

Metallosupramolecular complexes can enantioselectively target the central hydrophobic α/β discordant stretch of Aβ.
Abstract
Stereochemistry is vital for pharmaceutical development and can determine drug efficacy. Herein, 10 pairs of asymmetric triplex metallohelix enantiomers as a library were used to screen inhibitors of amyloid β (Aβ) aggregation via a fluorescent cell–based high-throughput method. Intriguingly, Λ enantiomers show a stronger inhibition effect than Δ enantiomers. In addition, the metallohelices with aromatic substituents are more effective than those without, revealing that these groups play a key role in the Aβ interaction. Fluorescence stopped-flow kinetic studies indicate that binding of the Λ enantiomer to Aβ is much faster than that of the Δ enantiomer. Furthermore, studies in enzyme digestion, isothermal titration calorimetry, nuclear magnetic resonance, and computational docking demonstrate that the enantiomers bind to the central hydrophobic α-helical region of Aβ13–23, although with different modes for the Λ and Δ enantiomers. Finally, an in vivo study showed that these metallohelices extend the life span of the Caenorhabditis elegans CL2006 strain by attenuating Aβ-induced toxicity. Our work will shed light on the design and screening of a metal complex as an amyloid inhibitor against Alzheimer’s disease.
INTRODUCTION
Supramolecular chemistry aims at building functional, highly complex chemical systems from components assembled by intermolecular forces (1). It offers an outstanding methodology for the design and synthesis of complex arrays at the molecular level (1, 2). Certain self-assembling multimetallic coordination complexes, named helicates, can have a similar size to α-helical peptides, particularly in terms of their diameter and charge (3, 4), and this can lead to specific target-binding affinity with biomolecules (5). Recently, the application of the supramolecular helical scaffold has received much attention, including studies on asymmetric catalysis (6), antimicrobials (3), anticancer (4, 7), and the targeting of amyloid β (Aβ) for the treatment of Alzheimer’s disease (AD) (5).
AD, the most prevalent age-related dementia, affects more than 30 million people worldwide, and it is estimated that, by 2050, some 106 million people will be afflicted with the disease (8, 9). However, the exact cause of AD, as well as the molecular mechanisms involved, has not been fully understood. Emerging evidence has shown that the origin of neurotoxicity is closely linked to the aggregate process of Aβ monomers to oligomers and then subsequently to fibrils in the pathogenesis (10, 11). Therefore, immense attention has been paid toward inhibiting Aβ aggregation (12–16) as an effective preventive and therapeutic method for the treatment of AD. Although considerable achievements have been made in screening inhibitors of Aβ aggregation and toxicity, the complex interactions involved in Aβ aggregation dynamics and morphology (17) are difficult to address using conventional small molecules. Therefore, chiral recognition of the supramolecular structures may be key to developing an understanding of the disease and discovering new treatments.
Recent studies have demonstrated that forming α-helical intermediates facilitates fibrillization (18–20). An α-helical structure in the 13 to 23 segment of Aβ is predicted to become a β strand identified as an α/β discordance (21). Because an Aβ analog that lacks residues 13 to 23 and results in deficiency of the α/β discordant stretch does not form detectable amyloid fibrils (22), there may be a causal connection between α/β discordance and fibril structure. Emerging evidence suggested that stabilization of helical motif(s) may be a suitable target for screening inhibitors of Aβ aggregation (5, 23). Therefore, chiral supramolecules that are capable of binding the α-helical form may exhibit enantioselectivity on inhibition of Aβ aggregation.
As previously reported, chiral helicates, which are synthesized by symmetric rigid ligands AB-BA (Fig. 1A), can be potential therapeutic agents (8) for enantioselectively inhibiting Aβ aggregation. However, this symmetric system offers little scope to adjust the structure to interact with Aβ or to create a library for screening Aβ aggregation inhibitors resulting from the high symmetry and modest functionality of the system. Recently, we developed a new strategy whereby directional ligands AB-CD were used to synthesize optically pure head-to-head-to-tail (HHT) asymmetric constitutions in the absence of head-to-head-to-head (HHH) isomers (Fig. 1B). Herein, we strategically prepared a highly stereoselective self-assembly of diverse, functionalized metallohelices with this HHT structure to enantioselectively inhibit Aβ aggregation. These metallohelices have amphipathic topology and high, structure-dependent, selective biological activity. Using a cell-based high-throughput screening system, these asymmetric triplex metallohelices enantioselectively inhibited Aβ aggregation. Our work shows that these metallosupramolecules can act as Aβ inhibitors, specifically targeting the central hydrophobic α/β discordant stretch.
Fig. 1. The structures of metallohelices.
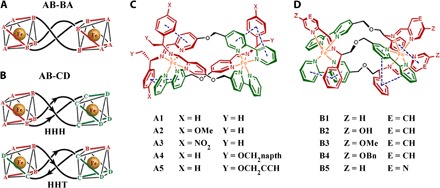
(A) Symmetric metallohelices based on rigid ditopic bidentate ligands AB-BA yield D3-symmetric enantiomers [Fe2(AB-BA)3]. (B) The directional ligands AB-CD lead to C1-symmetric HHH and HHT “triplex” architectures. (C and D) Metallohelices self-assembled from various components of a range of functionalized helices with HHT structure. Me, methyl; napth, naphthyl; Bn, benzyl.
RESULTS AND DISCUSSION
Ten pairs of HHT asymmetric metallohelix enantiomers (Fig. 1, C and D) were synthesized and characterized according to our previously reported methods (3, 4) and used to screen inhibitors of Aβ aggregation by a cell-based high-throughput screening method (Fig. 2A) (5). The high-throughput screen system is constructed by a fusion of Aβ42 to enhanced cyan fluorescent protein (Aβ-ECFP). In general, Aβ42 in the Aβ-ECFP fusion system aggregates rapidly, which hinders ECFP folding into its native structure to emit cyan fluorescence. In the presence of Aβ aggregation inhibitors, ECFP in the Aβ-ECFP fusion system folds into its native structure and emit cyan fluorescence. ECFP, a non-Aβ fusion system, was used as a control in our experiments. Almost no fluorescence enhancements were observed for the non-Aβ fusion system with the incubation of metallohelices (fig. S1). Screening results are shown in Fig. 2 (B and C), and the inhibition ability of the complex can be estimated by the related fluorescence value of Aβ-ECFP/ECFP. Intriguingly, ΛA1 and ΔA1 showed remarkable enantiomeric differences on inhibition of Aβ aggregation, whereas ΛB4 and ΔB4 had the greatest inhibition effect. Therefore, we selected these two pairs of A1 and B4 as examples for further study.
Fig. 2. The inhibition ability of metallohelices on Aβ.
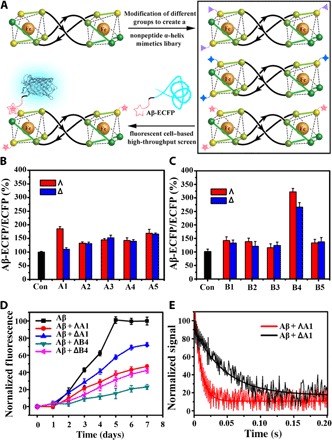
(A) The Aβ-ECFP fusion system used in screening Aβ aggregation inhibitors. (B and C) Screening results of metallohelices A and B from the Aβ-ECFP fusion screen system. (D) Aggregation kinetics of Aβ40 monitored by ThT assay with or without the incubation of A1 and B4. (E) The stopped-flow kinetic study of Aβ and A1. Values show means ± SD, and independent experiments were performed three times.
