

Abstract
Tan-67 is a selective non-peptidic δ-opioid receptor (DOR) agonist that confers neuroprotection against cerebral ischemia/reperfusion (I/R)-caused neuronal injury in pre-treated animals. In this study, we examined whether post-ischemic administration of Tan-67 in stroke mice is also neuroprotective and whether the treatment affects expression, maturation and processing of the amyloid precursor protein (APP). A focal cerebral I/R model in mice was induced by middle cerebral artery occlusion for 1 hour (h) and Tan-67 ( 1.5, 3 or 4.5 mg/kg) was administered via the tail vein at 1 h after reperfusion. Alternatively, naltrindole, a selective DOR antagonist (5 mg/kg), was administered 1 h before Tan-67 treatment. Our results showed that post-ischemic administration of Tan-67 (3 mg/kg or 4.5 mg/kg) was neuroprotective as shown by decreased infarct volume and neuronal loss following I/R. Importantly, Tan-67 improved animal survival and neurobehavioral outcomes. Conversely, naltrindole abolished Tan-67 neuroprotection in infarct volume. Tan-67 treatment also increased APP expression, maturation and processing in the ipsilateral penumbral area at 6 h but decreased APP expression and maturation in the same brain area at 24 h after I/R. Tan-67-induced increase of APP expression was also seen in the ischemic cortex at 24 h following I/R. Moreover, Tan-67 attenuated BACE-1 expression, β-secretase activity and the BACE cleavage of APP in the ischemic cortex at 24 h after I/R, which was abolished by naltrindole. Our data suggest that Tan-67 is a promising DOR-dependent therapeutic agent for treating I/R-caused disorder and that Tan-67-mediated neuroprotection may be mediated via modulating APP expression, maturation and processing, despite an uncertain causative relationship between the altered APP and the outcomes observed.
Keywords: ischemic stroke, brain injury, neuroprotection, opioid receptor, Tan-67, amyloid precursor protein
Graphical Abstract
We proposed that post-ischemic administration of Tan-67, a δ-opioid receptor (DOR), agonist, inhibits neuronal injury caused by ischemic stroke. Tan-67-mediated neuroprotection is dependent on DOR activation and is associated with suppression of ischemic stroke-caused alterations of amyloid precursor protein (APP) expression, maturation and processing as well as β-secretase activity. Our results suggest Tan-67 as a promising therapeutic agent for treating ischemic stroke-caused disorder.
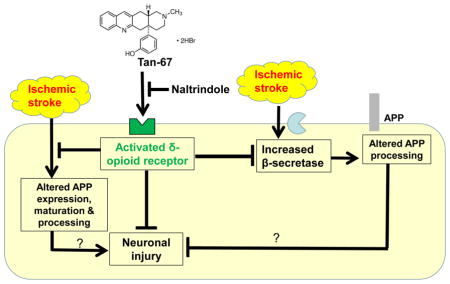
Introduction
Stroke causes high mortality and disability and is a major health problem worldwide. The majority of strokes are ischemic and occur when a blood clot blocks the blood supply to the brain, leading to neuronal injury and functional disability (O’Donnell et al. 2016). Given its public health importance, it is striking that tissue plasminogen activator (tPA), a protein that breaks down blood clots and reopens an occluded artery, is still the only Food and Drug Administration-approved drug for treating acute ischemic stroke in the United States (Liang et al. 2014). However, the utilization of tPA in ischemic stroke has some important limitations including narrow eligibility and treatment windows (Chapman et al. 2014). Therefore, additional therapeutic agents for treating ischemic stroke are in urgent need. Stroke survivors usually develop dementia and share some similar neuropathological features with Alzheimer’s disease (AD), containing altered amyloid precursor protein (APP) processing and β-amyloid (Aβ) accumulation (Badan et al. 2004, Shi et al. 2000). APP is a transmembrane glycoprotein that can be proteolytically cleaved by β- and γ-secretases to produce Aβ fragments and are implicated in AD (Panegyres 2001). It has been reported that APP is both N- and O-glycosylated (Saito et al. 1993, Pahlsson & Spitalnik 1996). The mature form (mAPP) of APP is an N-/O-glycosylated species concentrated in the trans-Golgi/plasma membrane. The immature form of APP (imAPP) is predominantly N-glycosylated and localized in the endoplasmic reticulum and cis-Golgi (Su et al. 2003). The majority of APP cleavage by α-, β-, and γ-secretases generate a soluble ectodomain (sAPP), carboxy terminal fragments (CTFs) and an APP intracellular C-terminal domain (AICD), Which occurs after O-glycosylation during APP transport through the Golgi complex or in the compartments subsequent to trans-Golgi of the APP secretory pathway (Tomita et al. 1998). The β-secretase cleavage generates β-CTFs fragments, C99/100 and C89 and α cleavage generates the fragment C83, while the γ cleavage gives rise to fragments C59-57, or AICD (Schettini et al. 2010).
Despite its key role in the molecular neuropathology of AD, increasing evidence indicate that APP is a neuroprotective factor in acute ischemic stroke (Hefter & Draguhn 2017). On the other hand, ischemic stroke also alters APP expression and processing (Hiltunen et al. 2009). Furthermore, ischemic stroke induces an increase in β-secretase activity and β-site APP cleaving enzyme (BACE) expression and thus inhibition of BACE may have a therapeutic effect on stroke (Wen et al. 2004a). Interestingly, inhibition of γ-secretase reduces ischemic stroke-caused brain injury and improves functional outcome (Arumugam et al. 2006). These studies suggest that modulation of APP expression and processing via appropriate agents may be a therapeutic strategy for stroke.
