

Abstract
Neutrophils contain high levels of chymotrypsin‐like serine proteases (NSPs) within their azurophilic granules that have a multitude of functions within the immune system. In response, the pathogen Staphylococcus aureus has evolved three potent inhibitors (Eap, EapH1, and EapH2) that protect the bacterium as well as several of its secreted virulence factors from the degradative action of NSPs. We previously showed that these so‐called EAP domain proteins represent a novel class of NSP inhibitors characterized by a non‐covalent inhibitory mechanism and a distinct target specificity profile. Based upon high levels of structural homology amongst the EAP proteins and the NSPs, as well as supporting biochemical data, we predicted that the inhibited complex would be similar for all EAP/NSP pairs. However, we present here evidence that EapH1 and EapH2 bind the canonical NSP, Neutrophil Elastase (NE), in distinct orientations. We discovered that alteration of EapH1 residues at the EapH1/NE interface caused a dramatic loss of affinity and inhibition of NE, while mutation of equivalent positions in EapH2 had no effect on NE binding or inhibition. Surprisingly, mutation of residues in an altogether different region of EapH2 severely impacted both the NE binding and inhibitory properties of EapH2. Even though EapH1 and EapH2 bind and inhibit NE and a second NSP, Cathepsin G, equally well, neither of these proteins interacts with the structurally related, but non‐proteolytic granule protein, azurocidin. These studies expand our understanding of EAP/NSP interactions and suggest that members of this immune evasion protein family are capable of diverse target recognition modes.
Keywords: neutrophil serine proteases, neutrophil elastase, S. aureus, protease inhibitor, protein interactions
Introduction
Neutrophils are the earliest acting cellular players in innate immunity, and are best known for their ability to destroy invading microorganisms. Although neutrophils circulate in blood in a quiescent state, they respond quickly to pathogen (e.g., fMLP) and danger/damage‐associated (e.g., C5a) molecular patterns via high‐affinity receptors on their cell surface. This allows them to be the first leukocytes to infiltrate sites of infection or damaged/healing tissue.1, 2, 3, 4 Activation of neutrophils results in remarkable changes in their morphology, and triggers mobilization and secretion of their cytosolic granules.4 Chief among these are the azurophilic granules, which contain critical components of the neutrophil's anti‐bacterial arsenal. Two of the most abudant components of azurophilic granules are the enzyme myeloperoxidase (MPO), which converts hydrogen peroxide (H2O2) into cytotoxic hypohalous acids (e.g., HOCl), and a series of chymotrypsin‐like serine proteases (NSPs), which can directly attack the pathogen cell and its contents.5, 6, 7 Though MPO and NSPs initially act inside the phagolysosome, they can also be secreted into the extracellular milieu. Here, they physically associate with webs of chromatin fibers known as Neutrophil Extracellular Traps (NETs). As their name implies, NETs ensnare bacteria, and help increase the local concentration of antimicrobial MPO and NSPs in the environment.8, 9 It is this concerted action of MPO and NSPs—both in phagolysosomes and in NETs—that forms a foundation of neutrophil‐mediated defense against bacteria.
Whereas neutrophils contribute to antibacterial defense in a beneficial context, it is now recognized that unregulated neutrophil activation also directly correlates with the severity of various inflammatory diseases. Under such circumstances, excessive neutrophil‐driven inflammation causes substantial damage to host cells and extracellular matrices.4 In this regard, the lung is particularly susceptible to damage by NSPs,4, 5 since neutrophils can be activated by acute (e.g., pneumonia) or persistent pulmonary infections (e.g., P. aeruginosa secondary to Cystic Fibrosis), or other underlying pathologies such as those seen in Acute Respiratory Distress Syndrome (ARDS). Environmental insults (e.g., tobacco smoke) are also known to trigger neutrophil activation, and NSP‐mediated destruction of lung elastin has been proposed as a major contributor to morbidity and mortality in those suffering from chronic obstructive pulmonary disease (COPD)/emphysema.5, 10 Even though the human body produces many physiological serine protease inhibitors (e.g., α1‐antitrypsin, secretory leukocyte protease inhibitor5), hereditary loss of α1‐antitrypsin results in early onset COPD/emphysema due to chronically elevated levels of NSP activity in the lung.11 Thus, targeted inhibition of NSP activity has been proposed as a treatment route for these diseases.5
We recently discovered that the pathogen Staphylococcus aureus secretes a family of previously unrecognized NSP inhibitors.12 These so‐called EAP domain proteins are collectively required for maximal bacterial virulence in murine models of inhalational pneumonia and intravenous infection,12 and expression of the genes that encode them is upregulated by S. aureus in response to neutrophil granule components.13 The EAP domain family is composed of three distinct molecules: the ∼50–70 kDa Extracellular Adherence Protein (Eap), and its homologs, EapH1 (∼12 kDa) and EapH2 (∼13 kDa).14 Each of these proteins is comprised of either one (e.g., EapH1 and EapH2) or multiple (e.g., Eap) repeats of an approximately 100 residue domain that adopts a β‐grasp type fold.14 A co‐crystal structure of the EapH1/NE complex determined at 1.85 Å limiting resolution indicates that the bacterially derived inhibitor binds across the catalytic cleft of the protease, and does so in a non‐covalent manner12 [Fig. 1(A)]. This observation strongly suggests that EapH1 blocks NE activity by preventing access of the enzyme to its various substrates. While this type of steric hindrance mechanism appears to be a common theme amongst serine protease inhibitors (e.g., serpins, elafin, etc.), the fact that EAP domains function non‐covalently and share no obvious sequence relationships with better understood regulators argues that they represent a novel class of physiological serine protease inhibitors.12
Figure 1.
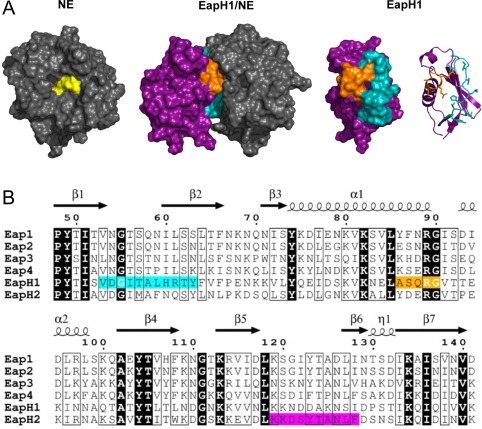
Structure/function relationships of EAP domains as deduced from the EapH1/NE structure. (A) (Left Panel) Crystal structure of NE depicted as a grey molecular surface, with the sidechains of the catalytic triad colored yellow. (Left Center Panel) Co‐crystal structure of EapH1/NE (PDB Code 4NZL) represented as a molecular surface. EapH1 is shown in purple, with the interfacial residues in cyan (site #1) and orange (site #2). Note that the orientation of NE has been rotated approximately 90° clockwise in the viewing plane to better show the relationship of EapH1 to the NE active site. (Right Center Panel) EapH1 alone is shown as a molecular surface with identical coloration as the left center panel. (Right Panel) Identical orientation to the right center panel, except that EapH1 is shown as a ribbon diagram. The sidechains found at the EapH1/NE interface are depicted as sticks. (B) Sequence alignment of EAP domains found in S. aureus strain Mu50. Identical residues are shown in reverse typeface, while residues conserved in a majority of sequences are highlighted in a box. NE‐interfacing residues are highlighted in cyan (site #1) and orange (site #2). Residues in EapH2 that are required for binding to NE are highlighted in magenta (see Fig. 5). The secondary structure diagram and sequence numbering reflects values specific to EapH1 throughout the text for simplicity.