To investigate the effect of the metallohelices on Aβ aggregation kinetics, we performed a thioflavin T (ThT) fluorescence assay. This is a widely used assay to study the formation of amyloid fibrils (24–26). Under our experimental conditions, metallohelices alone did not affect ThT fluorescence (fig. S2). Kinetic analysis of the ThT assay showed that ThT fluorescence did not increase in the early stages, thus demonstrating that the aggregation of Aβ40 and Aβ42 was greatly suppressed with the incubation of metallohelices (Fig. 2D and fig. S3). Free ligands La1 and Lb4, which are components of A1 and B4, showed little inhibition effect on Aβ40 aggregation (fig. S4). Fluorescence stopped-flow kinetic studies (Fig. 2E) showed that the process of ΛA1 binding to Aβ40 was completed about 0.04 s under our experimental conditions, which was five times faster than its enantiomer ΔA1 (about 0.2 s).
Herein, we compared the fibrillation inhibitors mentioned in Introduction with the metallohelices used in our study. In comparison with peptide–organic molecule coassemblies (12), the metallohelices in our study exhibited better inhibition effect. With 50 μM peptide–organic molecule coassemblies, the inhibition ratio was 70%. In our study, the inhibition ratio was up to 80% with 50 μM ΛB4. Among the fibrillation inhibitors discussed in Introduction, CdTe nanoparticles (NPs) (15) performed the best inhibition effect. The intensity of ThT can be decreased by 80% with only 2.5 μM CdTe NPs. With 25 μM inhibitors, the inhibition ratios were 63 and 80% with polyoxometalate (POM)/peptide composites (POM@P) (13) and POM-Dawson derivatives (14), respectively. On the basis of the inhibition efficiency, CdTe NPs and POM@P exhibited better inhibition effect than the metallohelices used in our study. This could be because one NP may offer many active sites for interacting with Aβ, whereas one metallohelix can only offer one active site per molecule. Although these NPs have better inhibition activity, NPs are easily aggregated and accumulated in the body because of their hard degradation. For metallohelices, they are stable and highly water-soluble compared to peptide–organic molecules and NPs, and the structures and stereochemistry of metallohelices are adjustable and optimized. They can specifically bind to the central hydrophobic α-helical region of Aβ13–23, showing the potential for AD treatment.
Median inhibitory concentration (IC50) values can also be estimated by ThT fluorescence assay (8). As shown in fig. S5, metallohelices A1 and B4 can inhibit Aβ40/Aβ42 aggregation in a dose-dependent manner. For Aβ40, IC50 values were 3.65 μM for ΛA1, 32.29 μM for ΔA1, 0.94 μM for ΛB4, and 2.55 μM for ΔB4 (table S1). For Aβ42, IC50 values were 6.76 μM for ΛA1, 69.82 μM for ΔA1, 1.66 μM for ΛB4, and 3.72 μM for ΔB4. These results demonstrated that ΛA1 and ΛB4 had stronger inhibition on Aβ40/Aβ42 aggregation than their enantiomers ΔA1 and ΔB4. Furthermore, the enantioselectivity of A1 was higher than that of B4. Therefore, there were two features for the metallohelices to inhibit Aβ aggregation. First, the Λ enantiomers showed higher inhibition effects than the Δ enantiomers. Second, the metallohelices with aromatic substituents are more effective than those without aromatic substituents.
To better compare the inhibitory effects of the asymmetric triplex metallohelices and the previously reported symmetric systems, we summarized their IC50 values in table S1. The asymmetric metallohelix ΛB4 showed a higher inhibition effect than the symmetric architectures, whereas the chiral discrimination of A1 is the most obvious. This may be due to the nature of the asymmetric fold of the triplex metallohelices, which creates a distinctly amphipathic architecture, reminiscent of α-helical peptides (4).
We also investigated the inhibition of Aβ40 aggregation by an SDS–polyacrylamide gel electrophoresis (PAGE) assay (27, 28). After the incubation of Aβ40 with metallohelices at 37°C, we separated the high–molecular weight Aβ40 aggregates and the low–molecular weight Aβ40 species by centrifugation. The aggregated peptide pellets were resuspended in sample buffer and then boiled with SDS for 10 min to destroy the secondary and tertiary structures. The total amounts of aggregated Aβ40 were estimated by the SDS-PAGE assay. After incubation with metallohelices A1 and B4, the amount of the resuspended peptide was decreased (fig. S6). SDS-PAGE results indicated that these metallohelices inhibited Aβ aggregation.
To further study the morphology of Aβ40 and Aβ42 aggregates affected by these metallohelices, we carried out an atomic force microscopy (AFM) study (29, 30). In samples of Aβ40 or Aβ42 alone, traditional nonbranched amyloid fibrils (helical filaments with diameters of about 10 nm and lengths of up to several micrometers) were observed (Fig. 3, control). After incubating Aβ40 or Aβ42 with the metallohelices, particularly ΛB4, numerous small, globular dots can be observed, suggesting the excellent efficacy of metallohelices in inhibiting the aggregation of Aβ40 and Aβ42 (Fig. 3).
Fig. 3. AFM images of Aβ40/Aβ42 with or without the incubation of metallohelices (area corresponding to 3 μm × 3 μm).

Control: 50 μM Aβ40/Aβ42 alone. ΛA1, ΔA1, ΛB4, and ΔB4: 50 μM Aβ40/Aβ42 with the incubation of 50 μM ΛA1, ΔA1, ΛB4, and ΔB4.
Aβ40/Aβ42 aggregation involves formation of β sheet–rich frameworks, which can be determined by circular dichroism (CD; a wide band near 217 nm). We calculated the proportional contribution of the β sheet structure in the samples of Aβ40 according to previous reports (31, 32). On average, the Aβ40 fibrils contained approximately 57 ± 5% of β sheet structure, which decreased to 29 ± 3%, 35 ± 4%, 19 ± 3%, and 23 ± 4% on incubation with ΛA1, ΔA1, ΛB4, and ΔB4, respectively (Fig. 4, A and B). As shown in fig. S7, the metallohelices had a similar effect on Aβ42. This indicates that the metallohelices inhibit significant structural change in Aβ40. To reveal the enantiomeric selectivity of metallohelix enantiomers, we performed dialysis experiments (33). Racemic mixtures of A1 and B4 were dialyzed against Aβ40. In addition, the enhanced sign of CD was attributed to the left enantiomer with lower binding affinity to Aβ40. As shown in Fig. 4 (C and D), the dialyzate was rich in ΔA1 and ΔB4, which unambiguously indicated that ΛA1 and ΛB4 preferentially bind to Aβ40 compared with their enantiomers.
Fig. 4. The influence of metallohelices on the structure of Aβ40 and the binding affinity of metallohelices to Aβ40.
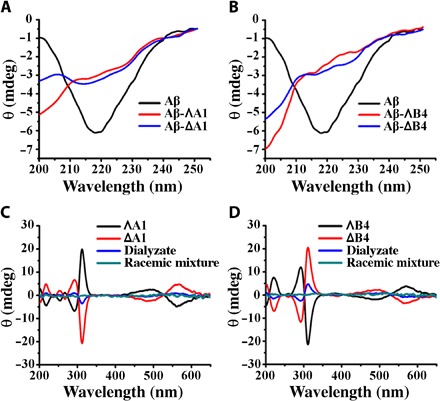
(A and B) CD spectra of Aβ40 without or with the incubation of A1 (A) and B4 (B). (C and D) CD spectra of dialyzate were used to discriminate the enrichment of the enantiomer with lower binding affinity to Aβ40 in competition dialysis experiments. The concentrations of metallohelices were 50 μM.
The four metallohelices showed different inhibiting capacities. To have a better understanding of the inhibiting ability of the metallohelices, we determined the Aβ40 binding affinity of metallohelices by the fluorescence titration method (14, 34). With increasing amounts of metallohelices, the fluorescence intensity of Aβ40 was decreased (fig. S8). The apparent binding constants (Ka) were extracted by a nonlinear least-squares procedure. As shown in table S2, Ka values of ΛA1 and ΛB4 were 8.9-fold and 2.3-fold higher than those of their Δ enantiomers. As previously reported, the Ka values of symmetric complexes Λ1 and Λ2 are only 3.9 and 5.3 times higher than those of their enantiomers (table S2). Together, symmetric complexes showed less chiral discrimination on Aβ aggregation than asymmetric ones. These results are in agreement with the IC50 value of the metallohelices against Aβ40 aggregation. Therefore, we conjectured that the distinct inhibition ability of these metallohelices mainly depended on the different affinities to Aβ40. Although both symmetric and asymmetric metallohelices have similar triple-helical structures, they presented different Aβ40 inhibition activities and binding affinities as a result of their different spatial structures. Notably, although the affinities of asymmetric metallohelices to Aβ40 were not as strong as those of the Aβ antibody (35, 36), the chiral recognition between these enantiomers was significant for targeting and inhibition of Aβ40 aggregation.