Opioid receptors (ORs) are widely distributed throughout the nervous system, which consist of three major types: δ-opioid receptor (DOR), μ-opioid receptor (MOR), and κ-opioid receptor (KOR) (Stein et al. 2003). Many pre-clinical studies have shown that opioid receptor agonists play a neuroprotective role in ischemic stroke. For instance, (D-Ala2, D-Leu5)-enkephalin, a selective DOR agonist, increases neuronal survival (Su et al. 2007, Wang et al. 2011), promote beneficial activation of astrocytes (Duan et al. 2011), regulate neurogenesis (Wang et al. 2016) and enhance antioxidative enzyme activity (Yang et al. 2009) following ischemia. BRL52537, kappa-opioid receptor (KOR) agonist, exerts neuroprotective effects partially through up-regulation of STAT3 activation and down-regulation of caspase-3 expression after stroke (Fang et al. 2013). Salvinorin A, a non-opioid kappa opioid receptors (KOR) agonist, protects against cerebral ischemia induced brain injury by modulating AQP4 expression (Xin et al. 2016) and preservation of cerebrovascular autoregulation (Wang et al. 2012). EM1 and EM2 are endogenous μ-opioid receptor (MOR) agonists, which enhance mitochondrial respiratory activity against oxidative stress during brain ischemia/reperfusion (I/R) (Feng et al. 2008). Biphalin, a highly potent, non-selective OR agonist, reduces glutamate neurotoxicity and oxidative stress following ischemia (Yang et al. 2015). These studies indicate that OR agonists may be valuable therapeutic agents for treating ischemic stroke caused problems.
Here we chose to study the therapeutic effect of Tan-67, a selective non-peptidic DOR agonist in ischemic stroke mice. Pre-treatment of Tan-67 is neuroprotective in both in vitro and in vivo models of ischemic stroke (Tian et al. 2013, Zhao et al. 2006). However, whether post-ischemic treatment of Tan-67 is neuroprotective remains unknown. We therefore examined the effect of post-ischemic Tan-67 treatment on cerebral I/R caused brain injury and on APP expression and processing as well as secretase activities in different brain regions (penumbral cortex and ischemic cortex) at different time points following stroke. Additionally, naltrindole, a selective δ-opioid receptor antagonist, was used to determine whether the neuroprotective effects of post-ischemic Tan-67 are due to activation of delta opioid receptor.
Materials and Methods
Animals
Adult C57BL/6J male mice (2–3 months of age, mean body weight 25 grams) were purchased from Jackson Laboratories (RRID IMSR_JAX:000664, Bar Harbor, ME, USA). Animals were randomly separated into the vehicle group, Tan-67 treatment group, or sham group using an online tool (http://www.graphpad.com/quickcalcs/). Mice were maintained in a 12 h light/dark cycle (lights on from 8 AM to 8 PM) in an air-conditioned (23°2°C) room with food and water available ad libitum. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of South Dakota (Permit Number: A-3207-01) and were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Transient middle cerebra artery occlusion (MCAO) ischemic stroke model
Focal cerebral ischemia was induced according to the method described in our previous study (Min et al. 2017). Isoflurane was used as the animal inhalation anesthetic agent in both short and lengthy procedures due to its short induction and recovery time and the reliability of its effects. It does not sensitize the myocardium to catecholamines, and it thus spares cardiac output more than other volatile agents (Gargiulo et al. 2012). Accordingly, anesthesia of mice was initially inhaled with 5% isoflurane (95% oxygen) and was then maintained at 1.5% isoflurane. Body temperature of the mice is maintained constant during surgery using a heating pad. To perform the surgery, a midline neck incision was made and the left common and external carotid arteries were isolated and ligated. A 20 mm-long 7-0 silicon-coated monofilament suture (7020910PK5Re; Doccol Corporation, USA) was introduced into the left internal carotid artery through the common carotid artery to block the origin of the MCA, Doppler blood flowmeter (Vasamed, Eden Prairie, MN, USA) was used to confirm MCA occlusion. After 60 minutes of ischemia, the suture was withdrawn and reperfusion was achieved. Mice showing < 20% of baseline regional cerebral blood flow in the MCA territory after occlusion and > 70% of baseline after reperfusion were considered as a successful reperfusion. In addition, when mice regained complete consciousness, their neurological deficit was evaluated using a 5-point scale as previously described (Atochin et al. 2003). Only the animals with scores between 2 and 3 were included in the experiments. Those mice with score 1, indicating unsuccessful MCAO, or 4, indicating less chance of survival, were euthanized and excluded from the experiments. Mice in the sham group were subjected to the same procedure except that the MCA was not occluded.
Drug Administration
Drug treatment experiments were carried out in a blinded manner such that the experimenters did not know the identity of each agent. A single dosage of Tan-67 (T5824; Sigma-Aldrich) (3 mg/kg) was administered via the tail vein at 1 hour (h) before ischemia (Figure 1A) or a different dosage of Tan-67 (1.5, 3 or 4.5 mg/kg) was administered 1 h after ischemia (Figure 1C). Alternatively, naltrindole (N115; Sigma-Aldrich, 5 mg/kg), a selective δ-opioid receptor antagonist, was administered via the tail vein at the beginning of reperfusion and 1 hour in prior to Tan-67 treatment (Figure 1E). The sham and vehicle groups received the same volume of saline as the treatment groups.
Figure 1.

Tan-67 reduces infarct volume in I/R-caused brain injury. A, Upper panel, Diagram of the experimental design. Tan-67 (3 mg/kg) was administered via the tail vein injection at 1 h in prior to ischemia. Mice were sacrificed at 48 h following ischemia and brains were isolated for TTC staining. Lower panel, Representative images of TTC-stained mouse brains pre-treated with Tan-67 or vehicle. B, Measurement of infarct volume. Data are shown as mean ± SEM; Vehicle group, n = 10 mice; Tan-67-treated group, n = 6 mice. ** p < 0.01. C, Upper panel, Diagram of the experimental design. Tan-67 (3 mg/kg) was administered via the tail vein injection at 1 h after ischemia. Animals were sacrificed at 48 h after ischemia and brains were then isolated for TTC staining. Lower, Representative images of TTC-stained mouse brains post-treated with Tan-67 or vehicle. D, Measurement of infarct volume. Data are shown as mean ± SEM; Vehicle group, n = 10 mice; Tan-67-treated group, n = 6 mice. * p < 0.05, ** p <0.01. E, Upper panel, Diagram of the experimental design. Naltrindole was administered intravenously 1 hour before the administration of Tan-67. Lower, Representative images of TTC-stained mouse brains post-treated with vehicle, Tan-67 alone, or naltrindole + Tan-67. F, Measurement of infarct volume. Data are shown as mean ± SEM; Vehicle group, n = 10 mice; Tan-67-treated group, n = 6 mice. naltrindole + Tan-67-treated group, n = 6 mice. * p < 0.05.
