

Abstract
Epigenome editing is a powerful method for life science research and could give rise to new therapies for diseases initiated or maintained by epigenetic dysregulation, including several types of cancers and autoimmune disorders. In addition, much is still unknown about the mechanisms by which histone-modifying proteins work in concert to properly regulate gene expression. To investigate and manipulate complex epigenetic interactions in live cells, we have developed a small molecule platform for specifically inducing gene repression and histone deacetylation at a reporter gene. We synthesized bifunctional ligands, or chemical epigenetic modifiers (CEMs), that contain two functional groups: a FK506 derivative capable of binding to a FKBP-Gal4 fusion transcription factor, and a histone deacetylase (HDAC) inhibitor that recruits HDAC-containing corepressor complexes. In our reporter cell line, which contains a GFP reporter allele upstream of a Gal4 DNA binding array in the murine Oct4 locus, our lead CEM repressed GFP expression by 50%. We also show that CEM recruitment of deacetylation activity causes marked deacetylation along our target loci. This system allowed us to detail the direct results of deacetylation to chromatin and measure the resulting gene expression in a chemically dependent and reversible manner. The CEMs system provides new insights into epigenetic gene regulation and has the potential to control disease-relevant gene regulation. The CEMs are derived from FDA-approved epigenetic modulator drugs, and use their pharmacology in a gene-specific way that avoids the toxicities and off-target effects caused by whole-cell application of these drugs.
Keywords: bifunctional molecules, chemical induced proximity histone deacetylase, gene repression chromatin regulation, epigenetics
Graphical Abstract
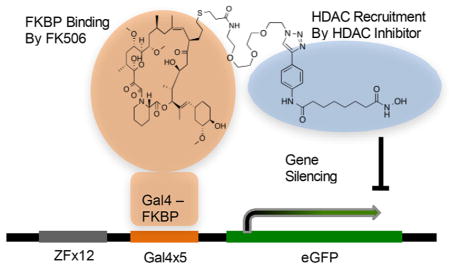
Specific control of gene expression programs is critical for proper mammalian development and allows for the creation of differentiated tissues from a pluripotent starting state. Much of this control comes from regulation at the level of chromatin, where the presence or absence of chemical marks on DNA and histone tails regulate expression of the associated gene by directing the binding of transcription factors, corepressors, coactivators, and other biomolecules.1,2 These epigenetic pathways are commonly disrupted in many human diseases including cancer.3 Thus, both to understand human biology and to develop new therapeutic strategies for treating diseases driven by epigenetic dysregulation, it is important to develop new technologies capable of synthetically modulating epigenetic processes at individual genes or sets of genes. Site-specific editing of these epigenetic marks can be accomplished with dCas9 or TALE mediated recruitment of epigenetic enzymes.4–6 In one example, a small guide RNA (sgRNA) is capable of recruiting a dCas9-p300 fusion protein to target genes, causing histone acetylation and transcription activation.7 These new approaches have proved to be extremely useful opening up new avenues for the study of epigenetics and could turn out to be a whole new class of therapeutic for personalized medicine.
One major drawback of using a genetic editing technology as a therapeutic or an in vivo tool, is off target or continuous recruitment resulting in permanent genetic mutation at undesirable loci.8 As an alternative, we considered a small molecule platform for epigenome editing. Small molecules have some advantages over genetic methods as a therapeutic and a research tool: treating cells with small molecules is technically easy and requires little optimization, they are typically easy to synthesize, they can be active in vivo, effects are often dose sensitive, and most importantly their effects are reversible. Disadvantages also exist for small molecules: their functions are typically more difficult to engineer, their cellular effects may be less profound, and they may have more off-target effects than genetic methods.
Using bivalent small molecules to direct proteins to DNA has been investigated.9 “Small molecule transcription factors” featuring hairpin polyamide DNA binding motifs linked to histone deacetylase inhibitors were used to activate transcription of DNA.10 Because polyamides are difficult to synthesize and the effects likely result from a combination of on-target and off-target effects, this approach is not ideal. Similar examples have been reported that use polyamides or transcription factor ligands as the DNA binding ligand and coactivator-binding molecules as a transcriptional activation domain.9 In another example, glucocorticoid receptor (GR) ligands tethered to FK506 were used to recruit a histone deacetylase (HDAC) 1–FKBP fusion protein to DNA, causing down-regulation of GR-controlled genes.11 Recently, Liszczak et al. used native chemical ligation to attach small molecules to dCas9, allowing CRISPR guided targeting of the molecules to DNA.12 This system did not cause any changes in gene expression unless the cells were cotransfected with a VP64-Brd4 fusion protein. Thus far none of these methods have been shown to specifically change both the epigenetic state and the expression of a target gene.
