

Abstract
Laboratory-evolved RNAs bind a wide variety of targets and serve highly diverse functions, including as diagnostic and therapeutic aptamers. The majority of aptamers have been identified using in vitro selection (SELEX), a molecular evolution technique based on selecting target-binding RNAs from highly diverse pools through serial rounds of enrichment and amplification. In vitro selection typically yields multiple distinct motifs of highly variable abundance and target-binding affinities. The discovery of new aptamers is often limited by the difficulty of characterizing the selected motifs, because testing of individual sequences tends to be a tedious process. To facilitate the discovery of new aptamers within in vitro selected pools, we developed Apta-Seq, a multiplex analysis based on quantitative, ligand-dependent 2′ acylation of solvent-accessible regions of the selected RNA pools, followed by reverse transcription (SHAPE) and deep sequencing. The method reveals, in a single sequencing experiment, the identity, structural features, and target dissociation constants for aptamers present in the selected pool. Application of Apta-Seq to a human genomic pool enriched for ATP-binding RNAs yielded three new aptamers, which together with previously identified human aptamers suggest that ligand-binding RNAs may be common in mammals.
Graphical Abstract
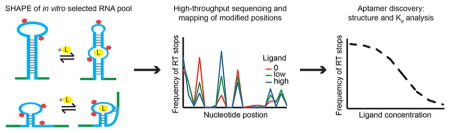
Functional RNAs play central roles in regulating gene expression and catalyzing essential cellular reactions.1 RNAs evolved in the laboratory serve equally diverse functions, including as diagnostic and therapeutic aptamers that bind a wide variety of targets.2 The vast majority of aptamers have been identified using in vitro selection (or SELEX), a molecular evolution technique based on selecting target-binding RNAs from highly diverse pools through serial rounds of enrichment and amplification.3,4 The RNA pools are transcribed from either synthetic (typically random) or genomic DNAs,5 and selections often yield multiple distinct motifs of highly variable abundance, target-binding affinities, and specificities.
The discovery of new aptamers is often hampered by the difficulty of identifying and characterizing the structural motifs that result from the selection process, because testing of individual sequences identified in selected pools tends to be a tedious process. Moreover, low copy number sequences, which may play key functional roles, can go undetected when only the dominant sequences are identified and tested individually. High-throughput sequencing can be applied to measure sequence diversity of selected pools and identify potential aptamers,6,7 but their structural and binding characteristics have to be established individually for each sequence, often making this the limiting step in the discovery of novel aptamers. Indeed, many functional aptamers may go uncharacterized due to the challenge associated with testing the structure and affinities of the majority of sequences within a selected pool. This handicaps the description of selected RNAs and is a key hurdle to overcome for the efficient discovery of functional aptamers that may have diverse and important functions.
One application of in vitro selections seeking to characterize all enriched RNAs, including low copy number sequences, is genomic8,9 and transcriptomic SELEX.10–13 These experiments reveal RNAs encoded by the genomes of predetermined species and are particularly important in the discovery of new instances of known functional RNAs, such as aptamers and ribozymes.14–16 In contrast to selections based on synthetic sequences, which are typically designed to identify a single (or a few) fittest functional RNA, genomic and transcriptomic selections are designed to map out all instances of a given function, such as binding of a target metabolite or protein. However, the relative abundance of in vitro selected RNAs is biased toward not only sequences that fulfill the selection criteria but also those that are highly amplifiable (sequences that transcribe, reverse-transcribe, and PCR amplify more efficiently than others),5,17 thus the resulting distribution of the functional RNAs in the selected pools may be strongly skewed by these two properties.
The ability to more quickly characterize selection pools for the binding function would be enabled if the structures of many aptamers and their physical interactions with their targets could be determined in a single experiment. RNA probing with chemical reagents is a robust method for analyzing the structures of RNAs and characterizing important conformational changes that can occur due to interaction with small molecules and proteins; however, such experiments are usually accomplished on single RNA molecules.18–20 Information about structure and ligand-binding sites in RNAs can be extracted from experiments based on partial hydrolysis (in-line probing) and chemical modification of RNAs, using selective 2′-hydroxyl acylation (SHAPE) or base modification (e.g., by dimethyl sulfate).18–20 The SHAPE method detects 2′-OH accessibility and reactivity to acylation, thereby reporting on which aptamer segments are more flexible and reactive in response to a ligand.21 Changes in structure can be probed under different environmental conditions or in the presence of varying concentrations of ligands, such as small molecules22 and proteins,23 and can be used to extract the dissociation constants for the RNA–ligand interactions.24 Several methods combine SHAPE with high-throughput sequencing (SHAPE-seq) to achieve single-nucleotide resolution of acylation reactivity on a diverse set of sequences simultaneously and couple the output to computational modules developed to yield genomic locations, intrinsic reverse transcriptase (RT) stops, SHAPE reactivities, and secondary structure models for each transcript.25–27 Although useful, most of these efforts have been largely descriptive with few examples of their use for novel biological discovery.
To overcome the limitations of single-sequence characterization of in vitro selected pools, we developed a multiplexed approach to couple RNA selection with structural and binding characterization of individual sequences within the selected pools. We marry selection with chemical probing of RNA structure to reveal the sequence, structural features, and ligand affinities of both dominant and minor species from the same pool. We use this technique, Apta-Seq, to discover and characterize both known and novel adenosine aptamers in the human genome. Our methodology not only increases the rate of novel aptamers discovery for our studied ligand (ATP) but also has the potential to be applied to any ligand-pool pair, thereby greatly enhancing the speed of aptamer discovery and structural characterization.