Infarct volume measurement
48 h following I/R, animals were perfused trans-cardially with phosphate-buffered saline (PBS) under deep anesthesia. The brains were removed and sectioned into 2 mm slices and were then immersed into 2% TTC (T8877; 2, 3, 5-triphenyltetrazolium chloride monohydrate, Sigma-Aldrich) solution at 37°C for 5 minutes (min), which was followed by incubation into 4% paraformaldehyde for overnight. The infarct volume was traced and analyzed using the Image J software (National Institute of Health) as described previously (Min et al. 2017).
Animal body weight and behavior assessment
To test whether Tan-67 promotes functional recovery after stroke, the mice were allowed to survive until day 7 and received daily Tan-67 injection (3 mg/kg). During this period of time, measurements of animal body weight, survival rate and a battery of behaviors (Chen et al. 2005, Chen et al. 2001) were performed at 1, 3, 5 and 7 days following MCAO by an investigator who was blinded to the experimental groups.
Nissl staining and cell counting
For minimizing the numbers of animals used, animals were perfused with PBS followed by 4% paraformaldehyde after they finished the behavior tests for 7 days following MCAO under deep anesthesia. The brains were removed for Nissl staining by following the standard protocol (Min et al. 2015). For each animal, 3–6 coronal sections were used for Nissl staining. Six random visual fields of the ischemic core region were photographed under a 40 × objective lens (RRID 490023-0007-000; Zeiss) in each section. The total number of Nissl stained cells in each field was counted using the Imaging-Pro-Plus software (Media Cybernetics, USA). The data were represented as the total number of cells per visual filed.
Western blot analysis
The brain samples (penumbral and ischemic cortex) were collected at 6 and 24 h following stroke and were used for Western blot analysis according to a previous study (Adegoke et al. 2017). Blots were probed with an anti-BACE (1:1000, 5606S, RRIDAB_1903900, Cell Signaling Technology), anti-Amyloid Precursor Protein (C-terminal) (1:1000, A8717, RRIDAB_258409, Sigma-Aldrich), or anti-actin (1:1000, sc-7210, RRID AB_2223518, Santa Cruz Biotechnology) antibody, or appropriate secondary antibodies conjugated with fluorescent dyes (1:5000, C-1003, LI-COR Inc., Lincoln, NE, USA). Protein band intensities were measured using an Odyssey scanner (LI-COR) and quantified using UN-SCAN-IT gel6.1 software (Silk Scientific Inc., Orem, Utah, USA). The total APP level (APPtot) was calculated as “APPtot = APPm + APPim” that was normalized to actin, while the ratios of APPm/APPim and APP CTF/APPtot normalized to actin were compared between each group.
β-secretase activity assay
The ipsilateral penumbral and ischemic cortex from fresh brain hemisphere were harvested in 20 volumes of PBS and were immediately frozen in dry ice. After addition of an equal volume of 2× lysis buffer (1×PBS containing 2% Nonidet P-40 and 1% sodium deoxycholate) to the frozen tissues, the tissues were homogenized and the homogenates were then incubated on ice for 60 min with intermittent vortexing. The extracts were centrifuged at 12,000 × g for 20 min and supernatant protein concentrations were determined by a Nano-Drop (ND-2000; ThermoFisher). 40 μg proteins of extracts were incubated at 37°C for 2 h in 200 μl of assay buffer containing 20 mmol/L HEPES, 10% glycerol, 2 mmol/L DTT and one microliters of fluorogenic substrates of β-secretase (Calbiochem, Darmstadt, Germany). Fluorescence was detected using a plate reader (2030-00020; Perkin–Elmer Victor) with an excitation wavelength of 355 nm and an emission wavelength of 460 nm. The background fluorescence from each sample was subtracted from the measurements in the final analysis.
Statistical Analysis
GraphPad Prism Statistical Software version 6.0 was used for statistical analysis and graphical display. The primary outcome for stroke experiments was infarct volume, which was used to determine the group size for each experiment based on our preliminary experiments. We planned a study of a continuous response variable from independent control and experimental subjects with one control per experimental subject. In a previous study, the response within each subject group was normally distributed with standard deviation 8.258034. If the true difference in the experimental and control means was 15.39912, we would need to study 6 experimental subjects and 6 control subjects to be able to reject the null hypothesis that the population means of the experimental and control groups were equal with probability (power) 0.8. The Type I error probability associated with this test of this null hypothesis was 0.05. All numerical data were presented as mean ± SEM. Significant differences between two groups were analyzed using Student’s t-test. Significant differences between more than two groups were analyzed using one-way ANOVA followed by Tukey’s post hoc analysis. P < 0.05 was considered statistically significant. We hypothesized that our data followed a normal distribution, and the hypothesis would be rejected if our evidence was against it. We did not perform a test for outliers on our data.
Results
Post-ischemic treatment of Tan-67 reduces the infarct volume following ischemic stroke
Since previous data have shown that Tan-67 (3 mg/kg) preconditioning decreases brain infarct volume after permanent MCAO (Zhao et al. 2006). To verify the neuroprotective effect of the compound on mouse brain in our ischemic stroke mouse model, we first administered the same dose of Tan-67 (3 mg/kg) via the tail vein injection at 1 h in prior to ischemia. Consistent with previous studies, we found that Tan-67 pre-treatment significantly reduced the infarct volume (p < 0.01, Figures 1A & 1B). Furthermore, to determine whether a post-ischemic treatment of Tan-67 is still beneficial to the brain, animals were treated with different doses of Tan-67. When the mice were treated with Tan-67 at a dose of 3 mg/kg or 4.5 mg/kg, they showed significant reduction of infarct volume (p < 0.05, p < 0.01, Figures 1C & 1D, respectively). As Tan-67 at 3 and 4.5 mg/kg showed a similar effect, we elected 3 mg/kg as the dosage for all other experiments. Conversely, the selective opioid receptor antagonist, naltrindole (5mg/kg), abolished the neuroprotective effect exerted by Tan-67 (Figures 1E & 1F). These results indicate that post-treatment of Tan-67 reduces infarct volume via activation of DOR following ischemic stroke.