Controlling chemically induced proximity between corepressor complexes and DNA-bound transcription factors requires a bivalent ligand capable of strong interactions with both the transcription factor and a corepressor complex. Components of corepressor complexes with high-affinity ligands include the histone deacetylase (HDAC) enzymes and EZH2.13,14 We chose HDAC inhibitors as the ligands for many reasons: they are potent, easy to synthesize, and tolerate the attachment of long linker groups while still being cell-permeable.15 Also, the isoform selectivity and residence time can be tuned by modifying the ligand.16,17 HDAC enzymes reverse the histone acetyl marks associated with open chromatin and active transcription.18,19 It may seem counterintuitive to use enzyme inhibitors to recruit a complex that enzymatically modifies histones, but HDACs exist in complexes that contain proteins capable of silencing genes through many different mechanisms, and some HDACs are catalytically inactive pseudoenzymes that still bind inhibitors.20,21 Furthermore, HDAC enzymes can exist in complexes with other HDAC isoforms that would not be inhibited by the small molecule.22 HDAC inhibitors bind to corepressor complexes such as CoREST, NuRD, and Sin3 with unique selectivity profiles, depending on the chemical structure and HDAC isoform bound.15,23 By tuning the inhibitor structure, it may be possible to recruit specific corepressor complexes to the target gene.
Additionally, these bifunctional molecules are derived from FDA-approved HDAC inhibitors that have been well studied and characterized. Suberanilohydroxamic acid (SAHA) and several other epigenetic inhibitors are widely used in the clinic.24,25 Though these drugs are effective modulators of certain cellular pathways, their cell-wide effects cause high toxicity and modulate the expression of many genes.26 We describe a targeted, gene-specific application of these molecules. This technology can control gene expression in a dose sensitive and reversible manner, allowing us to gain exquisite control over gene expression through modulation of acetylation levels at the target gene promoter.
RESULTS
Design of Chemical Epigenetic Modifiers
We used the previously described Chromatin in vivo assay (CiA) cell line to develop bivalent ligands capable of epigenome editing, which we call chemical epigenetic modifiers (CEMs).27 This cell line is derived from mouse embryonic stem cells (mESC). One copy of the Pou5F locus is modified to include a Gal4 DNA-binding array upstream of the transcriptional start site, and the downstream Oct4 gene is replaced with enhanced GFP (eGFP). Because the eGFP gene is highly expressed, this system is sensitive to the recruitment of gene silencing complexes, and the change in gene expression can be monitored by flow cytometry.
The mESCs were lentivirally infected with a plasmid expressing an FKBP-Gal4 fusion protein, which serves as our DNA-anchor to recruit the CEMs (Figure 1a). This was ideal for development of the CEM system because FK506, a natural product that forms a high affinity interaction with FKBP, can serve as the transcription factor ligand.28 Our control cell line expresses the unmodified Gal4 protein, removing the ability to bind and recruit FK506.
Figure 1.

Design and structure of chemical epigenetic modifiers (CEMs). (a) A Gal4-FKBP fusion binds to a 5X Gal4 binding array at the promoter of the CiA:Oct4 locus. A bivalent FK506-HDAC inhibitor binds to FKBP and recruits HDAC corepressor complexes. (b) Structures of CEM compounds.
The CEMs were constructed by attaching FK506 to an HDAC inhibitor through a short linker using click chemistry, so as not to disturb the stereocenters of FK506 (Figure 1b).28,29 HDAC inhibitors can be attached to linkers or solid supports through the solvent-exposed aryl region with little impact on potency. Kozikowski et al. reported click-chemistry compatible, triazole HDAC inhibitors with low-nM potency at HDACs 1, 2, 3, 6, and 10.30 We used this template to create CEM42 and CEM23. Both CEMs have the same HDAC-recruiting moiety, but we wanted to investigate if the linker would have an impact on CEM-efficiency. CEM42 contains a relatively long 3X PEG linker and CEM23 contains a propyl linker (Figure 1b).