The stoichiometry and binding constants between these new inhibitors and Aβ40 were estimated by isothermal titration calorimetry (ITC) (14). The ITC integrated heat data profiles were used to estimate the binding parameters between Aβ and ΛA1, between Aβ and ΔA1, between Aβ and ΛB4, and between Aβ and ΔB4. The binding was exothermic according to the net enthalpy change (fig. S9) (37) and gave the best fit to 1:1 binding. The thermodynamic parameters were summarized in table S3, including the binding constant (Ka), enthalpy change (ΔH), entropy change (ΔS), and Gibbs free energy change (ΔG) upon the interaction between Aβ and metallohelices. Binding constants of ΛA1-Aβ40, ΔA1-Aβ40, ΛB4-Aβ40, and ΔB4-Aβ40 were estimated to be 4.93 × 106, 5.36 × 105, 8.41 × 106, and 3.01 × 106, respectively. These analyses further indicated that ΛB4 had much stronger interactions to Aβ40 than its enantiomer ΔB4.
Aβ12–28 and Aβ25–35 fragments were used to explore the binding site on Aβ40. Given that the metallohelices have a similar size to short α-helical peptides, it was considered that they may bind to the Aβ13–23 region. Because the cleavage site of Aβ12–28 (lysine residues) was involved in the central hydrophobic region, we chose this fragment as the trypsin substrate for digestion experiments to verify our hypothesis (16). Figure S10 shows that the digestion of Aβ12–28 was prevented by the presence of metallohelices, suggesting that they bind to the α/β discordant stretch of Aβ13–23. The band of Aβ12–28 incubated with compound B4 remained even stronger than that incubated with A1, demonstrating that B4 bound to Aβ40 more tightly than A1. Furthermore, with the incubation of ΛA1, the band of Aβ12–28 was stronger than that with ΔA1, because the Λ enantiomer bound to Aβ40 more tightly than the Δ one. On the other hand, previous reports demonstrated that Aβ25–35 can also form fibrils as Aβ40 (38, 39). However, the inhibition effect of these metallohelices on Aβ25–35 was much weaker than that on Aβ40 (fig. S11). The aggregation process was not inhibited completely by the metallohelices even at a high concentration; that is, they bind weakly to Aβ25–35.
Nuclear magnetic resonance (NMR) spectroscopy was further used to study the binding site with Aβ40 (40–42). The 1H NMR signals of the aromatic rings (F19 and F20) (40, 41) of Aβ40 changed significantly upon the addition of A1, revealing that the triplex bound to the central hydrophobic region of Aβ40 (Fig. 5, A to D). Moreover, compared with the NMR spectrum of Aβ40 alone, obvious peak shifts were observed with the incubation of A1 in 2.95 and 2.52 parts per million owing to the amide protons of K16 and E22, respectively (Fig. 5, B and C) (42, 43). ΛA1 induced the peaks to move to downfield, whereas ΔA1 led to a shift to upfield, suggesting a binding difference between ΛA1 and ΔA1 with Aβ40.
Fig. 5. 1H NMR spectra of Aβ40 with or without treatment of A1.
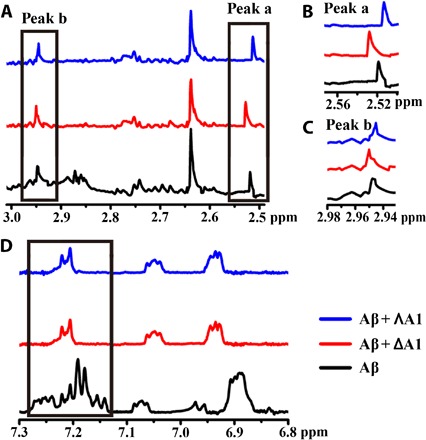
(A) The signals caused by amide protons of K16, F19, F20, and E22 of Aβ40. (B) Locally amplified 1H NMR spectra of peak a. (C) Locally amplified 1H NMR spectra of peak b. (D) The peaks caused by the protons of F19 and F20 (black box) were obviously reduced in intensity with treatment of A1. The chemical shift values of Aβ40 were assigned on the basis of previous studies (40–43).
Fourier transform infrared (FTIR) spectroscopy is one of the powerful methods to study the structure of peptides. Figure S12 shows the FT-IR spectra of Aβ40 under different conditions. The amide I and amide II regions (1750 to 1500 cm−1) play a key role in analyzing the second structure of the peptide. As shown in the FTIR spectra of the Aβ40 fibril, the signals at 1620 cm−1 can be assigned to antiparallel β sheets, whereas the band at 1680 cm−1 can be assigned to β turns, in agreement with previous reports (44, 45). For Aβ40 incubated with metallohelices, the signals at 1620 cm−1 slightly shift to lower frequencies. The band at 1650 cm−1 can be assigned to α helices, together with a small contribution from random coils and unstructured conformations. As for the Aβ40 monomer, the band around 1440 cm−1 can be assigned to His residues (46). After the conversion of the Aβ40 monomer to fibril, His residues became involved in the β sheet structure and their vibrations became limited. Therefore, its vibrational band can hardly be observed. With the incubation of metallohelices (ΛA1 and ΔA1), the vibrational band of His residues is observable, demonstrating that the metallohelices (ΛA1 and ΔA1) can inhibit Aβ to form β sheet structures and fibrils.
The above results revealed a prominent enantioselective interaction between Aβ40 and the chiral metallohelices. Arene units at the terminus of the metallohelices featured parallel π-stacking interactions and formed strong π surfaces. F19 or F20 in the central hydrophobic region of Aβ40 bound to the arene rings through hydrophobic interactions and π-π stacking. π-π stacking plays an important role in Aβ40 and affects the physical and chemical behaviors. In addition, the interactions between Aβ and the metallohelices may also involve His residues (His13 and His14). The chirality of the α-helical structure and l–amino acids of the peptides would result in different hydrophobic and π-π interactions between Aβ40 and the Δ/Λ enantiomers. Furthermore, the positive charge of the metallohelices also strengthens the binding affinity to negatively charged Aβ13–16 by electrostatic interactions.
To better understand and visualize the interactions responsible for such binding of triplex metallohelices, we performed docking studies based on the reported NMR structure of the Aβ40 monomer [Protein Data Bank (PDB) 2LFM] in aqueous solution using AutoDock Vina (8, 47, 48). Under these conditions, the Aβ40 peptide comprises a 310 helix structure in the 13 to 23 region (fig. S13) (43). Figure 6A and fig. S14 show the resultant low-energy conformations of the Aβ40-metallohelix interactions. ΛB4 was parallel to the α helix, and the binding was further stabilized by π-π stacking between F19 and ΛB4 (Fig. 6, C and D). Although ΔB4 also formed π-π stacking with residue F19 (Fig. 6, E and F), it was perpendicular to the α helix, and the involved π-π stacking moiety was smaller than that of ΛB4. This is at least consistent with the weaker binding and less potent inhibition observed in the above ITC and ThT studies. The docking studies reported here are also in agreement with the NMR observation that the triplex metallohelices bind to the hydrophobic region.
Fig. 6. The interaction of Aβ40 and B4 by docking study.
