

Abstract
Background
Mesenchymal stromal/stem cells (MSCs) are broadly used for many diseases, but the efficacy of MSC engraftment is very low due to low viability and high cell death rate under a stressful microenvironment. The present study aimed to investigate whether microRNA-34a (miR-34a), which is a downstream target of P53, is involved in H2O2-induced MSC cell death.
Material/Method
Human bone marrow MSCs (hMSCs) were purchased from Lonza and were cultured as previously described. hMSCs were transfected with miR-34a inhibitor and exposed to H2O2. Cell proliferation assay was used to assess the survival rate of hMSCs. Real-time PCR and Western blot analysis were used to examine proliferation and survival ability of hMSCs.
Results
H2O2 exposure significantly increased miR-34a expression in human bone marrow MSCs. H2O2 challenge induced massive MSC cell death along with reduction of expression of proliferation marker Ki67 and survival-related genes Bcl-2 and Survivin. Transfection of miR-34a inhibitor anti-34a led to a significant protective effect and rescued MSC cell death triggered by H2O2 exposure by 50%. Moreover, anti-34a dramatically increased Bcl-2 and Ki67 mRNA expression levels by over 10-fold compared to the mock control group under H2O2 exposure. The protein levels of Bcl-2 and Survivin were also rescued by anti-34a treatment by 50%.
Conclusions
Our results suggest that miR-34a plays a key role in oxidative stress-induced MSC cell death, and targeting miR-34a might be a promising strategy to enhance the survival rate of engrafted stem cells, which may improve therapeutic outcome.
MeSH Keywords: Cell Survival, Mesenchymal Stromal Cells, MicroRNAs, Oxidative Stress
Background
Mesenchymal stromal/stem cells (MSCs) are a group of multipotent stromal cells with capacity of self-renewal and multilineage potential that can differentiate into osteoblasts, chondrocyte, myocytes, and adipocytes [1–3]. MSCs has been broadly used and proven safe and effective in clinical cell therapy trials for many diseases, such as stroke, neurodegenerative diseases, myocardial infarction, diabetes, and kidney injury [4–7]. There are also reports of adverse effect of MSC in clinical studies [8] and in clinical trials [9], suggesting MSCs must be used with extreme caution in clinical experiments and in clinical trials [8,10]. However, it is well accepted that the therapeutic outcomes of MSCs are attributed to direct cell differentiation and paracrine effects that promote endogenous tissue regeneration, anti-inflammation, and anti-apoptosis [11]. Although MSCs therapies have shown promising effectiveness, the efficacy of MSC transplantation is far from satisfactory due to low viability and high cell death rate of transplanted MSCs [12–14].
A highly stressful microenvironment is the main factor that induces apoptosis in engrafted MSCs [15,16]. In ischemia tissue (e.g., cardiac muscle or brain infarction), increased reactive oxygen species (ROS) induces massive cell death of donor MSCs and thus shortens its effective time to only a few days [6]. Prevention of MSC apoptosis from ROS challenge is urgently needed for improving the therapeutic outcome of MSCs therapy.
MicroRNAs (miRNAs) are small non-coding RNAs (approximately 22 nucleotides in length) which play vital roles in post-transcriptional regulation of gene expression. miRNAs can base-pair with complementary sequences with target mRNAs and silence the protein translation process by cleavage or destabilization of target mRNA and stop protein translation. Among the various miRNAs, miR-34a is a direct transcriptional target of p53 [17,18], which is considered as the gatekeeper protein that determinates if a cell should undergo cell-cycle arrest, repair, or apoptosis under ROS and oxidative stress [19,20]. The p53 pathway is involved in MSC apoptosis, and platelet-derived growth factor treatment decreased the expression of p53 and protected MSCs from apoptosis under hypoxia condition [21,22]. p53 activation induces expression of miR-34a both in vivo and in vitro. In unrelated cell lines, such as cancer cell lines, inactivation of miR-34a strongly inhibits p53-mediated apoptosis in a cell stress condition, while overexpression of miR-34a increases apoptosis and cell-cycle arrest [23]. In our previous work, we demonstrated that miR-34a is involved in stem cell fate determination and regeneration in the nervous system [5]. We also found that miR-34a expression was negatively correlated with cell survival rate in rat bone marrow-derived dedifferentiated MSCs [5]. Emerging evidence indicates that miRNAs are actively involved in MSC differentiation [5,24], but there has been scant study of the role of miR-34a in the apoptosis of MSCs under stressful conditions.