RESULTS AND DISCUSSION
In order to establish the identity, secondary structure, and binding properties of aptamers in a single experiment, we combined in vitro selection, SHAPE-seq, and StructureFold into a pipeline (Figure 1), which provides all the information required for aptamer discovery. We applied this method to an in vitro selected pool derived from the human genome and enriched for ATP-binding aptamers, as described previously.15 We chose adenosine/ATP as the target of the selection, because it has been used extensively over the past two decades, and as such, it is an excellent model system for the development of new in vitro selection and analysis methods. Adenosine aptamers with a conserved motif consisting of an 11-nucleotide binding loop and an opposing bulging guanosine, flanked by two helices (the Sassanfar–Szostak motif), were initially isolated from synthetic, random pools by in vitro selections targeting ATP,28 nicotinamide adenine dinucleotide,29 S-adenosyl methionine,30 and S-adenosyl homocysteine.31 More recently, a genomic SELEX experiment revealed the same motif in two distinct loci in the human genome:15 the FGD3 aptamer resides in an intron of the FGD3 gene, and the ERV1 aptamer maps antisense to a junction between an ERV1 LTR repeat and its 3′ insertion site. These were the only two aptamers revealed by traditional cloning; however, this approach tests only a small number of sequences and can miss low copy number aptamers, we therefore reanalyzed the pool using high-throughput sequencing and SHAPE to uncover other genomic aptamers.
Figure 1.
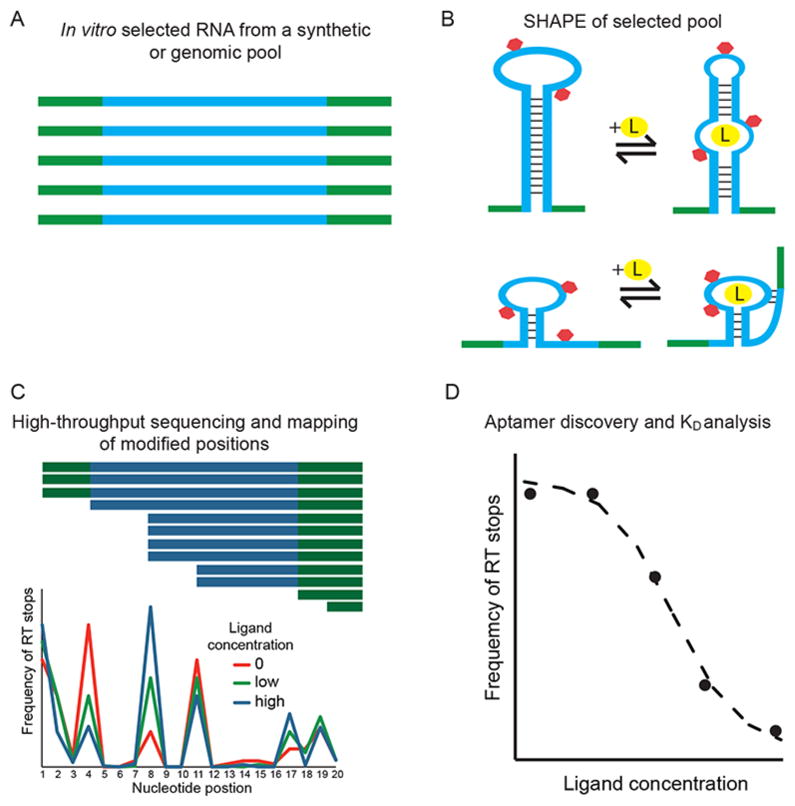
Apta-Seq scheme. The pipeline consists of (A) an in vitro selection, (B) SHAPE analysis of selected pool, and (C) high-throughput sequencing for determination of sequence identity, secondary structure, and (D) binding isotherms of individual aptamers.
To test these potential aptamers, we adapted the SHAPE-Seq analysis coupled to StructureFold to the selected human genomic RNA pool at varying ATP concentrations. The pool was transcribed and purified as during the SELEX experiment, divided into 10 fractions, and incubated with ATP at concentrations ranging from 3 μM to 10 mM, with two fractions set aside for control reactions (no-SHAPE and no-ligand SHAPE controls). The samples were acylated by 2-(azidomethyl)nicotinic acid acyl imidazole (NAI-N3),32 reverse transcribed, and ligated to yield circular single-stranded DNAs, in which the ligation positions correspond to the reverse transcriptase termination points. These occur either due to acylation of the RNA template on the nucleotide immediately upstream of the termination point or at boundaries of RNA structural elements, such as paired segments. The no-SHAPE control reaction reveals the natural RT stops for each sequence in the pool, whereas the SHAPE experiments reveal additional RT stops corresponding to the acylated positions in the RNA, some of which change upon ligand binding (Figure 1B). Amplification of the ssDNA by PCR and barcoding each experimental condition in the ligand titration allow one-pot sequencing of the entire population. Subsequent barcode-based demultiplexing of the sequences reveals all sequences and SHAPE-dependent RT stops for each experimental condition. Nucleotide positions of all detected sequences were analyzed for RT termination and expressed as RT-stop frequency, which is defined as a fraction of total reads (per experiment) for a given sequence (Figure 1C).
The frequency of RT stops was used to obtain SHAPE profiles in the presence of increasing concentration of ATP-Mg (Figure 1C). The RT stops were resolved with single-nucleotide resolution, and the ATP dependence of SHAPE profiles allowed us to determine the apparent KD for each aptamer at several positions. For the FGD3 aptamer,15 two positions (G46 and U15, numbered by position in the pool sequence and shown in Figures 2D and S1A) yielded a KD ~700 μM (Figure 2B, Table 1). The Apta-Seq data show that the adenosine-binding loop of the Sassanfar–Szostak motif becomes more reactive toward the SHAPE reagent at high concentrations of ATP at the third adenosine (A60, Figure 2D) of the binding loop (seen in the ATP-dependent increase of the A60 peak in Figure 2A). Previous in-line probing data for the FGD3 and other ATP-binding aptamers have also demonstrated that the adenosine-binding loop becomes more susceptible to in-line attack at the third adenosine (equivalent to A60).15,18 In the solution structures of the in vitro selected aptamers bound to AMP, the nucleotide equivalent to A60 makes direct contacts with the ligand through stacking,33,34 and the 2′-OH of the A60 equivalent appears partially solvent-exposed and hydrogen-bonded to the adjacent phosphate, which likely activates it for acylation.35 The sugar–phosphate backbone of this nucleotide in the adenosine-bound conformation is thus highly sensitive to modification, and in the case of the FGD3 aptamer, the binding loop must undergo a significant conformational change upon ligand binding, because the same position is weakly acylated in the absence of the ligand (Figure 2A, low-ATP traces, shown in shades of red).
Figure 2.

