

Abstract
Malate dehydrogenase (MDH) and citrate synthase (CS) are two pacemaking enzymes involved in the tricarboxylic acid (TCA) cycle. Oxaloacetate (OAA) molecules are the intermediate substrates that are transferred from the MDH to CS to carry out sequential catalysis. It is known that, to achieve a high flux of intermediate transport and reduce the probability of substrate leaking, a MDH‐CS metabolon forms to enhance the OAA substrate channeling. In this study, we aim to understand the OAA channeling within possible MDH‐CS metabolons that have different structural orientations in their complexes. Three MDH‐CS metabolons from native bovine, wild‐type porcine, and recombinant sources, published in recent work, were selected to calculate OAA transfer efficiency by Brownian dynamics (BD) simulations and to study, through electrostatic potential calculations, a possible role of charges that drive the substrate channeling. Our results show that an electrostatic channel is formed in the metabolons of native bovine and recombinant porcine enzymes, which guides the oppositely charged OAA molecules passing through the channel and enhances the transfer efficiency. However, the channeling probability in a suggested wild‐type porcine metabolon conformation is reduced due to an extended diffusion length between the MDH and CS active sites, implying that the corresponding arrangements of MDH and CS result in the decrease of electrostatic steering between substrates and protein surface and then reduce the substrate transfer efficiency from one active site to another.
Keywords: Krebs cycle, Brownian dynamics simulation, electrostatic property, association rate constant
Introduction
The tricarboxylic acid (TCA) cycle, also known as the citric acid cycle (CAC) or the Krebs cycle, is the second stage of cellular respiration in aerobic organisms. The process involves a cascade of chemical reactions to release stored energy through breaking down organic fuel molecules in the presence of oxygen and the reducing power of nicotinamide adenine dinucleotide (NADH) to form adenosine triphosphate (ATP).1, 2 Several enzymes, such as mitochondrial malate dehydrogenase (MDH), citrate synthase (CS), and aconitase (ACO), participate in the TCA cycle and directly contribute to the metabolic process.3 It is believed that supramolecular complexes, called metabolons, form along the metabolic pathway by noncovalent interactions, leading to enzyme self‐assembly to enhance the transport of reactants from one enzyme active site to another.4, 5 Thus, the substrate channeling within metabolons plays an essential role in maintaining high enzyme efficiency and improving selectivity in catalytic performance.6
In one of the steps of the TCA cycle, MDH converts malate into oxaloacetate (OAA), while NAD+ is reduced to form NADH. The protein complex of MDH and CS is formed to channel the OAA from the MDH to the CS active site. Then, the four‐carbon molecule, OAA, joining with acetyl coenzyme A (CoA) in the CS, forms a six‐carbon molecule, citrate, and then releases a CoA group (Fig. 1).2, 7 The reaction cascade is particularly interesting since a high concentration of free OAA molecules in the solution may result in an unfavorable equilibrium of the MDH reaction in the forward direction.8 The OAA channeling between MDH and CS not only leads to a high flux of OAA along a pathway to overcome the unfavorable thermodynamics but also protects the reactant from the competition of other enzymes in the bulk environment.
Figure 1.
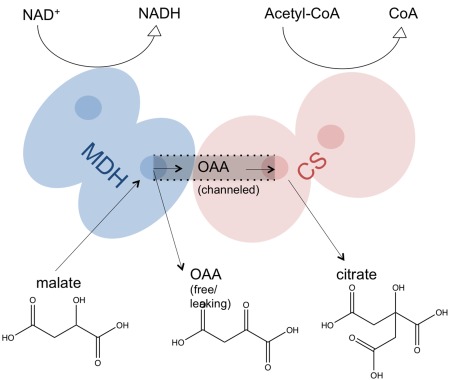
Scheme of MDH‐CS catalysis. In the MDH active sites, malate is oxidized to OAA with a reversible chemical reaction of NADH. Then, the OAA intermediate is channeled along the protein complex surface from the MDH to CS active site. Finally, the OAA and acetyl‐CoA are converted into citrate and CoA in the CS active sites.