In the present study, we sought to determine if miR-34a is involved in apoptosis of human bone marrow MSCs under oxidative stress induced by H2O2 and to provide a new target to improve MSC survival in cell therapies in multiple diseases.
Material and Methods
Cell culture
Human bone marrow MSCs were purchased from Lonza (Walkersville, MD, USA). These cells have been tested for MSC surface marker expression and multilineage differentiation ability. Briefly, cells were cultured in alpha-MEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen) at 37°C in a humidified 5% CO2 incubator, and media were changed every 2–3 days. After reaching to 70–80% confluence, cells were washed with phosphate-buffered saline (PBS), detached with 0.25% trypsin-EDTA (Invitrogen), then centrifuged and expanded at 1: 3 split ratios.
Transfection of miR-34a inhibitor (anti-34a)
MSCs were transfected with anti-34a as previously described [5,4]. Briefly, MSCs were seeded at a density of 2×105 cells per well in 6-well plates in culture medium without antibiotics. Cells were transfected with 200 nM anti-34a and negative control (Ambion, USA) using Lipofectamine 2000 (Invitrogen) at 70% confluence following the manufacturer’s instructions. Transfection efficiency was determined by quantitative real-time PCR at day 1 and day 2 after transfection.
Detection of miR-34a expression
Total RNA was extracted using the mirVana miRNA isolation kit (Applied Biosystems/Ambion, Austin, TX) according to the manufacturer’s instructions. We converted 40 ng total RNA to cDNA using a TaqMan microRNA Reverse Transcription kit (Applied Biosystems) with specific primers for miR-34a and RNU48. Real-time PCR was carried out on the ABI 7500 real-time PCR machine (Applied Biosystems) in reaction volumes of 20 μl containing 10 μl TaqMan 2× Universal PCR Master Mix, no AmpErase UNG, 1 μl 20× TaqMan microRNA Assay Mix (Applied Biosystems) and 1.33 μl template cDNA. Thermal cycling conditions were 40 cycles at 95°C for 15 s and 60°C for 60 s. RNU48 was used as an endogenous control. Relative expression of miR-34a was calculated using the comparative delta-delta Ct value method.
Cell proliferation assay
Cell proliferation was determined using Cell Titer96® Aqueous cell proliferation assay (Promega, Madison, WI). Briefly, MSCs were seeded in triplicates in 96-well plates at a density of 1×103 cells/well in 100 μl full culture medium and transfected with anti-34a, as described above. One day post-transfection, cells were treated with 100 μM H2O2 for 1 h. Cells were incubated with 20 μl Cell Titer96 Aqueous One solution 24 h later, according to the manufacturer’s protocol. Viable cells were spectrophotometrically measured at a wavelength of 490 nm.
SYBR Green-based real-time PCR for other target genes
SYBR Green-based real-time PCR was employed to measure relative mRNA expression levels of Bcl-2, Survivin, and Ki-67. Total mRNA was extracted using Trizol (Invitrogen) according to the manufacturer’s instructions and then quantified using a Nano-drop ND-2000 spectrophotometer. We reverse-transcribed 500 ng mRNA using the High-Capacity cDNA synthesis kit (Applied Biosystems). All qPCR was performed in triplicate using the SYBR Green PCR Master Mix kit (Life Technologies) on an ABI 7500 real-time PCR machine (Applied Biosystems) starting at a hold stage of 95°C for 10 min, and 40 cycles of denaturation at 95°C for 15 s, annealing/extension at 60°C for 60 s followed by disassociation melt curve analysis. Negative controls (no template) were also conducted in triplicate for all target genes. Beta-actin expression was used as an internal control. Relative mRNA levels were expressed as fold-change compared to untreated cells.