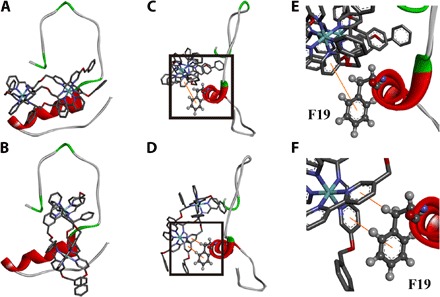
(A and B) Energy-minimized average models of ΛB4 (A) and ΔB4 (B) with Aβ40 interactions. (C and D) Hydrophobic interaction between Aβ40 and ΛB4 (C) and between Aβ40 and ΔB4 (D). (E) Locally amplified image of the black box in (C). (F) Locally amplified image of the black box in (D).
It has been proposed that Aβ aggregation–induced overgeneration of reactive oxygen species (ROS), and the subsequent detrimental reactions with biomacromolecules including proteins, lipids, DNA, and RNA in the cortex, is a factor in AD pathogenesis (49–51). Superoxide dismutase (SOD) enzymes are promising pharmaceuticals in the treatment of AD. In addition, it is well known that catalase and SOD activity can be achieved by certain metal-containing model compounds, such as iron porphyrins (52). Although the Fe(II) center in our metallohelices is highly stabilized in this oxidation state and fully encapsulated in the structure, we know that imine complexes of redox-active metals can nevertheless be radical scavengers (53, 54). Therefore, we conjectured that the metallohelices used here may be delivering Fe ions in such a form as to display SOD-like activity or are otherwise depleting harmful ROS.
The SOD activities of these metallohelices were estimated by a modified nitro blue tetrazolium (NBT) assay system, 2,2-azinobis(3-ethylbenzothiozoline)-6-sulfonic acid (ABTS) methodology, and cyclic voltammetry (CV) (8, 55–57). With the irradiation of light, NBT can be photochemically oxidized by the superoxide anion produced by riboflavin. These metallohelices inhibit this process by reducing the superoxide anion radical. Owing to the distinct absolute configurations, the metallohelices showed different SOD mimic activities (fig. S15A). Using the ABTS methodology (56), the results also demonstrated that the metallohelices scavenged ROS effectively (fig. S15, B to F). In addition, we also used CV to assess the reactivity of metallohelices with superoxide (57). As shown in fig. S16, after the addition of the metallohelix solution, the oxidation peak current of O2•− (Ipa: anodic current; oxygen regeneration) decreased, whereas the reduction current (Ipc: cathodic current; O2•− formation) was nearly unchanged. These data indicated that the metallohelices reacted with O2•−; that is, they scavenged O2•−. Next, we further studied the effects of the metallohelices on superfluous ROS induced by the aggregation of Aβ40 in PC12 cells. Our data revealed that the metallohelices effectively suppressed the Aβ40-induced ROS production (fig. S17). On the basis of these results, the metallohelices could serve as SOD mimics or otherwise as free radical scavengers.
On the basis of the capacities of the metallohelices to inhibit Aβ40 assembly, these complexes might be helpful in protecting cells from Aβ-mediated toxicity. Therefore, we performed MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay to probe the cellular metabolism of differentiated PC12 cells, which have been proven to be more susceptible to neurotoxicity of Aβ40 aggregations than normal PC12 cells (29). Before the following experiments, the stability of the complex should be studied under physiological conditions. Upon incubation in phosphate-buffered saline (PBS), the absorption wavelength and the intensity of the metal-to-ligand charge transfer bands of the metallohelices did not change (fig. S18), demonstrating that the metallohelices were stable under our experimental conditions. Further studies showed that A1 or B4 alone had little influence on PC12 cell viability (fig. S19). As shown in fig. S20, aged Aβ40 and Aβ42 caused a decrease in cell viability. With the addition of A1 or B4, cell death can be suppressed in a dose-dependent manner. As a result of the different binding affinities to Aβ40 and Aβ42, these metallohelices showed chiral discrimination even in the culture medium. Although B4 displayed higher binding affinity to Aβ40, it only showed a lightly stronger protection effect than A1, suggesting that Aβ-mediated cytotoxicity was complicated when Aβ40 and the metallohelices were incubated together.
The transgenic Caenorhabditis elegans (C. elegans) strain CL2006, which expresses an Aβ protein fragment involved in the development of AD, is widely used in studying and developing drug effects (58, 59). The strain shows a phenotype of Aβ expression and aggregation in muscle, leading to progressive paralysis. In untreated control C. elegans, the worms were almost completely paralyzed within 12 days; by comparison, the paralysis rate induced by Aβ was obviously postponed in CL2006 worms with the treatment of metallohelices. Complete paralysis occurred in 13 days with the incubation of ΔA1, whereas the worms were completely paralyzed within 15 days with ΛA1 treatment (Fig. 7A). In addition, both ΛB4 and ΔB4 can also delay the paralysis of CL2006 worms to about 16 days (Fig. 7B). These data show that metallohelix treatment led to an increased life span in this strain.
Fig. 7. The effect of A1 and B4 on the life span and Aβ plaques of the transgenic strain CL2006.
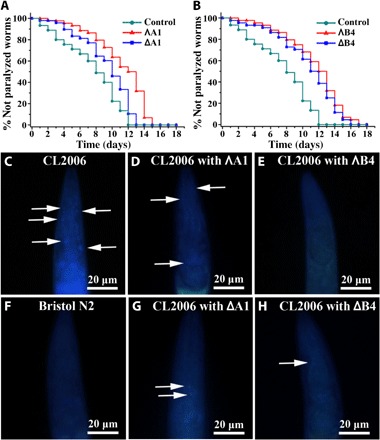
(A and B) Kaplan-Meier survival curves of the transgenic strain CL2006 treated with or without metallohelices. Plots are representative of three independent experiments. (C to H) Representative images of Aβ plaques in the worm’s head region. Arrows indicate Aβ deposits. (C) Transgenic worm CL2006. Transgenic worm CL2006 with the incubation of ΛA1 (D) and ΛB4 (E). (F) Bristol N2 worm (wild type). Transgenic worm CL2006 with the incubation of ΔA1 (G) and ΔB4 (H).
Moreover, to investigate whether life-span extension was related to Aβ accumulation, thioflavin S (ThS) staining was performed to estimate the amyloid levels in the CL2006 strain (59). N2 wild-type strain worm was used as a control, which did not express Aβ transgene or undergo paralysis. The fluorescent images of the head region are shown in Fig. 7 (C to H). The images showed that metallohelix treatment greatly inhibited Aβ aggregation and deposition in the CL2006 strain compared with the untreated CL2006 worms. These findings demonstrated that metallohelices extend the life span of C. elegans by attenuating Aβ-induced toxicity.
As potential therapeutic agents for the treatment of AD, these metallohelices should cross the blood-brain barrier (BBB). Metallohelix permeability of the BBB is evaluated by the quantity of Fe in the mouse brain through inductively coupled plasma mass spectrometry (ICP-MS). Compared to the control mouse, a higher level of Fe can be detected in the cerebrospinal fluid (CSF) of metallohelix-treated mouse. About 1.68 and 0.82% for A1 and B4, respectively, can be accumulated in the brain effectively, suggesting that these metallohelices had the ability to cross the BBB. Briefly, these metallohelices can be potential candidates for AD treatment. Moreover, compared with B4, A1 exhibited a stronger capability to cross the BBB.
CONCLUSION
In summary, asymmetric triplex metallohelices have been demonstrated to provide a new generation of chiral Aβ inhibitors. Through binding to α/β discordant stretches, these architecturally chiral species show enantioselectivity in the inhibition of Aβ42 aggregation, evidenced by fluorescent cell–based screening systems and multiple biophysical and biochemical techniques. In addition, the compounds can cross the BBB and block Aβ-mediated cellular toxicity. In vivo study proved that these metallohelices extend the life span of the C. elegans CL2006 strain by attenuating Aβ-induced toxicity. The modular nature of the synthesis of these triplex systems will allow us to further tune the chiral multivalent interactions we have begun here to characterize. Our work indicates that these new generations of asymmetric chiral supramolecular complexes are viable as Aβ inhibitors against AD.