Through a series of functional studies, we observed that Eap, EapH1, and EapH2 all inhibit the three most abundant NSPs [i.e., neutrophil elastase (NE), cathepsin G (CG), and proteinase‐3 (PR3)].12 Other more distantly related chymotrypsins found in the bloodstream, including plasmin, thrombin, and plasma kallikrein, are not inhibited by EAP domain proteins.12 To our knowledge, the NSP‐selective activity profile of EAP domain proteins is unprecedented among biological serine protease inhibitors. Such selectivity is even more surprising considering that all individual EAP domains potently inhibit NE even though they share only 26–83% sequence identity as a group.12, 14
Examination of the EapH1/NE interface reveals that the contacts between the inhibitor and protease arise from two distinct regions within the EapH1 protein. Despite the fact that these regions are comprised of only 11 and five residues, respectively12 [Fig. 1(B)], they exhibit an unexpectedly low level of sequence identity across EAP domains (3/16 positions are invariant). This lack of strict conservation is particularly noteworthy in those four positions that correspond to Ala58–Arg61 of EapH1, which together contribute greater than half of the surface area buried in the EapH1/NE interface (427 Å2 of 830 Å2). This piece of information raises questions about the quantitative contributions of various EapH1 residues in forming the NE inhibitory complex, and whether or not the corresponding residues in other EAP domain proteins are equally important.
To address these and related issues, we designed a series of studies to (i) investigate binding of EapH1, EapH2, and various site‐directed mutants thereof to the prototypical NSP, NE, and (ii) correlate observed binding affinities with the ability of these EAP domain variants to inhibit NE activity. The results we present here establish a direct relationship between the affinities of EAP domains for NE and their previously determined Ki values in NE activity assays. While the outcome of our experiments validates the functional importance of EapH1 residues found at the NE interface, we found that loss of residues at a distal site was required to diminish EapH2 function. Together, our results imply that EapH1 and EapH2 recognize NE through distinct binding modes, and suggest that EAP domains as a whole are capable of diverse physical interactions with NSPs. These studies provide fundamental information that may be valuable for future design of novel NSP‐targeted protease inhibitors using the naturally occurring EAP domain as a template.
Results
EapH1 and EapH2 form kinetically stable complexes with NE and CG
We previously demonstrated that the multi‐domain Eap protein, as well as EapH1 and EapH2, inhibit NE, CG, and PR3 activity in assays using fluorescent substrates specific for each protease.12 The Ki values for each inhibitor/NSP pair that could be determined with statistical confidence ranged between 0.2 and 20 nM, and were indicative of high‐affinity binding between EAP domain proteins and NSPs. Consistent with this prediction, we used isothermal titration calorimetry (ITC) to determine an affinity of 25 nM for the EapH1/NE complex as a prelude to co‐crystallography studies.12 Unfortunately, the costly nature of the ITC approach, particularly in terms of material, precluded a more detailed analysis of the remaining inhibitor/NSP pairs.
To circumvent the practical limitations of ITC, we devised a surface plasmon resonance (SPR) approach for obtaining quantitative insight into the stability of EAP domain/NSP complexes. We first analyzed the EapH1/NSP interaction, since we had pre‐existing ITC data on this system to use as a comparator. Since the NE protein lacks lysine residues, we oxidized its N‐linked glycans with metaperiodate to generate NE‐borne aldehyde groups, derivatized these with a bifunctional amine (ethylenediamine), and reduced the Schiff Base adduct with cyanoborohydride to produce a stable primary amine‐containing NE, which was then successfully immobilized on a biosensor surface. Subsequent injections of an EapH1 dilution series over the NE surface showed concentration‐dependent binding, which fit to a KD value of 2.95 nM when analyzed by single‐site kinetic model [Fig. 2(A) and Table 1]. An analogous dilution series of EapH2 injected over the same surface likewise produced dose‐dependent binding, and fit to a KD value of 24.7 nM when analyzed similarly [Fig. 2(B) and Table 1]. The affinity parameters we derived from kinetic fitting are in good agreement with the KD previously obtained for EapH1/NE by ITC (KD = 25 nM).12 They are also consistent with the experimentally observed Ki values for EapH1 (1.9 nM) and EapH2 (4.5 nM) in NE activity assays.12 Importantly, the trend of EapH1 forming a higher‐affinity complex with NE than EapH2 was also reflected in these results.
Figure 2.

Characterization of EapH1 and EapH2 binding to NE and CG by surface plasmon resonance. Binding of EapH1 (A, C) and EapH2 (B, D) to surfaces derivatized with NE or CG, respectively, was assessed by SPR. The reference subtracted sensorgrams are shown in black, while the results of curve fitting to a Langmuir binding model are shown in red. Curve fitting parameters are found in Table I.
Table 1.
Surface Plasmon Resonance and Functional Assessment of EapH1, EapH2, and Various Site‐Directed Mutants' Interactions with Neutrophil Elastase and Cathepsin G
| Analytea | KD (nM) | k on (M−1s−1) | error k on (M−1s−1) | k off (s−1) | error k off (s−1) | χ2 | IC50 (nM)b |
|---|---|---|---|---|---|---|---|
| EapH1 | 2.95 | 1.27*105 | 1.60*102 | 3.75*10−4 | 4.30*10−6 | 2.210 | 5.7 |
| EapH1c | 0.191 | 3.67*105 | 2.23*103 | 6.44*10−5 | 1.51*10−7 | 0.212 | n/a |
| EapH1d | 9.84 | 3.48*104 | 2.89*103 | 3.42*10−4 | 8.72*10−6 | 0.108 | n/a |
| EapH1Δ56–57 | 0.835 | 2.49*105 | 3.59*103 | 2.08*10−4 | 6.93*10−6 | 0.486 | 6.3 |
| EapH1Δ59–61 | 1.65 | 5.55*105 | 8.58*103 | 9.17*10−4 | 8.14*10−6 | 0.392 | 6.5 |
| EapH1Δ56–61 | 6.75 | 9.99*104 | 2.30*103 | 6.74*10−4 | 9.95*10−6 | 0.300 | 6.5 |
| EapH1Δ89,94–95 | 14,200 | 6.09*103 | 4.20*102 | 8.64*10−2 | 5.70*10−3 | 0.207 | >1,000 |
| EapH1Δ119–128 | 43.3 | 6.80*103 | 7.70*100 | 2.95*10−4 | 5.60*10−6 | 0.079 | 19 |
| EapH2 | 24.7 | 6.03*104 | 9.30*101 | 1.49*10−3 | 3.90*10−6 | 0.118 | 6.3 |
| EapH2c | 2.38 | 3.75*105 | 3.27*103 | 4.74*10−4 | 2.96*10−6 | 0.441 | n/a |
| EapH2d | 3.25 | 2.95*105 | 7.52*103 | 9.59*10−4 | 8.18*10−6 | 0.232 | n/a |
| EapH2Δ56–61 | 0.643 | 5.66*105 | 1.10*103 | 3.64*10−4 | 6.66*10−6 | 1.260 | 6.6 |
| EapH2Δ89,94–95 | 4.97 | 2.16*105 | 6.30*102 | 1.07*10−3 | 4.10*10−6 | 0.043 | 6.4 |
| EapH2Δ119–128 | 32,100 | 1.57*102 | 1.90*100 | 5.05*10−3 | 8.70*10−5 | 0.006 | >1,000 |
Unless otherwise denoted, these values represent binding to immobilized Neutrophil Elastase (NE).