Western blot analysis
Cells were lysed in RIPA buffer (150 mM NaCl; 50 mM Tris-Cl, pH 8.0; 1% NP-40; 0.5%; DOC; 0.1% SDS), and total protein extracts were collected. Protein concentration was determined using bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL) and 30 μg of total protein was loaded into 10% SDS-PAGE gel. Following electrophoresis, proteins were transferred onto the nitrocellulose membranes (Millipore, USA). The membranes were blocked for non-specific binding with 5% nonfat dry milk in Tris-buffered saline containing 0.05% Tween 20 (TBST) for 1 h at room temperature and then incubated with specific primary antibodies at 4°C overnight. After washing 3 times with TBST, membranes were incubated with HRP-conjugated secondary antibodies (Cell Signaling Technology, 1: 10 000) for 1 h at room temperature. Later, the signals were detected by chemiluminescence using ECL kit (Pierce, USA). Primary antibodies used in this study were: anti-Bcl-2 (Santa Cruz, 1: 200), anti-Survivin (Cell Signaling Technology, 1: 500), and anti-beta-Actin (Sigma, 1: 2000).
Immunostaining
Cells were cultured on chamber slides and treated with H2O2 (0, 50,100, 200, and 500 μM) for 1 h. Then, 24 h later, cells were fixed with 4% paraformaldehyde in PBS, blocked in 5% normal serum at room temperature for 1 h, followed with primary antibody for Ki67 (Cell Signaling Technology) at 4°C overnight. Then, cells were washed with PBS and incubated with secondary antibodies (Alexa Fluor568 goat anti-rabbit IgG, Invitrogen). Samples were counterstained with DAPI (49,6-diamidino-2-phenylindole; Invitrogen), examined with an Olympus Eclipse 80i Upright Microscope, and photographed using a SPOT RT-SE6 1.4MP Slider camera system (Diagnostic Instruments, Inc.).
Statistical analysis
The data were processed using Prism 5.0 software. Data are reported as means ±SEM if not indicated otherwise. Comparisons among values for all groups were performed by one-way ANOVA or Student’s t-test. A “P” value of less than 0.05 was considered statistically significant.
Results
H2O2 challenge increased miR-34a levels in MSCs
To determine whether oxidative stress can change miR-34a level in MSCs, we used 100 μM H2O2 to treat the cells for 1 h in normal culture conditions. Mild ROS is provided by 100 μM H2O2 and can directly produce free radicals, including hydroxyl radical, which induces DNA injury and membrane lipid peroxidation [14,25]. After 1 h of H2O2 treatment, the expression level of miR-34a was dramatically increased compared to the control group, indicating that 100 μM H2O2 stimulation activates miR-34a expression in MSCs (Figure 1).
Figure 1.
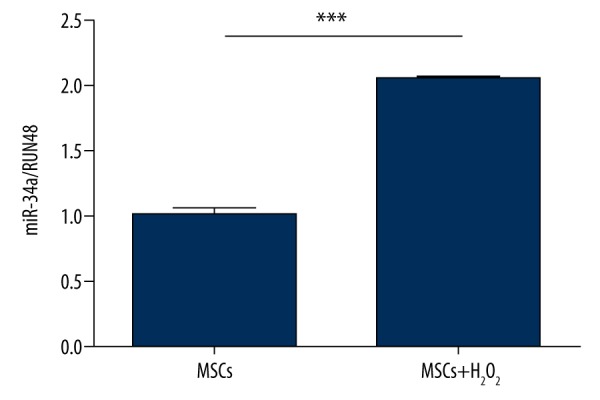
Increased miR-34a expression in MSCs under H2O2 stress. Cultured human bone marrow MSCs were challenged by 100 μM H2O2 for 1 h. miR-34a levels were measured by real-time PCR. There was an approximately 2-fold increase of miR-34a expression in MSCs after H2O2 exposure. Results were representative of 3 independent experiments. *** P<0.001.
Anti-34a successfully reduced miR-34a level by 70% in MSCs
To determine if miR-34a can be down-regulated by anti-34a, 200 nM anti-34a were transfected into MSCs by using Lipofectamine 2000. After 24 h of transfection, the miR-34a level was significantly down-regulated by about 70% (Figure 2). This result indicates that the level of miR-34a in MSCs can be manipulated using anti-34a.
Figure 2.