Recruitment of HDAC Activity Can Efficiently Repress the CiA:Oct4 Locus
To serve as a positive control for HDAC-mediated repression, we developed fusion proteins between Gal4 and 8 different HDAC isozymes. HDAC3-Gal4 caused repression in 38.8% of the cells (Figure 2a), while the other fusions did not have any repressive effects.
Figure 2.

CEM23 reversibly and specifically represses GFP expression. (a) Flow cytometry data for ES cells expressing either Gal4 or Gal4-HDAC3. (b) ES cells expressing Gal4 were treated with either 0 or 100 nM CEM23 for 48 h. (c) ES cells expressing Gal4-FKBP were treated with CEM23 over 72 h. (d) ES cells treated with 100 nM CEM23 were sorted to keep only GFP(−) cells. These cells were treated with either 0 or 100 nM CEM23 for 5 additional days, and analyzed on days 5 or 7. (e) CEM23 treated, GFP(−) sorted cells were washed out for 5 days, treated with either 0 or 100 nM CEM23 for 2 days, and analyzed by flow cytometry. All histograms are representative plots of multiple experiments.
Dose response experiments identified the optimal concentration of CEM23. We observed repressive activity with 10 nM of CEM23. The level of repression peaked around 100 nM. Doses went as high as 10 μM, after which significant cell death occurred and an accurate level of repression was not obtainable (Supporting Information, Figure S1a). We followed FKBP-Gal4 cells treated with 100 nM CEM23 for 72 h. The cell population reached a maximum of 48% GFP(−) after 48 h, and sustained this level through 72 h (Figure 2c). In cells transfected with Gal4, which cannot recruit the CEMs, 100 nM of CEM23 did not cause a significant increase in the GFP(−) population (Figure 2b). The Gal4 control experiment proves that any changes in gene expression are not due to the general gene-silencing effect of the HDAC inhibitors, but are only possible when the inhibitor is localized at the target gene. It also shows that the gene silencing is not caused by Gal4 blocking progression of RNA polymerase. We next exposed the cells to CEM23 with and without an excess of FK506 (the component of CEM23 that binds to FKBP).
To demonstrate the stability and reversibility of CEM23 effects, we sorted CEM23-treated cells using fluorescence activated cell sorting (FACS) (Figure S2). After 48 h of 100 nM CEM23 treatment, the cells were separated into positive and negative populations. The GFP(−) was plated with or without continuous CEM23 exposure and maintained for evaluation. The negatively sorted cells were assessed with flow cytometry 3 days after FACS. The cells that were not treated with CEM23 regained almost full GFP expression (92.3% GFP+) and 71.8% of the CEM23 treated populated remained GFP(−) (Figure 2d).
To investigate the ability of the CEMs to be washed out and dosed back into the cells, we re-exposed a portion of the negatively sorted cells that were not given postsort CEM-treatment and had regained GFP-expression. After 48 h post reintroduction of 100 nM CEM23, 78% of the population repressed GFP (Figure 2e). These results demonstrate the specificity and reversibility of our system to tune gene expression in a chemically depend manner through endogenous HDAC recruitment. CEM42, featuring a longer linker region, behaved similarly to CEM23 in the FKBP-Gal4 cells. At 100 nM, after 48 h, CEM42 causing 41.1% of the cells to reside in the GFP(−) state, with no significant effects on transcription for the Gal4 control cells (Figure S1b).
Knowing that recruitment of HDAC3 was likely to be responsible for gene repression, we synthesized CEM36, based on a different chemotype reported to be selective for HDAC1/331 (Figure 1b). CEM36 features a benzamide zinc chelating group that is known to have a very long residence time at the enzyme relative to the hydroxamic acid zinc chelating groups of CEM23 and CEM42, but is less potent. After treatment of FKBP-Gal4 cells with 100 nM CEM36 for 2 days, 29.9% of cells were GFP(−), with no effect in Gal4 control cells (Supplementary Figure S1c).
To confirm that the bifunctional CEMs repress GFP, rather than one of the components alone, we tested the CEMs with FK506. Our results show that with a 10× excess of FK506, the repressive effects of CEM23 are removed, so FK506 is competing with CEM23 for the same FKBP binding site (Figure S1d). We also tested the effect that the individual components have on GFP expression. FK506 alone did not alter expression at 100 nM and 1000 nM concentrations. SAHA did have a slightly repressive effect at 1000 nM, but no repression was observed at 100 nM (Figure S1e).