Apta-Seq analysis of the human FGD3 and ERV1 adenosine aptamers. (A) Graph of acylation positions at the Sassanfar–Szostak motif of the FGD3 aptamer and varying concentrations of ATP (dark red through dark blue, for ATP concentrations ranging from 3 μM to 10 mM). The adenosine-binding loop is underlined in red. (B) Binding isotherms of the aptamer extracted from positions U15 (black circles) and G46 (gray circles). Both positions reveal a KD ~ 700 μM. (C) PAGE analysis of a purified FGD3 aptamer clone showing that RT stops for position C72 strongly increase at high concentrations of ATP, validating the SHAPE-Seq data shown in A. (E) Secondary structure of the FGD3 aptamer with ATP binding loop in red. Boxed nucleotides indicate aptamer positions that show an increase (black outlines) or decrease (gray) in acylation with increasing ATP. (E) Predicted secondary-structure change accompanying ATP binding by the ERV1 aptamer. Positions with ATP-dependent acylation changes are indicated as in D. The boundary of the ERV1 retrotransposon is indicated with a blue arrow.
Table 1.
Relative Abundance and Binding Affinity of Selected Aptamersa
| aptamer | rel. abundance (%) | KD (mM) |
|---|---|---|
| FGD3 | 0.3839 | 0.7 ± 0.1 |
| ERV1 | 0.4893 | 1.8 ± 0.6 |
| PRR5 | 0.1742 | 1.7 ± 0.6 |
| L1PA16 | 0.0848 | 1.3 ± 0.1 |
| THE1B | 0.1191 | 1.1 ± 0.4 |
Relative abundance for each aptamer was calculated out of 1 533 526 sequences. Reported errors are standard errors of the mean of KDs calculated from binding curves with R2 > 0.5.
To validate the results obtained from Apta-Seq of the pool with the reactivity of the purified FGD3 aptamer, we performed a SHAPE analysis on an isolated clone of the aptamer. One of the positions with the most prominent ATP-dependent changes in SHAPE reactivity is C72 (Figures 2A and S1A), which maps to a domain adjacent to the adenosine binding loop (Figure 2D). We confirmed this result by conventional (PAGE-based) SHAPE analysis of the aptamer clone, which also revealed a strong, ATP-dependent increase in reverse transcription termination at position C72 (Figure 2C), reaffirming that the method reveals comparable data for the same aptamer within a highly heterogeneous pool of sequences. These results demonstrate that our high-throughput sequencing approach parallels more traditional structural analysis normally reserved to single clone analyses.
For the ERV1 aptamer, we derived the SHAPE reactivity using the Reactivity Calculation module of StructureFold, with RT stops from the no-ligand SHAPE data set as the negative control for the reactivity profile of the 10 mM ATP experiment (Figure S1B), and carried out structure predictions using the RNAprobing server of the Vienna RNA Package.36 In the absence of ATP, the ERV1 structure is not predicted to form the ATP-binding motif, because the binding loop is sequestered within a stem flanked by two bulges. However, in the presence of ATP, the structure rearranges to form the classical ATP binding loop (Figure 2E). The binding loop showed similar acylation trends to those of the FGD3 aptamer, but shifted by one nucleotide: the third guanosine (G16; Figure 2E) of the ATP binding loop becomes more accessible with increasing concentration of ATP, analogous to the change of A60 in the FGD3 aptamer (Figure 2D). Altogether, the data are consistent with previous findings and structure predictions for the FGD3 and ERV1 aptamers,15 and the KDs we extracted from the Apta-Seq data (Table 1) are within 2-fold of the KDs determined previously by in-line probing of individual aptamers. The FGD3 and ERV1 aptamers are the dominant aptamers in the selected pool (Table 1), with a relative abundance of 0.38% and 0.49%, respectively, to all sequences. The sequences represent only a small fraction of all reads obtained from the Apta-Seq experiment, because the high-throughput sequencing output is dominated by sequences corresponding to short primer extensions of the reverse primers and are thus difficult to map to the genome uniquely.
The Apta-Seq pipeline also yielded novel, less abundant sequences not found in previous analyses. Our previously reported aptamers were discovered in an ATP column binding analysis of individual clones to find potential candidates to undergo structural analysis; Apta-Seq condenses the process to gain a large-scale analysis of the pool in one experiment in solution. Surprisingly, three new sequences also contained the Sassanfar–Szostak motif. The PRR5 aptamer maps to the second intron of the PRR5 gene or the first intron of the PRR5-ARHGAP8 fusion protein (Figure 3A). PRR5 codes for a protein that is part of the mTORC2 complex37 and, like the FGD3, plays important roles in pathways that regulate cell growth, but the significance of the ATP/adenosine-binding aptamers in their introns remains unknown. The PRR5 aptamer bound to ATP-agarose beads and eluted in the presence of free ATP (Figure 3C) and Apta-Seq data revealed a KD of ~1.7 mM (Figure 3B and D, Table 1) at a relative abundance of 0.17% among the mapped sequences. Interestingly, mutations (including a 14-nt insertion) of the aptamer sequence throughout the genomes of primates preserve the aptamer structure, suggesting that it may be a functional RNA in primates (Figure 3E and F).
Figure 3.

A novel adenosine aptamer, mapping to the PRR5 gene in primates. (A) Location of the aptamer (red line) in the PRR5 intron. The aptamer also appears in the first intron of a PRR5–ARHGAP8 fusion. (B) Trace of acylation for the PRR5 aptamer with varying ATP concentrations, ranging from 0 (dark red) to 10 mM (dark blue). The conserved adenosine-binding motif is underlined in red, and positions with ATP-dependent changes in RT stops are indicated. (C) Graph of column binding fractions of the PRR5 sequence amplified out of the selected pool. The graph displays 18% of RNA eluting with free 5 mM ATP. (D) Binding isotherms of the aptamer modeled from the change in RT stops at positions indicated in the legend, yielding an average KD of 1.7 ± 0.6 mM (average deviation). (E) Predicted secondary structure of the PRR5 aptamer with ATP binding loop shown in red. Boxed nucleotides indicate aptamer positions that show an increase (black outlines) or decrease (gray) in acylation with increasing ATP. Black triangle indicates sequence-insertion site for some primates. (F) Aptamer sequence conservation among primates. All mutations within the aptamer motif are conservative with respect to base-pairing interactions of the proposed structure: U49C mutation leads to a G•U wobble pair to be replaced with a canonical G–C pair in helix 5 (h5). Insertion at A54 (black triangle) extends the sequence into the loop of h5 and potentially extends the helical domain by four base-pairs, and the A81G mutation in h4 creates a G•U wobble pair from an A–U canonical base-pair.
