment and the potentially unique functions of cells in the various microenvironments, using neurons and microglia from the same region in a co-culture system would be preferred. The decision to use microglia isolated from the cerebral cortex to study effects on cerebellar neurons was mainly, if not solely, practical. It is commonly experienced that obtaining high numbers of primary microglia is challenging, for a number of reasons. First, the number of microglia in the brain is relatively low compared to other cell types. Second, microglia are differentially distributed between brain regions, with the percentage of microglia ranging from 5% to 17% (Block et al., 2007). The substantia nigra has the highest number of microglia relative to other regions, with the relative numbers in the cortex higher than that in the cerebellum (Block et al., 2007; Yang et al., 2013; Lawson et al., 1990). If the co-culture system is to be used to investigate specific regional effects, consideration should be given to whether using cells of the same brain region would be the more appropriate study design, given these issues. Using the protocol to isolate microglia presented here, we have successfully cultured primary cerebellar microglia. In parallel experiments, we have found that the effect on neuronal survival after treatment with diesel exhaust particles was not different when neurons were cultured in the presence of primary cerebellar or cortical microglia (Roqué et al., 2016).
Critical Parameters
When working with a co-culture system, planning becomes an important part of the experimental design. Since the timing of two distinct culture systems, each requiring different tasks at different times, must be “aligned”, careful planning of the experiments at the start is crucial. In particular, the timing of microglia isolation may present the most difficulty, since the yield can be the limiting factor. To ensure sufficient numbers of microglia, we have prepared multiple mixed glial cell preparations per week. Since each flask is expected to provide two passages of microglia, weekly preparations of mixed glia cultures may allow combining microglia from two independent preparations.
For the health and survival of the mixed glia cultures, medium changes are performed at 8 and at 15 days. Waiting too long, without replacing the nutrients necessary for the glial survival, especially at the later medium change when the cultures contain more cells in a confluent monolayer, can cause cell death and the astrocytic layer may begin to lift from the surface of the flask. To gain familiarity with the typical development of the mixed glia cultures and the morphology of the primary microglia when they are sub-cultured, consistent observation of the cultures is key, especially when first working with this protocol. In our experience, maximum proliferation is usually reached by 15 days after initial establishment of mixed glia cultures; however, at times sufficient numbers of microglia may be available as early as 13 days. Having knowledge of the rate of proliferation and a qualitative understanding of how the visualized number of microglia on the monolayer relates to cell counts when isolated will help estimating the most appropriate time within this window to isolate microglia. This also allows for recognition of cultures that may veer from typical. For example, abnormally high number of microglia outside of what is typically seen could be indicative of contamination within the flask.
Because it is well understood that primary microglia in culture are generally in a slightly more activated phenotype at baseline compared to highly ramified microglia in vivo (Witting and Möller, 2011; Flode and Combs, 2007; Szabo and Gulya, 2013), becoming familiar with the morphology of microglia sub-cultures is important. Figure 2 shows representative images of primary microglia prepared using this protocol. While cells are plated in 6-well plates, the images provide visualizations of microglia morphology in various states, and include control wells, an atypical, over-activated control well, and wells treated with DEP and lipopolysaccharide (LPS) for 24 hours. Microglia in inserts can and should also be visualized before and after treatment, using a light microscope.
Figure 2.
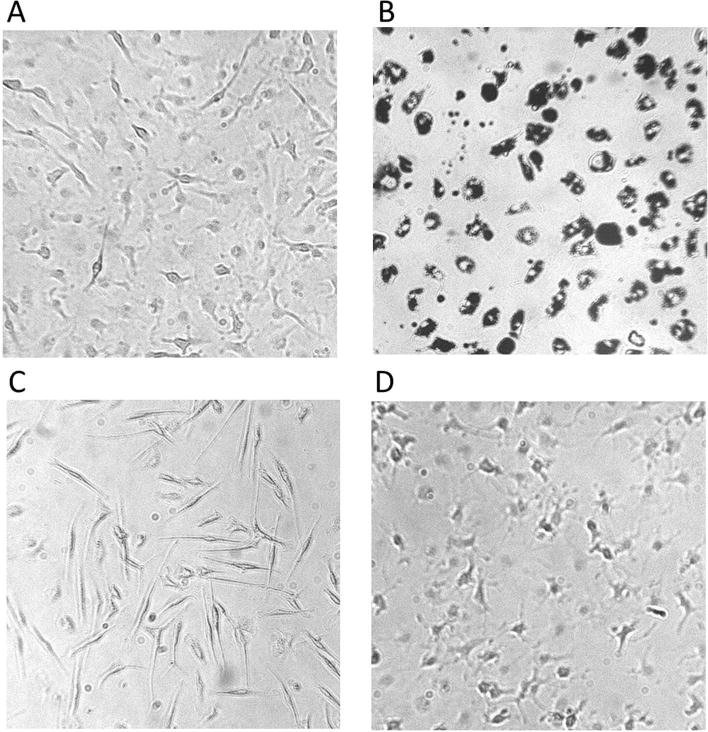
Sub cultured microglia morphology post-24 hour treatment (control (A), DEP (50 μg/2 cm2) (B), LPS (500 ng/mL) (D), and atypical, activated control (C)
In the co-culture system presented here, microglia are sub-cultured two days prior to co-culture. This timing allows microglia, which are functionally reactive inherently, to acclimate after the initial plating. While a longer period of time may be reasonable, our laboratory had not explored alternative timing, so proper maintenance protocols may need to be developed if a longer time frame is used. Again, it is important to remain consistent with the timing of medium changes and microglia plating so that the microglia remain in a mostly similar activation state between experiments to ensure reproducibility. Finally, one should always remember to work using sterile practices at all stages of the co-culture procedures.
