

Abstract
The mitogen-activated protein kinases (MAPKs) participate in a multitude of processes that control hepatic metabolism. The liver regulates glucose and lipid metabolism and under pathophysiological conditions, such as obesity, type 2 diabetes mellitus, and non-alcoholic fatty liver disease these processes become dysfunctional. Stress responses activate the hepatic MAPKs which, is thought to impair insulin action and lipid metabolism. The MAPKs also activate the MAPK phosphatases which, oppose their actions. How the MAPK/MKP balance is controlled in liver metabolism and how perturbations in these activities contribute to metabolic disease remains unclear. A discussion of recent insights into the MAPK/MKP signaling role in hepatic metabolic function and disease will be the focus of this review.
Keywords: Mitogen-activated protein kinase, liver, metabolism, signal transduction, obesity, type 2 diabetes
Overview: Mitogen-activated protein kinases in liver metabolism
Obesity predisposes to the development of type 2 diabetes mellitus (T2DM) [1]. In the liver, T2DM manifests as insulin resistance, altered glucose homeostasis and the accumulation of fat (hepatosteatosis) [2]. The progressive increase in hepatic fat content leads to non-alcoholic fatty liver disease (NAFLD) which, can subsequently be accompanied by hepatic inflammation leading to non-alcoholic steatohepatitis (NASH) [3]. The rising obesity epidemic has resulted in an increased incidence of the transition from hepatosteatosis to NASH which, represents a significant risk factor for the development of liver cancer [4]. Therefore, understanding the underlying mechanisms that govern the development of hepatosteatosis to NASH will play an important role towards identifying new modalities of therapeutic intervention.
In the liver, the mitogen-activated protein kinases (MAPKs) play a prominent role in processes that regulate metabolism [5–7]. Mammalian MAPKs comprise of 3 major sub-groups that are classified based on sequence similarity, differential activation by agonists and substrate specificity. These include the extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun NH2-terminal kinases 1, 2 and 3 (JNK1/2/3) and p38α/β/δ/γ MAPKs [8–11]. Because obesity and the associated inflammatory state in insulin-responsive tissues activate the stress-responsive MAPKs, such as the p38 MAPKs and JNKs, the notion that these MAPKs drive hepatic metabolic dysfunction has become an entrenched working hypothesis [3, 12]. As such, the stress-responsive MAPKs, namely p38α/β MAPK and JNK1/2, have figured prominently as critical effectors of physiological and pathophysiological hepatic metabolism. In contrast, evidence for ERK1/2 in hepatic metabolism, which is activated preferentially by growth factors rather than stressors associated with obesity, has been less well developed. The MAPK kinases (MKKs) phosphorylate the MAPKs on regulatory threonine and tyrosine residues resulting in MAPK activation [13, 14]. Conversely, the MAPKs are inactivated by direct dephosphorylation of their regulatory threonine and tyrosine residues. The inactivation of the MAPKs is catalyzed by MAPK dephosphorylation by a group of dual-specificity protein tyrosine phosphatases (DUSPs) called MAPK phosphatases (MKPs) [15]. It is the balance between the actions of the upstream MAPK activators (MKKs) and downstream MAPK inactivators (MKPs), that sets the cellular strength of downstream MAPK signaling [16]. Indeed, mathematical modeling of the MAPK signaling network indicates that the MKPs play an exquisite rate-limiting role in controlling the magnitude and temporal kinetics of MAPK activity [17]. Thus, modest alterations in the regulation of the MKPs are likely to result in more profound and deleterious outcomes on the fidelity of MAPK signaling. In this review, we will discuss current evidence supporting the role of MAPK signaling in hepatic metabolism and incorporate studies on the MKPs that dephosphorylate them in order to fully illustrate the complexity of MAPK regulation in liver metabolism. It will become evidently clear that despite the fact that a substantial amount of work on the role of the MAPKs has been accomplished in this area, an understanding of whether the MAPKs are definitive causal drivers of hepatic metabolic disease in some cases remains to be resolved. Furthermore, how the MAPKs are in balance with the MKPs in hepatic metabolic function still presents as a challenging avenue that requires resolution. Here, we will not attempt to reconcile these complexities but we will merely attempt to illustrate the key issues and questions of functional conflicts of the hepatic MAPK/MKP signaling axis.
JNK in hepatic metabolism and pathophysiology
There are three major JNK isoforms, JNK1 (Mapk8), JNK2 (Mapk9) and JNK3 (Mapk10). JNK3 is not expressed in the liver and thus, studies on hepatic JNKs have focused on JNK1 and JNK2. Here, we will examine the more recent findings of the JNKs in liver metabolism, suffice to say, earlier insights into the actions of JNK and metabolism that emerged through the earlier work of Hotamisligil and others will not be covered, but are discussed thoroughly elsewhere [18].
JNK has been shown to be activated in the liver of rodent models fed a high-fat diet (Table 1). In humans, JNK activation in the liver has also been observed during states of obesity and NASH (Table 2). However, other reports suggest little difference in either the expression of JNK or its phosphorylation levels in the livers patients with NASH (Table 2). One explanation for the increased levels of JNK activity at least under conditions of high-fat feeding has been attributed directly to saturated free fatty acids that accumulate in obesity which, has been shown to activate JNK [19]. Pro-inflammatory cytokines that are overexpressed in states of obesity activate JNK through the actions of the MKPs that are inactivated as a result of increased levels of reactive oxygen species [20]. Nonetheless, there appears to be a limited data set on the activation status of JNK in the liver during situations of obesity, T2DM and NASH/NAFLD in both rodents and humans (Table 1 and Table 2). Despite the well-documented involvement of JNK in hepatic metabolism, discussed below, the contribution of JNK hyperactivation per se as a driver of hepatic metabolic dysfunction in states of obesity, T2DM and NASH/NAFLD still requires further study.