Tan-67 improves neurological outcomes
Following surgery procedure, the body weight of Tan-67- and vehicle-treated mice was monitored from day 0 to day 7. As shown in Figure 2A, both Tan-67 and vehicle groups of animal rapidly lost their body weights following I/R. There was no significant difference between the two groups within 7 days following stroke. However, Tan-67 treatment promoted animal survival compared with the vehicle treatment (Figure 2B). To evaluate whether Tan-67 enhances animal functional recovery, animals were assessed using a modified neurological score system that assesses both animal motor and sensory deficits following MCAO (Chen et al. 2005, Chen et al. 2001). Mice treated with Tan-67 showed more rapid functional recovery than the control group: Tan-67 significantly enhanced functional recovery after 3 days, which persisted until 7 days (p < 0.05, Figure 2C). Thus, post-treatment of Tan-67 enhances animal survival and functional recovery following ischemic stroke.
Figure 2.

Tan-67 treatment improves survival and neurobehavioral performance after I/R. A, Body weight of Tan-67- and vehicle-treated mice. Data are shown as mean °SEM; Vehicle group, n = 6; Tan-67-treated group, n = 8. B, Tan-67-treated mice show improved survival rate compared to the vehicle-treated mice. Vehicle group, n = 6/15; Tan-67-treated group, n = 8/12. C, Tan-67-treated mice show more rapidly functional recovery than the vehicle-treated mice. Data are shown as median ± SEM; Vehicle group, n = 6 mice; Tan-67-treated group, n = 8 mice. * p < 0.05.
Tan-67 treatment reduces neuronal cell death
Neuronal cell loss in the ipsilateral ischemic cortex was identified by Nissl staining 7 days after I/R. As expected, MCAO procedure significantly reduced the number of cells and resulted in shrinkage and irregularly arranged neurons compared with the contralateral cortex (Figures 3A). Cell loss caused by MCAO was rescued, at least partially, by Tan-67 treatment (p < 0.01, Figures 3A & 3B), indicating post-treatment of Tan-67 reduces neuronal loss following the procedure.
Figure 3.

Tan-67 treatment attenuates neuronal death following I/R. A, Nissl staining of damaged ischemic cortex in coronal sections of the ipsilateral or contralateral brains from Tan-67-treated and vehicle-treated mice at 7 days after MCAO. B, Quantitation of Nissl-stained neurons. Data are shown as mean ± SEM; n = 3 mice. ** p < 0.01.
Tan-67 increases both total APP and mature APP (APPm) levels and APP processing at an early time point (6 h) but decreases their levels at a late time point (24 h) in the penumbral cortex following MCAO
We next asked whether post-ischemic treatment of Tan-67 affects APP expression, maturation, and processing following MCAO. Accordingly, we examined the levels of both mature (APPm) and immature (APPim) APP as well as the C-terminal fragment (CTF) of APP at 6 and 24 h following MCAO in the penumbral cortex (or peri-ischemic cortex) based on TTC staining. APP expression was reflected by the total APP level (APPm plus APPim). MCAO induced remarkable reduction of both total APP level and APPm/APPim ratio in the penumbral cortex at 6 h after MCAO (Figures 4A & 4B). However, APPm/APPim ratio and total APP level were significantly increased in the penumbral cortex, which was associated with increased CTF levels (normalized against total APP levels), in the Tan-67-treated MCAO mice compared with vehicle-treated mice. In contrast, Tan-67 induced an increase of APPm/APPim ratio, total APP and CTF levels was reversed by naltrindole (p < 0.05, Figures 4A & 4B).
Figure 4.

Effects of Tan-67-mediated activation of DOR on APP expression and processing and BACE-1 level in the ipsilateral penumbral cortex of MCAO mice at 6 and 24 h after MCAO. A, Western blot analysis of APPm, APPim, as well as APP CTF levels at 6 h after MCAO. B, Quantitation of Western blot results shown in (A). C, Western blot analysis of BACE-1 level at 6 h following MCAO. D, Quantitation of BACE-1 level shown in (C). E, Western blot analysis of APPm, APPim, as well as APP CTF levels at 24 h after MCAO. F, Quantitation of Western blot results shown in (E). G, Western blot analysis of BACE-1 level at 24 h following MCAO. H, Quantitation of BACE-1 level shown in (G). Molecular weight markers are indicated as kDa on the right and actin was used as a loading control. All numerical data are shown as mean° SEM; n = 6 mice. * p < 0.05, ** p < 0.01, ***p < 0.001.
At 24 h, APPm/APPim ratio and total APP level were significantly decreased in Tan-67 treated group compared with vehicle group and naltrindole plus Tan-67-treated group following MCAO (p < 0.05, Figures 4E & 4F). However, Tan-67 and naltrindole treatment did not alter BACE-1 expression at any time points examined (Figures 4C, 4D, 4G & 4H). Thus, post-ischemic treatment of Tan-67 increases APP expression, maturation and processing at an early time point, while it reduces their levels at a late time point in the penumbral cortex following I/R.