The second aptamer (Figures 4A and S2) was found in a long interspersed element (LINE), L1PA16, and the third (Figures 4B and S3) maps antisense to a repeat element THE1B, which is derived from a subfamily of ERV Mammalian apparent LTR-retrotransposons (ERV-MaLR), and may be stabilized by fortuitous base-pairing with a part of the sequence derived from the forward primer of the pool (Figure 4B). The KDs derived from Apta-Seq (Figure 4C and D) were 1.3 mM and 1.1 mM (Table 1); however, the SHAPE profile of the THE1B aptamer afforded only a single peak (G32; Figures 4D and S3A) that yielded a KD model with a good fit (R2 ~ 0.95), whereas the L1PA16 aptamer exhibited several peaks from which the dissociation constants could be derived (Figures 4C and S2A). When transcribed from individual DNA templates and purified using PAGE, both aptamers bound to ATP-agarose beads and eluted in the presence of free ATP (Figures 4C and 3D). Mutations of key residues (Figure 4A and B) in the binding loops of these aptamers abolish binding to ATP columns (Figures S2C and S3C). These aptamers, together with the previously discovered adenosine15 and GTP16 aptamers, indicate that ligand-binding RNAs are likely common in higher eukaryotes.
Figure 4.
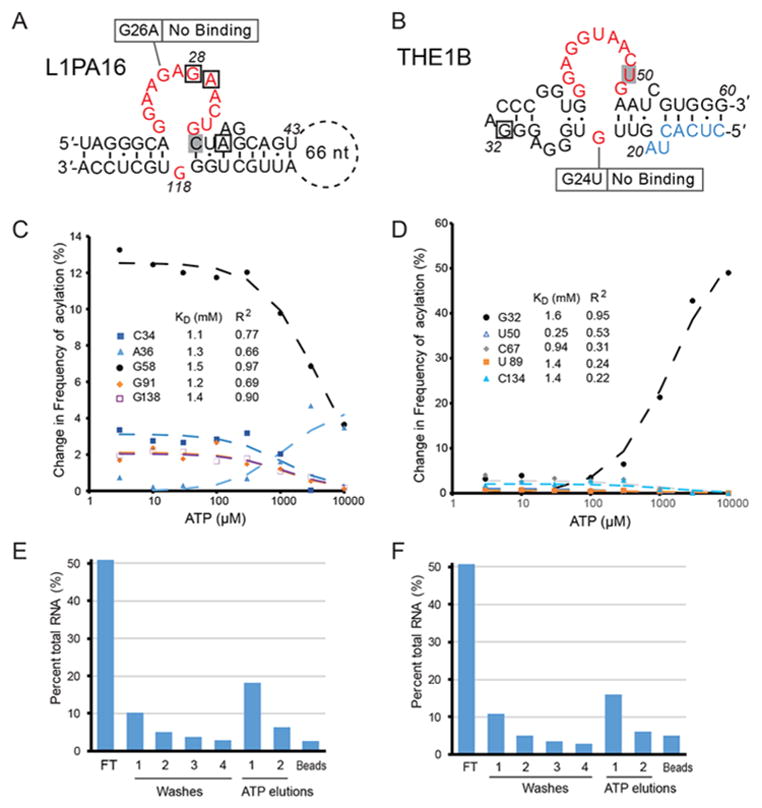
Novel adenosine aptamers revealed by Apta-Seq in human retrotransposons. (A) The Sassanfar–Szosak motif mapping to the L1PA16 LINE1 element, with ATP-dependent acylation positions indicated by squares and annotated as in Figures 2 and 3. Mutation of a key binding site residue (G26A) results in no ATP binding. (B) An aptamer mapping to a THE1B retrotransposon. Part of the 5′ sequence that originated from the forward primer-binding region of the DNA pool is shown in light blue. A mutation of an essential binding site residue (G24U) abolished ATP binding. Binding isotherms of the L1PA16 (C) and THE1B (D) aptamers modeled from the change in acylation at positions indicated in the legend, yielding a KD of 1.3 ± 0.1 mM and 1.1 ± 0.4 mM, respectively. Graph of ATP column binding of the L1PA16 (E) and THE1B (F) sequences amplified out of the selected pool, showing robust activity of the purified sequences and validating the Apta-Seq data. Full SHAPE-Seq profiles and secondary structure models of these aptamers are presented in Supporting Information Figures S2 and S3.
To analyze these Sassanfar–Szostak motifs for potential binding in the context of the human transcriptome, we isolated total RNA from four cell culture experiments (SHSY-5Y cells incubated with DMSO or DMSO + 10 mM adenosine and OV90 and MCF7 cells) and treated it in the same manner as during the SELEX experiment. The total RNA samples were annealed in the binding buffer, introduced to the ATP beads, washed with five column volumes of the binding buffer, and eluted with 5 mM ATP-Mg. RNAs isolated from the last wash fraction and ATP elution were reverse-transcribed using primers specific for the five aptamers described above and analyzed using nested primers by qPCR. Two of the aptamers, FGD3 and THE1B, were detected at higher levels in the elution fractions than in the last washes in the SHSY-5Y cells. FGD3 aptamer exhibited more robust binding to ATP beads and the levels of the aptamer appear insensitive to the presence of adenosine in the tissue culture medium (Figure S1B). In contrast, the THE1B aptamer showed significantly higher levels in the ATP elution fraction only in RNA extracted from SHSY-5Y cells incubated with 10 mM adenosine (Figure S3D and E). These experiments suggest that at least two of the aptamers described herein have the capacity to bind ATP within the context of their endogenous transcripts and the human transcriptome, and that their expression or activity can be modulated by exogenous adenosine.