Although many efforts have been made to uncover structures and arrangements of the above metabolon molecules, we still have little understanding of enzyme‐enzyme interactions in their natural form. This can be attributed to the experimental difficulty in crystallizing and purifying the metabolons, since a small disturbance may give rise to the dissociation of the weakly bound complexes. However, recently the Minteer group combined in vivo chemical cross‐linking, mass spectrometry and protein docking to identify the interfacial interactions within MDH, CS, and ACO and establish a low‐resolution structure of the MDH‐CS‐ACO supercomplex in the TCA cycle.9 The structure showed that the active sites of the MDH and CS dimer are relatively close in the complex with an average distance of 35 Å, reducing the diffusion distance of OAA. The charge distribution of the MDH‐CS conformations includes positive charges not only around the active sites but also between these sites, which could facilitate the diffusion of the negatively charged OAA along the channel.
Later on, the group continued to characterize a synthetic metabolon formed in vitro from three different enzyme sources: native tissue enzymes isolated from intact bovine mitochondria (native bovine), commercially available porcine wild‐type enzymes (wild‐type porcine), and recombinantly produced porcine enzymes (recombinant porcine).10 The structural and kinetic study of that work showed that the MDH‐CS associated conformations from recombinant porcine enzyme have similar kinetic properties to their native bovine counterparts in vitro. Different orientations of the MDH‐CS complex structures would affect the channeling kinetics of reaction intermediates. The charge distribution along the channel and OAA transfer efficiency between MDH and CS of the three enzyme metabolons were not quantitatively studied, but transient times for the intermediate provide experimental indication of channeling.10
A computational technique, Brownian dynamics (BD) simulation, used to describe the motion of molecules in solution, has been widely applied to study free diffusion models in biomolecular systems, especially the understanding of electrostatically driven association processes.11, 12 The method is typically applied to simulate the association of two molecules, by fixing a protein at the center of the simulation system while allowing a substrate to freely move in the solution. The BD method has recently been used to show that electrostatics directly promotes the association rate and substrate transfer efficiency in the system of signaling protein, phosphodiesterase‐3′‐5′‐cyclic adenosine monophosphate (PDE‐cAMP) and in the noncovalent enzyme complex, dihydrofolate reductase‐thymidylate synthase (DHFR‐TS), respectively.13, 14 Elcock et al. also performed BD simulations to investigate a possible role for electrostatic channeling in transferring substrate between MDH and CS.15, 16 However, the latter model of the MDH‐CS complex was generated by docking tools. Although the multiple energy minimizations during docking could reduce the uncertainty of the resulting structures, the orientations of the proteins within the complex structure were necessarily somewhat speculative. As a result, in this work, we explored the substrate channeling of MDH‐CS metabolons with initial complex structures determined by experimental refinement.
Results
Electrostatic and kinetic properties of the CS dimer
Before studying the channeling efficiency of the MDH‐CS metabolon, we first inspected the electrostatic and kinetic properties of the CS dimer protein to construct BD parameters, such as reaction radii, for the later calculations. The electrostatic map from APBS calculations shows that the total charge of the CS dimer is +10 e– and both OAA active sites of the protein are positively charged (Fig. 2).17, 18 This can be attributed to a Lys and three Arg residues within 10 Å of the bound OAA molecule. Since the OAA contains −2 e– charges, we could expect the main driving force of the association reaction between CS dimer and OAA molecule to be an electrostatic interaction.
Figure 2.
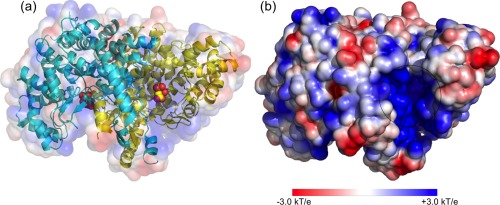
Electrostatic potential map of CS dimer. (a) The CS dimer is composed of two chains, which are colored in yellow and cyan. OAA molecules in the two active sites are shown in a bead representation. (b) One of the OAA active sites in CS dimer with highly positive charges is circled.