Troubleshooting
Additional considerations
Anticipated Results
Microglia Cultures
In our experience, the first passage of microglia from each flask of mixed glia from 4–6 pups will yield approximately 0.5 × 106 to 0.6 × 106 per flask; thus, one typical mouse litter of 8–12 pups will be generate 2 to 3 flasks, or approximately 1 – 1.2 × 106 microglia by the end of 15 days in culture. With cells plated into inserts at a density of 60,000 cells per insert, one flask should allow plating of 8–10 inserts per flask, or enough for three treatment groups in triplicate. Cell numbers are reduced during the second passage of microglia, thus the isolation of microglia from more than two flasks is suggested. The dissociation process of cortical mixed glia may result in initial cultures that have high amounts of cellular debris visible in the medium and on the surface of the flask; this is typical. The first wash, and changing medium one day after seeding will remove a significant amount of this debris, although some will remain.
CGN cultures
The protocol described for isolating CGNs produces a relatively high number of cells. With practice one should expect to obtain 2.5–3.0 × 106 neurons per pup. When plating in triplicate, one treatment group would then require 1.8 × 106 neurons (6 × 105 per well × 3), thus even a small litter of mice is usually sufficient. At the first medium change, one hour after plating, the majority of cells adhere to the surface of the well, but a significant number that die and are floating in the medium. This is also the case at the DIC4 medium change. This is not atypical, and washing the cells at these steps will remove the dead cells. By DIC10, the neurons will appear well developed and will be connected by extensive networks.
Time Considerations
An important aspect of the neuron-microglia co-culture system is significant planning, so that the maturation of the CGN cultures and sufficient microglia numbers at the time of isolation coincide. Neurons and microglia are co-cultured after CGNs have been grown for 10 days, while the time from the preparation of mixed glial cultures to the isolation of primary microglia cultures is about 14–15 days, and enough microglia must be available for seeding into inserts two-days prior to co-culturing (see Critical Parameters). The time necessary for individual tasks varies and will most likely change and improve as experience with the procedure increases. The dissection and preparation of primary cells is the task that takes the longest and requires a dedicated block of time. From set-up to dissection and dissociation, a good estimate of the CGN preparation is between 2.5–3 h, with the main variables affecting that estimate being the number of pups dissected and one’s experience with dissecting the tissue. There is one-half hour wait time for enzymatic tissue digestion which is included in this time frame. One hour should be added to this estimate to account for the change of medium after initial plating of the cells, although this is time that can be used for other tasks. Generally, because there are fewer steps in the protocol for isolating and preparing the mixed glia cultures and there is no cell counting step, the time to complete the cortical glia preparation is slightly less than the time to prepare CGNs. Sub-culturing microglia into inserts can be completed in approximately one hour once medium is warmed and if the inserts have been pre-coated with PDL. In both the case of neurons and microglia, more time should be allotted if animals are separated either by sex or genotype, since distinct groups will require additional trituration and cell counting steps. The time necessary for the co-culturing of microglia and neurons is not that extensive, ranging from 1 to 2 h, and is influenced mostly by the complexity of the treatment paradigms planned, the number of treatment groups, and the number or wells. These steps are straight forward and include warming the medium, preparing the treatment solutions, washing neurons and microglia and ultimately transferring the inserts to the neuronal culture and adding the medium.