Values correspond to inhibition of NE activity in a proteolytic assay.
Values were obtained by single‐cycle kinetic analysis.
These values represent binding to immobilized Cathepsin G (CG).
Since no direct binding data were available for EAP domain interactions with other NSPs, we next prepared a biosensor surface modified by random amine‐immobilized CG. Injections of an EapH1 dilution series over this surface yielded a series of sensorgrams that fit to a KD of 9.84 nM when analyzed by a single‐site kinetic model [Fig. 2(C) and Table 1]. Injections of a corresponding dilution series of EapH2 yielded sensorgrams that fit to a KD of 3.25 nM [Fig. 2(D) and Table 1]. Similarly to the study of EapH1 and EapH2 binding to NE, these observations are in agreement with the previously determined Ki value of EapH1 in a CG activity assay (4.2 nM).12 The Ki of EapH2 for CG was not confidently determined in that previous investigation due to a low signal‐to‐noise ratio of the specific experimental series.
In addition to providing information on the affinity of EapH1 and EapH2 for NE and CG, our SPR studies also revealed important details regarding the kinetics of EAP domain association/dissociation with these NSPs. Of particular interest were the dissociation rates (k off) for these interactions (Table 1). We found that each of EAP domain/NSP pair is characterized by a dissociation rate constant on the order of ∼10−4 s−1, which is indicative of a long‐lived complex. Dissociation rates in this range can be difficult to measure accurately via traditional kinetic analyses, however, because the change in such sensorgrams during a typical SPR dissociation phase (i.e., 3–5 min) is quite small with respect to time. To investigate these interactions through an independent approach, we measured the binding of EapH1 and EapH2 to immobilized NE using single‐cycle kinetic methods. By following the dissociation phase for a time period of 60 min, sufficient data points were gathered to arrive at a more accurate determination of the dissociation rate constants for these high‐affinity interactions (Fig. S1 and Table 1). Replicate single‐cycle injections of EapH1 over an NE surface yielded a KD of 0.191 nM, and a k off of 6.44 × 10−5 s−1. A similar experimental series of EapH2 over an NE surface yielded a KD of 2.38 nM, and a k off of 4.74 × 10−4 s−1. Thus, in light of these alternative determinations of the dissociation rate constants, our data suggest that the KD values for both EapH1/NE and EapH2/NE may be up to one order of magnitude higher in affinity than originally suggested by ITC12 or conventional SPR.
Site‐directed mutagenesis identifies a critical region for EapH1 function
Analysis of the EapH1/NE co‐crystal structure predicted the involvement of two specific groups of EapH1 residues in binding to NE12 (Fig. 1). To directly investigate the importance of these residues, we expressed a series of site‐directed mutants in EapH1, and characterized these proteins in terms of their ability to bind NE and inhibit its proteolytic activity. Since EapH1 residues Ile56–Arg61 account for a majority of the buried surface area in the EapH1/NE co‐crystal, we constructed three mutants wherein these positions were substituted for alanine: EapH1Δ56–57 (as position 58 is an alanine already), EapH1Δ59–61, and EapH1Δ56–61. CD spectra of each mutant were essentially identical with that of wild‐type EapH1, and confirmed that there were no global changes in protein folding as a result of the mutagenesis procedure (Fig. S2). Surprisingly, we found that none of these mutants displayed any substantial defect in NE binding, as judged by SPR [Fig. 3(A–C) and Table 1]. This lack of change in NE binding properties was reflected in an NE activity assay, which revealed that each of these mutants had an IC50 practically indistinguishable from that of wild‐type EapH1 [Fig. 3(E) and Table 1].
Figure 3.
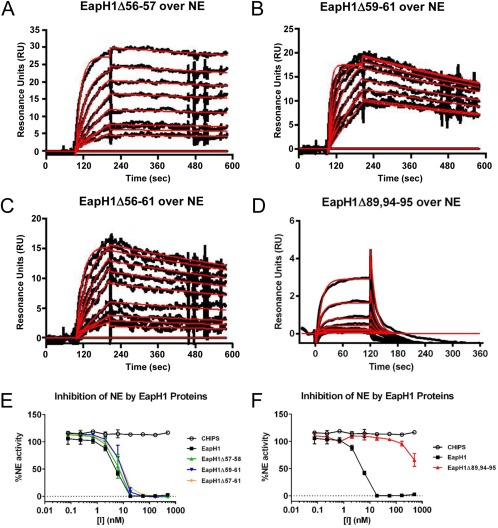
Residues Arg89, Glu94, and Lys95 are required for EapH1 function. The contributions of residues found at the EapH1/NE interface were probed by a combination of site‐directed mutagenesis, NE‐binding measurements, and functional assays. Binding of EapH1 mutants EapH1Δ56–57 (A), EapH1Δ59–61 (B), EapH1Δ56–61 (C), and EapH1Δ89,94–95 (D) to an NE surface was assessed by SPR. The reference subtracted sensorgrams are shown in black, while results of curve fitting to a Langmuir binding model are shown in red. (E, F) The impact of site‐directed mutations in EapH1 on NE activity was monitored by a fluorometric activity assay. All data points were measured in duplicate, and error bars depict SD. A legend is inset.
EapH1 positions Ala86–Gly90 are also found at the NE interface (Fig. 1). Although we did not pursue mutations in residues Ala86–Gln88, since they are found in the long α1‐helix of the EAP domain and changes here could potentially disrupt folding, we noticed that several residues following this stretch are also oriented toward the NE surface in the EapH1/NE co‐crystal. To probe this general region, we constructed an additional mutant where positions Arg89, Glu94, and Lys95 were simultaneously mutated to alanine (i.e., EapH1Δ89,94–95). After confirming its structural integrity by CD (Fig. S2), we analyzed the NE‐binding properties of EapH1Δ89,94–95 by SPR. Interestingly, the NE‐binding activity of this mutant was diminished by nearly 104‐fold (KD=14.2 µM) when compared to wild‐type EapH1 [Fig. 3(D) and Table 1]. Consistent with this observation, the IC50 of EapH1Δ89,94–95 could not be accurately determined, since the highest concentrations of protein included the NE activity assay resulted in only ∼50% inhibition of proteolysis [Fig. 3(F) and Table 1]. Together, these results indicate that the residues at and following the C‐terminus of the α1‐helix contribute to inhibition of NE by EapH1.
Evidence for an alternative NE‐binding mode in EapH2
Although all individual EAP domain proteins inhibit multiple NSPs, only a single structure of an EAP‐inhibited NSP has been determined thus far (i.e., EapH1/NE).12 This has raised questions as to whether all EAP domains recognize NSPs simiarly, or whether structural diversity exists within EAP‐inhibited NSPs. To begin addressing this issue, we constructed a panel of site‐directed mutants in EapH2 that altered residues corresponding to those previously investigated for EapH1. As with EapH1Δ56–61, we found that EapH2Δ56–61 was well‐folded (Fig. S2), but displayed no defect in either NE binding or inhibition of NE proteolytic activity [Fig. 4(A,C) and Table 1]. In contrast to EapH1Δ89,94–95 however, the EapH2Δ89,94–95 mutant actually bound NE modestly better than wild‐type EapH2 (4.97 vs. 24.7 nM, respectively) [Fig. 4(B) and Table 1]. Since EapH2Δ89,94–95 inhibited NE activity to an apparently identical extent to the wild‐type protein [Fig. 4(C) and Table 1], these results suggested that EapH2 might recognize NE differently from EapH1.