Next, we focused on the association rate and reaction probability of OAA binding to CS dimer. The BD simulations were carried out to obtain OAA association rate constants (kon) as a function of reaction radii. Because each CS monomer protein has an OAA active site, as long as an OAA molecule can reach one of the active sites, we considered the association to be complete. Figure 3 shows that high kon values are observed when the hypothetical reaction surface is ∼85 Å away from the protein. In this region, since the substrates are far away from the CS dimer, the effect of the forces from the protein on the OAA motions is small, giving a case of random diffusion. Stronger interactions between the OAA and CS form with the reduction of the reaction radii. A rapid decrease of the kon value is also noted, due to the decreasing area of the reaction surface. Starting at about 20 Å from the protein, the motions of the OAA can be influenced by both positive and negative charges from the CS dimer. Thus, the substrates spend more time interacting with the protein and searching for potential binding spots, which results in a slow change of the kon value. After passing this region, around ∼15 Å, the OAA molecules displacements are governed by attractions of the strong positive charges from the CS active sites, and the kon value becomes constant over a wide range; here, the OAA molecules are committed to encounter. Under this reaction radius, we considered the binding between substrates and protein as having completed. Therefore, the reaction radii, 3 and 7 Å, were chosen for the following BD studies. This analysis did not account for the subsequent catalytic step, which is slower for this system.
Figure 3.
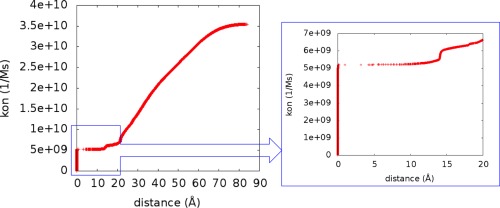
Association rate constant (kon) of OAA binding to CS dimer for different reaction radii. (Left) The kon changes with reaction radii. (Right) An enlarged figure that shows the kon changes with the reaction radii from 0 to 20 Å.
Figure 4 shows OAA association rate and reaction probability to CS dimer at different solution ionic strengths and reaction radii. It is clear that, without charges on the OAA molecules, almost no binding events could happen. However, the −2 e– charged OAA molecules are attracted by the positive potential around the active sites of CS dimer, indicating that the electrostatic interactions drive the substrate toward the binding site and enhance the association process of OAA binding to the CS dimer. The association rate of OAA increases at low ionic strength, because the ions in solution screen the enzyme‐substrate interactions and slow down the binding process. The reaction probability here is defined by the percentage of OAA molecules that can successfully diffuse to the CS active sites before escaping. Figure 4 shows that the binding probability can be enhanced through reducing the ionic strength, although only 30% of the trajectories result in successful binding events at 0.05 M. The above results are consistent at both reaction radii, 3 and 7 Å.
Figure 4.
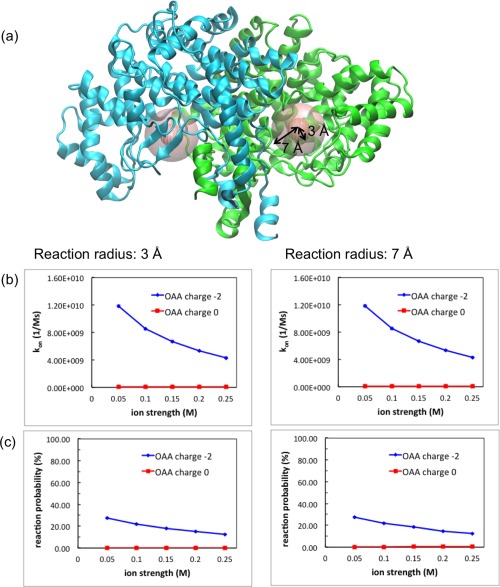
Association rate constant (kon) and reaction probability of OAA binding to CS dimer for different ionic strengths. (a) Two reaction radii, 3 and 7 Å, applied in BD simulations, are shown as spheres. (b) The kon changes with ionic strengths in the presence and absence of OAA charges. (c) The OAA reaction probability changes with ionic strengths with charged and uncharged OAA molecules.
Electrostatic potential of MDH‐CS metabolons
Both MDH and CS enzymes are dimers composed of identical subunits. The structure of MDH‐CS complex is formed by a MDH dimer and a CS dimer, which includes four chains in total, labeled from chain A to D in Figure 5. Three different structures of MDH‐CS metabolon from the sources of native bovine, wild‐type porcine and recombinant porcine enzymes were used to construct electrostatic maps. The three metabolons share the same sequence but with different MDH and CS orientations. The different structural models correspond to different sample preparations and cross‐linking conditions. The total charge of native bovine, wild‐type porcine and recombinant porcine metabolon is +18, +15, and +17 e–, respectively, through PROPKA calculations.19 Figure 5 shows that the MDH‐CS metabolon from both native bovine and recombinant porcine display an electrostatic channel; a continuously positive potential extends across the protein surface that bridges active sites, one from MDH dimer and the other from CS dimer. The positively charged residues, Arg and Lys, around the protein surface play an important role in the construction of this electrostatic channel. However, the charged channel is not visibly formed in the metabolon formed from the wild‐type porcine enzyme in the cross‐linking experiment, because both OAA active sites in its MDH dimer face to the solution, away from the OAA active sites in the CS dimer.