MATERIALS AND METHODS
ThT assay
After incubation for different times, samples of 50 μM Aβ40 with or without 50 μM metallohelices were diluted 50-fold with 0.01 M Hepes buffer containing 0.15 M NaCl (pH 7.3). A JASCO FP-6500 spectrofluorometer was used for the ThT fluorescence assay. [ThT] = 10 μM, λex = 444 nm, and λem = 482 nm.
Aβ-ECFP fusion system
The ECFP and Aβ1–42 coding sequences were connected through a short linker DNA. The vector with Aβ-linker-ECFP or linker-ECFP transformed to Escherichia coli strain BL21 (DE3) and cultured in LB medium with ampicillin (50 μg/ml) at 37°C. Metallohelices with different concentrations were mixed to the culture medium for 30 min. Then, isopropyl-β-d-thio-galactoside (1 mM) was added to the culture medium to induce the expression of ECFP. The recombinant proteins were expressed at 20°C for 3 hours. After measuring the optical density at 600 nm (OD600) of all the samples, they were diluted to OD600 = 0.1. Fluorescence measurements were performed with a JASCO FP-6500 spectrofluorimeter. λex = 433 nm.
CD studies
A JASCO J-810 spectropolarimeter was used to collect CD spectra. The experiments were performed at 37°C using a quartz cell with a path length of 1 mm. The spectra of the samples were an average of three scans with a wavelength of 200 to 250 nm and a speed of 5 nm/min. The experimental parameters were set as follows: response, 4 s; interval, 0.1 nm. The samples of Aβ and metallohelices were incubated in 0.01 M Hepes buffer with 0.15 M NaCl (pH 7.3) at 37°C for 7 days.
AFM studies
Aβ40 with or without equimolar A1 or B4 treatment was incubated at 37°C for 7 days. The Aβ40 samples were diluted 50-fold to obtain a concentration of 1 μM with deionized H2O. Then, 0.05 ml of the sample was dropped on a newly prepared mica substrate. After incubating for 30 min, the substrate was washed twice with deionized H2O and dried before AFM studies. Under ambient conditions, tapping mode was used to obtain the images. [Aβ40] = 50 μM.
Trypsin digestion of Aβ12–28
Aβ12–28 (20 μM, 10 μl) with or without the incubation of A1 and B4 (Aβ12–28:[A1 or B4] = 1:1) was reacted with trypsin (0.1 mg/ml) at 37°C for 1 hour. Then, all peptide samples were mixed with SDS-PAGE reducing sample buffer and boiled for 10 min. The SDS-PAGE gels were run in tris-tricine buffer. Finally, the SDS-PAGE gel was stained by silver.
Sedimentation and PAGE
Samples of 50 μM Aβ40 peptides were treated with or without 50 μM metallohelices at 37°C for 7 days. The samples were centrifuged at 13,500 rpm for 20 min to separate the aggregated Aβ40 peptide. The pellets were resuspended and heated to 100°C after mixing the sample buffer with SDS. Samples were run on a tris-tricine SDS gel (12%) for 1 hour at 100 V. Finally, the gel was stained by silver.
NMR spectroscopy
In NMR measurement, peptide samples were prepared in 0.01 M tris buffer (pH 7.3, containing 0.15 M NaCl) with 10% D2O added. The concentration of Aβ40 samples was 0.1 mM. The Aβ40 samples were treated with metallohelices ΛA1 and ΔA1 at 37°C for 2 hours. An NMR study was performed on a Bruker AVANCE NMR 600 MHz spectrometer equipped with a triple-channel cryoprobe at 5°C. [Metallohelices] = 0.1 mM.
FT-IR measurement
The concentration of the Aβ40 peptide was 6 mg/ml in D2O. The concentration of metallohelices was 1.4 mM in D2O. FT-IR spectra were obtained by using a Bio-Rad FTS-40 spectrometer with a HgCdTe detector. Solvent correction was performed by subtracting a spectrum of D2O.
Docking studies
We obtained the structure of the Aβ40 from the PDB (NMR structure in aqueous solution, PDB 2LFM). Specifically, the structures of Aβ40 and the metallohelices (ΛA1, ΔA1, ΛB4, and ΔB4) were prepared by using AutoDock Tools. The torsion angles of these metallohelices were regarded as flexible. Binding calculations were carried out between Aβ40 and the metallohelices by AutoDock Vina. In the docking study, the exhaustiveness for the docking runs was 400. The starting positions of the metallohelices were randomly determined, and 20 modes of docking were obtained during docking. Docked models of the metallohelices with Aβ40 were visualized using Discovery Studio.
SOD activity measurement by NBT
The percent inhibition of the photoreduction of NBT can be used to evaluate the SOD activities of the metallohelices. The solutions containing 0.075 mM NBT, 13 mM methionine, 0.02 mM riboflavin, and metallohelices of various concentrations were prepared in 0.05 M PBS (pH 7.8). The samples were irradiated with a lamp for 10 min at room temperature. Immediately, the absorbance of the samples was measured at 560 nm after irradiation. The reaction mixture in an identical tube was kept in the dark, serving as a blank control. The inhibition ability was assessed using the following formula: inhibition (%) = [(A0 – A)/A0] × 100, where A0 is the absorbance of the blank control and A is the absorbance of the sample.
Cyclic voltammetry
The electrolytic medium was dimethyl sulfoxide (DMSO) in 100 mM tetrabutylammonium hexafluorophosphate. CV studies were performed with a stationary glassy carbon electrode as the working electrode and platinum wire as the auxiliary electrode. The reference electrode was a Ag/AgCl electrode in saturated KCl solution. Before and after each measurement, we polished the surface of the electrode with alumina powder (0.3 to 0.05 μm). The O2/O2•− was measured at 0.1 V s−1 by using different metallohelices (20 μM) at 25°C. We bubbled the O2 gas directly into the cell with DMSO for 20 min. During the CV experiment, O2 was flushed over the cell solution using an apparatus consisting of two flowmeters (Cole-Parmer 316 SS) for O2 and N2.
Fluorescence titrations
The experimental conditions were listed as follows: [Aβ40] = 3 μM, [metallohelices] = 0.5 to 7 μM, λex = 278 nm, and λem = 306 nm. The binding constants were calculated according to the 1:1 binding stoichiometric equation. To achieve the appropriate intensity values of fluorescence, the inner filter effect needs to be corrected. Attenuation of the excitation beam and emission signal causes the inner filter effect, because the absorption by fluorophore resulted in a decrease in fluorescence intensity. The inner filter effect can be corrected with the absorbance values. Using the following equation, the fluorescence data of the system can be corrected
where Fcorr and Fobs are the corrected and observed intensity of fluorescence, respectively, and Aex and Aem are the absorbance at the excitation and emission wavelength, respectively.
ITC studies
Titrations were carried out in 10 mM Hepes buffer (pH 7.4, containing 150 mM NaCl). An ITC study was carried out on a Nano ITC System (TA Instruments Inc.). Injections of 10-μl 0.45 mM metallohelices (ΛA1, ΔA1, ΛB4, and ΔB4) were mixed by a computer-controlled microsyringe at an interval of 10 min into the 0.020 mM Aβ40 solution, with stirring at 400 rpm at a constant temperature of 25°C. NanoAnalyze software was used to analyze the experimental data (TA Instruments Inc.). A blank model was used to correct the heat of dilution. In addition, the experimental data were acquired by 25 injections of 0.45 mM metallohelices into 1400 μl of Aβ at a constant temperature of 25°C. As a result of each 10-μl metallohelix injected into the Aβ40 aqueous solution, a heat-burst curve was obtained.
Stopped-flow experiments
Fluorescence stopped-flow experiments were performed by using an SX20 Stopped-Flow Spectrometer (Applied Photophysics Limited). The final concentration of Aβ and metallohelices (ΛA1 and ΔA1) was 25 μM. Fluorescence changes were monitored at 306 nm with an excitation wavelength of 278 nm.