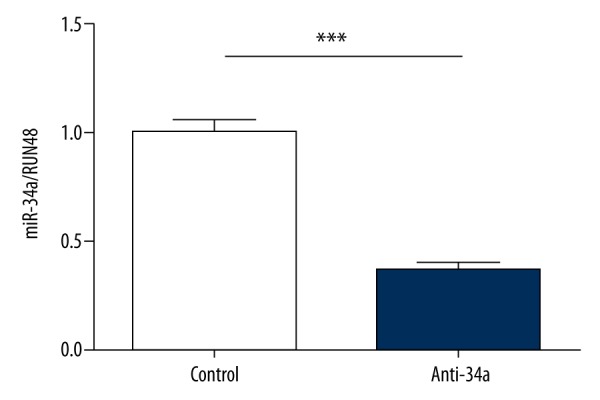
Anti-34a effectively decreased miR-34a expression in MSCs. We transfected 200 nM anti-34a into human bone marrow MSCs for 24 h, and the expression level of miR-34a was measured by real-time PCR. *** P<0.001.
Anti-34a rescued H2O2 -induced cell death in MSCs
We next performed cell proliferation assay to determine if H2O2-induced cell death of MSCs is mediated by miR-34a. First, we did a pre-test to verify that the effects of anti-34a still existed after 2 days of transfection followed by H2O2 exposure. Anti-34a reduced miR-34a level by 50% at 48 h after transfection (Figure 3A). One day post-transfection, cells were treated with 100 μM H2O2 or vehicle for 1 h, and the number of viable cells was measured 24 h later. As shown in Figure 3B, H2O2 treatment significantly decreased the number of living cells by 50–60% compared with the control group. MSCs pre-transfected with anti-34a exhibited a markedly higher number of viable cells under H2O2 stress, indicating inhibition of miR-34a in MSCs enhanced cell survival against H2O2 stress.
Figure 3.

Anti-34a rescued H2O2 -induced cell death in MSCs. (A). miR-34a level was effectively suppressed by anti-34a under H2O2 stress. (B). H2O2 challenge significantly reduced the number of viable MSCs, and anti-34a partially rescued the effect of H2O2 challenge. * P<0.05; ** P<0.01.
Anti-34a rescued Bcl-2, Survivin, and Ki67 levels reduced by H2O2 exposure
To explore the underlying mechanism of the effect of anti-34a against oxidative stress induced by H2O2, we hypothesized that anti-34a modulates the balance between cell proliferation and cell death. It has been reported that H2O2 represses anti-apoptotic gene Bcl-2 expression in many cell lines [15,26]. Hence, we measured the mRNA levels of Bcl-2 and a proliferation marker Ki67 to see whether anti34a could rescue the down-regulation of Bcl-2 and Ki67 induced by H2O2 treatment. As shown in Figure 4A, H2O2 significantly reduced mRNA expression of Bcl-2 in MSCs. Anti-34a not only rescued Bcl-2 mRNA decrease, but, more interestingly, augmented Bcl-2 mRNA to a level even higher than that of the normal control group. The protein expression level of Bcl-2 was totally eliminated in H2O2 treatment groups, whereas anti-34a could also partially restore Bcl-2 protein from H2O2 stress (Figure 4B). Likewise, Survivin, another important survival-related protein, showed a similar expression trend. The proliferation marker Ki67 was also dramatically increased in the anti-34a group (Figure 4A, Supplementary Figure 1), which suggested that inhibition of miR-34a promotes cell proliferation. These results demonstrated that enhanced expression of Bcl-2, Survivin, and Ki67 contributed to the protective effect of anti-34a in MSCs.
Figure 4.

Anti-34a protected MSCs against H2O2 by enhancing expression of Bcl-2, Survivin, and Ki67. (A). The mRNA levels of anti-apoptotic genes Bcl-2, Survivin, and proliferation marker Ki67 were significantly higher in MSCs transfected with anti-34a under H2O2 challenge. * P<0.05; ** P<0.01; *** P<0.001. (B). Consistent with mRNA expression, Bcl-2 and Survivin protein levels were partially rescued by anti-34a. *** P<0.001.