CEM-Mediated Repression of eGFP Occurs in a Variegated and Whole-Colony Fashion
mESCs are an adherent, colony-forming cell line, thus we hypothesized that these characteristics were influencing the ability of cells to uptake the CEMs and thereby limited the level of overall gene repression. To characterize the mode of the repression, we took fluorescent microscopy images of the HDAC3-Gal4 and CEM-treated cell line. In an unbiased, blinded manner we counted −200 mESC colonies for each line and classified them into three categories: GFP-Off, GFP-On, and GFP-Variegated. Colonies were labeled as “GFP-Off” if all cells within the colony were off or expressing background-level GFP expression. Colonies in which all the observable cells were expressing GFP relatively brightly were categories as “GFP-On”. “GFP-Variegated” colonies had cells within the colonies that maintained high GFP expression, in an otherwise GFP(−) colony. Examples of cells in these categories are shown in Figure S3a. Cell lines were grown and counted in duplicate, then averaged (Figure S3b).
The Gal4-expressing and FKBP-Gal4 expressing cell lines were treated with 100 nM of CEM23, CEM36, or CEM42 for 48 h. Images were taken throughout areas of the wells using a high-content fluorescent microscope (Figure 3a,b). HDAC3-Gal4 cell lines served as a positive control for HDAC3-recruitment. In the HDAC3-Gal4 cells, an average of 43% of cells were GFP-On, 25% were GFP-Off, and 32% were variegated. As expected, the GFP expression of the Gal4-infected colonies was not significantly affected by CEM treatment. Of the FKBP-Gal4 cells treated with CEM23, 43% of the colonies were GFP-On, 32% were GFP-Off, and 25% were variegated, similar to the HDAC3-Gal4 cell line. The standard deviation between replicates was low when comparing the percentage of GFP-off colonies, whereas the percentages varied more between GFP-On and GFP-Variegated. This could be due to differences in what was determined to be fully on versus a variegated phenotype. Nevertheless, some colonies remain fully on or fully off, which does not support the hypothesis that CEM-to-cell accessibility is limited by colony formation, resulting in an incomplete suppression of GFP expression. To validate cell morphology, representative images of the cell lines and treatment condition representative images are shown with a fluorescent microscope, which allowed phase microscopy of the colonies (Figure 3c and Figure S3c).
Figure 3.

Fluorescent microscopy images and colony categorization for CEM-treated cells. Fluorescence images were taken of ES cells, and colonies were characterized as being GFP-On, GFP-Off, or a variegated state (b). Representative images (c).
H3K27ac Levels Decrease upon CEM-Recruitment
To examine the effects of CEM-mediated HDAC recruitment on the chromatin environment, we performed chromatin immunoprecipitation (ChIP) followed by quantitative RT-PCR (qPCR). H3K27ac is a histone mark associated with active promoters and is regulated in part by HDAC3 activity.32 We tested for differences in H3K27ac at the Oct4 locus of the HDAC3-Gal4 cell line, the Gal4 cell line, and the FKBP-Gal4 cell line with and without 48 h of 100 nM CEM23 exposure (Figure 4a).
Figure 4.

H3K27Ac chromatin immunoprecipitation at the Cia:Oct4 locus. Quantitative RTPCR was performed at four sites at the nucGFP locus. Quantitative RT-PCR compared Gal4 to HDAC3-Gal4 cell lines (b) and Gal4-FKBP cells to Gal4-FKBP with 100 nM CEM23 (c) using an H3K27ac antibody. Results at all four regions are summarized (d). Significance was calculated using Student’s t test with n = 4 for B and n = 5 for C. (*) p > 0.05; (**) p > 0.005.
The normalized level of H3K27ac enrichment was significantly higher in the Gal4 cell line compared to that in the HDAC3-Gal4 cell line when tested at regions 489 base pairs (bp) and 738 bp downstream from the transcriptional start site (TSS) ( * = p < 0.05, n = 4) (Figure 4b). We next tested the H3K27ac levels at the same location in the FKBP-Gal4 cell lines with and without CEM23 treatment. Our results show that CEM23 decreased H3K27ac levels at 489, 738, and 1199 bp downstream of the TSS (Figure 4c, * = p < 0.05; * * = p < 0.005; n = 5). The primer set at 169 bp upstream of the TSS did not show a significant change between the Gal4 cells and HDAC3-Gal4 cells, nor between the FKBP-Gal4 ± CEM23 cells (Figure S4a,b). This is not surprising, as regions this close to the TSS are typically largely devoid of nucleosomes. The results of H3K27ac levels in the four cell lines at −169, 489, 738, and 1199 bp from the TSS are summarized in Figure 4d. These data support our hypothesis that our CEM-mediated gene repression occurs through endogenous HDAC recruitment.