Figure 4.
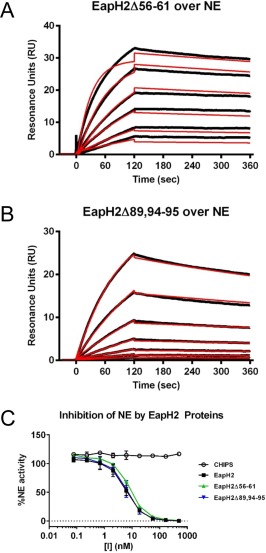
Residues Arg89, Glu94, and Lys95 are not required for EapH2 function. The contributions of EapH2 residues corresponding to positions required for EapH1 inhibition of NE were probed by a combination of site‐directed mutagenesis, NE‐binding measurements, and functional assays. Binding of EapH2 mutants EapH2Δ56–61 (A) and EapH2Δ89,94–95 (B) to an NE surface was assessed by SPR. The reference subtracted sensorgrams are shown in black, while the results of curve fitting to a Langmuir binding model are shown in red. (C) The impact of site‐directed mutations in EapH2 on NE activity was monitored by a fluorometric activity assay. All data points were measured in duplicate, and error bars depict SD. A legend is inset.
Because there is no EapH2/NE co‐crystal structure, we sought an alternative approach for gaining physical insight into the nature of this complex. We used the ClusPro server to generate plausible docking models of NE bound to EapH1 or EapH2.15, 16 The highest scoring model of EapH1/NE correctly identified the Val53–Tyr63 loop that contributes a majority of the buried surface area in EapH1/NE co‐crystal structure, although the orientation of the EAP domain in this model was flipped relative to the protease [Fig. 5(A)]. By contrast, the highest scored model of EapH2/NE suggested that an altogether different region of EapH2 was involved in inhibiting NE [Fig. 5(B)]. This region, which connects the β5 and β6 strands, lies on the opposite side of the EAP domain, and consists of residues Lys119–Ile128 in EapH1 and Lys119–Phe128 in EapH2 [Fig. 1(B)].
Figure 5.
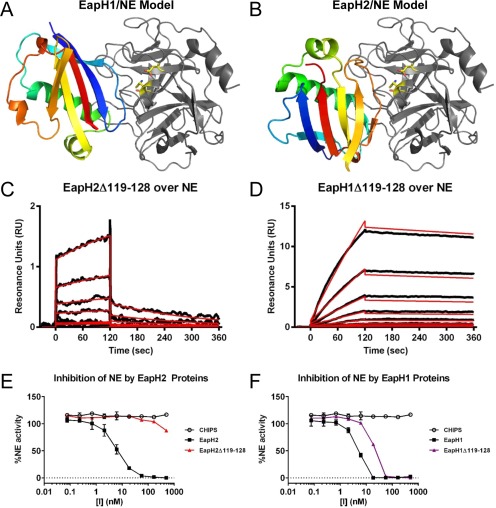
Residues 119–128 are required for EapH2 but not EapH1 function. The ClusPro server was used to generate plausible models for the EapH2/NE complex. (A) The highest scored ClusPro model for EapH1/NE is shown with EapH1 in an indexed color scheme (N‐terminus in blue, C‐terminus in red) and NE in the same orientation as Fig. 1A. Note that the loop corresponding to EapH1 residues 56–61 is correctly identified as interfacing with NE in the ClusPro model, although the orientation of EapH1 protein is flipped when compared to the EapH1/NE structure in Fig. 1A. (B) The highest scored ClusPro model for EapH2/NE is shown with EapH2 in an indexed color scheme (N‐terminus in blue, C‐terminus in red) and NE in the same orientation as Fig. 1A. Note that the loop from positions 119–128 is implicated in forming contacts with NE in this model. Binding of mutants EapH2Δ119–128 (C) and EapH1Δ119–128 (D) to an NE surface was assessed by SPR. The reference subtracted sensorgrams are shown in black, while the results of curve fitting to a Langmuir binding model are shown in red. The impact of the EapH2Δ119–128 (E) and EapH1Δ119–128 (F) mutants on NE activity was monitored by a fluorometric activity assay. All data points were measured in duplicate, and error bars depict SD. A legend is inset.
To test whether this β5‐β6 loop might contribute to EapH2 function, we constructed a site‐directed mutant where the entire Lys119–Ile128 sequence had been replaced with alanine (i.e., EapH2Δ119–128). A CD spectrum of this mutant confirmed that no structural aberrations were induced by these changes (Fig. S2). Injections of a dilution series of EapH2Δ119–128 over an NE surface revealed a >103‐fold decrease in affinity (KD = 32.1 µM) when compared to wild‐type EapH2 [Fig. 5(C) and Table 1]. Similarly, EapH2Δ119–128 was greatly diminished in its ability to inhibit NE activity [Fig. 5(E) and Table 1]. In this case, an IC50 value was not determined as only ∼10% inhibition was observed at the highest concentrations of EapH2Δ119–128 tested.
To determine if the β5–β6 region has a similar impact on EapH1 function, we expressed and purified the corresponding EapH1Δ119–128 poly‐alanine mutant. Again, the CD spectrum of EapH1Δ119–128 was in agreement that of wild‐type EapH1 (Fig. S2). Interestingly, injections of a dilution series of EapH1Δ119–128 over an NE surface revealed a slight change in its affinity for the protease when compared to wild‐type EapH1 (KD = 43.3 nM) [Fig. 5(D) and Table 1]. The IC50 of this mutant in a functional assay was measured at 19 nM, which compares favorably to that of wild‐type EapH1 (i.e., 5.7 nM) [Fig. 5(F) and Table 1]. When considered together, these results indicate that EapH1 and EapH2 recognize NE through different binding modes, and rely on unique regions within their structures to inhibit NE activity.
Sequence differences explain why EAP domains fail to bind azurocidin
In addition to the NSPs, the azurophilic granules of neutrophils contain significant quantities of azurocidin (AZU). AZU adopts a chymotrypsin‐like fold,17 and sequence analysis reveals that AZU shares a high level of identity with NE, CG, and PR3. In contrast to NSPs, however, AZU lacks proteolytic activity because alanines have replaced the histidine and serine residues that are normally found in the catalytic triad. Nevertheless, AZU has been shown to elicit important immune and inflammatory activities. Among these are to directly kill Gram‐negative bacteria18, 19 and to function as a chemotactic signal for monocytes and macrophages.20 Given these pro‐inflammatory functions, it seemed reasonable that S. aureus cells might gain a competitive advantage within their host(s) by binding AZU and blocking its action. In this regard, the high affinity of EapH1 and EapH2 for other NSP family members (Fig. 2 and Table 1) suggested that EapH1 or EapH2 might bind AZU.