Discussion
MSCs have been extensively used in clinical trials as stem cell therapy for a wide range of diseases in the past 10 years due to their clinical safety, multipotency, low immunogenicity, immunosuppressive properties, and production of cytokines and growth factors [11,27,28]. MSC based cell therapy has been proven effective in many diseases, including neurodegenerative disease [4], stroke [29], heart disease [7], and kidney injury [30]. Although significant progress has been made in MSC therapy, there are still several major hurdles limiting its widespread use. Low viability of transplanted MSCs is one of the major obstacles. ROS might play a crucial role in limiting the survival of engrafted MSCs. Increased ROS by ischemia induces neuronal or myocardial necrosis and infarction. Moreover, high levels of ROS in the ischemic tissue (i.e., brain or myocardium) are also harmful to the engrafted MSCs [31,32]. ROS accumulation is an important effector that activates the p53 pathway, which in turn induces cell apoptosis [19]. ROS inhibitor ameliorates senescence or apoptosis in cancer cells and stem cells [33–35]. These findings suggest that ROS is a priming effector that induces cell apoptosis, especially in ischemic tissue where ROS is highly elevated due to massive death cell. Therefore, the engrafted MSCs in the ischemic area thus might sustain oxidative stress from high levels of ROS in the microenvironment, resulting in MSC apoptosis and limitation of their therapeutic effect. Enhancing the survival of engrafted MSCs has been an attractive strategy to improve MSC therapeutic outcomes in the past few years. Several ex vivo genetic modification of MSCs targeting Bcl-2, TLR4, Survivin, and CCR1/CXCR2 has been proposed and proven effective in enhancing MSC survival [36]. Li et al. found that overexpression of Bcl-2 in MSCs reduced MSC apoptosis by 32% in vitro, and surviving MSCs were about 1–2 fold higher than that of control MSCs at different time points after cell transplantation in a rat myocardial infarction model [13]. We found that MSCs that have undergone a dedifferentiation-reprogramming improved cell survival in an H2O2 oxidative stress model and an in vivo rat hypoxia-ischemic brain damage model, and these improvements were partly attributed to increased Bcl-2 expression and decreased miR-34a in the dedifferentiation-reprogrammed MSCs [5].
Interestingly, a recent study showed hypoxia induced regulation of miR-34a in colorectal cancer cells, providing evidence of the link between miR-34 and ROS [37]. In the present study, we first confirmed that the miR-34a level in MSCs was significantly increased under mild H2O2-mediated oxidative stress. Anti-34a successfully repressed miR-34a expression in MSCs under control conditions. These data indicate that miR-34a is constantly expressed in MSCs, consistent with another report that miR-34a is functionally expressed in mouse neural stem cells [24]. H2O2 induced massive MSCs cell death, and pre-transfection of anti-34a could partially rescue cell death induced by H2O2. Bcl-2 is a 26-kDa anti-apoptotic protein serving as a critical regulator of the apoptotic pathway to inhibit cell death [38]. Several lines of evidence indicate that miR-34a directly targets Bcl-2 mRNA and represses its expression [39]. Consistent with previous findings, we found that H2O2 treatment severely reduced Bcl-2 mRNA and protein levels in MSCs. Pre-transfection of anti-34a into MSCs dramatically increased Bcl-2 mRNA level and partially restored its protein level. The proliferation marker Ki67 showed a similar trend as that of Bcl-2. Most interestingly, anti-34a treatment not only restored the mRNA level of Bcl-2 and Ki67, but achieved even higher levels than that of the control group. This finding indicates that miR-34a is constantly expressed in MSCs and is functionally involved in the regulation of cell proliferation and cell death.
Conclusions
In summary, our study revealed that miR-34a plays a key role in ROS-induced MSC apoptosis. Repressing miR-34a expression by transfection of anti-34a increased MSC survival under oxidative stress. These findings offer a new insight into the importance of miR-34a in MSC apoptosis and suggest that manipulation of miR-34a expression may be a promising strategy to improve survival of transplanted MSCs in clinical stem cell therapies.
Supplementary materials
Immunostaining of Ki67 in MSCs after H2O2 treatment. After H2O2 treatment at different dosages (0, 50,100, 200, and 500 μM), Ki67 expression was detected by immunostaining. Left lane: DAPI; middle lane: Ki67; right lane: the merged image of DAPI and Ki67.
Footnotes
Competing interests
The authors have no conflict of interest to disclose.