In summary, we describe an efficient process of multiplex analysis of in vitro selected RNA pools. An in vitro selection experiment is combined with SHAPE-Seq and StructureFold analysis to efficiently and quantitatively analyze the selected pools at a single-nucleotide resolution. A number of high-throughput methods, many of which can be adapted for application in aptamer discovery, have recently been developed to analyze RNA–protein interactions;38–40 however, all of these techniques require immobilization of the nucleic acid on the surface of a sequencing chip and a labeled target molecule. Another recent application of high-throughput analysis of in vitro selected pools (of mRNA-displayed peptides) provides binding parameters for the target interactions, but the method requires the target molecule to be immobilized on solid support and relatively slow binding and dissociation kinetics.41 None of these techniques reveal information about conformational changes associated with target binding but can provide secondary structure constraints thorough covariation analysis of the active RNAs. Moreover, these techniques derive equilibrium binding constants from kinetics and presume an absence of rate-limiting conformational changes associated with target binding, whereas Apta-Seq detects target binding under equilibrium conditions. Apta-Seq makes it significantly easier to discover, sort, and characterize aptamers by measuring structural changes in RNAs in solution, providing a straightforward way to measure affinity for the target molecules. Importantly, Apta-Seq is a powerful enabling technological pipeline that is sure to expedite the transition from aptamer selection to the unraveling of their structure and binding affinity and thus their biological or biotechnological relevance. Furthermore, because Apta-Seq is performed in solution, the method can be used with a large number of targets, providing a label-free approach to studying specificity of the selected sequences. In contrast, high-throughput methods based on immobilization of the RNAs or their binding partners either require modification (immobilization or fluorescent labeling) of each target to measure the binding kinetics or only measure the rates of association, but not dissociation, when unlabeled off-target molecules are introduced in competition with labeled targets. Our results show that ATP binding by the Sassanfar–Szostak motif is in some cases coupled to significant remodeling of the RNA structure in adjacent domains. The method thus reveals not just ligand binding but also concomitant large-scale conformational changes, facilitating multiplexed experimental discovery of potential riboswitches.
MATERIALS AND METHODS
Transcription
RNAs were transcribed for 2 h at 37 °C in 400 μL of a solution containing 40 mM tris chloride; 10% dimethyl sulfoxide (DMSO); 10 mM dithiothreitol (DTT); 2 mM spermidine; 5 mM each rCTP, rGTP, rUTP, and rATP; 20 mM MgCl2; one unit of T7 RNA polymerase; and ~0.5 μM DNA template. Transcripts were purified by 7% polyacrylamide gel electrophoresis (PAGE) under denaturing conditions (7 M urea). RNA was eluted from the gel into 400 μL of 400 mM KCl and precipitated by adding 800 μL of 100% ethanol at −20 °C.
Primer Phosphorylation
Primer labeling was prepared in a total volume of 20 μL. Then, 20 μM of primer, 1× T4 Polynucletoide Kinase (PNK) ligase buffer (NEB), 1 unit of T4 PNK (NEB), and 0.5 μCi [γ-32P]ATP was incubated at 37 °C for 1 h then purified from denaturing PAGE.
Synthesis of the SHAPE Reagent
The SHAPE reagent, 2-(azidomethyl)nicotinic acid acyl imidazole, was synthesized following a previously described protocol.32
Selective 2′-Hydroxyl Acylation and Primer Extension (SHAPE)
SHAPE reactions were prepared in a total volume of 10 μL. RNAs (pools or individual aptamer sequences) were resuspended in water and heated to 70 °C for 3 min. Then, 1 μM of purified RNA was added to a buffer containing 140 mM KCl, 10 mM NaCl, 10 mM tris chloride at pH 7.5, and 5 mM MgCl2. A dilution series of 5′-adenosine triphosphate (ATP) was prepared using a 1:1 stock of ATP:Mg2+. Then, 1 μM to 10 mM ATP was aliquoted to the RNA in a buffer and incubated at RT (~23 °C) for 1 min. Then, 50 mM 2-(azidomethyl)nicotinic acid acyl imidazole was added to the mixture, and the reaction was incubated for 45 min at RT. Reactions containing no SHAPE reagent were substituted with 10% DMSO. Reactions were precipitated with 10 μL 3 M KCl, 1 μL glycoblue, 89 μL H2O, and 300 μL 98% ethanol.
Primer Extension
The RNA pellet was reconstituted in a 10 μL reaction volume containing 0.1 μM 5′-[32P]-radiolabeled reverse transcription DNA primer, 2 μL of 5 × M-MuLV Reverse Transcriptase Reaction Buffer (New England Biolabs), 1 unit of M-MuLV enzyme, and 500 μM each of deoxyribonucleotide triphosphate. Extensions were performed at 42 °C for 15 min. 400 mM NaOH was added, and the reaction was incubated at 95 °C for 5 min to hydrolyze the RNA. Reactions were precipitated with 10 μL of 3 M KCl, 1 μL of glycoblue, 89 μL of H2O, and 300 μL of 98% ethanol. Complementary DNA (cDNA) was resolved using 12% denaturing PAGE or amplified for high-throughput analysis.
KD Analysis
RT stops were normalized by dividing peak intensities in SHAPE profiles by the total number of reads per aptamer and experimental conditions. These normalized profiles (presented as overlays in Figures 2 and 3 as well as S1–S3) were analyzed by plotting peak intensities for each position in the aptamers as a function of ATP concentration (such as in Figures 2B, 3D, and 4C and D) and modeled with a dissociation constant equation for the ligand
where “Range” corresponds to the range of values above baseline that a peak in the SHAPE profile can assume. The model was fit to the data using a linear least-squares analysis and the Solver module of Microsoft Excel to extract ATP KD for each peak. Because some of the apparent binding constants are near the maximum of the titration and in many cases the rising SHAPE signal did not level off at the highest ATP concentration, we predominantly used the positions where the SHAPE signal decreased with ATP concentration and approached zero for KD modeling.
SHAPE-Seq Library and Primer Design
Round 6 of an in vitro selection for an ATP aptamer from a human genomic library15 was used as the library for Apta-Seq. Libraries were given individual barcodes based on concentration of the ligand used during SHAPE. Ten libraries in total were made using a ligand titration of 1–10 000 μM, no-ligand control, and no-SHAPE (only DMSO, as described above) control.