Figure 5.
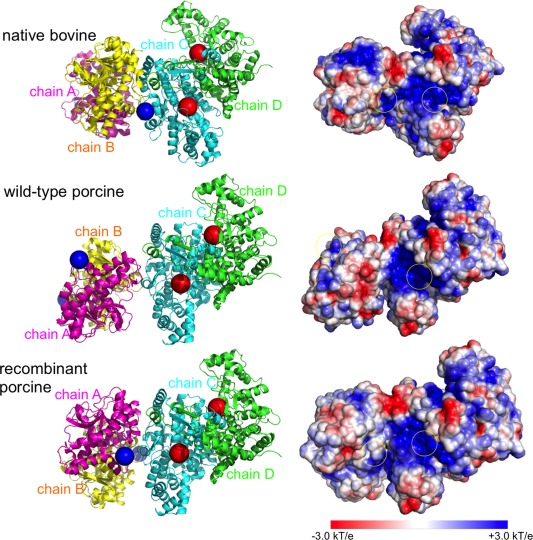
Structure orientations and electrostatic maps of MDH‐CS metabolons. (left) Three MDH‐CS complex structures are taken from native bovine, wild‐type porcine and recombinant porcine enzyme. The structures share the same sequence but with different MDH and CS orientations. Magenta and yellow cartoons represent the MDH dimer; green and cyan cartoons represent the CS dimer. Blue beads show the starting positions, where OAA molecules were initially placed in BD simulations, and red beads show the OAA active sites in the CS dimer. (right) Protein surface color of red and blue represent negative and positive electrostatic potential, respectively. The yellow circles indicate the OAA active sites in the MDH and CS dimer.
Channeling efficiency of MDH‐CS metabolons
The OAA transfer efficiency from the active site of MDH to CS in the three metabolons was quantitatively calculated by BD simulations. Since MDH in the metabolon is formed as a dimer structure, OAA molecules were released from both of the active sites; that is, we diffused OAA molecules from both chain A and B (Fig. 5). Once the OAA molecules reached the active site in either chain C or D of the CS dimer (Fig. 5), we considered the reaction to be complete and the BD simulation was terminated. Figure 6 shows the OAA channeling efficiency defined by the percentage of OAA molecules that successfully associated to a CS active site from the MDH surface before escaping into the surrounding bulk solution. The transfer probability of charged OAA molecules is much higher than neutral OAA. The diffusion of the negatively charged OAA molecules is strongly affected by the positive electrostatic potential from the metabolon surface. The substrate tends to be confined to the metabolon surface, enhancing the capture of diffusing OAA molecules.
Figure 6.
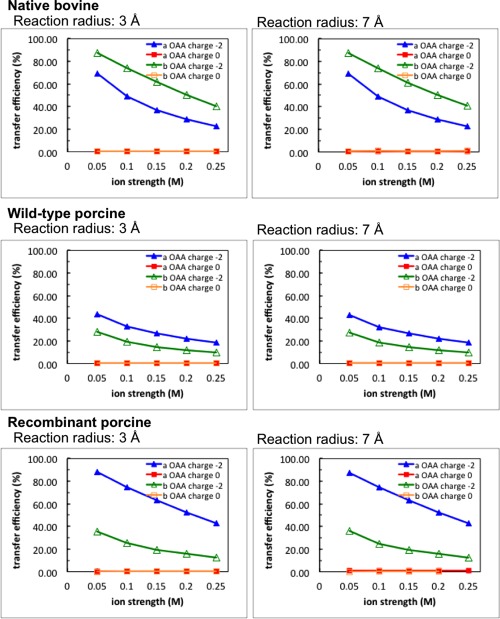
The transfer efficiency of OAA molecules within three different metabolon models. OAA transfer efficiency from the MDH to CS active sites changes with ionic strengths. Blue and red lines indicate the transfer probability that the OAA molecules were diffused from MDH chain A with and without negative charges on the OAA, respectively. Green and orange lines indicate the charged and uncharged OAA molecules were diffused from MDH chain B, respectively. Left and right column show the simulations with reaction radius 3 and 7 Å, respectively.