Table 1.
Recent studies using genetic animal models to investigate the role of the MAPKs in the liver
| MAPK | Genetic manipulation/other strategies | Phenotype in knock-out mouse models | Hepatic MAPK expression in obesity | References |
|---|---|---|---|---|
| JNK1/2 | Liver-specific deletion of JNK1 and JNK2 in mice | Protected from diet-induced obesity and exhibit enhanced insulin sensitivity. Activation of hepatic PPARα and its target gene FGF21. | Undetermined | [23] |
| JNK1 | Role of JNK1 signaling in autophagy and insulin resistance in NAFLD rat model | Rats fed a high-fat diet exhibit impaired glucose tolerance and hepatic injury, hepatic insulin resistance and enhanced autophagy. | Increase expression of phosphorylated JNK1 in obesity | [27] |
| JNK1 | Liver-specific deletion of JNK1 in mice | Reduced hepatic stellate cells derived extracellular matrix deposition and fibrosis. | Undetermined | [55] |
| p38α MAPK | Liver-specific p38α MAPK knock-out mice | Decreased fasting glucose and impaired gluconeogenesis associated with enhanced AMPKα phosphorylation. | Enhanced expression of p38 MAPK in obesity | [40] |
| p38α MAPK | Ob/ob whole body knock-out mice | Activation of p38 MAPK enhanced nuclear translocation of Xbp1s, decreased ER stress and maintained euglycemia in obese and diabetic mice. | Reduced hepatic p38 MAPK phosphorylation in ob/ob and HFD fed obese mice | [43] |
| p38α MAPK | Effect of p38 MAPK inhibition on free fatty acid-induced insulin resistance in rats | Inhibition of p38 MAPK blocked lipid-induced hepatic insulin resistance and is associated with attenuated activation of ATF2 | Undetermined | [45] |
| ERK1 | Ob/ob/ERK1 whole body double knock-out mice | Enhanced whole body insulin sensitivity, partially protected against hepatosteatosis | Undetermined | [54] |
| ERK2 | ERK2 liver-specific knock-out mice | High-fat/high-fructose diet fed ERK2 liver-specific KO mice exhibit hepatic steatosis, glucose intolerance and reduced insulin sensitivity. | No increase in hepatic ERK phosphorylation in obesity | [48] |
| ERK1 | ERK1 whole body knock- out mice | Decreased adiposity and are resistant to diet-induced obesity and insulin resistance. | No increase in hepatic ERK phosphorylation in obesity | [46] |
| ERK1 | ERK1 whole body knock- out | Increased weight gain and increased insulin resistance in diet- induced obesity | Increase in ERK2 phosphorylation in obesity | [47] |
Table 2.
Recent studies assessing hepatic expression of MAPKs in obese humans
| MAPK | Metabolic disorder/disease | Hepatic MAPK expression in obesity and NASH/NAFLD | References |
|---|---|---|---|
| JNK1/2 | Liver samples from patients with steatosis and NASH | No difference in hepatic JNK phosphorylation in NASH patients | [56] |
| JNK1 | Liver biopsies from obese patients with NASH | Elevated hepatic JNK phosphorylation in obese NASH patients | [57] |
| JNK1/2 | Liver biopsies from obese patients with hepatic steatosis/NASH | Increased hepatic JNK phosphorylation in obese NASH patients | [58] |
| p38δ MAPK | Liver biopsies from obese patients with hepatic steatosis/NAFLD | Increased hepatic expression of p38δ in obese patients with NAFLD | [44] |
Much of the advancement in our understanding of the roles of JNK in hepatic metabolism has been elegantly demonstrated by the Davis group. This group, have generated a variety of liver-specific JNK-deficient mouse models [21–23] (Table 1). Mice with hepatic deficiency of JNK1 exhibit increased insulin resistance [21] whereas, a compound deletion of both JNK1 and JNK2 in the liver, results in enhanced glucose and insulin tolerance, increased hepatic insulin action and reduced fasting blood glucose when these mice are fed a high-fat diet [23]. Interestingly, hepatic deletion of JNK2 alone recapitulates the enhanced insulin sensitivity of the JNK1/JNK2 hepatic deletion, suggesting that JNK2 rather than JNK1, contributes to the progression of obesity-induced hepatic insulin sensitivity (Figure 1). These results highlight an unappreciated level of complexity amongst the JNK isoforms where there appear to be differences in the substrate specificity between hepatic JNK1 and JNK2. Although there is evidence to suggest a molecular basis for the presumed differential substrate specificity between JNK1 and JNK2 [24], the precise substrates that are differentially phosphorylated in the liver to evoke these unique effects on hepatic glycemic control remain to be determined. Furthermore, this raises the question of why these isoforms might have different substrate selectivity’s, to begin with. Given the fact that both JNK1 and JNK2 are targets of nutritional stress that are registered in the liver, how these seemingly distinct responses to glycemic control are reconciled physiologically is unclear. The unique nature of the behavior of these two JNK isoforms highlights an apparently challenging problem relating to the potential therapeutic targeting of the JNKs for the control of glucose homeostasis in states of T2DM. Targeting of JNK2 would clearly be of benefit but concerns relating to inhibition of hepatic JNK1 would warrant careful consideration. Moreover, it has not been shown whether JNK2 is preferentially responsive to excess nutrient loads that are associated with T2DM. Nevertheless, it would be important to understand how these two hepatic JNK isoforms are integrated under physiological conditions as well as under conditions of obesity and T2DM in the liver.