Apta-Seq primers contain a reverse primer sequence for the pool of interest and flanking Illumina primers in order to barcode and sequence primer extensions by Illumina Sequencing. Apta-Seq primers for reverse transcription primer extension were designed 5′ to 3′ with the following components:
Illumina forward primer reverse-complement, NotI digestion site, Illumina reverse primer, reverse primer for RNA of interest
5′-AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTGCGGCCGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCCTGAGCTTGACGCA-3′
Primers for amplification were designed 5′ to 3′ with the following components: Forward primer containing Illumina forward adapter and primer.
5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3′
Reverse primer containing Illumina reverse adapter, barcode, and Illumina reverse primer.
5′-CAAGCAGAAGACGGCATACGAGAT [barcode] GTGACTGGAGTTCAGACGTGTGCTCTTCCG-3′
Primer Extension for Apta-Seq
Primer extension was carried out with the Apta-Seq primer as described above. cDNA was self-ligated in a 20 μL reaction using CircLigaseII Reaction Buffer (Epicenter), 2.5 mM MnCl2, 50 μM ATP, and five units of CircLigase ssDNA Ligase. Reactions were precipitated with 10 μL of 3 M KCl, 1 μL of glycoblue, 89 μL of H2O, and 300 μL of 98% ethanol, and cDNA was reconstituted in 20 μL of H2O. A polymerase chain reaction (PCR) was performed using 1 μM each of forward and reverse primers, cDNA template, and DreamTaq Master Mix (Thermo Fisher) and amplified for 16 cycles (denaturing 94 °C, 30 s, annealing 55 °C, 30 s, and elongation 72 °C, 30 s). Amplicons were sequenced on Illumina HiSeq 2500 at the UCI Genomics Facility.
SHAPE-Seq Reactivity Mapping
Galaxy (https://usegalaxy.org/) and the StructureFold module27 were used to map and determine the SHAPE reactivity of aptamers. Forward reads of libraries were used to analyze RT stops. Adapters and primers (CAATGCGTCAAG) were clipped using Clip adapter sequences on Galaxy. Default settings were used except for a minimum sequence length of 10 and an output of both clipped and nonclipped sequences. Clipped libraries were then processed using StructureFold, a series of Web-based programs to characterize RNA that have undergone SHAPE modifications. Aptamers were mapped to selected libraries using Iterative Mapping on Galaxy. Default settings for mapping were used except for a minimum read length of 12 nucleotides and three mismatches allowed (-v 3). RT stop counts were calculated using Get RT Stop Counts on Galaxy. RT stop counts were derived using mapped files from Iterative Mapping and aptamers as the reference sequences. Output from the Get RT Stop Counts module was tabulated and normalized to the total stop counts for each aptamer. The percentage of counts for each position was derived by dividing each position by the total number of RT counts for each aptamer.
The new aptamers were mapped to the following sequences obtained from Illumina sequencing:
>L1PA16
ctcactacgtGGCTTAGGGCAGGAAGAGAACTGCTAAGGC-AGTTTCTCCTAGAAGATGAGACCTGCAGCCAGGTCCAGCT-TGGTGACCTAGAACTGGTCTGCATGTGTCATTGCTGGGTG-CTCCACCCTGCTCCCCTGAGATCATGTTGag
>THE1B
ggggcagacgtgcctcactaGTTGTGGGAGGGACCCGGTG-GGAGGTAACTGAATCgTGGGATGAGTCTTTCCCGTACTGTc-GTCATGATAGTGAATAAGTCTCATGAGgTCTGATGGtTT-TTATATGGGGGAGcTTTCCCTTCgCAAACTCTag
>PRR5
gggtGGAGGAGCCTGGATGCTGCCTGCAGG-ACCTCAGGCTGTGCCTGCTGGGCAAAGgCCCTGGGCAG-GGAAGGAACTGCAGCCTCCACAGAGGGTGGA-TaTGGTGGAGAGGTGGGAGGCCAGCTCCTGTCATCCGAGG-TCCAGGCAAGCCAG
Lowercase letters are mutations acquired during the in vitro selection or library construction, as compared with the reference genome. The Sassanfar–Szostak adenosine-binding loop is highlighted in bold.
Structure Prediction
The Reactivity Calculation module on Galaxy was used to obtain SHAPE reactivities by using the output from the Get RT Stop Counts module. The RT stop counts for 10 mM ligand concentration was used as the (+) library and no ligand for the (−) library, both in the presence of the SHAPE reagent. Default settings were used except for nucleotide specificity, which was changed to AUCG. To predict structure, the RNAprobing Web server was used. All default settings were used according to the Washietel et al. SHAPE method.36
Total RNA Extraction
Human cell lines OV90 and MCF7, ovary-and breast-derived adenocarcinoma, respectively, were thawed from cryo-preservation and seeded onto separate T-75 culture plates with DMEM media containing 10% Fetal Bovine Serum (FBS), 10% amphotericin B, and 10% penicillin/streptomycin. Similarly, SHSY-5Y neuroblastoma cells were thawed and seeded in two separate T-75 flasks with DMEM/F12 culture media containing 10% FBS, 10% amphotericin B, and 10% penicillin/streptomycin. All cell cultures were passaged appropriately to achieve 80% confluency on the day of the experiment. In one SHSY-5Y experiment, 2% DMSO with 10 mM adenosine was added to adhered confluent SHSY-5Y cells 1 day prior to total RNA extraction.
Total RNA was harvested from all cell types using TRIzol Reagent (Ambion). All total RNA isolation steps were performed according to the user manual. Briefly, adhered cells were collected by washing each culture dish with 750 μL of TRIzol Reagent and collected into a fresh 1.5 mL microcentrofuge tube. A total of 200 μL of chloroform was added to each sample to allow for phase separation. Extraction of the aqueous later was followed by RNA precipitation using 100% isopropanol. The total RNA was pelleted by centrifugation and washed with 75% ethanol prior to resuspension in RNase-free H2O. Once resuspended, total RNAs were treated with DNase I to remove genomic DNA. The TRIzol extraction procedure was then repeated for all DNase I-treated total RNA samples to remove protein and residual DNA contaminants before column binding and RT-qPCR.