To confirm and further characterize the observed changes in histone tail acetylation, FKBP-Gal4 cells treated with 100 nM of CEM23 for 48 h were separated into positive and negative populations by FACS. We expected that the decreased acetylation levels observed in the CEM23-treated cells were due to the cells with decreased GFP expression (as observed by flow cytometry and microscopy). We measured the H3K27ac levels in the GFP(+) and GFP(−) population from three separate sorting experiments. We compared H3K27ac enrichment levels between the GFP(+) and GFP(−) populations. As expected, H3K27ac levels in the GFP(+) population were significantly higher than the GFP-negative population. Additionally, H3K27ac levels in the untreated FKBP-Gal4 cells were not significantly different from that in the FKBP-Gal4 + CEM23 positively sorted cells, and, H3K27ac levels in the FKBP-Gal4 CEM23-treated cells were not significantly different from that in the FKBP-Gal4 + CEM23 negatively sorted cells (Figure S4c). It should be expected that the sorted GFP(+) population has acetylation levels similar to that in the unsorted FKBP-Gal4 control cells, and that the sorted GFP(−) population has acetylation levels similar to that in the unsorted FKBP-Gal4 CEM23 treated cells. For all ChIP reactions, qPCR was done with primer pairs along the Oct4 locus (Figure S4d). H3K27ac enrichment levels were normalized to primers recognizing an intergenic region (IGR). These data confirm what we expected and show that within the CEM23-treated cells, the GFP(−) cells have lower H3K27ac levels than the GFP(+) cells.
DISCUSSION
CEM compounds cause specific, targeted gene suppression by modifying the local chromatin. The mechanism of action for these compounds is direct recruitment of HDAC-containing corepressor complexes to the FKBP-Gal4 transcription factor. Evidence supporting this claim is the known ability of similar HDAC inhibitors to bind HDAC-containing corepressor complexes,23 the reduction of transcription and H3K27 acetylation at the target gene, and the recapitulation of the drug-induced phenotype by a Gal4-HDAC3 fusion protein.
Suberanilohydroxamic acid (SAHA) derivatives like CEM23 and CEM42 are known to bind to multiple corepressor complexes, and have particularly high affinity toward CoREST, Sin3, and the HDAC3-containing nuclear receptor corepressor complex (NCOR).23,33 HDAC3 does not need to be catalytically active to cause gene repression in cells, and represses transcription independent of direct deacetylation, dependent on association with NCOR.34 When a CEM recruits HDAC3/NCOR to a gene, HDAC3/NCOR can suppress transcription even while HDAC3’s catalytic activity is blocked. HDAC3 is the only HDAC enzyme known to reside in the NCOR complex,35 so it is unclear how deacetylation takes place while the NCOR HDAC3 enzyme is engaged by an inhibitor. Because we observe the associated deacetylation in proximity to our FKBP-Gal4 DNA anchor, we are likely recruiting uninhibited HDACs to the gene locus. This could be happening through indirect recruitment of inhibited HDACs that our CEMs are recruiting, through direct recruitment whereby the reversible HDAC-CEMs interaction is allowing for previously inhibited HDACs to be active, or a mixture of both conditions. We hypothesize that we generate a cloud of HDACs in the proximity of the gene that are actively deacetylating and altering the chromatin environment in a chemically dependent manner.
Less selective, hydroxamic acid-containing HDAC inhibitors like CEM23 were most effective in our assays. Slow dissociating benzamide compounds like CEM36 were not as effective, perhaps because these compounds have less affinity for the enzyme.15,16 Future medicinal chemistry campaigns will more exhaustively explore alternative chromatin modification pathways by changing the chromatin activity portion of the bifunctional CEMs to include chemical probes that bind other complexes, for example polycomb repressive complex factors or heterochromatin pathway proteins, since the direct tethering of either pathway has been linked to gene repression.27,36
The CEM compounds could be used in other cell lines that contain Gal4 binding arrays at a gene of interest, or could be adapted for use with other genetic engineering systems. dCas9-CRISPR is currently the most programmable system for epigenome editing, and we are working to adapt dCas9 to work with the CEMs. Because the dose of small molecules can be precisely controlled, the CEMs could have value as part of a CRISPR-directed epigenome editing platform.