By checking the OAA transfer efficiency in the three different MDH‐CS metabolons (Fig. 6), one finds that metabolons cross‐linked from the native bovine and recombinant enzyme show higher OAA channeling probability than that from the wild‐type porcine enzyme. At low ionic strength, 0.05 M, up to 90% of OAA molecules effectively reach the CS active sites before escaping in the native bovine and recombinant porcine metabolons. However, about 50% probability can be detected in the metabolon from wild‐type porcine.
Discussion
Substrate channeling in the MDH‐CS metabolon systems not only helps to reduce the escape of OAA molecules into solution but also reduces the transient time of OAA molecules transferred from one active site to another. An unassembled or detached system will prevent the channeling process. The transient time, describing the lifetime of the OAA intermediates, has been predicted by the work from Bulutoglu et al. The measurement of the OAA transient time in native bovine, wild‐type porcine and recombinant porcine metabolons is 40, 290, and 30 ms, respectively.10 Note that the transient time of wild‐type porcine is almost one order of magnitude higher than native bovine and recombinant porcine. Although we could not directly measure the OAA transient time through the BD calculations, the transfer efficiency of OAA molecules in wild‐type porcine enzymes is significantly lower than the other two metabolons. This might result from the extended distance between the MDH and CS active sites in wild‐type porcine metabolons causing a higher chance of OAA escaping after release from the MDH active sites; thus, the longer OAA transient time of wild‐type porcine metabolons was observed. Hence, the proximity between MDH and CS active sites and the shortness of the electrostatic channel of native bovine and recombinant porcine enzyme reduce the intermediate transient time and increase the transfer efficiency. In addition, the similarity of the results of channeling probability between native bovine and recombinant porcine metabolons from our simulations agrees with the transient measurements and previous experiments that metabolons from native bovine and recombinant porcine enzymes basically share similar channeling characteristics.10
The OAA channeling within a cross‐linked MDH‐CS fusion protein was also studied by Elcock et al.15 Both Elcock's results and our work agree that the OAA transfer probability is lower than 1% in the absence of electrostatic forces and the transfer efficiency increases with the reduction of ionic strengths. However, the calculated transfer efficiency of OAA molecules in our simulations is 6‐fold higher than the results from Elcock's work. For example, the OAA transfer efficiency at 0.05 M ionic strength is 90% and 15%, from our and Elcock's study, respectively. The difference could be attributed to the construction of the initial enzyme and substrate model. First, the model of the fusion structure in Elcock's work was built by simply docking MDH and CS so that the C‐terminal of the CS dimer is closer to the MDH N‐terminal, and the MDH and CS active sites were separated by ∼60 Å. However, the two proteins in our study were oriented in a different way, shortening the OAA channel, which increases the substrate transfer efficiency. Second, in Elcock's work, all Glu and Asp residues were simply assigned with a −1 e‐ charge and His were all in neutral forms; thus, the total charges of the MDH‐CS metabolon was +4 e–. However, in our charge model, three Glu residues along the OAA channel were assigned as a protonated form, which enhances the positive charges of the channel. Thus, the different charge models of the protein residues would alter the electrostatic distribution on the metabolon surface and cause differences in channeling behavior. Third, an OAA molecule was modeled as a sphere of radius 2 Å in the previous work, while a larger value was used here (see Methods). We were able to rely on more detailed experimental structures, and our results confirm that electrostatic steering plays a key role in substrate channeling and the spatial proximity of MDH and CS active sites enhances the OAA transfer efficiency.