Column Binding Assay Using Total RNA Extracts
Prior to RNA binding, 10 μL of C8-linked ATP-agarose (1 mg mL−1; Sigma-Aldrich) was pre-equilibrated on a spin-filter with binding buffer (BB) containing 140 mM KCl, 10 mM NaCl, 5 mM MgCl2, and 20 mM tris-HCl, at pH 8.0. For each tissue culture source (OV90, MCF7, SHSY-5Y, and SHSY-5Y incubated with 10 mM adenosine), 500 ng of total RNA was annealed in the presence of BB at 72 °C for 1 min and allowed to cool to RT over 5 min before addition onto ATP-agarose beads. A total of 11 fractions were collected starting with the flow-through, which was collected after a 30 min incubation. After flow-through collection, a total of four washes were collected over a span of 10 min using 10 μL of BB. After washing, four 30 min elution fractions were collected using an elution buffer (EB) containing 5 mM ATP, 140 mM KCl, 10 mM NaCl, 10 mM MgCl2, and 20 mM tris-HCl, at pH 8.0. To remove any remaining RNAs attached to the column, one 10 μL denaturing wash was collected using 7 M urea following the elution fractions. Each fraction was precipitated using 300 mM KCl and 100% ethanol, pelleted by centrifugation, washed with 75% ethanol, and resuspended in ddH2O for reverse transcription.
RT-qPCR
Pellets from the last (fourth) wash and the elution fractions were resuspended in a 1× reverse transcription master mix containing 500 nM of aptamer-specific RT primers, 5 mM dNTPs, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, and 50 mM tris-HCl at pH 8.3. Resuspended RNAs were heat-treated at 90 °C for 30 s, followed by a 50 min incubation at 50 °C after the addition of superscript III reverse transcriptase. cDNA products were diluted 10-fold with nuclease-free H2O, and quantitative PCR was performed on a BioRad CFX Connect system using 1 μL of cDNA product, 500 nM of gene-specific primers, and BioRad iTaq supermix. The reverse primers were 3′-extended versions of the RT primers to ensure gene-specific amplification.
Supplementary Material
Acknowledgments
The authors would like to thank A. R. Chamberlin for use of his laboratory to synthesize the acylation reagents and D. Chan for help with the SHAPE protocol. This project/publication was made possible through the support of a grant from the John Templeton Foundation. The opinions expressed in this publication are those of the author(s) and do not necessarily reflect the views of the John Templeton Foundation. The authors also gratefully acknowledge the financial support from the Pew Charitable Trusts and the NSF (MCB 1330606). R.C.S. is supported by startup funds through UC Irvine, the National Institutes of Health Director’s New Innovator Award (1DP2GM119164), and 1R01MH109588. This work was made possible, in part, through access to the Genomic High Throughput Facility Shared Resource of the Cancer Center Support Grant (CA-62203) at the University of California, Irvine and National Institutes of Health shared instrumentation grants 1S10RR025496-01, 1S10OD010794-01, and 1S10OD021718-01.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.7b00001.
Supporting Figures S1–S3 (PDF)
References
- 1.Sharp PA. The centrality of RNA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Jijakli K, Khraiwesh B, Fu W, Luo L, Alzahmi A, Koussa J, Chaiboonchoe A, Kirmizialtin S, Yen L, Salehi-Ashtiani K. The in vitro selection world. Methods. 2016;106:3–13. doi: 10.1016/j.ymeth.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 4.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 5.Pobanz K, Luptak A. Improving the odds: Influence of starting pools on in vitro selection outcomes. Methods. 2016;106:14–20. doi: 10.1016/j.ymeth.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alam KK, Chang JL, Burke DH. FASTAptamer: A Bioinformatic Toolkit for High-throughput Sequence Analysis of Combinatorial Selections. Mol Ther–Nucleic Acids. 2015;4:e230. doi: 10.1038/mtna.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoinka J, Przytycka T. AptaPLEX - A dedicated, multithreaded demultiplexer for HT-SELEX data. Methods. 2016;106:82–85. doi: 10.1016/j.ymeth.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Singer BS, Shtatland T, Brown D, Gold L. Libraries for genomic SELEX. Nucleic Acids Res. 1997;25:781–786. doi: 10.1093/nar/25.4.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorenz C, von Pelchrzim F, Schroeder R. Genomic systematic evolution of ligands by exponential enrichment (Genomic SELEX) for the identification of protein-binding RNAs independent of their expression levels. Nat Protoc. 2006;1:2204–2212. doi: 10.1038/nprot.2006.372. [DOI] [PubMed] [Google Scholar]
- 10.Fujimoto Y, Nakamura Y, Ohuchi S. HEXIM1-binding elements on mRNAs identified through transcriptomic SELEX and computational screening. Biochimie. 2012;94:1900–1909. doi: 10.1016/j.biochi.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Yun SW, Seto J, Liu W, Toth M. The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience. 2003;120:1005–1017. doi: 10.1016/s0306-4522(03)00406-8. [DOI] [PubMed] [Google Scholar]
- 12.Dobbelstein M, Shenk T. In vitro selection of RNA ligands for the ribosomal L22 protein associated with Epstein-Barr virus-expressed RNA by using randomized and cDNA-derived RNA libraries. J Virol. 1995;69:8027–8034. doi: 10.1128/jvi.69.12.8027-8034.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terasaka N, Futai K, Katoh T, Suga H. A human microRNA precursor binding to folic acid discovered by small RNA transcriptomic SELEX. RNA. 2016;22:1918–1928. doi: 10.1261/rna.057737.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salehi-Ashtiani K, Lupták A, Litovchick A, Szostak JW. A genomewide search for ribozymes reveals an HDV-like sequence in the human CPEB3 gene. Science. 2006;313:1788–1792. doi: 10.1126/science.1129308. [DOI] [PubMed] [Google Scholar]