Figure 1.
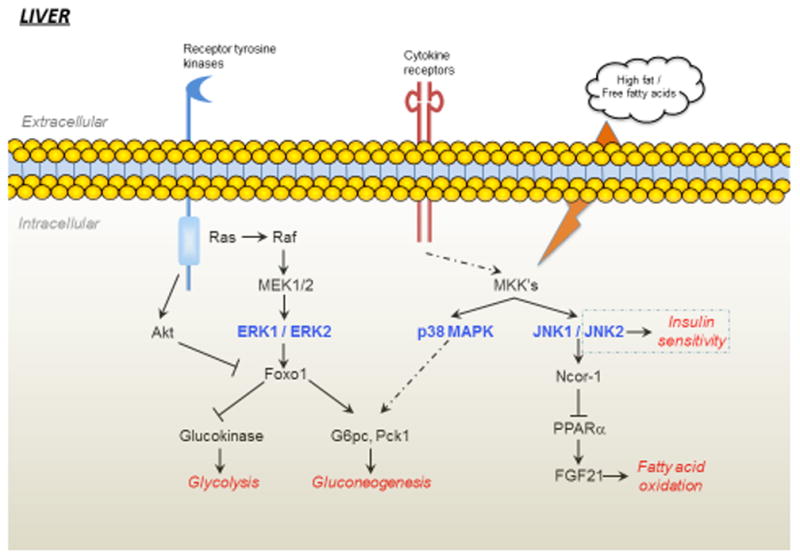
Growth factor receptors, cytokine receptors and free fatty acids can stimulate the activity of ERK, JNK and p38 MAPK in the liver. ERK is activated through the Ras-Raf-MEK cascade and can influence gluconeogenesis by phosphorylating Foxo1. Stress conditions such as in obesity can lead to increased cytokine activity and increased free fatty acid levels can generate reactive oxygen species resulting in the activation of p38 MAPKs and JNKs which, can lead to dysregulation of hepatic gluconeogenesis and fatty acid synthesis. In the liver, JNK2 appears to be mainly responsible for the regulation of insulin sensitivity. Whilst the activity of JNK1/2 maintains NCor1 expression which inhibits PPARα-dependent inhibition of FGF21 leading to reduced fatty acid oxidation.
The availability of lipids through the regulation of lipogenesis, fatty acid oxidation and triglyceride esterification and secretion are controlled in the liver. Postprandial increases in insulin levels which, serves to shut down the synthesis and storage of lipids, has been reported to be accompanied by a decrease in JNK activation [22]. In mice lacking the expression of hepatic JNK1 and JNK2 the expression of the peroxisome proliferator-activated receptor-alpha (PPARα) and its target genes involved in oxidative metabolism that enhances hepatic β-oxidation are activated [23] thereby, increasing lipid breakdown under conditions of reduced nutritional load. Interestingly, the pro-metabolic hormone fibroblast growth factor 21 (FGF21) is positively regulated by PPARα in the liver [23]. Feeding stimulates JNK activation concomitant with the downregulation of FGF21 and ablation of hepatic JNK abrogates this nutritional responsiveness. Hence, it is proposed that hepatic JNK mediates the fasting and feeding cues of circulating FGF21 expression [23]. In contrast to the studies by Vernia et al [22, 23], it has also been reported that p38α/β MAPK, rather than JNK, is involved in the regulation of FGF21 [25]. Mice lacking the expression of MKP-1 in the liver exhibit decreased levels of circulating FGF21 under conditions in which both hepatic p38α/β MAPK and JNK are elevated [25]. Activated mutants of the upstream MAPK kinases for JNK failed to have an effect on a minimal FGF21 promoter but rather a negative regulatory effect on p38α /β MAPK to control FGF21 through a PGC-1α-dependent pathway [25]. Hence, hepatic MKP-1, and its effects on p38α /β MAPK appears to play an integral role in the regulation of FGF21 in the liver. It certainly will be important to determine why there is an apparent difference in the MAPK targets involved in the regulation of FGF21 in the liver. It is also conceivable that inactivation of JNK interferes with the actions of p38α/β MAPK signaling in the liver which, could lead to the regulation of FGF21 expression. These possibilities will need to be investigated so as to further define how JNK in the liver regulates FGF21 expression.
Autophagy plays an essential role in hepatic lipid metabolism [26]. Using diet-induced obesity in rats Yan et al. examined the role of JNK1-mediated autophagy in the development of insulin resistance [27]. They found that inhibition of JNK reduced autophagy and improved hepatic insulin resistance suggesting that overexpression of JNK1 in obesity promotes NAFLD [27]. One aspect of JNK function on hepatic metabolism that must also be considered is its effect on hepatic metabolism as a result of its expression in other tissues. For example, it has been shown that deletion of JNK1 in skeletal muscle increases the susceptibility to hepatosteatosis in response to high-fat feeding [28]. Additionally, high-fat diet feeding of mice with a deletion of JNK1 in macrophages results in protection against the development of hepatic insulin resistance [29]. These examples, implicate JNK as engaging in complex tissue cross-talk that influences hepatic metabolism. On another level of cross-talk, JNK has been shown to integrate with other MAPK family members, such as p38α /β MAPK and ERK pathways [30, 31]. However, whether p38α /β MAPK and ERK activities are altered in the liver-specific JNK-deficient mice is unclear, and if so, how do these MAPKs in a JNK-deficient context influence hepatic metabolism? It certainly will be important to ascertain the relative contribution and the mechanistic relationship of p38α /β MAPK, and possibly ERK, in hepatic JNK-deficient mice as it relates to hepatic metabolism.