We used both ITC and SPR approaches to test if either EapH1 or EapH2 could bind to AZU purified from human neutrophils. Surprisingly, neither assay provided any evidence of an interaction between these proteins (Data Not Shown). Given the significant sequence homology between AZU and NSPs, we sought a potential structural explanation for this observation. We first superimposed the structure of AZU (17) on that of NE as observed in the EapH1/NE complex12 [Fig. 6(A,B)]. Consistent with the 46% identity between AZU and NE across their serine protease domains, 174 of the aligned Cα positions superimposed with a root–mean–square deviation (RMSD) of only 0.558 Å2. We then mapped the known EapH1 binding surface on NE [Fig. 6(C)], the sequence differences between NE and AZU [Fig. 6(D)], and combined these to identify the differences between AZU and NE that impact the EapH1 binding surface [Fig. 6(E, F)]. Significantly, of the 36 NE residues found at the EapH1 binding site, 20 of these are changed in AZU. These changes modify nearly 63% of the surface area at the EapH1 binding site, and alter two residues of NE (Asn74 and Asn162) whose sidechains participate in hydrogen bonds with EapH1. In light of this extensive alteration at a known EAP domain binding site on an NSP, it seems reasonable that neither EapH1 nor EapH2 can bind to AZU.
Figure 6.
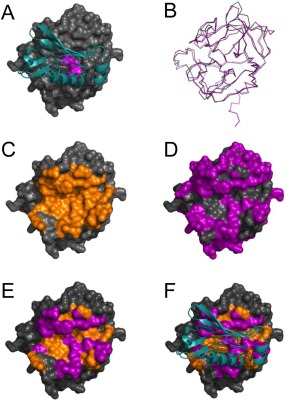
Sequence differences explain the failure of EapH1 and EapH2 to bind the NSP homolog, azurocidin. (A) Representation of the EapH1/NE complex where EapH1 is drawn as a teal ribbon and NE is shown as a grey molecular surface. Residues of the NE catalytic triad are colored magenta for the purposes of orientation. (B) Structural superposition of NE and AZU, where NE is drawn a grey wire and AZU as a purple wire. Note the extensive structural homology shared by the chymotrypsin fold. (C) NE shown as a molecular surface, where the EapH1 interfacing residues are colored in orange. (D) NE shown as a molecular surface, where the residues that differ between NE and AZU are colored in purple. (E) NE shown as a molecular surface where the residues that differ between NE and AZU are colored purple, while those residues that are the same and are found at the EapH1 interface are colored orange. (F) An identical representation to panel E, except that EapH1 is shown in cyan as it lies in the EapH1/NE complex.
Discussion
In this study, we further analyzed the physical properties and sequence features that drive inhibition of NSPs by staphylococcal EAP domain proteins. Given that S. aureus strain Mu50 expresses six distinct EAP domains (Fig. 1), and human neutrophils contain four different NSPs, we chose to focus our efforts here on a comparative analysis of EapH1 and EapH2. Our initial experiments investigated the affinity and kinetics of association/dissociation of these two staphylococcal inhibitors for the canonical NSP, NE, and its homolog, CG (Fig. 2 and Table 1). Using SPR approaches, we observed that both EapH1 and EapH2 bind NE and CG with low to sub‐nanomolar affinities (KD ∼0.2–20 nM). This is consistent with the potent inhibition by EAP domains previously observed in NE‐ and CG‐specific proteolytic assays.12 Furthermore, each of these complexes is characterized by a slow dissociation rate in the range of 10−4–10−5 s−1, depending on the experimental system used to derive the measurement (Fig. 2 and Table 1). It is interesting that dissociation rates in a range similar to these have been observed in both functional kinetic experiments and SPR studies of other protease/inhibitor interactions, most notably those of NSPs with the endogenous protease inhibitor, elafin.21, 22 That being said, it is equally important to note that while the dissociation rates of these complexes were low, they were still measurable. This observation is in line with our previous crystallographic analysis of EapH1/NE,12 which indicated that NSPs are inhibited by EAP domains in a non‐covalent manner.
Work by others has shown that the genes encoding Eap and EapH1 are induced following S. aureus exposure to neutrophil granule components.13 This suggests to us that the primary location for inhibition of NSPs by EAP domain proteins lies within the neutrophil's maturing phagolysosome. In this compartment, the concentrations of antimicrobial granule proteins are extremely high and have been suggested to reach levels of ∼1 mM for MPO.23, 24, 25 This makes it likely that EAP domain proteins find their NSP target almost immediately following secretion, and inhibit these immune proteases almost irreversibly. Nevertheless, the high affinity of EapH1 and EapH2 for their targets also raises the possibility that these staphylococcal immune evasion proteins can function extracellularly where the concentration of NSPs would be expected to be much lower. In such scenarios, EapH1 and EapH2 could act on NSPs associated with NETs, or proteases that have simply been released into the inflammatory environment following neutrophil degranulation or death. Additional experiments might carefully examine and distinguish amongst these possibilities, perhaps through the use of established mouse models of infectious or inflammatory disease. In this regard, our discovery that murine NSPs are also inhibited by EAP domain proteins provides a solid starting point for future investigations.12
We used the EapH1/NE co‐crystal structure as a basis for examining the functional importance of residues found at the EapH1/NE interface (Fig. 1).12 Curiously, we observed that loss of the side chains from Ile56 to Arg61 did not change the affinity of EapH1 for NE, nor its ability to inhibit NE activity (Fig. 3 and Table 1). This result was unexpected, as these six residues of EapH1 contribute a majority of the buried surface area in the EapH1/NE complex. They also form several side chain mediated hydrogen bonds with NE.12 Consistent with this observation, however, we found that a linear synthetic peptide corresponding to residues Gly55–Tyr63 of EapH1 could only inhibit NE at concentrations some 106‐fold greater than needed for EapH1 (Data Not Shown). This suggests that merely having a sequence that can associate with NE is not enough to bind NE and inhibit its activity. Instead, it seems likely that such a sequence must be presented to the protease in the appropriate structural context.
In contrast to residues Ile56–Arg61, we found that loss of Arg89, Glu94, and Lys95 had a dramatic impact on EapH1 function (Fig. 3 and Table 1). The physical basis for this observation is not immediately obvious from the EapH1/NE structure, however. Sequence alignment of EAP domains from S. aureus strain Mu50 reveals that Arg89 is invariant among in these proteins, while Glu94 and Lys95 are shared between EapH1 and EapH2 (Fig. 1). Remarkably, loss of these sidechains in EapH2 had no effect on the ability of this protein to bind and inhibit NE (Fig. 4 and Table 1). The paradoxical nature of this result prompted us to model the structure of EapH2 bound to NE (Fig. 5). Interestingly, this model suggested that an altogether different region of EapH2 could be responsible for binding to NE. Indeed, loss of the EapH2 side chains from Lys119 to Phe128 abrogated EapH2 function, while loss of corresponding side chains in EapH1 had no effect (Fig. 5 and Table 1). Based upon the results of these mutagenesis experiments, we conclude that EapH1 and EapH2 must form structurally distinct complexes with NE, and presumably other NSPs as well. This outcome was unexpected, given that EapH1 and EapH2 have similar functions and share ∼50% identity.
Even though our mutagenesis data confirmed the importance of residues Lys119–Phe128 in EapH2 function, the accuracy of the structural model that led us to test the involvement of this region remains uncertain. As an independent test of the method used, we also constructed a model of EapH1/NE (Fig. 5). Comparison of this model with the EapH1/NE structure determined by X‐ray crystallography12 indicated that the computational approach correctly identified the NE‐binding site of EapH1, albeit in an incorrect orientation. Whether a similar limitation exists in our EapH2/NE model is difficult to know, mostly since the results of the Lys119–Phe128 mutational analysis would not be expected to change much if this were true. Nevertheless, there is precedent for this specific β5–β6 loop region of β‐grasp proteins (a superfamily to which EAP domains belong) being a critical binding determinant. Specifically, a crystal structure of the staphylococcal β‐grasp superantigen SEC2 bound to a T‐cell receptor β‐chain shows extensive interactions made by the SEC2 β5–β6 loop.14, 26 These data, along with the mutagenesis and functional data presented herein demonstrate that EAP domains are capable of structurally divergent modes of ligand recognition. Further structure/function analyses of EAP domains' complexes with NSPs, and other known ligands such as complement component C4b,27, 28 should only serve to broaden our understanding of the many interactions formed by this diverse family of immune evasion proteins.