Table 1.
| Problem | Possible Cause | Solutions |
|---|---|---|
|
| ||
| Low CGN yield: | ||
| High neuronal death | Dissection of cerebellum too long | Work quickly, aim for 1–1.5 minutes per pup |
| Trituration too harsh, too long | Work quickly and triturate gently | |
|
| ||
| Tissue or cells lost during preparation | Incomplete dissociation of tissue to cells so tissue held in filter; | Ineffective DNase; reduce freeze-thaw numbers; make fresh DNase; Add additional DNase |
| Cells lost during aspiration steps | Aspirated carefully to not disturb the pellet; remove supernatant with 5 ml serological pipette | |
|
| ||
| Low microglia yield: | Cells lost during preparation of mixed glia cultures | Aspirate carefully or remove supernatant with a 5 ml serological pipette. These cells proliferate, so culture may be established, but delayed. |
| At microglia passage | Cells lost at aspiration due to no visible pellet | Be cautious when aspirating cells. Leave at least 1 ml medium remaining before resuspension. |
| Too few flasks of mixed glia | Increase number of preparations or number of pups per preparations | |
Significance.
Glial cells make unique and important contributions to normal and pathological neuronal development and function. Microglia, the resident immune cells of the brain, have been implicated in a number of neurodegenerative and neurodevelopmental diseases. Using an experimentally flexible and controlled in vitro system, where primary neurons and microglia are first grown separately and then co-cultured, allows for the design of experiments to investigate the contribution of microglia to the effect of toxicants on neuronal function and survival. Comparing the effect on neurons alone with that occurring in the presence of microglia can provide an understanding of the contributions of microglia to neuronal function or viability, offering opportunities to develop ways to treat and prevent negative CNS effects.
Acknowledgments
Work by the authors is supported by grants from NIEHS (R01ES22949, P30ES07033, P42ES04696)
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Literature Cited
- Banker G. Culturing Nerve Cells. 2nd. Massachusetts Institute of Technology; Boston, MA: 1998. pp. 9–36. [Google Scholar]
- Block ML, Wu X, Pei Z, Li G, Wang T, Qin L, Wilson B, Yang J, Hong JS, Veronesi B. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: the role of microglia, phagocytosis, and NADPH oxidase. FASEB Journal. 2004;18:1618–1620. doi: 10.1096/fj.04-1945fje. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Flode AM, Combs CK. Microglia repetitively isolated from in vitro mixed glial cultures retain their initial phenotype. Journal of Neuroscience Methods. 2007;164:218–224. doi: 10.1016/j.jneumeth.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Costa LG. Primary neurons in culture and neuronal cell lines for in vitro neurotoxicological studies. Methods in Molecular Biology (Clifton, N.J.) 2011;758:13–27. doi: 10.1007/978-1-61779-170-3_2. [DOI] [PubMed] [Google Scholar]
- Giordano G, White CC, McConnachie LA, Fernandez C, Kavanagh TJ, Costa LG. Neurotoxicity of domoic Acid in cerebellar granule neurons in a genetic model of glutathione deficiency. Molecular Pharmacology. 2006;70:2116–2126. doi: 10.1124/mol.106.027748. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. Clinical & Developmental Immunology. 2013;2013:208093. doi: 10.1155/2013/208093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nature Neuroscience. 2016;19:504–516. doi: 10.1038/nn.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Li W, Rodriguez ML. Role of microglia in CNS inflammation. FEBS letters. 2011;585:3798–3805. doi: 10.1016/j.febslet.2011.08.033. [DOI] [PubMed] [Google Scholar]
- Hilgenberg LGW, Smith MA. Preparation of Dissociated Mouse Cortical Neuron Cultures. Journal of Visualized Experiments : JoVE. 2007;10 doi: 10.3791/562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Giordano G, Costa LG. Comparative cytotoxicity and intracellular accumulation of five polybrominated diphenyl ether congeners in mouse cerebellar granule neurons. Toxicological Sciences. 2010;114:124–132. doi: 10.1093/toxsci/kfp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nature Protocols. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Harry GJ. Features of Microglia and Neuroinflammation Relevant to Environmental Exposure and Neurotoxicity. International Journal of Environmental Research and Public Health. 2011;8:2980–3018. doi: 10.3390/ijerph8072980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lee HY, Greene LA, Mason CA, Manzini MC. Isolation and culture of post-natal mouse cerebellar granule neuron progenitor cells and neurons. Journal of Visualized Experiments: JoVE. 2009;23 doi: 10.3791/990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás RR, Walton KD, Lang EJ. Cerebellum. In: Shepherd GM, editor. The Synaptic Organization of the Brain. Oxford University Press; 2004. pp. 271–310. [Google Scholar]
- Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Translational Neurodegeneration. 2012;1:9. doi: 10.1186/2047-9158-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnet-Tschudi F, Defaux A, Braissant O, Cagnon L, Zurich MG. Methods to assess neuroinflammation. Current Protocols in Toxicology. 2011;50:12.19.1–12.19.20. doi: 10.1002/0471140856.tx1219s50. [DOI] [PubMed] [Google Scholar]
- Ni M, Aschner M. Neonatal rat primary microglia: isolation, culturing, and selected applications. Current Protocols in Toxicology. 2010;43:12.17.1–12.17.16. doi: 10.1002/0471140856.tx1217s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerster J. Isolation of cerebellar granule cells from neonatal rats. Current Protocols in Toxicology. 2001;12:12.7.1–12.7.10. doi: 10.1002/0471140856.tx1207s09. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends in Neurosciences. 2006;29:547–553. doi: 10.1016/j.tins.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Qian L, Flood PM. Microglial cells and Parkinson’s disease. Immunologic Research. 2008;41:155–164. doi: 10.1007/s12026-008-8018-0. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Cardona AE. The myeloid cells of the central nervous system parenchyma. Nature. 2010;468:253–262. doi: 10.1038/nature09615. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annual Review of Immunology. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Roqué PJ, Dao K, Costa LG. Microglia mediate diesel exhaust particle-induced cerebellar neuronal toxicity through neuroinflammatory mechanisms. Neurotoxicology. 2016;56:204–214. doi: 10.1016/j.neuro.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roqué PJ, Guizzetti M, Costa LG. Synaptic structure quantification in cultured neurons. Current Protocols in Toxicology. 2014;60:12.22.1–12.22.32. doi: 10.1002/0471140856.tx1222s60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roqué PJ, Guizzetti M, Giordano G, Costa LG. Quantification of synaptic structure formation in cocultures of astrocytes and hippocampal neurons. Methods in Molecular Biology (Clifton, N.J.) 2011;758:361–390. doi: 10.1007/978-1-61779-170-3_25. [DOI] [PubMed] [Google Scholar]
- Schitine C, Nogaroli L, Costa MR, Hedin-Pereira C. Astrocyte heterogeneity in the brain: from development to disease. Frontiers in Cellular Neuroscience. 2015;9:76. doi: 10.3389/fncel.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvakumar T, Kilpatrick DL. Culturing mouse cerebellar granule neurons. Methods in Molecular Biology (Clifton, N.J.) 2013;1018:49–59. doi: 10.1007/978-1-62703-444-9_5. [DOI] [PubMed] [Google Scholar]
- Spijker S. Dissection of Rodent Brain Regions. In: Li KW, editor. Neuroproteomics Neuromethods. Humana Press; 2011. pp. 13–26. [Google Scholar]
- Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–266. [PubMed] [Google Scholar]
- Szabo M, Gulya K. Development of the microglial phenotype in culture. Neuroscience. 2013;241:280–295. doi: 10.1016/j.neuroscience.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Takano T. Role of Microglia in Autism: Recent Advances. Developmental Neuroscience. 2015;37:195–202. doi: 10.1159/000398791. [DOI] [PubMed] [Google Scholar]
- Tamashiro TT, Dalgard CL, Byrnes KR. Primary Microglia Isolation from Mixed Glial Cell Cultures of Neonatal Rat Brain Tissue. Journal of Visualized Experiments : JoVE. 2012:e3814. doi: 10.3791/3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of Synapse Number by Glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Viviani B. Coculturing neurons and glial cells. Current Protocols in Toxicology. 2003;15:12.10.1–12.10.17. doi: 10.1002/0471140856.tx1210s15. [DOI] [PubMed] [Google Scholar]
- Viviani B. Preparation and coculture of neurons and glial cells. Current Protocols in Cell Biology. 2006;32:2.7.1–2.7.21. doi: 10.1002/0471143030.cb0207s32. [DOI] [PubMed] [Google Scholar]
- Witting A, Möller T. Microglia cell culture: a primer for the novice. Methods in Molecular Biology (Clifton, N.J.) 2011;758:49–66. doi: 10.1007/978-1-61779-170-3_4. [DOI] [PubMed] [Google Scholar]
- Yang TT, Lin C, Hsu CT, Wang TF, Ke FY, Kuo YM. Differential distribution and activation of microglia in the brain of male C57BL/6J mice. Brain Structure & Function. 2013;218:1051–1060. doi: 10.1007/s00429-012-0446-x. [DOI] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature Neuroscience. 2014;17:400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]