p38 MAPK in hepatic metabolism and pathophysiology
The p38 MAPK family is comprised of four isoforms encoded by Mapk14 (p38α) Mapk11 (p38β), Mapk12 (p38γ) and Mapk13 (p38δ). p38 MAPKs are stress-responsive MAPKs that are activated by a variety of stresses in addition to cytokines and growth factors [10, 32, 33]. The p38 MAPK signaling module consists of MAPKKK-MKK3/4/6-p38MAPK [10]. Expression patterns differ depending on the isoform, p38α and p38β MAPKs, are widely expressed and are the most extensively studied [34]. In contrast, p38γ MAPK is expressed mainly in skeletal muscle, whereas p38δ MAPK is expressed predominately in the small intestine, pancreas, testis, and kidney. In addition, the level of expression of these isoforms as well as their upstream activators, MKK3 and MKK6, differ across cell types [35]. In the liver, the p38α/β MAPK isoforms are expressed predominately. Previous studies have demonstrated a role for p38α/β MAPK in the promotion of energy expenditure, glucose homeostasis and lipid metabolism [36–38]. Here, we will discuss primarily the actions of hepatic p38α/β MAPK function in the maintenance of energy homeostasis and the role of p38 MAPK in T2DM and obesity.
Several studies have demonstrated a role for hepatic p38α/β MAPK in the control of glucose homeostasis in animal models (Table 1). During states of obesity in rodent models, reports of upregulation as well as downregulation of p38α/β MAPK activity have been observed (Table 1). In humans, the expression of only p38δ MAPK has been shown to be elevated in the livers of obese patients with NAFLD (Table 2). With regards to the actions of hepatic p38α/β MAPK, it has been shown that p38α/β MAPK stimulates hepatic gluconeogenesis [39]. Several lines of evidence demonstrate that p38α/β MAPK drives activation of gluconeogenic genes. In mice, one study utilized hepatic deletion of p38α MAPK and showed that these mice exhibited reduced fasting glucose and impaired gluconeogenesis that was associated with increased AMPKα phosphorylation [40]. This study demonstrates that p38α MAPK regulates gluconeogenesis in an AMPK-dependent manner and suggests a possible mechanism for the development of hepatic insulin resistance. Further evidence for a role for p38α/β MAPK in gluconeogenesis is derived from work studying MKP-1. MKP-1 liver-specific knockout mice exhibit enhanced G6pc and Pck1, suggesting that MKP-1 negatively regulates gluconeogenesis by opposing p38α/β MAPK- and/or JNK-mediated activation of the gluconeogenic program [25]. These studies establish an important role for hepatic MKP-1 and hence the MAPKs in the regulation of gluconeogenesis (Table 3). Furthermore, studies utilizing genetic approaches in mice demonstrate that MKP-1 plays an important role in the dephosphorylation of p38α/β MAPK to negatively regulate hepatic triglyceride metabolism and management of lipid homeostasis [41, 42] (Figure 2). With regards to the role of MKP-1 in obesity, it has been observed that MKP-1 is overexpressed in mice fed a high-fat diet and this correlates with the development of hepatic steatosis [25], suggesting that hepatic p38α/β MAPK declines in states of obesity. Indeed, it has been reported that hepatic p38α/β MAPK activity decreases in mice fed a high-fat diet [43]. Therefore, the pro-metabolic actions of hepatic p38α/β MAPK activity become blunted in obesity which is, in part, related to the upregulation of MKP-1. This suggests a re-thinking of the role for at least p38α/β MAPK in liver-related metabolic dysfunction where it seems that reduced rather than increased p38α/β MAPK signaling may be responsible for hepatic metabolic dysfunction. It is conceivable that other p38 MAPK isoforms, such as p38δ MAPK which is increased in obesity [44] (Table 2), likely engages in distinct signaling effects as compared with the p38α/β MAPK isoforms.
Table 3.
Recent studies using genetic animal models to investigate the role of MKPs in hepatic metabolism
| Gene/Prote in | Subcellular Localization | Substrate Specificity | Genetic manipulation/Other strategies | Phenotype in knock-out mouse models | Hepatic MKP expression in obesity | References |
|---|---|---|---|---|---|---|
| MKP-1 (DUSP1) | Nuclear | JNK, p38α/β MAPK | Leptin receptor-deficient (db/db) mice lacking MKP-1 | db/db;mkp-1−/− mice exhibit enhanced hepatic β-oxidation and are protected from hepatic steatosis. | Undetermined | [41] |
| MKP-1 (DUSP1) | Nuclear | JNK, p38 α/β MAPK | MKP-1 liver- specific knock-out mice | MKP-1 liver- specific KO mice exhibit hepatic insulin resistance and are protected from the development of hepatic steatosis by MKP-1 to regulating FGF21. | Overexpression of MKP-1 in obesity | [25] |
| MKP-3 (DUSP6) | Cytosolic | ERK | MKP-3 whole-body knock-out mice | Resistant to diet-induced obesity, protection from hepatic steatosis, increased energy expenditure, improved systemic insulin sensitivity | Undetermined | [49] |
Figure 2.