Conclusions
In this work, we performed electrostatic potential calculations and BD simulations to study substrate channeling of metabolons in the TCA cycle by first inspecting the electrostatics and kinetic properties of a CS dimer, and then exploring the surface charge arrangements and OAA transfer efficiency of the three different MDH‐CS metabolons from native bovine, wild‐type porcine and recombinant porcine enzyme. Our electrostatic predictions show that the OAA active sites in a CS dimer are highly positively charged. Upon MDH‐CS association, a continuous surface with positive potential connects the MDH and CS active sites, forming an electrostatic channel. The electrostatic steering between negatively charged OAA molecules and the positive electrostatic channel guides OAA channeling and enhances substrate transfer efficiency. Due to the vicinity and relative orientation between MDH and CS active sites of native bovine and recombinant porcine enzyme, we identified a clear electrostatic channel that directs the substrates and leads to a high channeling efficiency. However, the wild‐type porcine metabolons reveal lower transfer probabilities of intermediate molecules because of the extended diffusion length from the MDH to CS enzyme. Our results qualitatively agree with experimental measurements of OAA transient time. Also, all of these structural orientations of the three MDH‐CS metabolons demonstrate higher OAA transfer efficiency compared to the calculated results from Elcock et al.15
Materials and Methods
Protein structures
The modeled structures of MDH‐CS metabolons were taken from the work published by Bulutoglu et al.10 In that study, the authors described cross‐linked metabolons in which MDH and CS exhibit different orientations, but all structures share the same sequence. The three different metabolons are from the sources of native bovine, wild‐type porcine and recombinant porcine enzyme. We chose these three structures to study enzyme‐substrate channeling efficiency through BD simulations. The structure of CS dimer to calculate the OAA association rate constant and reaction probability was extracted from the MDH‐CS metabolon of the native enzyme in the bovine mitochondrial case.
Before running electrostatic potential calculations and BD simulations, proper parameter setting of the proteins was necessary. First, the static protonation states of the proteins at physiological pH 7.0 were assigned through the PROPKA program version 3.0.19 Then, the software PDB2PQR version 1.820 was applied to generate the charges and radius of each atom with the Amber force field.21 Dummy atoms, without charge or radius, were placed at the center of the OAA active site in the CS dimer protein. These atom coordinates can be viewed as the locations where the substrate binds, which helps to define the positions where the enzyme‐substrate reactions occur.
Electrostatic potential calculations
To calculate electrostatic force fields and visualize electrostatic maps of the protein systems, the Adaptive Poisson–Boltzmann Solver (APBS) program version 1.4 was used to evaluate the properties of a CS dimer and three MDH‐CS metabolons.17, 18 The APBS input information, including atom coordinates, charges and radii of the systems, was obtained by the PDB2PQR program.20 Multiple ionic strengths, ranging from 0.05 to 0.25 M, were assigned in the APBS program to construct the electrostatic potential field. This method was also applied to generate the electrostatic grid in the following BD simulations.
Brownian dynamics simulations
The BrownDye package was used to perform Brownain dynamics simulations and calculate substrate association rate constant and channeling efficiency.22 The software allows the users to compute the second order reaction rate and substrate reaction probability of the encounter of two rigid bodies moving according to BD. In the simulations, the proteins were constructed in an all‐atom model. A ghost atom of 2 Å radius without charges was placed near Lys423 in a CS dimer to eliminate the artificial substrate “sticking” of trajectories in the initial models of the complexes due to the strong positive charges generated by surrounding Lys and Arg amino acids. Also, the OAA molecule was modeled as a single sphere of radius 3.62 Å and charge −2 e–, where the OAA bead radius was determined by the Stokes–Einstein equation based on the calculations of diffusion coefficient from the Hydropro program.23 In the BD simulations of a CS dimer, OAA molecules randomly diffused around the protein, starting from a distance of 85∼105 Å. However, in the simulations of MDH‐CS metabolons, the OAA started to travel from the two active sites of the MDH dimer under the influence of the hydrodynamic and electrostatic forces between the enzymes and substrate. The BD simulations were terminated when either the reaction occurred or the substrate escaped. If the OAA molecules reached a spherical region within 3 or 7 Å away from one of the center (dummy atom) of the CS active sites, we considered the reaction to have occurred. The substrate escape was defined by the travel of the OAA to a spherical boundary around the metabolon, large enough so that the forces between the substrate and complex were spherically symmetric, thereby allowing computation of the probability of a complete escape or subsequent return to a slightly smaller spherical surface.22 The maximum number of steps in the BD simulations was set to 1,000,000. Also, 50,000 copies of the BD trajectories for each system were used to calculate the substrate association rate constant and reaction probability.
Acknowledgments
We thank Fei Wu for all discussion of experimental details. We are grateful to Robert Konecny for helpful discussion of electrostatic calculations. This work was supported by the NIH, HHMI, NBCR, and NSF supercomputer centers.