Drugs that target the addition, removal, or recognition of histone modifications, such as HDAC inhibitors, do this across the genome. The HDAC inhibitors SAHA and depsipeptide have been shown to change the expression of up to 22% of genes.26 HDAC inhibitors also modulate acetylation of many nonhistone proteins.37 This lack of selectivity causes side effects, and thus, most of these drugs are reserved for use in cancers for which there are not many other therapeutic options.38 By tethering epigenetic modulator drugs to a DNA-targeting group, the pharmacology of these drugs is constrained to the target locus, providing a robust effect at the target gene with minimal off-target effects.
The success of this work, and other results,11 raises the possibility that purely small molecule epigenome editing is possible. An HDAC inhibitor could be tethered to a glucocorticoid or androgen receptor (AR) ligand, and be used to repress genes bound by these receptors. An AR receptor ligand – HDAC inhibitor conjugate could be a therapy for androgen-independent prostate cancer, where the AR inappropriately activates target genes with changes to the epigenome.39 Because of the difficulties in delivering proteins or genes in vivo, bivalent small molecules may be the easiest way to achieve targeted in vivo epigenome editing.
METHODS
Cell Lines and Infection
The CiA mouse embryonic stem cells, previously described in Hathaway et al., 2012,27 were grown on 0.1% gelatin coated tissue culture dishes in high-glucose DMEM (Corning, 10-013-CV) supplemented with FBS serum (Gibo, 26140-079), 10 mM HEPES (Corning, 25-060-Cl), NEAA (Gibco, 11140-050), Pen/Strep, 55 μM 2-Mercaptoethanol, and 1:500 LIF conditioned media produced from LIF-1C α (COS) cells. Low passage (22–30) mESC were used. Cells were typically passaged at a density of (3–4) × 106 cells per 10 cm plate, fed every day, and split every 2–3 days.
Lentivirus production of mESC infection was done using 293T LentiX cells (Clontech). Low passage cells were plated onto 15 cm cells such that they were 80% confluent 24 h later. Each plate was transfected with 18 μg of the plasmid of interest, 13.5 μg of the Gag-Pol expressing plasmid, and 4.5 μg of the VSV-G envelope expressing plasmid. PEI transfection was done and 60 h after transfection, the virus was spun down at 20 000 rpm for 2 ½ h and then added to the mESCs in combination with 5 μg/mL Polybrene (Santa Cruz, sc-134220). The selection of lentiviral constructs was done with either puromycin (1.5 μg/mL) or blasticidin (7.5 μg/mL).
CEM and FK506 were diluted in DMSO (Sigma D2650) and kept at −20 °C; they were added to the cells during cell passaging, and the cells were given fresh media and compound daily.
Construct Design
The plasmids expressing the Gal4-DBD control and FKBP-Gal4 were previously constructed and can be found on Addgene (44176 and 44245, respectively). HDAC3-Gal4 was constructed by stitching PCR. The HDAC3 was amplified from mESC cDNA and the Gal4 was amplified from Addgene #44176. Both pieces were stitched into a lentivirus backbone with EF1-α promoter driving HDAC3-Gal4 and a PGK promoter driving a resistance gene.
| AC044_H3_F | HDAC3-Gal4 fusion | tgaggatccgcggccgcgccaccatggccaagaccgtggcg |
| AC045_H3_MF | HDAC3-Gal4 fusion | cgacaaggaaagtgatgtggagattatgaagctactgtcttctatcgaac |
| AC046_H3_MR | HDAC3-Gal4 fusion | gttcgatagaagacagtagcttcataatctccacatcactttccttgtcg |
| AC047_H3_R | HDAC3-Gal4 fusion | agagccggcgcggccgcctacgatacagtcaactgtctttgacc |
Flow Cytometry and FACS
Flow cytometry analysis and fluorescence activated cell sorting were done with the University of North Carolina Flow Facility. The cells were washed, trypsinized, and plated into a 96 well format (at a density of about 2000 cells/μL) for analysis in the Intellicyt iQue screener PLUS. UNC Flow Core Staff conducted the FACS with the FACSAria II; 107 cells were harvested for ChIP immediately after the sorted populations were acquired.