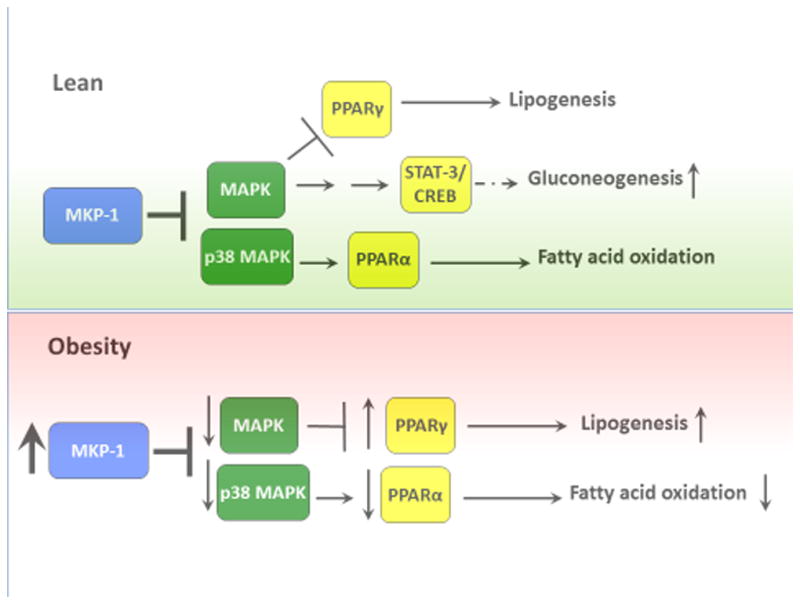
Regulation of glucose and lipid metabolism by MKP-1 through p38 MAPK and JNK. Under lean conditions MKP-1 modulates MAPK-dependent signals that regulate PPARα and PPARγ activities. Whereas p38 MAPK promotes PPARα activation and subsequently fatty acid oxidation, MAPK phosphorylation of PPARγ inhibits its activity thereby reducing lipogenesis. In addition, MKP-1 negatively engages p38 MAPK-dependent pathways to regulate gluconeogenesis. In obesity, MKP-1 is overexpressed leading to reduced MAPK activities in the nucleus. Decreased MAPK-dependent phosphorylation of PPARα and PPARγ impairs fatty acid oxidation and enhances lipogenesis, respectively.
Given the role of free fatty acids in the development of diet-induced insulin resistance, efforts have been made towards understanding the effects of p38α/β MAPK in mediating excess saturated fatty acid content in the liver. Using a lipid infusion model in rats Pereira et al. demonstrated that p38α/β MAPK inhibition attenuated hepatic insulin resistance [45]. These studies suggest that in obesity, inhibition of p38α/β MAPK could be of benefit towards improving hepatic insulin resistance. In contrast, it has been suggested that activation of p38α/β MAPK may be of therapeutic value towards the treatment of T2DM. In their study, Lee et al. reported that in obesity, hepatic p38α/β MAPK phosphorylates the spliced form of X-box binding protein 1 (Xbp1s) thereby stimulating its nuclear translocation, decreasing ER stress and maintaining euglycemia [43]. These studies imply that reduced p38α/β MAPK would promote the development of ER stress induced-hepatic insulin resistance. However, mice lacking MKP-1, which show increased p38α/β MAPK activities, fail to show any significant differences in the unfolded protein response in the absence of MKP-1 suggesting that at least p38α/β MAPK may not be sufficient to drive hepatic ER stress-responsiveness [25]. These apparent discrepancies once again, highlight the complexities of how the MAPKs, and here hepatic p38α/β MAPK, are involved in regulating glucose homeostasis and lipid management in physiological and pathophysiological states.
ERK in hepatic metabolism and pathophysiology
The extracellular signal-regulated kinases (ERKs) consist of at least six isoforms (ERK1–5 and ERK7/8) [26, 35]. The most studied among these are ERK1 and ERK2 which, are activated by a pair of closely related mitogen-activated protein kinase kinases (MEKs; MEK1 and MEK2). Both ERK1/2 are ubiquitously expressed [11]. The signaling events activating ERK1/2 are usually initiated via receptor tyrosine kinases, G-protein coupled receptors and ion channels, in part, through the Ras-Raf-MEK1/2-ERK cascade [11]. Despite the fact that ERK1/2 has been implicated in hepatic metabolism and that it is increased in states of obesity, other reports indicate that ERK1/2 activity is unaltered (Table 1). Moreover, whether ERK1/2 activity is altered in obese humans that progress towards NAFLD or NASH has yet to be established.
Initial reports showed that whole body ERK1-deficient mice are resistant to diet-induced obesity and are insulin sensitive [46]. However, more recently ERK1-deficient mice were shown to have the opposite phenotype, exhibiting increased sensitivity to diet-induced obesity [47]. No apparent explanation for these opposing phenotypes is evident although it could potentially be attributable to the composition of the diet. Interestingly, it was shown that in ERK1-deficient mice that showed increased sensitivity to obesity that the levels of phosphorylated ERK2 in the livers of these mice was elevated as compared with wild type mice [47]. While these studies have shown that the ERK pathway plays a role in the maintenance of body mass, and hepatic metabolism, it is unclear why there is an apparent difference for the requirement of ERK1 in these animal models.