Contributor Information
Yu‐ming M. Huang, Email: yuh155@ucsd.edu
Gary A. Huber, Email: gahuber95@gmail.com
References
- 1. Fernie AR, Carrari F, Sweetlove LJ (2004) Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol 7:254–261. [DOI] [PubMed] [Google Scholar]
- 2. Kornberg HL (1987) Krebs citric‐acid cycle: half a century and still turning—introductory. Biochem Soc Symp 54:1–2. [Google Scholar]
- 3. Robinson JB, Srere PA (1985) Organization of Krebs tricarboxylic‐acid cycle enzymes in mitochondria. J Biol Chem 260:800–805. [PubMed] [Google Scholar]
- 4. Schoffelen S, van Hest JCM (2012) Multi‐enzyme systems: bringing enzymes together in vitro. Soft Matter 8:1736–1746. [Google Scholar]
- 5. Srere PA, Mosbach K (1974) Metabolic compartmentation: symbiotic, organellar, multienzymic, and microenvironmental. Annu Rev Microbiol 28:61–83. [DOI] [PubMed] [Google Scholar]
- 6. Wheeldon I, Minteer SD, Banta S, Barton SC, Atanassov P, Sigman M (2016) Substrate channeling as an approach to cascade reactions. Nat Chem 8:299–309. [DOI] [PubMed] [Google Scholar]
- 7. Robinson JB, Srere PA (1985) Organization of Krebs tricarboxylic‐acid cycle enzymes. Biochem Med 33:149–157. [DOI] [PubMed] [Google Scholar]
- 8. Beeckmans S, Kanarek L (1981) Demonstration of physical interactions between consecutive enzymes of the citric‐acid cycle and of the aspartate‐malate shuttle: a study involving fumarase, malate‐dehydrogenase, citrate synthase and aspartate‐aminotransferase. Eur J Biochem 117:527–535. [DOI] [PubMed] [Google Scholar]
- 9. Wu F, Minteer S (2015) Krebs cycle metabolon: structural evidence of substrate channeling revealed by cross‐linking and mass spectrometry. Angew Chem Intl Ed 54:1851–1854. [DOI] [PubMed] [Google Scholar]
- 10. Bulutoglu B, Garcia KE, Wu F, Minteer SD, Banta S (2016) Direct evidence for metabolon formation and substrate channeling in recombinant TCA cycle enzyme. ACS Chem Biol 11:2847–2853. [DOI] [PubMed] [Google Scholar]
- 11. Davis ME, Madura JD, Luty BA, McCammon JA (1991) Electrostatics and diffusion of molecules in solutions: simulations with the university‐of‐Houston‐Brownian dynamics program. Comput Phys Commun 62:187–197. [Google Scholar]
- 12. Zhou HX (1990) Kinetics of diffusion: influenced reactions studied by Brownian dynamics. J Phys Chem 94:8794–8800. [Google Scholar]
- 13. Huang Y‐mM, Huber G, McCammon JA (2015) Electrostatic steering enhances the rate of cAMP binding to phosphodiesterase: Brownian dynamics modeling. Protein Sci 24:1884–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang N, McCammon JA (2016) Substrate channeling between the human dihydrofolate reductase and thymidylate synthase. Protein Sci 25:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elcock AH, McCammon JA (1996) Evidence for electrostatic channeling in a fusion protein of malate dehydrogenase and citrate synthase. Biochemistry 35:12652–12658. [DOI] [PubMed] [Google Scholar]
- 16. Elcock AH, Huber GA, McCammon JA (1997) Electrostatic channeling of substrates between enzyme active sites: comparison of simulation and experiment. Biochemistry 36:16049–16058. [DOI] [PubMed] [Google Scholar]
- 17. Holst MJ, Saied F (1995) Numerical solution of the nonlinear Poisson–Boltzmann equation: developing more robust and efficient method. J Comput Chem 16:337–364. [Google Scholar]
- 18. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bas DC, Rogers DM, Jensen JH (2008) Very fast prediction and rationalization of pK(a) values for protein–ligand complexes. Proteins 73:765–783. [DOI] [PubMed] [Google Scholar]
- 20. Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 35:522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huber GA, McCammon JA (2010) Browndye: a software package for Brownian dynamics. Comput Phys Commun 181:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ortega A, Amoros D, de la Torre JG (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic‐ and residue‐level models. Biophys J 101:892–898. [DOI] [PMC free article] [PubMed] [Google Scholar]