ROS measurement of cells
PC12 cells from a rat pheochromocytoma (obtained from the American Type Culture Collection) were cultured on 24-well plates for 24 hours. Then, samples of Aβ40 and metallohelices were added to the culture medium for 12 hours, and PC12 cells were treated with 0.050 mM DCFH-DA (2′,7′-dichlorofluorescin diacetate). After incubating for 30 min, PC12 cells were washed three times with PBS. Intracellular esterases catalyzed DCFH-DA to anionic DCFH to trap the probe in the cells. The reaction of DCFH with ROS yielded DCF. The fluorescence intensity of DCF was measured by flow cytometry.
MTT assay
PC12 cells from a rat pheochromocytoma were obtained from the American Type Culture Collection. They were cultured in Dulbecco’s minimum essential medium (Gibco) with 10% heat-inactivated horse serum and 5% fetal bovine serum. PC12 cells were cultured in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Differentiated PC12 cells were obtained by adding nerve growth factor (50 μg/ml; Invitrogen). The PC12 cells were seeded at 3 × 104 cells per well on 96-well plates for 24 hours. Aβ40 (5 μM) aged in the absence or presence of metallohelices was added into the cultured medium of differentiated PC12 cells, and the cells were cultured for another 24 hours at 37°C. Cytotoxicity was evaluated using the MTT method. Absorbance values were measured with an automatic plate reader at 490 nm. Three independent experiments were carried out for each group.
C. elegans strains
In our study, two kinds of nematodes were used, including the transgenic CL2006 strain and the N2 ancestral strain. Transfected CL2006 worms produce Aβ3–42 in the body wall muscles constitutively. The N2 strain is a wild-type strain. The nematode strains were fed with E. coli (OP50) and propagated at 20°C on solid nematode growth medium. To obtain age-synchronized worms, they were transferred to fresh NGM plates after reaching maturity at 3 days. Then, they laid eggs overnight. At day 1, isolated hatchlings were cultured on new NGM plates at 20°C.
C. elegans paralysis assay
Strains (the N2 and CL2006 worms) were cultured at 20°C on new NGM plates (35 × 10 mm culture plates, 50 worms per plate) after egg synchronization. The strains were fed with OP50 E. coli. In the experimental group, the strains were fed with metallohelices (100 μM, 100 μl per plate) at the larval stage (including L1, L2, or L3), and paralysis was estimated at the L4 larval stage. When strains only moved their head or did not move at all when gently touched with a platinum loop, we classified them as paralyzed. Three independent trials were performed.
Fluorescence staining of Aβ deposits
For strain CL2006, synchronized populations were cultured in S-basal liquid medium containing metallohelices (100 μM, 100 μl per plate) at 20°C. CL2006 worms were fixed in PBS (pH 7.4, containing 4% paraformaldehyde) for 24 hours at 4°C and rendered permeable in permeabilization buffer [1% Triton X-100, 5% β-mercaptoethanol, and 0.125 M tris (pH 7.4)] for another 24 hours at 37°C. After two washings with PBS, the nematodes were immersed in 50% ethanol with 0.125% ThS for 2 min, destained in 50% ethanol, mounted on slides, and analyzed by fluorescence images. At least three independent trials were performed.
Handling of mice
Eight- to 12-week-old Wistar rats (sex-randomized) were used as test animals. All animal experiments were carried out according to the principles and procedures in “Regulations for the Administration of Affairs Concerning Laboratory Animals” by the National Council of China and “The National Regulation of China for Care and Use of Laboratory Animals” by the National Science and Technology Commission of China. Protocols were approved by the Committee of Jilin University Institutional Animal Care and Use. Wistar rats were dissected after intraperitoneal injection of metal complexes for 4 hours. After anesthesia and exposure of the cisterna magna, CSF was obtained using a glass-pulled micropipette, ensuring that the CSF was not contaminated with blood. About 10 μl of the CSF was obtained. The CSF was immediately diluted 10-fold in 1% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) in PBS with protease inhibitors (Roche Diagnostics). The iron content of the samples was measured by ICP-MS (Varian 720-ES).
Supplementary Material
Acknowledgments
Funding: This work was supported by the 973 Project (2012CB720602), the National Natural Science Foundation of China (21210002, 21431007, 21402183, and 21533008), and the Chinese Academy of Sciences (QYZDJ-SSW-SLH052). We thank the China Scholarships Council and University of Warwick for supporting H.S. Author contributions: Y.G. performed the experiments and wrote the paper. Z.D. performed the stopped-flow kinetic and in vivo experiments. N.G. performed the docking study and ITC experiments. Y.C. and X.W. performed the in vivo experiments. P.S. and H.S. prepared the metallohelices. J.R. and X.Q. conceived, designed, and performed the experiments and wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaao6718/DC1
fig. S1. Influence of these metal complexes on the fluorescence of ECFP (a non-Aβ fusion system).
fig. S2. The influence of these metallohelices on the fluorescence of ThT.
fig. S3. Aggregation kinetics of Aβ42 monitored by ThT assay in the absence or presence of A1 and B4.
fig. S4. Aggregation kinetics of Aβ40 monitored by ThT assay in the absence or presence of the ligands of A1 and B4.
fig. S5. The inhibition effect of A1 and B4 on Aβ40/Aβ42 fibrillogenesis at different concentrations.
fig. S6. The inhibition effect of the metallohelices on Aβ40 aggregation measured by SDS-PAGE.
fig. S7. The influence of A1 and B4 on the second structures of Aβ42 monitored by CD.
fig. S8. Fluorescence titration of Aβ40 (3 μM) with various concentrations of metallohelices in 20 mM tris buffer.
fig. S9. ITC data for the Aβ40 titrations with metallohelices.
fig. S10. SDS-PAGE analysis of the effect of metallohelices on tryptic digests of Aβ12–28.
fig. S11. The aggregation kinetics of Aβ25–35 was monitored by the fluorescence of ThT in the absence or presence of A1 and B4.
fig. S12. FTIR spectra of Aβ40 in different conditions.
fig. S13. Structures of Aβ40 and metallohelices used for docking study.
fig. S14. Energy-minimized average models of ΔA1 and ΔA1 with Aβ40 interactions.
fig. S15. A1 and B4 scavenging ROS monitored by NBT and ABTS methods.
fig. S16. Cyclic voltammograms corresponding to the O2/O2• redox couple.
fig. S17. Effect of the metallohelices on ROS production in PC12 cells.
fig. S18. Absorption spectra of 5 μM metallohelices in water and PBS.
fig. S19. Effect of A1 and B4 on PC12 cell viability determined by MTT.
fig. S20. Protection effects of metallohelices on Aβ40- and Aβ42-induced cytotoxicity of PC12 cells.
table S1. IC50 values of metallohelices A1 and B4 for the inhibition of fibril formation and destabilization of the preformed fibrils.
table S2. Analysis of fluorescence titration and ITC data.
table S3. Enthalpy (ΔH), entropy (ΔS), and Gibbs free energy (ΔG) of the binding of Aβ with metallohelices at pH 7.3.