- 15.Vu MM, Jameson NE, Masuda SJ, Lin D, Larralde-Ridaura R, Luptak A. Convergent Evolution of Adenosine Aptamers Spanning Bacterial, Human, and Random Sequences Revealed by Structure-Based Bioinformatics and Genomic SELEX. Chem Biol. 2012;19:1247–1254. doi: 10.1016/j.chembiol.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curtis EA, Liu DR. Discovery of Widespread GTP-Binding Motifs in Genomic DNA and RNA. Chem Biol. 2013;20:521–532. doi: 10.1016/j.chembiol.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takahashi M, Wu X, Ho M, Chomchan P, Rossi JJ, Burnett JC, Zhou J. High throughput sequencing analysis of RNA libraries reveals the influences of initial library and PCR methods on SELEX efficiency. Sci Rep. 2016;6:33697. doi: 10.1038/srep33697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soukup GA, Breaker RR. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999;5:1308–1325. doi: 10.1017/s1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merino EJ, Wilkinson KA, Coughlan JL, Weeks KM. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE) J Am Chem Soc. 2005;127:4223–4231. doi: 10.1021/ja043822v. [DOI] [PubMed] [Google Scholar]
- 20.Ding Y, Kwok CK, Tang Y, Bevilacqua PC, Assmann SM. Genome-wide profiling of in vivo RNA structure at single-nucleotide resolution using structure-seq. Nat Protoc. 2015;10:1050–1066. doi: 10.1038/nprot.2015.064. [DOI] [PubMed] [Google Scholar]
- 21.Wang B, Wilkinson KA, Weeks KM. Complex ligand-induced conformational changes in tRNA(Asp) revealed by single-nucleotide resolution SHAPE chemistry. Biochemistry. 2008;47:3454–3461. doi: 10.1021/bi702372x. [DOI] [PubMed] [Google Scholar]
- 22.Stoddard CD, Montange RK, Hennelly SP, Rambo RP, Sanbonmatsu KY, Batey RT. Free state conformational sampling of the SAM-I riboswitch aptamer domain. Structure. 2010;18:787–797. doi: 10.1016/j.str.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu Y, Deiorio-Haggar K, Soo MW, Meyer MM. Bacterial RNA motif in the 5′ UTR of rpsF interacts with an S6:S18 complex. RNA. 2014;20:168–176. doi: 10.1261/rna.041285.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wakeman CA, Winkler WC. Analysis of the RNA backbone: structural analysis of riboswitches by in-line probing and selective 2′-hydroxyl acylation and primer extension. Methods Mol Biol. 2009;540:173–191. doi: 10.1007/978-1-59745-558-9_13. [DOI] [PubMed] [Google Scholar]
- 25.Lucks JB, Mortimer SA, Trapnell C, Luo S, Aviran S, Schroth GP, Pachter L, Doudna JA, Arkin AP. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc Natl Acad Sci U S A. 2011;108:11063–11068. doi: 10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loughrey D, Watters KE, Settle AH, Lucks JB. SHAPE-Seq 2.0: systematic optimization and extension of high-throughput chemical probing of RNA secondary structure with next generation sequencing. Nucleic Acids Res. 2014;42:e165. doi: 10.1093/nar/gku909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang Y, Bouvier E, Kwok CK, Ding Y, Nekrutenko A, Bevilacqua PC, Assmann SM. StructureFold: genome-wide RNA secondary structure mapping and reconstruction in vivo. Bioinformatics. 2015;31:2668–2675. doi: 10.1093/bioinformatics/btv213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sassanfar M, Szostak JW. An RNA motif that binds ATP. Nature. 1993;364:550–553. doi: 10.1038/364550a0. [DOI] [PubMed] [Google Scholar]
- 29.Burgstaller P, Famulok M. Isolation of RNA Aptamers for Biological Cofactors by in-Vitro Selection. Angew Chem, Int Ed Engl. 1994;33:1084–1087. [Google Scholar]
- 30.Burke DH, Gold L. RNA aptamers to the adenosine moiety of S-adenosyl methionine: structural inferences from variations on a theme and the reproducibility of SELEX. Nucleic Acids Res. 1997;25:2020–2024. doi: 10.1093/nar/25.10.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gebhardt K, Shokraei A, Babaie E, Lindqvist BH. RNA aptamers to S-adenosylhomocysteine: kinetic properties, divalent cation dependency, and comparison with anti-S-adenosylho-mocysteine antibody. Biochemistry. 2000;39:7255–7265. doi: 10.1021/bi000295t. [DOI] [PubMed] [Google Scholar]
- 32.Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, Chang HY. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–490. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dieckmann T, Suzuki E, Nakamura GK, Feigon J. Solution structure of an ATP-binding RNA aptamer reveals a novel fold. RNA. 1996;2:628–640. [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang F, Kumar RA, Jones RA, Patel DJ. Structural basis of RNA folding and recognition in an AMP-RNA aptamer complex. Nature. 1996;382:183–186. doi: 10.1038/382183a0. [DOI] [PubMed] [Google Scholar]
- 35.McGinnis JL, Dunkle JA, Cate JH, Weeks KM. The mechanisms of RNA SHAPE chemistry. J Am Chem Soc. 2012;134:6617–6624. doi: 10.1021/ja2104075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Washietl S, Hofacker IL, Stadler PF, Kellis M. RNA folding with soft constraints: reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res. 2012;40:4261–4272. doi: 10.1093/nar/gks009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo SY, Kim DH, Jun CB, Kim YM, Haar EV, Lee SI, Hegg JW, Bandhakavi S, Griffin TJ, Kim DH. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J Biol Chem. 2007;282:25604–25612. doi: 10.1074/jbc.M704343200. [DOI] [PubMed] [Google Scholar]
- 38.Buenrostro JD, Araya CL, Chircus LM, Layton CJ, Chang HY, Snyder MP, Greenleaf WJ. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat Biotechnol. 2014;32:562–568. doi: 10.1038/nbt.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tome JM, Ozer A, Pagano JM, Gheba D, Schroth GP, Lis JT. Comprehensive analysis of RNA-protein interactions by high-throughput sequencing-RNA affinity profiling. Nat Methods. 2014;11:683–688. doi: 10.1038/nmeth.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell. 2014;54:887–900. doi: 10.1016/j.molcel.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jalali-Yazdi F, Huong Lai L, Takahashi TT, Roberts RW. High-Throughput Measurement of Binding Kinetics by mRNA Display and Next-Generation Sequencing. Angew Chem, Int Ed. 2016;55:4007–4010. doi: 10.1002/anie.201600077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.