Tan-67 also increases both the total APP and APPm levels but reduces BACE-1 expression at a late time point (24 h) in the ischemic core following MCAO
At 6 h following ischemic stroke, compared to vehicle treatment and naltrindole plus Tan-67-treated group, Tan-67 treatment significantly decreased APPm/APPim ratio and total APP in the ischemic core (p < 0.05, p < 0.001 Figures 5A & 5B). Tan-67 treatment did not alter BACE-1 expression level at 6 h after MCAO (Figures 5C & 5D). At 24 h following MCAO, the total APP level in Tan-67-treated mice increased compared to that in vehicle-treated and naltrindole plus Tan-67-treated mice in the ischemic core (p < 0.01, Figures 5E & 5F). Interestingly, Tan-67-treated mice showed a significant lower BACE-1 level in ischemic cortex compared with the vehicle-treated and naltrindole plus Tan-67-treated mice at 24 h after MCAO (p < 0.01, p < 0.001 Figures 5G & 5H). These results indicate that activation of DOR increases total APP levels in a delayed manner in ischemic core following ischemic stroke.
Figure 5.

Effects of Tan-67-mediated activation of DOR on APP expression and processing and BACE-1 level in the ipsilateral ischemic core of MCAO mice at 6 and 24 h after MCAO. A, Western blot analysis of APPm, APPim, as well as APP CTF levels at 6 h after MCAO. B, Quantitation of Western blot results shown in (A). C, Western blot analysis of BACE-1 level at 6 h following MCAO. D, Quantitation of BACE-1 level shown in (C). E, Western blot analysis of APPm, APPim, as well as APP CTF levels at 24 h after MCAO. F, Quantitation of Western blot results shown in (E). G, Western blot analysis of BACE-1 level at 24 h following MCAO. H, Quantitation of BACE-1 level shown in (G). Molecular weight markers are indicated as kDa on the right and actin was used as a loading control. All numerical data are shown as mean° SEM; n = 6 mice. * p < 0.05, ** p < 0.01, *** p < 0.001.
Post-treatment of Tan-67 reduces β-secretase activity in ischemic cortex at 24 h following ischemic stroke
Finally, we assessed the β- and γ-secretase activities in the ipsilateral penumbral and ischemic cortex at 6 and 24 h after MCAO in each group of animal. β-secretase activity was not significantly altered by Tan-67 treatment in the ipsilateral penumbral cortex at different time points (Figure 6A). However, Tan-67 treatment remarkably reduced β-secretase activity in the ipsilateral ischemic cortex at 24 h after MCAO compared with the vehicle treatment (Figure 6B). This effect was significantly attenuated by naltrindole (p < 0.05, Figure 6B). Intriguingly, γ-secretase activity in the Tan-67 treated animals did not differ from other groups of mice either in penumbral cortex or in ischemic core (Data not shown). Thus, Tan-67 reduces of β-secretase activity in ischemic core at a late time point.
Figure 6.
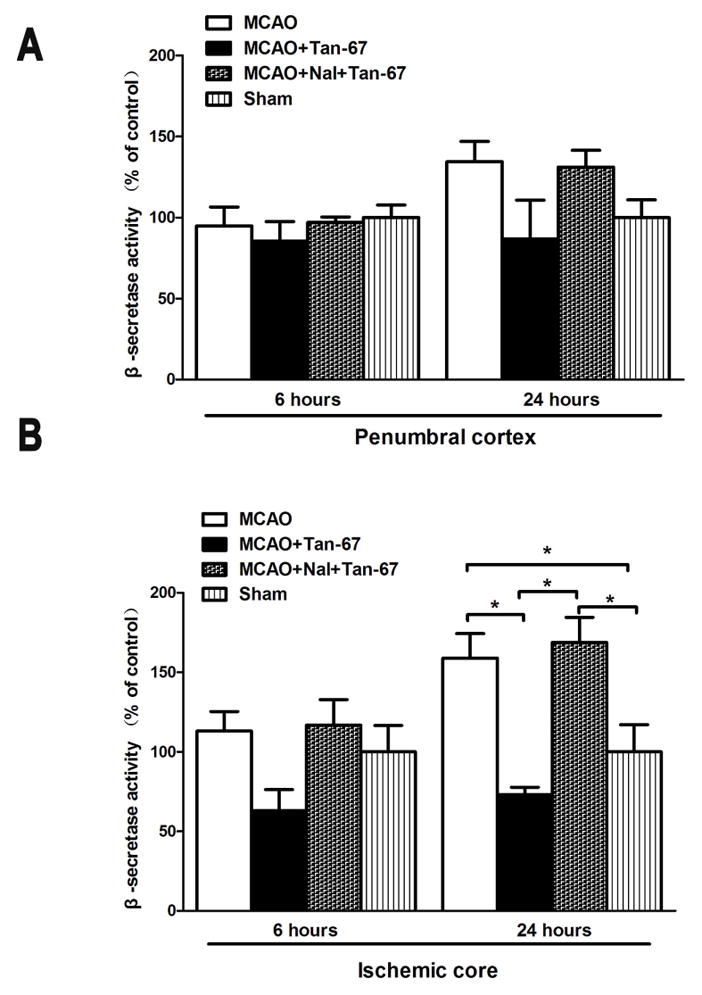
Effect of Tan-67-mediated activation of DOR on β-secretase activity in the ischemic cortex after MCAO. A, β-secretase activity did not show significant difference in the ipsilateral penumbral cortex between each group of mice at 6 and 24 h after MCAO. B, β-secretase activity was significantly increased in the ipsilateral ischemic cortex of vehicle-treated and naltrindole plus Tan-67-treated mice compared with that of sham surgery mice at 24 h following MCAO, but Tan-67-treatment inhibited the increase. Data are shown as mean ± SEM; n = 3 mice. * p < 0.05.
Discussion
Using a mouse stroke model, we here demonstrated that pre-ischemic treatment of mice with a selective non-peptidic δ-opioid receptor agonist, Tan-67, reduced cerebral infarct size induced by MCAO. This result is in accordance with two previous observations that injection of the δ-opioid receptor (DOR) agonist Tan-67, into the lateral cerebroventricle 30 min in prior to MCAO significantly alleviated neuronal injury in MCAO rats (Tian et al. 2013), and that pre-treatment of hippocampal slice cultures with Tan-67 increased ischemic tolerance (Zhao et al. 2006). More importantly, we also demonstrated that post-ischemic treatment of MCAO mice with Tan-67 (3 mg/kg) administered peripherally was also neuroprotective as seen by decreased neuronal injury, reduced animal mortality, and enhanced animal functional recovery. However, Tan-67 mediated neuroprotection was completely abolished by administration of a selective δ-opioid receptor antagonist, naltrindole, in the study. Therefore, the data strongly suggest that when administered post-ischemically, Tan-67 can exert potent neuroprotection through the activation of DOR. Thus, our data provide evidence that Tan-67 is a potential therapeutic agent for ischemic stroke.