One of the most direct studies demonstrating a role for ERK1/2 in liver metabolism was determined in mice in which ERK2 was specifically deleted from the liver. Kujiraoka et al demonstrated that despite the fact that ERK2 liver-specific knockout mice were not sensitive to weight gain they did exhibit elevated levels of fasting blood glucose when fed a high-fat/high sucrose diet as compared with wild-type controls [48]. Further analysis of glucose homeostasis and insulin sensitivity showed that ERK2 liver-specific knockout mice exhibited increased blood glucose levels and reduced insulin responsiveness [48]. This study suggests that hepatic ERK2 deficiency promotes impairment of glucose metabolism and insulin resistance. Additionally, when fed a high-fat/high-sucrose diet, mice deficient in hepatic ERK2 developed increased levels of triglycerides in the liver leading to the development of hepatic steatosis [48]. As discussed earlier, the MAPKs can engage in cross-talk amongst other family members and in fact when the phosphorylation levels of JNK were determined in the livers of mice deficient for the expression of ERK2 it was found that JNK levels were increased [48]. Therefore, it is possible that the increased levels of insulin resistance could at least, in part, be due to enhanced JNK activity in the in the liver of hepatic ERK2 null mice. Interestingly, loss of hepatic ERK2 also triggered the development of ER stress. These results would suggest that the progression of hepatic glucose and lipid dysfunction, as a result of ER stress in obesity, would be promoted by decreased levels of ERK2 activity. However, the activity of either ERK1 and/or ERK2 in states of obesity show either an increase or no increase at all (Table 1) raising the question of what the precise contribution of ERK1/2 is in mediating hepatic dysfunction.
Are there clues to be uncovered about the regulation of the ERKs in obesity and T2DM that can be inferred by looking at the MKPs? There are several MKPs that can dephosphorylate the ERKs, however, one of the major ERK phosphatases is MKP-3. MKP-3 is located in the cytoplasm and specifically dephosphorylates ERK1/2 [16]. Obese mice express increased levels of MKP-3 and lean mice overexpressing MKP-3 in the liver have significantly enhanced blood glucose levels [49]. Whereas, mice with reduced expression of hepatic MKP-3 levels have decreased plasma blood glucose. These observations would predict that loss of ERK2 activity would promote dysfunction of glucose homeostasis and insulin action in the liver. However, as discussed others have shown that ERK1/2 may either be unaltered or increased in its level of activity in the liver (Table 1). Interestingly, Wu et al showed that MKP-3 dephosphorylates and interacts with FOXO1 [49, 50]. Overexpression of MKP-3 correlates with increased dephosphorylation of FOXO1 on Ser256, and when dephosphorylated, FOXO1 translocates to the nucleus to activate gluconeogenic gene expression. Though MKP-3 can interact with FOXO1 [50], whether MKP-3 directly dephosphorylates FOXO1 on Ser256 remains to be clearly demonstrated. It has been reported that MKP-3 degradation is stimulated by insulin through an ERK1/2 pathway, wherein ERK1/2 phosphorylates MKP-3, and targets it for degradation [51]. It is possible that upon over-expression, MKP-3 dephosphorylates and inactivates ERK1/2 leading to reduced ERK1/2-mediated FOXO1 phosphorylation [52]. This raises the possibility that MKP-3 indirectly regulates FOXO1 via ERK1/2 in the control of hepatic gluconeogenesis. In subsequent studies using a diet-induced obesity model, Feng et al utilized MKP-3-deficient mice and showed that these mice were resistant to diet-induced obesity [53]. Consistent with this, high-fat diet fed MKP-3-deficient mice exhibited fasting blood glucose and fasting plasma insulin concentrations that were significantly lower in MKP-3 knockout mice compared with wild-type mice [53]. The livers of MKP-3 deficient mice were also protected from the development of hepatosteatosis [53]. These studies are provocative as they suggest a potential role for a non-MAPK substrate for MKP-3 in metabolism. However, whether all the functions of these animals are solely due to just the direct actions on FOXO1 or on other ERK targets, has yet to be fully evaluated.
The relative contributions of the ERK isoforms, ERK1 and ERK2, in the liver have yet to be studied. Khan et al. examined the role of ERK1 in obesity, and found that ERK1 knockout mice fed a high-fat diet were not only susceptible to diet-induced obesity but were more obese than wild-type controls showing increased levels of fat and liver mass [47]. In addition, these mice exhibit enhanced lipogenesis, low fatty acid oxidation and high hepatic triglyceride levels that are associated with the development of hepatic steatosis [47]. This study suggests that ERK1 confers protection from the development of fatty liver opposite to that of the role of ERK2 in the liver. A study by Jager et al showed that ERK1 promotes the development of diet-induced hepatic steatosis suggesting that upregulation of the ERK1 pathway contributes to hepatic dysfunction associated with obesity [54]. Although further studies are required, these data indicate that ERK1 and ERK2 have different functions in the liver. While hepatic ERK2 is implicated in the maintenance of glucose and lipid metabolism, ERK1 is involved in the development of fatty liver. Future work will require metabolic analysis of the ERK isoform-deficient mice in hepatic pathophysiology in order to understand the relative contributions and roles these isoforms play in hepatic glucose and lipid homeostasis.
Concluding Remarks and Future Perspectives
While there is strong evidence for an important role for JNK, p38 MAPK and ERKs in liver physiology and pathophysiology the precise details of the relative contributions of each of their isoforms in hepatic metabolism still remains a major knowledge gap. Indeed, in some cases, hepatic MAPK isoforms appear to act antagonistically raising the question of what are the essential isoforms of the MAPKs in the liver. Despite the fact that we have accumulated a wealth of understanding of MAPK substrate selectivity, we as yet do not know the full repertoire of MAPK substrates engaged by the MAPKs in the liver that participates in metabolic regulation. This information will be important to establish since it will lead the way towards a more in-depth mechanistic understanding of the complexity of the MAPKs. Here we have focused on the actions of the MAPKs within the liver but it is also clear that the function of the liver is influenced by external, non-hepatocyte, MAPK functions from other tissues such as macrophages, skeletal muscle, brain and adipose tissue. Understanding the role of the MAPKs in terms of inter-tissue communication will also be important. The MAPKs must be tightly regulated in order for them to achieve such high levels of signal specificity. As such, deconvoluting how the MAPKs exert pathway cross-talk between family members and target downstream MKPs that serve to inactivate them will provide valuable insight into their actions as regulators of liver function in healthy metabolic situations as well as those that manifest in liver disease.