Source of support: This work was supported by the Natural Science Foundation Project of CQ CSCT (No. 2011BB5138) and the National Natural Science Foundation of China (No. 81100475 and 81271385)
References
- 1.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276:71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 2.Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–47. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 3.Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–49. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 4.Joyce N, Annett G, Wirthlin L, et al. Mesenchymal stem cells for the treatment of neurodegenerative disease. Regen Med. 2010;5:933–46. doi: 10.2217/rme.10.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Jiang X, Zhang X, et al. Dedifferentiation-reprogrammed mesenchymal stem cells with improved therapeutic potential. Stem Cells. 2011;29:2077–89. doi: 10.1002/stem.764. [DOI] [PubMed] [Google Scholar]
- 6.Wen Z, Zheng S, Zhou C, et al. Repair mechanisms of bone marrow mesenchymal stem cells in myocardial infarction. J Cell Mol Med. 2011;15:1032–43. doi: 10.1111/j.1582-4934.2010.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen Z, Zheng S, Zhou C, et al. Bone marrow mesenchymal stem cells for post-myocardial infarction cardiac repair: microRNAs as novel regulators. J Cell Mol Med. 2012;16:657–71. doi: 10.1111/j.1582-4934.2011.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ning H, Yang F, Jiang M, et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical study. Leukemia. 2008;22:593–99. doi: 10.1038/sj.leu.2405090. [DOI] [PubMed] [Google Scholar]
- 9.Kuriyan AE, Albini TA, Townsend JH, et al. Vision loss after intravitreal injection of autologous “stem cells” for AMD. New Engl J Med. 2017;376:1047–53. doi: 10.1056/NEJMoa1609583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ankrum J, Karp JM. Mesenchymal stem cell therapy: Two steps forward, one step back. Trends Mol Med. 2010;16:203–9. doi: 10.1016/j.molmed.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yagi H, Soto-Gutierrez A, Parekkadan B, et al. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010;19:667–79. doi: 10.3727/096368910X508762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song H, Kwon K, Lim S, et al. Transfection of mesenchymal stem cells with the FGF-2 gene improves their survival under hypoxic conditions. Mol Cells. 2005;19:402–7. [PubMed] [Google Scholar]
- 13.Li W, Ma N, Ong L-L, et al. Bcl-2 Engineered MSCs inhibited apoptosis and improved heart function. Stem Cells. 2007;25:2118–27. doi: 10.1634/stemcells.2006-0771. [DOI] [PubMed] [Google Scholar]
- 14.Zeng W, Xiao J, Zheng G, et al. Antioxidant treatment enhances human mesenchymal stem cell anti-stress ability and therapeutic efficacy in an acute liver failure model. Sci Rep. 2015;5:11100. doi: 10.1038/srep11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohammadzadeh M, Halabian R, Gharehbaghian A, et al. Nrf-2 overexpression in mesenchymal stem cells reduces oxidative stress-induced apoptosis and cytotoxicity. Cell Stress Chaperones. 2012;17:553–65. doi: 10.1007/s12192-012-0331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trouche E, Mias C, Seguelas MH, et al. Characterization of monoamine oxidases in mesenchymal stem cells: Role in hydrogen peroxide generation and serotonin-dependent apoptosis. Stem Cells Dev. 2010;19:1571–78. doi: 10.1089/scd.2009.0353. [DOI] [PubMed] [Google Scholar]
- 17.Tarasov V, Jung P, Verdoodt B, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 18.Liao JM, Cao B, Zhou X, et al. New insights into p53 functions through its target microRNAs. J Mol Cell Biol. 2014;6:206–13. doi: 10.1093/jmcb/mju018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macip S, Igarashi M, Berggren P, et al. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol. 2003;23:8576–85. doi: 10.1128/MCB.23.23.8576-8585.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karawajew L, Rhein P, Czerwony G, et al. Stress-induced activation of the p53 tumor suppressor in leukemia cells and normal lymphocytes requires mitochondrial activity and reactive oxygen species. Blood. 2005;105:4767–75. doi: 10.1182/blood-2004-09-3428. [DOI] [PubMed] [Google Scholar]