One question raised from this study is how Tan-67 exerts its neuroprotection against ischemic stroke-induced neuronal injury. As we discovered that the neuroprotective effect of Tan-67 was associated with its modulation of APP expression, maturation and processing, it is possible that Tan-67-mediated neuroprotection may through its upregulation of APP expression and processing. Indeed, APP participates in a number of important functions in the brain, including synaptic function and cellular response to stress (Panegyres 2001). Numerous studies have suggested that APP is a neuroprotective factor in acute neuronal insults, including acute hypoxia-ischemia, traumatic brain injury, and excitotoxicity (Hefter & Draguhn 2017). Our data demonstrated that MCAO induced evident decrease of APP level compared with the sham-control at 6 h, while Tan-67 treatment attenuated MCAO-induced APP decrease and this effect could be reversed by naltrindole. As the altered intracellular neuroprotective molecules in early phase (such as 6 h) following MCAO may be critical in order to achieve neuroprotection, Tan-67-mediated upregulation of total APP, APPm and APP processing may play a significant role in its induced neuroprotection observed here. Given the fact that increased APP level protects neurons against excitotoxic and ischemic damage both in vitro (Goodman & Mattson 1994) and in vivo (Bowes et al. 1994, Masliah et al. 1997, Clarke et al. 2007), Tan-67 mediated DOR-dependent neuroprotection following I/R may be, at least partially, via modulation of APP expression/processing. However, whether the APP expression/processing alterations caused by Tan-67 treatment is causal to the outcomes observed remains to be investigated.
At 24 h, we observed MACO-induced increase of APP level in the penumbral cortex. This was in accordance with previous reports showing that APP protein or mRNA expression is increased after 1 or 2 days following ischemic stroke (Hiltunen et al. 2009, Kim et al. 1998, Shi et al. 2000). The observation showing that activation of DOR by Tan-67 reduced total APP and APPm levels at 24 h indicates the possibility that a high APP level is no longer required for neuroprotection in late phase (such as after 24 h) following MCAO. On the contrary, after 24 h, a persistent high level of APP or APPm may be harmful for neuronal recovery and survival in the penumbral region, as they can produce increased level of toxic Aβ peptides to induce neuronal death.
It is also possible that Tan-67-mediated neuroprotection is through modulation of BACE-1 expression and β-secretase activity. There was a tendency toward increase in APP CTF either in vehicle or naltrindole plus Tan-67-treated group compared to Tan-67 alone treated group in the ischemic core at 24h following MCAO. Additionally, we also observed that I/R upregulated BACE-1 expression and β-secretase activity both in vehicle and naltrindole plus Tan-67-treated groups, while Tan-67 alone appeared to suppress BACE-1 upregulation and β-secretase activity. Taken together, these data suggest that BACE-1 cleavage of APP was at least partially inhibited by Tan-67. These observations are also in accordance with previous studies showing that BACE-1 protein and β-secretase activity were increased in the ischemic cortex, and BACE-1 was strongly associated with apoptotic cells (Wen et al. 2004b). Moreover, activation of BACE-1 enhances caspase activation and DNA fragmentation, as observed in AD brains (Fukumoto et al. 2002, Hugon et al. 2000, Rohn et al. 2002). Thus, suppression of BACE-1 and β-secretase activity by Tan-67 is neuroprotective in late phase following I/R, although whether β-secretase activity change caused by Tan-67 is causative to the altered APP expression/processing remains uncertain.
In summary, our data indicate that post-ischemic administration of Tan-67, a non-peptide δ-opioid receptor agonist, confers DOR-dependent neuroprotection, as shown by reduced infarct size and cell death, improved animal survival, as well as enhanced functional recovery. Moreover, we also provide data showing that Tan-67 treatment is associated with altered APP expression, maturation and processing following ischemic stroke. These results indicate that Tan-67 is likely a therapeutic agent for treating ischemic stroke, and that the neuroprotective role of Tan-67 may link to its modulation of APP expression and processing.
Acknowledgments
We would like to thank Dr. J. Scott Pattison for assistance in microscopy. This work was supported by the National Institute of Neurological Disorders and Stroke under research grants R01NS088084.
Abbreviations
- I/R
ischemia/reperfusion
- APP
amyloid precursor protein
- APPm
mature APP
- APPim
immature APP
- CTF
carboxy terminal fragment
- sAPP
soluble ectodomain
- AICD
APP intracellular C-terminal domain
- tPA
tissue plasminogen activator
- Aβ
β-amyloid
- BACE
β-site APP cleaving enzyme
- OR
Opioid receptors
- DOR
δ-opioid receptor
- MOR
μ-opioid receptor
- KOR
κ-opioid receptor
- AD
Alzheimer’s disease
- MCAO
middle cerebra artery occlusion
- TTC
2, 3, 5-triphenyltetrazolium chloride monohydrate
- PBS
phosphate-buffered saline
Footnotes
conflict of interest disclosure
The authors declare no conflict of interest. This study was not pre-registered.