Aside from identifying distinct modes of NSP binding between EapH1 and EapH2, our current studies have also expanded upon the concept that EAP domains can discriminate between a number of closely related binding partners. Whereas previous studies across a cohort of six chymotrypsins found in human blood had suggested that EAP domain proteins are selective for NSPs, we found here that neither EapH1 nor EapH2 bind to AZU. Although these observations can ultimately be rationalized on the basis of sequence/structure relationships (Fig. 6), the results themselves are still intriguing when considered in their biological context. In this regard, the failure of EapH1 and EapH2 to recognize AZU—which is not only a structural homolog of the NSPs, but also resident in the very same subcellular granules of neutrophils—is noteworthy. Despite the fact that AZU is associated with diverse antibacterial and inflammatory functions,18, 19, 20 and has been reported to serve as a specific opsonin for S. aureus,29 the structural basis for these various activities and how/whether this relates to AZUs NSP‐like fold remains poorly understood. Thus, while EAP domain proteins initially appeared to be obvious candidates as AZU‐targeted evasion molecules, our results here suggest that any AZU inhibitor from S. aureus (assuming one exists) is most likely not a member of the EAP domain family.
Detailed knowledge of the minimally inhibitory EAP sequence/structure is required to develop clinical NSP inhibitors based on the non‐covalent EAP/NSP interaction. Moreover, to avoid the potential of undesirable side effects, a full understanding of the proteases that can be inhibited by EAP domains is essential. Because neutrophils' azurophilic granules also contain a fourth NSP, termed PRSS5730 or NSP‐4,31 in addition to the more well‐studied NE, CG, and PR3, we tested whether any EAP domain proteins might be able to inhibit NSP‐4. Curiously, while neither EapH1 nor EapH2 inhibited NSP‐4, we observed that full‐length Eap appeared to be a rather potent inhibitor NSP‐4 activity (Fig. S3). Although this experiment did not investigate the NSP‐4‐inhibitory capacity of the individual repeats of Eap, we believe it is consistent with our studies here on AZU (Fig. 6) because it suggests that bona fide target selectivity for various NSPs and NSP‐like proteins exists on the part of S. aureus EAP domains. Additional structure/function analyses will clearly be needed to explain why certain EAP repeats are able to bind and inhibit NSP‐4 whereas others cannot.
Classification of immune proteases is typically made based on the cell type in which they are primarily expressed (e.g., neutrophils vs. mast cells), yet the proteases themselves are very similar in terms of both their primary and tertiary structures. To determine if the selectivity of EAP domains might extend beyond NSPs, we examined the phylogeny of all human members of the MEROPS S1A class of chymotrypsin‐like immune proteases (Fig. S3).32 Those proteases most closely related to the NSPs included cytotoxic lymphocyte granzymes A, B, K, H, and M,33 along with chymase (MCC), which is most typically associated with the granules of mast cells. Intriguingly, we found that both EapH1 and EapH2 inhibit MCC (Fig. S3). While this result was unexpected, our observation that S. aureus secretes several high‐affinity inhibitors of MCC suggests a potentially important contribution of mast cells in the innate immune response against S. aureus infection. Indeed, roles in both phagocytic34 and non‐phagocytic killing of bacteria by mast cells have been described.35, 36 In the future, additional work may provide the proper biological context for understanding MCC inhibition by staphylococcal EAP domain proteins and its impact on bacterial virulence and survival within the host.
Materials and Methods
Human proteases and proteins
Natively expressed NE (Elastin Products Corp. #SE563) and CG (Elastin Products Corp. #SG623) were isolated from purulent human sputum using chromatographic methods. Chymase isolated from human mast cells was obtained from Sigma‐Aldrich (#C9612). NSP‐4 was a kind gift from Prof. Dieter Jenne (Helmholtz Zentrum München). Azurocidin isolated from human neutrophils was obtained from Athens Research & Technology (#16‐14‐012621).
Chemical modification of Asn‐linked glycans to amine‐bearing derivatives was used to facilitate immobilization of NE on biosensor surfaces. A solution of NE (0.1 mg/mL) was first prepared in oxidation buffer [0.1 M sodium acetate (pH 5.5)]. Sodium metaperiodate was then added to a final concentration of 10 mM, followed by a 30 min incubation at RT. Excess oxidant was removed by dialysis against PBS (pH 7.4) overnight at 4°C. Reductive amination was carried out by incubating the oxidized NE with 500 µM ethylenediamine (Sigma) and 50 mM sodium cyanoborohydride (Sigma) at RT for 6 h. The reaction was quenched with 100 mM ethanolamine for 30 mins at RT, prior to exchanging the amine‐bearing NE into PBS (pH 7.4) for downstream use.
Construction of EapH1 and EapH2 mutants
Mutations were introduced into expression vectors for wild‐type EapH1 and EapH2 through a sequence of three PCR reactions. First, one reaction was started with a forward primer on the vector and the reverse primer encoding the mutations in its 3′‐overhang. The second reaction was started with a forward primer encoding the mutations in its 5′‐overhang and a reverse primer on the vector. Both reaction products were used as starting material in the third step with the non‐mutagenic forward and reverse primers to obtain the final DNA product. Following a restriction digest with Sal1 and Not1, the mutated DNA was subcloned into the prokaryotic expression vector, pT7HMT.37 The presence of desired mutations was confirmed by standard DNA sequencing methods.
Protein expression and purification
EapH1, EapH2, and various site‐directed mutants thereof were expressed in E. coli strain BL21(DE3) according to the methods described in previous publications.14, 37
Circular dichroism analyses
Spectropolarimetry was used to assess the secondary structure content of EapH1, EapH2, and various mutants. Samples were dissolved in 20 mM HEPES (pH 7.4), 140 mM NaCl at a concentration of 1 mg/mL (∼100 μM). A buffer control was also collected. Spectra were collected across a 190–260 nm range, at 50 nm min−1, using 0.5 nm pitch, 1 s response time, and a bandwidth of 1 nm. All data were collected on a Jasco J‐815 instrument using a cylindrical small volume quartz cuvette (1 mm path length) (Starna Cells, Inc., Atascadero, CA). CD signals at wavelengths below 200 nm should be considered unreliable, as the instrument was not equipped with the hardware necessary to evacuate the sample chamber.
Surface plasmon resonance
SPR measurements were collected using either a Biacore 3000 or T‐200 instrument at 25°C. Experiments were carried out with a flow rate of 20 μL/min in a mobile phase of HBS‐T (20 mM HEPES (7.4), 140 mM NaCl, 0.005% (v/v) Tween‐20). Amine‐bearing NE and Cathepsin G were immobilized to ∼600 RU via standard NHS/EDC coupling to either CM‐5 (GE Healthcare) or CMD‐200M sensor chips (Xantec Bioanalytics, GmbH). A reference surface was generated by activation and ethanolamine quenching of a separate flow cell.