Image Acquisition and Quantification
The images taken for quantification were taken with the GE IN Cell Analyzer 2200 24 and 48 h after compound exposure. Ten images, randomly dispersed throughout the well were taken per cell line. For each cell line treatment condition, 147–213 colonies were counted and characterized as GFP-On, GFP-Off, or GFP-Variegated. The percentage of each category was determined. This was done in duplicate with freshly infected and selected cell lines. Percentages for each category were averaged. The brightness/contrast of the brightfield images was uniformly adjusted in ImageJ FIJI. The background artifact in the GPF-fluorescent images was uniformly removed in FIJI with a sliding paraboloid with a rolling ball radius of 10 pixels.
Confirmatory images were taken of the cells 24 and 48 h after compound exposure with the Leica Olympus IX71. Phase images were not edited. Fluorescent images were edited in FIJI with a sliding paraboloid with a rolling ball radius of 20 pixels. Five images were taken per replicate per condition, and representative images are shown.
ChIP-qPCR
For each sample, cells were trypsinized for 8–10 min, trypsin was quenched with 10 mL of ES media, and 107 cells were obtained. Cells were spun down, washed with 10 mL PBS, fixed for 12 min in a mix of formaldehyde (to a final concentration of 1%) and Fix Buffer (50 mM HEPES pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl), and then quenched by glycine (final concentration of 0.125 M). Cells were incubated on ice, and then spun down at 1000g for 5 min. Nuclei were prepared by consecutive washes with Rinse 1 Buffer (50 mM HEPES pH 8.0, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP40, 0.25% Triton X100), Rinse 2 Buffer (10 mM Tris pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl), and Shearing Buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris HCl pH 8). After a final spin down, cells were resuspended in 100 μL of Shearing Buffer and 1× protease inhibitor cocktail (Calbiochem) nanodroplets (generous gift from Samantha Pattenden),40 and sonicated in an E110 Covaris Sonicator for 4 min or until DNA was sheared to between 70bp and 500bp (as confirmed by agarose gel).
After sonication, the rest of the protocol was performed with a ChIP-IT High Sensitivity Kit (Active Motif, 53040), and an H3K27ac antibody was used for the pull down (Abcam, ab4729).
Supplementary Material
Acknowledgments
We would like to thank the Hathaway and Jin laboratories for discussions that were helpful in developing this project. This work was in part supported by the State of North Carolina’s University Cancer Research Fund (UCRF), an IBM Junior Faculty Development Award, and Grant R01GM118653 from the U.S. National Institutes of Health (to N.A.H.); and by Grants R01GM122749, R01CA218600, and R01HD088626 from the U.S. National Institutes of Health (to J.J.). Additional funding from an American Cancer Society postdoctoral fellowship PF-14-021-01-CDD (to K.V.B.) and a T-32 GM007092 (A.M.C). We also thank the UNC Flow Cytometry Core Facility, that is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center and by a North Carolina Biotech Center Institutional Support Grant 2015-IDG-1001.
Footnotes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00295.