References
- Adegoke OO, Qiao F, Liu Y, Longley K, Feng S, Wang H. Overexpression of Ubiquilin-1 Alleviates Alzheimer’s Disease-Caused Cognitive and Motor Deficits and Reduces Amyloid-beta Accumulation in Mice. J Alzheimers Dis. 2017 doi: 10.3233/JAD-170173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam TV, Chan SL, Jo DG, et al. Gamma secretase-mediated notch signaling worsens brain damage and functional outcome in ischemic stroke. Nature medicine. 2006;12:621–623. doi: 10.1038/nm1403. [DOI] [PubMed] [Google Scholar]
- Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL. Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke; a journal of cerebral circulation. 2003;34:1299–1303. doi: 10.1161/01.STR.0000066870.70976.57. [DOI] [PubMed] [Google Scholar]
- Badan I, Dinca I, Buchhold B, Suofu Y, Walker L, Gratz M, Platt D, Kessler CH, Popa-Wagner A. Accelerated accumulation of N- and C-terminal beta APP fragments and delayed recovery of microtubule-associated protein 1B expression following stroke in aged rats. European Journal of Neuroscience. 2004;19:2270–2280. doi: 10.1111/j.0953-816X.2004.03323.x. [DOI] [PubMed] [Google Scholar]
- Bowes MP, Masliah E, Otero DAC, Zivin JA, Saitoh T. Reduction of Neurological Damage by a Peptide Segment of the Amyloid Beta/A4 Protein-Precursor in a Rabbit Spinal-Cord Ischemia Model. Experimental neurology. 1994;129:112–119. doi: 10.1006/exnr.1994.1152. [DOI] [PubMed] [Google Scholar]
- Chapman SN, Mehndiratta P, Johansen MC, McMurry TL, Johnston KC, Southerland AM. Current perspectives on the use of intravenous recombinant tissue plasminogen activator (tPA) for treatment of acute ischemic stroke. Vasc Health Risk Manag. 2014;10:75–87. doi: 10.2147/VHRM.S39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke; a journal of cerebral circulation. 2001;32:2682–2688. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- Chen JL, Zhang CL, Jiang H, Li Y, Zhang LJ, Robin A, Katakowski M, Lu M, Chopp M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cerebr Blood F Met. 2005;25:281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J, Thornell A, Corbett D, Soininen H, Hiltunen M, Jolkkonen J. Overexpression of APP provides neuroprotection in the absence of functional benefit following middle cerebral artery occlusion in rats. European Journal of Neuroscience. 2007;26:1845–1852. doi: 10.1111/j.1460-9568.2007.05807.x. [DOI] [PubMed] [Google Scholar]
- Duan YL, Wang SY, Zeng QW, Su DS, Li W, Wang XR, Zhao Z. Astroglial Reaction to Delta Opioid Peptide [D-Ala2, D-Leu5] Enkephalin Confers Neuroprotection against Global Ischemia in the Adult Rat Hippocampus. Neuroscience. 2011;192:81–90. doi: 10.1016/j.neuroscience.2011.06.067. [DOI] [PubMed] [Google Scholar]
- Fang SD, Xu H, Lu JR, Zhu YS, Jiang H. Neuroprotection by the Kappa-Opioid Receptor Agonist, BRL52537, is Mediated via Up-Regulating Phosphorylated Signal Transducer and Activator of Transcription-3 in Cerebral Ischemia/Reperfusion Injury in Rats. Neurochemical research. 2013;38:2305–2312. doi: 10.1007/s11064-013-1139-4. [DOI] [PubMed] [Google Scholar]
- Feng Y, Lu YW, Lin X, Gao YF, Zhao QY, Li W, Wang R. Endomorphins and morphine limit anoxia-reoxygenation-induced brain mitochondrial dysfunction in the mouse. Life sciences. 2008;82:752–763. doi: 10.1016/j.lfs.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol-Chicago. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Gargiulo S, Greco A, Gramanzini M, Esposito S, Affuso A, Brunetti A, Vesce G. Mice Anesthesia, Analgesia, and Care, Part I: Anesthetic Considerations in Preclinical Research. Ilar Journal. 2012;53:E55–E69. doi: 10.1093/ilar.53.1.55. [DOI] [PubMed] [Google Scholar]
- Goodman Y, Mattson MP. Secreted Forms of Beta-Amyloid Precursor Protein Protect Hippocampal-Neurons against Amyloid Beta-Peptide-Induced Oxidative Injury. Experimental neurology. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- Hefter D, Draguhn A. APP as a Protective Factor in Acute Neuronal Insults. Front Mol Neurosci. 2017;10:22. doi: 10.3389/fnmol.2017.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen M, Makinen P, Peraniemi S, Sivenius J, van Groen T, Soininen H, Jolkkonen J. Focal cerebral ischemia in rats alters APP processing and expression of A beta peptide degrading enzymes in the thalamus. Neurobiology of disease. 2009;35:103–113. doi: 10.1016/j.nbd.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Hugon J, Esclaire F, Terro F, Yardin C. Apoptosis and Alzheimer’s disease: contribution of cellular and transgenic models. Rev Neurol. 2000;156:123–125. [PubMed] [Google Scholar]
- Kim HS, Lee SH, Kim SS, Kim YK, Jeong SJ, Ma J, Han DH, Cho BK, Suh YH. Post-ischemic changes in the expression of Alzheimer’s APP isoforms in rat cerebral cortex. Neuroreport. 1998;9:533–537. [PubMed] [Google Scholar]
- Liang X, Hu Q, Li B, McBride D, Bian H, Spagnoli P, Chen D, Tang J, Zhang JH. Follistatin-like 1 attenuates apoptosis via disco-interacting protein 2 homolog A/Akt pathway after middle cerebral artery occlusion in rats. Stroke. 2014;45:3048–3054. doi: 10.1161/STROKEAHA.114.006092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L. Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience. 1997;78:135–146. doi: 10.1016/s0306-4522(96)00553-2. [DOI] [PubMed] [Google Scholar]