For conventional kinetic dose/response studies, a dilution series of EapH1, EapH2, and various mutants was injected across the NE experimental and control surface for 2 min, followed by a 4 min dissociation phase. Regeneration of the NE surface was achieved with a single, 30 s injection of 7.5 mM glycine (pH 2.2), 1.5 M NaCl.
Regeneration of the CG surface could not be achieved without loss of its activity. As a consequence, experimental injections over the CG flow cell were done using a single‐cycle method. EapH1 and EapH2 concentrations ranging from 1.95 to 500 nM were injected over both the CG and control surfaces for 2 min, followed by a 6 min dissociation phase. This was immediately followed by the next injection at a higher analyte concentration until the entire series was complete. A similar approach was used to investigate the dissociation rate of the NE complexes of both EapH1 and EapH2, with the exception that each injection phase was for 2 min and the final dissociation phase was for 60 min.
Kinetic analysis of each reference subtracted injection series was performed using BIAEvaluation software (GE Healthcare). All sensorgram series were fit to a 1:1 (Langmuir) binding model and a local value for R max.
Protease activity assays
NE activity was measured in a total volume of 100 µl using a reaction buffer of PBS (pH 7.4) with 0.05% (v/v) Igepal‐CA‐630 (Sigma) to promote solubility of the reaction components. The final composition of each reaction was 5 nM NE (Elastin Products Corp.), 50 µM AAPV‐AMC substrate (Sigma), and various concentrations of EAP domain proteins as inhibitors, or a structurally related, negative control protein, CHIPs.38 NE was incubated for 15 min at room temperature with various inhibitors across a concentration series prior to addition of AAPV‐AMC. Residual proteolytic activity was measured at 37°C using a Fluostar Omega microplate reader. Only those data points reflecting linear substrate conversion were used to determine relative protease activity. IC50 values were obtained by fitting the data to a sigmoidal curve without constraints.
CG activity was measured in a similar manner to NE, except that the final concentration of protease in each reaction was 15 nM and the substrate AAPF‐AMC (Genecust, Luxembourg) was included at 500 µM.12 Chymase activity assays contained 6.3 nM protease and 500 µM substrate AAPF‐AMC.
NSP‐4 activity was assessed in a volume of 100 µL, with 10 nM protease and 5 µM substrate [Mca‐GIKPRSRP‐Lys(Dnp)‐rr (r = D‐Arg)]. The assay buffer was either tris‐buffered saline (20 mM tris (pH 7.5), 150 mM NaCl) or PBS (pH 7.5), supplemented with 0.05% (v/v) Igepal‐CA‐630 (Sigma). Fluorescence was measured in a FluoStar Omega microplate reader with excitation at 320 nm and emission at 420 nm.
Structural modeling & analyses
The ClusPro server was used to generate a structural model for the EapH2/NE complex.16 An analogous model of EapH1/NE was constructed as a benchmark, and compared to the EapH1/NE co‐crystal structure (PDB code 4NZL)12 as a means to gauge the accuracy of the computational approach. Briefly, the structures of EapH1 (PDB code 1YN414), EapH2 (PDB code 1YN514), and NE (PDB code 1HNE39) were used as the inputs without experimentally derived restraints on modeling calculations. The highest scored model was selected in each case for further analysis.
Calculation of buried surface areas and identification of hydrogen bonds were performed by the EBI‐PISA web server (http://www.ebi.ac.uk/msd-srv/prot_int/). All structural superpositions and figure renderings were carried out using PyMol (Schrodinger).
Phylogenetic tree
Human serine proteases from the S1A clade were selected from the MEROPS database.32 Protein sequences were aligned using Clustal Omega. The boundaries of the serine protease domain were taken from the sequence of NE, and spanned from Ile30 to the C terminus. The phylogenetic tree of serine proteases was built using these protease domain sequences in Clustal Omega and visualized using EvolView.40
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions
D.A.C.S. designed and performed experiments, analyzed the data, and wrote the manuscript.
J.L.W. designed and performed experiments, analyzed the data, and wrote the manuscript.
F.J.M. performed experiments and analyzed the data
A.T.T. performed experiments and analyzed the data
A.A.v.B. performed experiments and analyzed the data
W.C.d.G. performed experiments and analyzed the data
S.C.B. performed experiments and analyzed the data
N.M.W. performed experiments and analyzed the data
S.H.M.R. designed the overall scope of the study, analyzed the data, and wrote the manuscript.
B.V.G. designed the overall scope of the study, analyzed the data, and wrote the manuscript.
Supporting information
The supplementary material consists of three supplemental figures and their corresponding legends in a single .pdf file: Stapels_etal_Supplemental.pdf
Acknowledgments
The authors gratefully acknowledge Prof. Dieter Jenne for contributing purified NSP‐4 and its substrate to this study. This work was supported by European Research Council Starting Grant 639209 (to S.H.M.R.), US National Institutes of Health Grants AI071028, AI111203, and GM121511 (to B.V.G.), and American Heart Association Predoctoral Fellowship 15PRE25750013 (to J.L.W.).
Statement: Serine proteases found within neutrophil granules form a cornerstone of our body's innate response to invading microorganisms. The results we present here further our understanding of how the pathogen Staphylococcus aureus disrupts function of these proteases by secreting two small inhibitory proteins, known as EapH1 and EapH2. While EapH1 and EapH2 share significant homology with one another, our data surprisingly suggest that they form structurally distinct complexes with neutrophil serine proteases.