Synthetic methods for CEM compounds (PDF)
References
- 1.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 2.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–99. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao Y, Xiong X, Wong S, Charles EJ, Lim WA, Qi LS. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat Methods. 2016;24:2016. doi: 10.1038/nmeth.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kungulovski G, Jeltsch A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016;32:101–113. doi: 10.1016/j.tig.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–7. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanphier E, Urnov F, Haecker SE, Werner M, Smolenski J. Don’t edit the human germ line. Nature. 2015;519:410–411. doi: 10.1038/519410a. [DOI] [PubMed] [Google Scholar]
- 9.Højfeldt JW, Van Dyke AR, Mapp AK. Transforming ligands into transcriptional regulators: building blocks for bifunctional molecules. Chem Soc Rev. 2011;40:4286–94. doi: 10.1039/c1cs15050b. [DOI] [PubMed] [Google Scholar]
- 10.Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. A Cell-Permeable Synthetic Transcription Factor Mimic. Angew Chem, Int Ed. 2007;46:2865–2868. doi: 10.1002/anie.200604485. [DOI] [PubMed] [Google Scholar]
- 11.Højfeldt JW, et al. Bifunctional ligands allow deliberate extrinsic reprogramming of the glucocorticoid receptor. Mol Endocrinol. 2014;28:249–59. doi: 10.1210/me.2013-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liszczak GP, et al. Genomic targeting of epigenetic probes using a chemically tailored Cas9 system. Proc Natl Acad Sci U S A. 2017;114:681–686. doi: 10.1073/pnas.1615723114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim W, et al. Targeted disruption of the EZH2–EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–650. doi: 10.1038/nchembio.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 15.Becher I, et al. Chemoproteomics reveals time-dependent binding of histone deacetylase inhibitors to endogenous repressor complexes. ACS Chem Biol. 2014;9:1736–1746. doi: 10.1021/cb500235n. [DOI] [PubMed] [Google Scholar]
- 16.Wagner FF, et al. Kinetically Selective Inhibitors of Histone Deacetylase 2 (HDAC2) as Cognition Enhancers. Chem Sci. 2015;6:804–815. doi: 10.1039/c4sc02130d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalin JH, Bergman JA. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J Med Chem. 2013;56:6297–6313. doi: 10.1021/jm4001659. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H, Gao L, Anandhakumar J, Gross DS. Uncoupling Transcription from Covalent Histone Modification. PLoS Genet. 2014;10:e1004202. doi: 10.1371/journal.pgen.1004202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenet. 2012;4:5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lahm A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–40. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris LG, et al. Evidence for a non-canonical role of HDAC5 in regulation of the cardiac Ncx1 and Bnp genes. Nucleic Acids Res. 2016;44:3610–3617. doi: 10.1093/nar/gkv1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly RDW, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41:741–9. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- 23.Bantscheff M, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29:255–65. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 24.Marks P. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 25.Zwergel C, Stazi G, Valente S, Mai A. Histone Deacetylase Inhibitors: Updated Studies in Various Epigenetic -Related Diseases. Expert Opin Ther Pat. 2016;2:1–15. [Google Scholar]
- 26.Peart MJ, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hathaway Na, et al. Dynamics and memory of heterochromatin in living cells. Cell. 2012;149:1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo ZF, Zhang R, Liang FS. Facile functionalization of FK506 for biological studies by the thiol–ene ‘click’ reaction. RSC Adv. 2014;4:11400. [Google Scholar]
- 29.Hong V, Presolski SI, Ma C, Finn MG. Analysis and optimization of copper-catalyzed azide-alkyne cyclo-addition for bioconjugation. Angew Chem, Int Ed. 2009;48:9879. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, et al. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J Med Chem. 2008;51:3437–3448. doi: 10.1021/jm701606b. [DOI] [PubMed] [Google Scholar]
- 31.Rai M, Soragni E, Chou CJ, Barnes G, Jones S, Rusche JR, Gottesfeld JM, Pandolfo M. Two new pimelic diphenylamide HDAC inhibitors induce sustained frataxin upregulation in cells from Friedreich’s ataxia patients and in a mouse model. PLoS One. 2010;5:e8825. doi: 10.1371/journal.pone.0008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.You SH, et al. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013;20:182–187. doi: 10.1038/nsmb.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Kalkum M, Chait BT, Roeder RG. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol Cell. 2002;9:611–623. doi: 10.1016/s1097-2765(02)00468-9. [DOI] [PubMed] [Google Scholar]
- 34.Sun Z, et al. Deacetylase-Independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell. 2013;52:769–782. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urvalek AM, Gudas LJ. Retinoic acid and histone deacetylases regulate epigenetic changes in embryonic stem cells. J Biol Chem. 2014;289:19519–19530. doi: 10.1074/jbc.M114.556555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bintu L, et al. Dynamics of epigenetic regulation at the single-cell level. Science (Washington, DC, U S) 2016;351:720–724. doi: 10.1126/science.aab2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seto E, Yoshida M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harbor Perspect Biol. 2014;6:a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals. 2010;3:2751–2767. doi: 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q, et al. Androgen Receptor Regulates a Distinct Transcription Program in Androgen-Independent Prostate Cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kasoji SK, Pattenden SG, Malc EP, Jayakody CN, Tsuruta JK, Mieczkowski PA, Janzen WP, Dayton PA. Cavitation enhancing nanodroplets mediate efficient DNA fragmentation in a bench top ultrasonic water bath. PLoS One. 2015;10:e0133014. doi: 10.1371/journal.pone.0133014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.