REFERENCES AND NOTES
- 1.Lehn J.-M., From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry. Chem. Soc. Rev. 36, 151–160 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Davis A. V., Yeh R. M., Raymond K. N., Supramolecular assembly dynamics. Proc. Natl. Acad. Sci. U.S.A. 99, 4793–4796 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Howson S. E., Bolhuis A., Brabec V., Clarkson G. J., Malina J., Rodger A., Scott P., Optically pure, water-stable metallo-helical ‘flexicate’ assemblies with antibiotic activity. Nat. Chem. 4, 31–36 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Faulkner A. D., Kaner R. A., Abdallah Q. M. A., Clarkson G., Fox D. J., Gurnani P., Howson S. E., Phillips R. M., Roper D. I., Simpson D. H., Scott P., Asymmetric triplex metallohelices with high and selective activity against cancer cells. Nat. Chem. 6, 797–803 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Yu H., Li M., Liu G., Geng J., Wang J., Ren J., Zhao C., Qu X., Metallosupramolecular complex targeting an α/β discordant stretch of amyloid β peptide. Chem. Sci. 3, 3145–3153 (2012). [Google Scholar]
- 6.Desmarchelier A., Caumes X., Raynal M., Vidal-Ferran A., van Leeuwen P. W. N. M., Bouteiller L., Correlation between the selectivity and the structure of an asymmetric catalyst built on a chirally amplified supramolecular helical scaffold. J. Am. Chem. Soc. 138, 4908–4916 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Brabec V., Howson S. E., Kaner R. A., Lord R. M., Malina J., Phillips R. M., Abdallah Q. M. A., McGowan P. C., Rodger A., Scott P., Metallohelices with activity against cisplatin-resistant cancer cells; does the mechanism involve DNA binding? Chem. Sci. 4, 4407–4416 (2013). [Google Scholar]
- 8.Li M., Howson S. E., Dong K., Gao N., Ren J., Scott P., Qu X., Chiral metallohelical complexes enantioselectively target amyloid β for treating Alzheimer’s disease. J. Am. Chem. Soc. 136, 11655–11663 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Brookmeyer R., Johnson E., Ziegler-Graham K., Arrighi H. M., Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 3, 186–191 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Hamley I. W., The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 112, 5147–5192 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Kosik K. S., Alzheimer’s disease: A cell biological perspective. Science 256, 780–783 (1992). [DOI] [PubMed] [Google Scholar]
- 12.Niu L., Liu L., Xi W., Han Q., Li Q., Yu Y., Huang Q., Qu F., Xu M., Li Y., Du H., Yang R., Cramer J., Gothelf K. V., Dong M., Besenbacher F., Zeng Q., Wang C., Wei G., Yang Y., Synergistic inhibitory effect of peptide–organic coassemblies on amyloid aggregation. ACS Nano 10, 4143–4153 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Li M., Xu C., Wu L., Ren J., Wang E., Qu X., Self-assembled peptide-polyoxometalate hybrid nanospheres: Two in one enhances targeted inhibition of amyloid β-peptide aggregation associated with Alzheimer’s disease. Small 9, 3455–3461 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Gao N., Sun H., Dong K., Ren J., Duan T., Xu C., Qu X., Transition-metal-substituted polyoxometalate derivatives as functional anti-amyloid agents for Alzheimer’s disease. Nat. Commun. 5, 3422 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Yoo S. I., Yang M., Brender J. R., Subramanian V., Sun K., Joo N. E., Jeong S.-H., Ramamoorthy A., Kotov N. A., Inhibition of amyloid peptide fibrillation by inorganic nanoparticles: Functional similarities with proteins. Angew. Chem. Int. Ed. 50, 5110–5115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geng J., Li M., Ren J., Wang E., Qu X., Polyoxometalates as inhibitors of the aggregation of amyloid β peptides associated with Alzheimer’s disease. Angew. Chem. Int. Ed. 50, 4184–4188 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Rubin N., Perugia E., Goldschmidt M., Fridkin M., Addadi L., Chirality of amyloid suprastructures. J. Am. Chem. Soc. 130, 4602–4603 (2008). [DOI] [PubMed] [Google Scholar]
- 18.De Carufel C. A., Quittot N., Nguyen P. T., Bourgault S., Delineating the role of helical intermediates in natively unfolded polypeptide amyloid assembly and cytotoxicity. Angew. Chem. Int. Ed. 54, 14383–14387 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Nath A., Miranker A. D., Rhoades E., A membrane-bound antiparallel dimer of rat islet amyloid polypeptide. Angew. Chem. Int. Ed. 50, 10859–10862 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knight J. D., Hebda J. A., Miranker A. D., Conserved and cooperative assembly of membrane-bound α-helical states of islet amyloid polypeptide. Biochemistry 45, 9496–9508 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Kallberg Y., Gustafsson M., Persson B., Thyberg J., Johansson J., Prediction of amyloid fibril-forming proteins. J. Biol. Chem. 276, 12945–12950 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Tjernberg L. O., Callaway D. J. E., Tjernberg A., Hahne S., Lilliehöök C., Terenius L., Thyberg J., Nordstedt C., A molecular model of Alzheimer amyloid β-peptide fibril formation. J. Biol. Chem. 274, 12619–12625 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Nerelius C., Sandegren A., Sargsyan H., Raunak R., Leijonmarck H., Chatterjee U., Fisahn A., Imarisio S., Lomas D. A., Crowther D. C., Strömberg R., Johansson J., α-Helix targeting reduces amyloid-β peptide toxicity. Proc. Natl. Acad. Sci. U.S.A. 106, 9191–9196 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cukalevski R., Yang X., Meisl G., Weininger U., Bernfur K., Frohm B., Knowles T. P. J., Linse S., The Aβ40 and Aβ42 peptides self-assemble into separate homomolecular fibrils in binary mixtures but cross-react during primary nucleation. Chem. Sci. 6, 4215–4233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi J.-S., Braymer J. J., Nanga R. P. R., Ramamoorthy A., Lim M. H., Design of small molecules that target metal-Aβ species and regulate metal-induced Aβ aggregation and neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 21990–21995 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arosio P., Michaels T. C. T., Linse S., Månsson C., Emanuelsson C., Presto J., Johansson J., Vendruscolo M., Dobson C. M., Knowles T. P. J., Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 7, 10948–10957 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bieschke J., Herbst M., Wiglenda T., Friedrich R. P., Boeddrich A., Schiele F., Kleckers D., Lopez del Amo J. M., Grüning B. A., Wang Q., Schmidt M. R., Lurz R., Anwyl R., Schnoegl S., Fändrich M., Frank R. F., Reif B., Günther S., Walsh D. M., Wanker E. E., Small-molecule conversion of toxic oligomers to nontoxic β-sheet–rich amyloid fibrils. Nat. Chem. Biol. 8, 93–101 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Szczepankiewicz O., Linse B., Meisl G., Thulin E., Frohm B., Sala Frigerio C., Colvin M. T., Jacavone A. C., Griffin R. G., Knowles T., Walsh D. M., Linse S., N-terminal extensions retard Aβ42 fibril formation but allow cross-seeding and coaggregation with Aβ42. J. Am. Chem. Soc. 137, 14673–14685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li M., Shi P., Xu C., Ren J., Qu X., Cerium oxide caged metal chelator: Anti-aggregation and anti-oxidation integrated H2O2-responsive controlled drug release for potential Alzheimer’s disease treatment. Chem. Sci. 4, 2536–2542 (2013). [Google Scholar]