References
- 1. Martin P (1997) Wound healing—aiming for perfect skin regeneration. Science 276:75–81. [DOI] [PubMed] [Google Scholar]
- 2. Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J Med 341:738–746. [DOI] [PubMed] [Google Scholar]
- 3. Frantz S, Vincent KA, Feron O, Kelly RA (2005) Innate immunity and angiogenesis. Circ Res 96:15–26. [DOI] [PubMed] [Google Scholar]
- 4. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489. [DOI] [PubMed] [Google Scholar]
- 5. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F (2010) Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human disease. Pharmacol Rev 62:726–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pham CTN (2006) Neutrophil serine proteases: Specific regulators of inflammation. Nat Rev Immunol 6:541–550. [DOI] [PubMed] [Google Scholar]
- 7. Stapels DA, Geisbrecht BV, Rooijakkers SH (2015) Neutrophil serine proteases in antibacterial defense. Curr Opin Microbiol 23:42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parker H, Albrett AM, Kettle AJ, Winterbourn CC (2012) Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukocyte Biol 91:369–376. [DOI] [PubMed] [Google Scholar]
- 9. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. [DOI] [PubMed] [Google Scholar]
- 10. Barnes PJ, Stockley RA (2005) COPD: current therapeutic interventions and future approaches. Eur Respir J 25:1084–1106. [DOI] [PubMed] [Google Scholar]
- 11. Stoller JK, Aboussouan LS (2005) Alpha1‐antitrypsin deficiency. Lancet 365:2225–2236. [DOI] [PubMed] [Google Scholar]
- 12. Stapels DAC, Ramyar KX, Bischoff M, von Kockritz‐Blickwede M, Milder FJ, Ruyken M, Eisenbeis J, McWhorter WJ, Herrmann M, van Kessel KPM, Geisbrecht BV, Rooijakkers SHM (2014) Staphylococcus aureus secretes a novel class of neutrophil‐serine‐protease inhibitors that promote bacterial infection. Proc Natl Acad Sci USA 111:13187–13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Palazzolo‐Ballance AM, Reniere ML, Braughton KR, Sturdevant DE, Otto M, Kreiswirth BN, Skaar EP, Deleo FR (2008) Neutrophil microbicides induce a pathogen survival response in community‐associated methicillin‐resistant Staphylococcus aureus . J Immunol 180:500–509. [DOI] [PubMed] [Google Scholar]
- 14. Geisbrecht BV, Hamaoka BY, Perman B, Zemla A, Leahy DJ (2005) The crystal structures of EAP domains from Staphylococcus aureus reveal an unexpected homology to bacterial superantigens. J Biol Chem 280:17243–17250. [DOI] [PubMed] [Google Scholar]
- 15. Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20:45–50. [DOI] [PubMed] [Google Scholar]
- 16. Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: a fully automated algorithm for protein‐protein docking. Nucleic Acids Res 32:W96–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iversen LF, Kastrup JS, Bjorn SE, Rasmussen PB, Wiberg FC, Flodgaard HJ, Larsen IK (1997) Structure of HBP, a multifunctional protein with a serine proteinase fold. Nat Struct Biol 4:265–268. [DOI] [PubMed] [Google Scholar]
- 18. Gabay JE, Scott RW, Campanelli D, Griffith J, Wilde C, Marra MN, Seeger M, Nathan CF (1989) Antibiotic proteins of human polymorphonuclear leukocytes. Proc Natl Acad Sci USA 86:5610–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shafer WM, Martin LE, Spitznagel JK (1984) Cationic antimicrobial proteins isolated from human neutrophil granulocytes in the presence of diisopropyl fluorophosphate. Infect Immun 45:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chertov O, Ueda H, Xu LL, Tani K, Murphy WJ, Wang JM, Howard OM, Sayers TJ, Oppenheim JJ (1997) Identification of human neutrophil‐derived cathpesin G and azurocidin/CAP37 as chemoattractants for mononuclear cells and neutrophils. J Exp Med 186:739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zani M‐L, Nobar SM, Lacour SA, Lemoine S, Boudier C, Bieth JG, Moreau T (2004) Kinetics of the inhibition of neutrophil proteinases by recombinant elafin and pre‐elafin (trappin‐2) expressed in Pichia pastoris. Eur J Biochem 271:2370–2378. [DOI] [PubMed] [Google Scholar]
- 22. Guyot N, Zani M‐L, Maurel M‐C, Dallet‐Choisy S, Moreau T (2005) Elafin and its precursor trappin‐2 still inhibit neutrophil serine proteinases when they are covalently bound to extracellular matrix proteins by tissue transglutaminase. Biochemistry 44:15610–15618. [DOI] [PubMed] [Google Scholar]
- 23. Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416:291–297. [DOI] [PubMed] [Google Scholar]
- 24. Stapels DAC, Kuipers A, von Köckritz‐Blickwede M, Ruyken M, Tromp AT, Horsburgh MJ, de Haas CJC, van Strijp JAG, van Kessel KPM, Rooijakkers SHM (2016) Staphylococcus aureus protects its immune‐evasion proteins against degradation by neutrophil serine proteases. Cell Microbiol 18:536–545. [DOI] [PubMed] [Google Scholar]
- 25. Winterbourn CC, Hampton MB, Livesey JH, Kettle AJ (2006) Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome. J Biol Chem 281:39860–39869. [DOI] [PubMed] [Google Scholar]
- 26. Fields BA, Malchiodi EL, Li H, Ysern X, Stauffacher CV, Schlievert PM, Karjalainen K, Mariuzza R (1996) Crystal structure of a T‐cell receptor b‐chain complexed with a superantigen. Nature 384:188–192. [DOI] [PubMed] [Google Scholar]
- 27. Woehl JL, Stapels DAC, Garcia BL, Ramyar KX, Keightley A, Ruyken M, Syriga M, Sfyroera G, Weber AB, Zolkiewski M, Ricklin D, Lambris JD, Rooijakkers SHM, Geisbrecht BV (2014) The extracellular adherence protein from Staphylococcus aureus inhibits the classal and lectin pathways of complement by blocking formation of the C3 pro‐convertase. J Immunol 193:6161–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Woehl JL, Ramyar KX, Katz BB, Walker JK, Geisbrecht BV (2017) The structural basis for inhibition of the classical and lectin complement pathways by S. aureus extracellular adherence protein. Protein Sci 26:1595–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heinzelmann M, Platz A, Flodgaard H, Miller FN (1998) Heparin binding protein (CAP37) is an opsonin for Staphylococcus aureus and increases phagocytosis in monocytes. Inflammation 22:493–507. [DOI] [PubMed] [Google Scholar]
- 30. Clark HF, Gurney AL, Abaya E, Baker K, Baldwin D, Brush J, Chen J, Chow B, Chui C, Crowley C, Currell B, Dueul B, Dowd P, Eaton D, Foster J, Grimaldi C, Gu Q, Hass PE, Heldens S, Huang A, Kim HS, Klimowski L, Jin Y, Johnson S, Lee J, Lewis L, Liao D, Mark M, Robbie E, Sanchez C, Schoenfeld J, Seshagiri S, Simmons L, Singh J, Smith V, Stinson J, Vagts A, Vandlen R, Watanabe C, Wieand D, Woods K, Xie MH, Yansura D, Yi S, Yu G, Yuan J, Zhang M, Zhang Z, Goddard A, Wood WI, Godowski P, Gray A (2003) The Secreted Protein Discovery Initiative (SPDI), a large‐scale effort to identify novel human secreted and transmembrane proteins: a bioinformatics assessment. Genome Res 13:2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Perera NC, Schilling O, Kittel H, Back W, Kremmer E, Jenne DE (2012) NSP4, an elastase‐related protease in human neutrophils with arginine specificity. Proc Natl Acad Sci USA 109:6229–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: The database of proteolytic enzymes, their substrates, and inhibitors. Nucleic Acids Res 40:D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Voskoboinik I, Whisstock JC, Trapani JA (2015) Perforin and granzymes: Function, dysfunction and human pathology. Nat Rev Immunol 15:388–400. [DOI] [PubMed] [Google Scholar]
- 34. Feger F, Varadaradjalou S, Gao Z, Abraham SN, Arock M (2002) The role of mast cells in host defense and their subversion by bacterial pathogens. Trends Immunol 23:151–158. [DOI] [PubMed] [Google Scholar]
- 35. von Kockritz‐Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby‐Teglund A, Rohde M, Medina E (2008) Phagocytosis‐independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111:3070–3080. [DOI] [PubMed] [Google Scholar]
- 36. Mollerherm H, von Kockritz‐Blickwede M, Branitzki‐Heinemann K (2016) Antimicrobial activity of mast cells: role and relevance of extracellular DNA traps. Front Immunol 7:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Geisbrecht BV, Bouyain S, Pop M (2006) An optimized system for the expression and purification of secreted bacterial proteins. Prot Expr Purif 46:23–32. [DOI] [PubMed] [Google Scholar]
- 38. de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, van Wamel WJ, Heezius EC, Poppelier MJ, van Kessel KP, van Strijp JA (2004) Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med 199:687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Navia MA, McKeever BM, Springer JP, Lin TY, Williams HR, Fluder EM, Dorn CP, Hoogsteen K (1989) Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84‐A resolution. Proc Natl Acad Sci USA 86:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang H, Gao S, Lercher MJ, Hu S, Chen W‐H (2012) EvolView, an online tool for visualizing, annotating, and managing phylogenetic trees. Nucleic Acids Res 40:W569–W572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The supplementary material consists of three supplemental figures and their corresponding legends in a single .pdf file: Stapels_etal_Supplemental.pdf