Outstanding Questions.
What is the activation status of the various MAPKs in hepatosteatosis and related conditions in obesity in humans?
What are the differences in metabolic effects between the various MAPK isoforms in the liver?
Is it clear that activation, rather than inactivation, of the MAPKs is important for the development of metabolic dysfunction in the liver?
What are the repertoire of MAPK substrates phosphorylated by each MAPK isoform that are critical for the pathogenesis of hepatic metabolic dysfunction?
Trends.
In the liver, the concerted activities of the MAPKs result in the phosphorylation of downstream targets that regulate lipid metabolism and glucose homeostasis.
Mouse models in which there is deficiency in the expression of the MAPKs, such as JNK, p38 MAPK and the ERKs, reveal a variety of liver-related metabolic defects that result either directly or indirectly from the loss of these MAPKs.
The balance of hepatic MAPK activity is critical for normal liver function. Inactivation of MAPK phosphatases, which oppose the MAPKs, also gives rise to altered hepatic lipid regulation and glucose production.
Both altered activity and expression of the MAPKs and MKPs, respectively appear to occur in the livers of obesity models. How the balance between the MAPK and MKPs evoke a net signaling flux in the pathogenesis of insulin resistance and hepatic metabolic dysfunction remains unclear.
Acknowledgments
A.M.B. is supported by grants from the National Institutes of Health grants P01 DK57751 and R01 AR66671.
Glossary of terms
- Glucose homeostasis
Is defined as the equilibrium between glucose uptake by skeletal muscle and adipose tissue and production by the liver.
- Gluconeogenesis
Is the synthesis of glucose from precursors such as alanine, glutamine, pyruvate, lactate and glycerol.
- Insulin sensitivity
Is defined as the ability of insulin to exert its action.
- Insulin resistance
Is an impairment of insulin signaling in the peripheral organs specifically skeletal muscle, liver, adipose and the heart.
- Lipogenesis
Is the synthesis of fatty acids from carbohydrates or proteins. Glucose and fructose stimulates the transcription of lipogenic genes.
- Type-2 diabetes
Is characterized by post-receptor insulin resistance combined with β-cell failure.
- Mitogen-activated protein kinase (MAPK)
A protein kinase that phosphorylates substrates specifically on serine and threonine residues in a sequence-specific context.
- Mitogen-activated protein kinase (MAPK) phosphatases
An enzyme that dephosphorylates the MAPKs on regulatory residues that are responsible for the activity of the MAPKs.
- Nonalcoholic fatty liver disease (NAFLD)
A chronic liver disease that is characterized by excessive intrahepatic lipid accumulation in the absence of significant alcohol ingestion or liver diseases.
- Nonalcoholic steatohepatitis (NASH)
A disease condition that is characterized by a variable degree of hepatic inflammation and fibrosis as well as hepatic steatosis (lipid accumulation in the liver).
- Triglycerides (TGs)
TGs are water-soluble lipids containing three fatty acids esterified to a glycerol backbone. They are synthesized in the liver and dietary fat is a major source of TGs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23:804–814. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Titchenell PM, et al. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends in endocrinology and metabolism: TEM. 2017;28:497–505. doi: 10.1016/j.tem.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samuel VT, Shulman GI. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2017 doi: 10.1016/j.cmet.2017.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma C, et al. Nonalcoholic fatty liver disease promotes hepatocellular carcinoma through direct and indirect effects on hepatocytes. FEBS J. 2017 doi: 10.1111/febs.14209. [DOI] [PubMed] [Google Scholar]
- 5.Manieri E, Sabio G. Stress kinases in the modulation of metabolism and energy balance. J Mol Endocrinol. 2015;55:R11–22. doi: 10.1530/JME-15-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gehart H, et al. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep. 2010;11:834–840. doi: 10.1038/embor.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bost F, et al. The role of MAPKs in adipocyte differentiation and obesity. Biochimie. 2005;87:51–56. doi: 10.1016/j.biochi.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 8.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 10.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 11.Roskoski R., Jr ERK1/2 MAP kinases: structure, function, and regulation. Pharmacol Res. 2012;66:105–143. doi: 10.1016/j.phrs.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 13.Derijard B, et al. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 14.Widmann C, et al. Mitogen-activated protein kinase: conservation of a three- kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 15.Boutros T, et al. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60:261–310. doi: 10.1124/pr.107.00106. [DOI] [PubMed] [Google Scholar]
- 16.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhalla US, et al. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science. 2002;297:1018–1023. doi: 10.1126/science.1068873. [DOI] [PubMed] [Google Scholar]
- 18.Pal M, et al. The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and insulin resistance. The Journal of physiology. 2016;594:267–279. doi: 10.1113/JP271457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holzer RG, et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamata H, et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 21.Sabio G, et al. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10:491–498. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vernia S, et al. Fibroblast Growth Factor 21 Mediates Glycemic Regulation by Hepatic JNK. Cell Rep. 2016;14:2273–2280. doi: 10.1016/j.celrep.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vernia S, et al. The PPARalpha-FGF21 Hormone Axis Contributes to Metabolic Regulation by the Hepatic JNK Signaling Pathway. Cell Metab. 2014 doi: 10.1016/j.cmet.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogoyevitch MA. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): differences revealed by gene targeting. Bioessays. 2006;28:923–934. doi: 10.1002/bies.20458. [DOI] [PubMed] [Google Scholar]