- 30.Bieschke J., Russ J., Friedrich R. P., Ehrnhoefer D. E., Wobst H., Neugebauer K., Wanker E. E., EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 7710–7715 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen S. W., Drakulic S., Deas E., Ouberai M., Aprile F. A., Arranz R., Ness S., Roodveldt C., Guilliams T., De-Genst E. J., Klenerman D., Wood N. W., Knowles T. P. J., Alfonso C., Rivas G., Abramov A. Y., Valpuesta J. M., Dobson C. M., Cremades N., Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. U.S.A. 112, E1994–E2003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J., Zhao C., Zhao A., Li M., Ren J., Qu X., New insights in amyloid beta interactions with human telomerase. J. Am. Chem. Soc. 137, 1213–1219 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Qu X., Trent J. O., Fokt I., Priebe W., Chaires J. B., Allosteric, chiral-selective drug binding to DNA. Proc. Natl. Acad. Sci. U.S.A. 97, 12032–12037 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu H., Ren J., Qu X., Different hydration changes accompanying copper and zinc binding to amyloid β-peptide: Water contribution to metal binding. Chembiochem 9, 879–882 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Morgado I., Wieligmann K., Bereza M., Rönicke R., Meinhardt K., Annamalai K., Baumann M., Wacker J., Hortschansky P., Malešević M., Parthier C., Mawrin C., Schiene-Fischer C., Reymann K. G., Stubbs M. T., Balbach J., Görlach M., Horn U., Fändrich M., Molecular basis of β-amyloid oligomer recognition with a conformational antibody fragment. Proc. Natl. Acad. Sci. U.S.A. 109, 12503–12508 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greiner E. R., Kelly J. W., Palhano F. L., Immunoprecipitation of amyloid fibrils by the use of an antibody that recognizes a generic epitope common to amyloid fibrils. PLOS ONE 9, e105433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xavier-Junior F. H., Rabello M. M., Hernandes M. Z., Dias M. E. S., Andrada O. H. M. S., Bezerra B. P., Ayala A. P., Santos-Magalhães N. S., Supramolecular interactions between β-lapachone with cyclodextrins studied using isothermal titration calorimetry and molecular modeling. J. Mol. Recognit. 30, e2646 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Fu Z., Luo Y., Derreumaux P., Wei G., Induced β-barrel formation of the Alzheimer’s Aβ25–35 oligomers on carbon nanotube surfaces: Implication for amyloid fibril inhibition. Biophys. J. 97, 1795–1803 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanekiyo T., Ban T., Aritake K., Huang Z.-L., Qu W.-M., Okazaki I., Mohri I., Murayama S., Ozono K., Taniike M., Goto Y., Urade Y., Lipocalin-type prostaglandin D synthase/β-trace is a major amyloid β-chaperone in human cerebrospinal fluid. Proc. Natl. Acad. Sci. U.S.A. 104, 6412–6417 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma G., Huang F., Pu X., Jia L., Jiang T., Li L., Liu Y., Identification of [PtCl2(phen)] binding modes in amyloid-β peptide and the mechanism of aggregation inhibition. Chemistry 17, 11657–11666 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Zhang X., Tian Y., Li Z., Tian X., Sun H., Liu H., Moore A., Ran C., Design and synthesis of curcumin analogues for in vivo fluorescence imaging and inhibiting copper-induced cross-linking of amyloid beta species in Alzheimer’s disease. J. Am. Chem. Soc. 135, 16397–16409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zagorski M. G., Barrow C. J., NMR studies of amyloid β-peptides: Proton assignments, secondary structure, and mechanism of an α-helix → β-sheet conversion for a homologous, 28-residue, N-terminal fragment. Biochemistry 31, 5621–5631 (1992). [DOI] [PubMed] [Google Scholar]
- 43.Scherzer-Attali R., Pellarin R., Convertino M., Frydman-Marom A., Egoz-Matia N., Peled S., Levy-Sakin M., Shalev D. E., Caflisch A., Gazit E., Segal D., Complete phenotypic recovery of an Alzheimer’s disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLOS ONE 5, e11101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaw C. P., Middleton D. A., Volk M., Lévy R., Amyloid-derived peptide forms self-assembled monolayers on gold nanoparticle with a curvature-dependent β-sheet structure. ACS Nano 6, 1416–1426 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Baldassarre M., Li C., Eremina N., Goormaghtigh E., Barth A., Simultaneous fitting of absorption spectra and their second derivatives for an improved analysis of protein infrared spectra. Molecules 20, 12599–12622 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barth A., The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 74, 141–173 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Vivekanandan S., Brender J. R., Lee S. Y., Ramamoorthy A., A partially folded structure of amyloid-beta(1–40) in an aqueous environment. Biochem. Biophys. Res. Commun. 411, 312–316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trott O., Olson A. J., AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou Y., Ghosh P., Wan R., Ouyang X., Cheng H., Mattson M. P., Cheng A., Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1–42 on neural progenitor cell proliferation. Neurobiol. Aging 35, 975–989 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Butterfield D. A., Reed T., Sultana R., Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic. Res. 45, 59–72 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Mattson M. P., Pathways towards and away from Alzheimer’s disease. Nature 430, 631–639 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pasternack R. F., Halliwell B., Superoxide dismutase activities of an iron porphyrin and other iron complexes. J. Am. Chem. Soc. 101, 1026–1031 (1979). [Google Scholar]
- 53.Knight P. D., Clarke A. J., Kimberley B. S., Jackson R. A., Scott P., Problems and solutions for alkene polymerisation catalysts incorporating Schiff-bases; migratory insertion and radical mechanisms of catalyst deactivation. Chem. Commun. 352–353 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Woodman P. R., Sanders C. J., Alcock N. W., Hitchcock P. B., Scott P., Schiff-base chemistry of niobium: Unexpected products of radical elimination and diastereoselective addition. New J. Chem. 23, 815–817 (1999). [Google Scholar]
- 55.Wu Y., Wang D., A new class of natural glycopeptides with sugar moiety-dependent antioxidant activities derived from Ganoderma lucidum fruiting bodies. J. Proteome Res. 8, 436–442 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Floegel A., Kim D.-O., Chung S.-J., Koo S. I., Chun O. K., Comparison of ABTS/DPPH assays to measure antioxidant capacity in popular antioxidant-rich US foods. J. Food Compos. Anal. 24, 1043–1048 (2011). [Google Scholar]
- 57.Salazar R., Navarrete-Encina P. A., Squella J. A., Camargo C., Núñez-Vergara L. J., Reactivity of C4-indolyl substituted 1,4-dihydropyridines toward superoxide anion O2•− in dimethylsulfoxide. J. Phys. Org. Chem. 22, 569–577 (2009). [Google Scholar]
- 58.Diomede L., Rigacci S., Romeo M., Stefani M., Salmona M., Oleuropein aglycone protects transgenic C. elegans strains expressing Aβ42 by reducing plaque load and motor deficit. PLOS ONE 8, e58893 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi Y., Pan T., Liao V., Monascin from Monascus-fermented products reduces oxidative stress and amyloid-β toxicity via DAF-16/FOXO in Caenorhabditis elegans. J. Agric. Food Chem. 64, 7114–7120 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/eaao6718/DC1
fig. S1. Influence of these metal complexes on the fluorescence of ECFP (a non-Aβ fusion system).
fig. S2. The influence of these metallohelices on the fluorescence of ThT.
fig. S3. Aggregation kinetics of Aβ42 monitored by ThT assay in the absence or presence of A1 and B4.
fig. S4. Aggregation kinetics of Aβ40 monitored by ThT assay in the absence or presence of the ligands of A1 and B4.
fig. S5. The inhibition effect of A1 and B4 on Aβ40/Aβ42 fibrillogenesis at different concentrations.
fig. S6. The inhibition effect of the metallohelices on Aβ40 aggregation measured by SDS-PAGE.
fig. S7. The influence of A1 and B4 on the second structures of Aβ42 monitored by CD.
fig. S8. Fluorescence titration of Aβ40 (3 μM) with various concentrations of metallohelices in 20 mM tris buffer.
fig. S9. ITC data for the Aβ40 titrations with metallohelices.
fig. S10. SDS-PAGE analysis of the effect of metallohelices on tryptic digests of Aβ12–28.
fig. S11. The aggregation kinetics of Aβ25–35 was monitored by the fluorescence of ThT in the absence or presence of A1 and B4.
fig. S12. FTIR spectra of Aβ40 in different conditions.
fig. S13. Structures of Aβ40 and metallohelices used for docking study.
fig. S14. Energy-minimized average models of ΔA1 and ΔA1 with Aβ40 interactions.
fig. S15. A1 and B4 scavenging ROS monitored by NBT and ABTS methods.
fig. S16. Cyclic voltammograms corresponding to the O2/O2• redox couple.
fig. S17. Effect of the metallohelices on ROS production in PC12 cells.
fig. S18. Absorption spectra of 5 μM metallohelices in water and PBS.
fig. S19. Effect of A1 and B4 on PC12 cell viability determined by MTT.
fig. S20. Protection effects of metallohelices on Aβ40- and Aβ42-induced cytotoxicity of PC12 cells.
table S1. IC50 values of metallohelices A1 and B4 for the inhibition of fibril formation and destabilization of the preformed fibrils.
table S2. Analysis of fluorescence titration and ITC data.
table S3. Enthalpy (ΔH), entropy (ΔS), and Gibbs free energy (ΔG) of the binding of Aβ with metallohelices at pH 7.3.