- Min JW, Hu JJ, He M, et al. Vitexin reduces hypoxia-ischemia neonatal brain injury by the inhibition of HIF-1 alpha in a rat pup model. Neuropharmacology. 2015;99:38–50. doi: 10.1016/j.neuropharm.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Min JW, Lu LH, Freeling JL, Martin DS, Wang HM. USP14 inhibitor attenuates cerebral ischemia/reperfusion-induced neuronal injury in mice. Journal of Neurochemistry. 2017;140:826–833. doi: 10.1111/jnc.13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell MJ, Chin SL, Rangarajan S, et al. Global and regional effects of potentially modifiable risk factors associated with acute stroke in 32 countries (INTERSTROKE): a case-control study. Lancet. 2016;388:761–775. doi: 10.1016/S0140-6736(16)30506-2. [DOI] [PubMed] [Google Scholar]
- Pahlsson P, Spitalnik SL. The role of glycosylation in synthesis and secretion of beta-amyloid precursor protein by Chinese hamster ovary cells. Archives of biochemistry and biophysics. 1996;331:177–186. doi: 10.1006/abbi.1996.0296. [DOI] [PubMed] [Google Scholar]
- Panegyres PK. The functions of the amyloid precursor protein gene. Reviews in the neurosciences. 2001;12:1–39. doi: 10.1515/revneuro.2001.12.1.1. [DOI] [PubMed] [Google Scholar]
- Rohn TT, Rissman RA, Head E, Cotman CW. Caspase activation in the Alzheimer’s disease brain: Tortuous and torturous. Drug News Perspect. 2002;15:549–557. doi: 10.1358/dnp.2002.15.9.740233. [DOI] [PubMed] [Google Scholar]
- Saito F, Yanagisawa K, Miyatake T. Soluble Derivatives of Beta A4 Amyloid Protein-Precursor in Human Cerebrospinal-Fluid Are Both N-Glycosylated and O-Glycosylated. Mol Brain Res. 1993;19:171–174. doi: 10.1016/0169-328x(93)90164-k. [DOI] [PubMed] [Google Scholar]
- Schettini G, Govoni S, Racchi M, Rodriguez G. Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: signal transduction and/or transcriptional role - relevance for Alzheimer pathology. Journal of neurochemistry. 2010;115:1299–1308. doi: 10.1111/j.1471-4159.2010.07044.x. [DOI] [PubMed] [Google Scholar]
- Shi J, Yang SH, Stubley L, Day AL, Simpkins JW. Hypoperfusion induces overexpression of beta-amyloid precursor protein mRNA in a focal ischemic rodent model. Brain Res. 2000;853:1–4. doi: 10.1016/s0006-8993(99)02113-7. [DOI] [PubMed] [Google Scholar]
- Stein C, Schafer M, Machelska H. Attacking pain at its source: new perspectives on opioids. Nature medicine. 2003;9:1003–1008. doi: 10.1038/nm908. [DOI] [PubMed] [Google Scholar]
- Su DS, Wang ZH, Zheng YJ, Zhao YH, Wang XR. Dose-dependent neuroprotection of delta opioid peptide [D-Ala2, D-Leu5] enkephalin in neuronal death and retarded behavior induced by forebrain ischemia in rats. Neuroscience letters. 2007;423:113–117. doi: 10.1016/j.neulet.2007.06.044. [DOI] [PubMed] [Google Scholar]
- Su Y, Ryder J, Ni BH. Inhibition of A beta production and APP maturation by a specific PKA inhibitor. FEBS letters. 2003;546:407–410. doi: 10.1016/s0014-5793(03)00645-8. [DOI] [PubMed] [Google Scholar]
- Tian XS, Guo JC, Zhu M, Li MW, Wu GC, Xia Y. delta-Opioid Receptor Activation Rescues the Functional TrkB Receptor and Protects the Brain from Ischemia-Reperfusion Injury in the Rat. PloS one. 2013:8. doi: 10.1371/journal.pone.0069252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Kirino Y, Suzuki T. A basic amino acid in the cytoplasmic domain of Alzheimer’s beta-amyloid precursor protein (APP) is essential for cleavage of APP at the alpha-site. Journal of Biological Chemistry. 1998;273:19304–19310. doi: 10.1074/jbc.273.30.19304. [DOI] [PubMed] [Google Scholar]
- Wang SY, Duan Y, Su DS, Li W, Tan J, Yang DQ, Wang WL, Zhao Z, Wang XR. Delta opioid peptide [d-Ala2, d-Leu5] enkephalin (DADLE) triggers postconditioning against transient forebrain ischemia. European journal of pharmacology. 2011;658:140–144. doi: 10.1016/j.ejphar.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Wang SY, Duan YL, Zhao B, Wang XR, Zhao Z, Zhang GM. Effect of delta opioid receptor activation on spatial cognition and neurogenesis in cerebral ischemic rats. Neuroscience letters. 2016;620:20–26. doi: 10.1016/j.neulet.2016.03.035. [DOI] [PubMed] [Google Scholar]
- Wang ZH, Ma N, Riley J, Armstead WM, Liu RY. Salvinorin A Administration after Global Cerebral Hypoxia/Ischemia Preserves Cerebrovascular Autoregulation via Kappa Opioid Receptor in Piglets. PloS one. 2012:7. doi: 10.1371/journal.pone.0041724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Onyewuchi O, Yang SH, Liu R, Simpkins JW. Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain research. 2004a;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Wen Y, Onyewuchi O, Yang SH, Liu R, Simpkins JW. Increased beta-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004b;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Xin JH, Zhang Y, He ZZ, Wang ZH. Highly selective non-opioid kappa opioid receptor (KOR) agonist salvinorin A protects against forebrain ischemia-induced brain injury in rats. Brain research. 2016;1637:168–176. doi: 10.1016/j.brainres.2016.02.024. [DOI] [PubMed] [Google Scholar]
- Yang L, Islam MR, Karamyan VT, Abbruscato TJ. In vitro and in vivo efficacy of a potent opioid receptor agonist, biphalin, compared to subtype-selective opioid receptor agonists for stroke treatment. Brain research. 2015;1609:1–11. doi: 10.1016/j.brainres.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YL, Xia XW, Zhang Y, Wang Q, Li L, Luo GH, Xia Y. delta-Opioid receptor activation attenuates oxidative injury in the ischemic rat brain. Bmc Biol. 2009:7. doi: 10.1186/1741-7007-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Huang YM, Zuo ZY. Opioid preconditioning induces opioid receptor-dependent delayed neuroprotection against ischemia in rats. Journal of neuropathology and experimental neurology. 2006;65:945–952. doi: 10.1097/01.jnen.0000235123.05677.4b. [DOI] [PubMed] [Google Scholar]