- 21.Lv B, Li F, Fang J, et al. Hypoxia inducible factor 1alpha promotes survival of mesenchymal stem cells under hypoxia. Am J Transl Res. 2017;9:1521–29. [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang JM, Feng FE, Wang QM, et al. Platelet-derived growth factor-BB protects mesenchymal stem cells (MSCs) derived from immune thrombocytopenia patients against apoptosis and senescence and maintains MSC-mediated immunosuppression. Stem Cells Transl Med. 2016;5:1631–43. doi: 10.5966/sctm.2015-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raver-Shapira N, Marciano E, Meiri E, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 24.Aranha MM, Santos DM, Sola S, et al. miR-34a regulates mouse neural stem cell differentiation. PLoS One. 2011;6:e21396. doi: 10.1371/journal.pone.0021396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Konat GW. H2O2-induced higher order chromatin degradation: A novel mechanism of oxidative genotoxicity. J Biosci. 2003;28:57–60. doi: 10.1007/BF02970132. [DOI] [PubMed] [Google Scholar]
- 26.Valks DM, Kemp TJ, Clerk A. Regulation of Bcl-xL expression by H2O2 in cardiac myocytes. J Biol Chem. 2003;278:25542–47. doi: 10.1074/jbc.M303760200. [DOI] [PubMed] [Google Scholar]
- 27.Mohammadian M, Shamsasenjan K, Lotfi Nezhad P, et al. Mesenchymal stem cells: New aspect in cell-based regenerative therapy. Adv Pharm Bull. 2013;3:433–37. doi: 10.5681/apb.2013.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei X, Yang X, Han ZP, et al. Mesenchymal stem cells: A new trend for cell therapy. Acta Pharmacol Sin. 2013;34:747–54. doi: 10.1038/aps.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diez-Tejedor E, Gutierrez-Fernandez M, Martinez-Sanchez P, et al. Reparative therapy for acute ischemic stroke with allogeneic mesenchymal stem cells from adipose tissue: A safety assessment: A phase II randomized, double-blind, placebo-controlled, single-center, pilot clinical trial. J Stroke Cerebrovasc Dis. 2014;23(10):2694–700. doi: 10.1016/j.jstrokecerebrovasdis.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Humphreys BD, Bonventre JV. Mesenchymal stem cells in acute kidney injury. Annu Rev Med. 2008;59:311–25. doi: 10.1146/annurev.med.59.061506.154239. [DOI] [PubMed] [Google Scholar]
- 31.Levraut J, Iwase H, Shao ZH, et al. Cell death during ischemia: Relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol. 2003;284:H549–58. doi: 10.1152/ajpheart.00708.2002. [DOI] [PubMed] [Google Scholar]
- 32.Carpi A, Menabo R, Kaludercic N, et al. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta. 2009;1787:774–80. doi: 10.1016/j.bbabio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Chen T, Wong YS. Selenocystine induces apoptosis of A375 human melanoma cells by activating ROS-mediated mitochondrial pathway and p53 phosphorylation. Cell Mol Life Sci. 2008;65:2763–75. doi: 10.1007/s00018-008-8329-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen Ngoc TD, Son YO, Lim SS, et al. Sodium fluoride induces apoptosis in mouse embryonic stem cells through ROS-dependent and caspase- and JNK-mediated pathways. Toxicol Appl Pharmacol. 2012;259:329–37. doi: 10.1016/j.taap.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan L, Wei S, Wang J, et al. Isoorientin induces apoptosis and autophagy simultaneously by reactive oxygen species (ROS)-related p53, PI3K/Akt, JNK, and p38 signaling pathways in HepG2 cancer cells. J Agric Food Chem. 2014;62:5390–400. doi: 10.1021/jf500903g. [DOI] [PubMed] [Google Scholar]
- 36.Lee S, Choi E, Cha M-J, et al. Cell adhesion and long-term survival of transplanted mesenchymal stem cells: A prerequisite for cell therapy. Oxid Med Cell Longev. 2015;2015:9. doi: 10.1155/2015/632902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Rokavec M, Jiang L, et al. Antagonistic effects of p53 and HIF1A on microRNA-34a regulation of PP1R11 and STAT3 and hypoxia-induced epithelial to mesenchymal transition in colorectal cancer cells. Gastroenterology. 2017 doi: 10.1053/j.gastro.2017.04.017. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 38.Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–36. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 39.Chen H, Wang J, Hu B, et al. MiR-34a promotes Fas-mediated cartilage endplate chondrocyte apoptosis by targeting Bcl-2. Mol Cell Biochem. 2015;406:21–30. doi: 10.1007/s11010-015-2420-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunostaining of Ki67 in MSCs after H2O2 treatment. After H2O2 treatment at different dosages (0, 50,100, 200, and 500 μM), Ki67 expression was detected by immunostaining. Left lane: DAPI; middle lane: Ki67; right lane: the merged image of DAPI and Ki67.