- 25.Lawan A, et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Mol Cell Biol. 2015;35:26–40. doi: 10.1128/MCB.00503-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan H, et al. Inhibition of JNK suppresses autophagy and attenuates insulin resistance in a rat model of nonalcoholic fatty liver disease. Mol Med Rep. 2017;15:180–186. doi: 10.3892/mmr.2016.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabio G, et al. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol. 2010:106–115. doi: 10.1128/MCB.01162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han MS, et al. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339:218–222. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hui L, et al. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 31.Shen YH, et al. Cross-talk between JNK/SAPK and ERK/MAPK pathways: sustained activation of JNK blocks ERK activation by mitogenic factors. J Biol Chem. 2003;278:26715–26721. doi: 10.1074/jbc.M303264200. [DOI] [PubMed] [Google Scholar]
- 32.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 33.Nandipati KC, et al. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem. 2017;426:27–45. doi: 10.1007/s11010-016-2878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci. 2008;65:3525–3544. doi: 10.1007/s00018-008-8170-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Somwar R, et al. A dominant-negative p38 MAPK mutant and novel selective inhibitors of p38 MAPK reduce insulin-stimulated glucose uptake in 3T3-L1 adipocytes without affecting GLUT4 translocation. J Biol Chem. 2002;277:50386–50395. doi: 10.1074/jbc.M205277200. [DOI] [PubMed] [Google Scholar]
- 37.Sumara G, et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell. 2009;136:235–248. doi: 10.1016/j.cell.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barger PM, et al. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem. 2001;276:44495–44501. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 39.Cao W, et al. p38 Mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J Biol Chem. 2005;280:42731–42737. doi: 10.1074/jbc.M506223200. [DOI] [PubMed] [Google Scholar]
- 40.Jing Y, et al. Hepatic p38alpha regulates gluconeogenesis by suppressing AMPK. Journal of hepatology. 2015;62:1319–1327. doi: 10.1016/j.jhep.2014.12.032. [DOI] [PubMed] [Google Scholar]
- 41.Flach RJ, et al. Loss of mitogen-activated protein kinase phosphatase-1 protects from hepatic steatosis by repression of cell death-inducing DNA fragmentation factor A (DFFA)-like effector C (CIDEC)/fat-specific protein 27. J Biol Chem. 2011;286:22195–22202. doi: 10.1074/jbc.M110.210237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu JJ, et al. Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to diet-induced obesity. Cell Metabolism. 2006;4:61–73. doi: 10.1016/j.cmet.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 43.Lee J, et al. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat Med. 2011;17:1251–1260. doi: 10.1038/nm.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez-Teran B, et al. p38gamma and p38delta reprogram liver metabolism by modulating neutrophil infiltration. EMBO J. 2016;35:536–552. doi: 10.15252/embj.201591857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pereira S, et al. Effect of a p38 MAPK inhibitor on FFA-induced hepatic insulin resistance in vivo. Nutrition & diabetes. 2016;6:e210. doi: 10.1038/nutd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bost F, et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54:402–411. doi: 10.2337/diabetes.54.2.402. [DOI] [PubMed] [Google Scholar]
- 47.Khan AS, et al. ERK1 and ERK2 activation modulates diet-induced obesity in mice. Biochimie. 2017;137:78–87. doi: 10.1016/j.biochi.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 48.Kujiraoka T, et al. Hepatic extracellular signal-regulated kinase 2 suppresses endoplasmic reticulum stress and protects from oxidative stress and endothelial dysfunction. J Am Heart Assoc. 2013;2:e000361. doi: 10.1161/JAHA.113.000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Z, et al. MAPK phosphatase-3 promotes hepatic gluconeogenesis through dephosphorylation of forkhead box O1 in mice. J Clin Invest. 2010;120:3901–3911. doi: 10.1172/JCI43250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiao P, et al. Mapping MKP-3/FOXO1 interaction and evaluating the effect on gluconeogenesis. PLoS One. 2012;7:e41168. doi: 10.1371/journal.pone.0041168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng B, et al. MEK/ERK pathway mediates insulin-promoted degradation of MKP-3 protein in liver cells. Mol Cell Endocrinol. 2012;361:116–123. doi: 10.1016/j.mce.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asada S, et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 53.Feng B, et al. Mitogen-activated protein kinase phosphatase 3 (MKP-3)-deficient mice are resistant to diet-induced obesity. Diabetes. 2014;63:2924–2934. doi: 10.2337/db14-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jager J, et al. Deficiency in the extracellular signal-regulated kinase 1 (ERK1) protects leptin-deficient mice from insulin resistance without affecting obesity. Diabetologia. 2011;54:180–189. doi: 10.1007/s00125-010-1944-0. [DOI] [PubMed] [Google Scholar]
- 55.Zhao G, et al. Jnk1 in murine hepatic stellate cells is a crucial mediator of liver fibrogenesis. Gut. 2014;63:1159–1172. doi: 10.1136/gutjnl-2013-305507. [DOI] [PubMed] [Google Scholar]
- 56.Lake AD, et al. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci. 2014;137:26–35. doi: 10.1093/toxsci/kft230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koliaki C, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 58.Ferreira DM, et al. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetologia. 2011;54:1788–1798. doi: 10.1007/s00125-011-2130-8. [DOI] [PubMed] [Google Scholar]