

Abstract
The human sinoatrial node (SAN) efficiently maintains heart rhythm even under adverse conditions. However, the specific mechanisms involved in the human SAN’s ability to prevent rhythm failure, also referred to as its robustness, are unknown. Challenges exist because the three-dimensional (3D) intramural structure of the human SAN differs from well-studied animal models, and clinical electrode recordings are limited to only surface atrial activation. Hence, to innovate the translational study of human SAN structural and functional robustness, we integrated intramural optical mapping, 3D histology reconstruction, and molecular mapping of the ex vivo human heart. When challenged with adenosine or atrial pacing, redundant intranodal pacemakers within the human SAN maintained automaticity and delivered electrical impulses to the atria through sinoatrial conduction pathways (SACPs), thereby ensuring a fail-safe mechanism for robust maintenance of sinus rhythm. During adenosine perturbation, the primary central SAN pacemaker was suppressed, whereas previously inactive superior or inferior intra-nodal pacemakers took over automaticity maintenance. Sinus rhythm was also rescued by activation of another SACP when the preferential SACP was suppressed, suggesting two independent fail-safe mechanisms for automaticity and conduction. The fail-safe mechanism in response to adenosine challenge is orchestrated by heterogeneous differences in adenosine A1 receptors and downstream GIRK4 channel protein expressions across the SAN complex. Only failure of all pacemakers and/or SACPs resulted in SAN arrest or conduction block. Our results unmasked reserve mechanisms that protect the human SAN pacemaker and conduction complex from rhythm failure, which may contribute to treatment of SAN arrhythmias.
INTRODUCTION
As the primary pacemaker of the human heart, the sinoatrial node (SAN) is responsible for generating spontaneous electrical impulses and conducting the impulses to the atria to initiate the heartbeat (1–5). The SAN is a collection of specialized cardiomyocytes located at the junction of the right atria and the superior vena cava (2, 6, 7). To efficiently keep the heart beating, the SAN requires a sophisticated ability to maintain uninterrupted function during internal or external perturbations, known as biological robustness (8). It has been proposed that the human SAN pacemaker and conduction complex is a specialized and heterogeneous intramural three-dimensional (3D) structure with multiple intranodal pacemakers (4) and several sinoatrial conduction pathways (SACPs), which are responsible for the transmission of electrical impulses to the right atrium (3, 5). However, the role of multiple intranodal SAN pacemakers and SACPs in the robust protection of human SAN rhythm and how this system fails, leading to sinoatrial node dysfunction (SND) (4, 9, 10), have remained an enigma.
Substantial gaps exist in data on the pacemaking and conduction functions of the human SAN (1, 11, 12). Most current understanding is inferred from animal models, which may not reproduce the human clinical phenomena (3, 13), or clinical electrogram recordings restricted to the atrial surface (5, 14–17). Consequently, studies of the ex vivo human heart (7, 18) provide an opportunity to study human cardiac disease by applying state-of-the-art intramural mapping techniques consisting of near-infrared optical mapping, 3D structural imaging (2), and molecular mapping (19) to resolve mechanisms of human SAN robustness, which are not possible in vivo.
The study of biological robustness requires the system to be challenged by external perturbations or stimuli (8). Adenosine, an endogenous metabolite of the heart (20), is known to contribute to the daily regulation of sinus rhythm (21) and is abundantly produced during ischemia and heart failure (22). In 1985, Watt hypothesized that increased endogenous adenosine production and/or hypersensitivity to adenosine could result in SND (21), and it was later shown that adenosine receptor blockers can treat SND (23, 24). Several independent clinical studies have observed that patients with SND exhibit an enhanced bradycardic response and atrial pauses (2 to 23 s) to adenosine bolus (25–27). Thus, adenosine represents a physiologically and clinically relevant tool for external perturbation of SAN robustness through excessive negative chronotropic depression, simulating an SND-like phenotype (28, 29). On the basis of animal studies, the negative chronotropic effects of adenosine may stem partially from activation of the adenosine A1 receptor (A1R) and its downstream inward rectifier potassium channel, IK.Ado, formed by G protein–coupled inwardly rectifying potassium channel subunits (GIRK1 and GIRK4) (30–32); however, the expression profiles of these key proteins are unknown in the human SAN.
We hypothesized that multiple intranodal pacemakers and SACPs with redundant functions have diverse sensitivities to physiologic and pathophysiologic perturbations, which are fundamental to maintaining robustness of the human SAN at the tissue level. Using our integrated ex vivo approach, we identified redundant and diverse pacemakers and SACPs within the human SAN complex, which ensure a fail-safe or backup mechanism for robust maintenance of human sinus rhythm when challenged by adenosine. SAN function collapses when all pacemakers or SACPs are inhibited. These unique features of the human SAN pacemaker-conduction complex may have implications for developing a robust “biological” pacemaker or treatments targeting impaired intranodal pacemakers or SACPs to restore SAN robustness in SND patients.
RESULTS
Intranodal leading pacemaker and conduction in the human SAN
Near-infrared optical mapping is currently the only reliable method to delineate activation and conduction within the intramural human SAN complex structure (12 to 27 mm long, 3 to 8 mm wide, and 1 to 3 mm deep) (1, 2, 33) that is obscured to clinical surface electrode mapping by the surrounding atria (2, 7). We optically mapped human hearts (n = 11, 34 to 69 years old, five females) from patients with a variety of recorded comorbidities, ranging from no diseases to chronic hypertension, diabetes, or acute heart failure (table S1). At baseline, all coronary-perfused human hearts exhibited stable intrinsic sinus rhythms [55 to 101 beats per minute (bpm); Table 1] with cycle lengths (CLs) of 819 ± 173 ms. These values are within the range of “intrinsic” rates measured in vivo during autonomic blockade with propranolol and atropine in adult human subjects with diseased hearts (range, 57 to 126 bpm) (34), although some fall outside the range previously reported for healthy patients (range, 69 to 128 bpm) (35). The SAN is defined by optical action potential (OAP) morphologies as the region of slow diastolic depolarization and slow upstroke preceding atrial fast excitation (7). Slow excitation from the SAN leading pacemaker conducts through preferential SACP(s) to initiate excitation at the earliest atrial activation sites (EASs) (Fig. 1A). Intramural optical mapping revealed that SAN activation originated from one of the intranodal leading pacemakers and conducted through one of the lateral SACPs in the crista terminalis (Fig. 1 and fig. S1). The distance (9.2 ± 5.6 mm; range, 3.5 to 23.2 mm) between the location of the intranodal leading pacemaker and the EAS exemplifies the inability of the EAS to reliably predict the intranodal location of the SAN leading pacemaker.
Table 1.
The effect of sequential drug applications on sinus rhythm (SR) in individual human hearts.
| Heart no. | Case no. | Baseline | Adenosine (10 μM) | Adenosine (100 μM) | Tertiapin | Adenosine (100 μM)* |
|---|---|---|---|---|---|---|
| 1 | 685884 | SR 59 bpm | SR 39 bpm | — | SR 63 bpm | SR 56 bpm |
| 2 | 481041 | SR 94 bpm | SR 75 bpm | Arrest 95 s | SR 95 bpm | — |
| 3 | 415217 | SR 81 bpm | SR 46 bpm | — | SR 70 bpm | — |
| 4 | 257102 | SR 71 bpm | SR 49 bpm | SR 52 bpm | — | — |
| 5 | 947202 | SR 90 bpm | SR 39 bpm | Arrest 6 min | SR 100 bpm | — |
| 6 | 118258 | SR 96 bpm | Exit block 30 s | Arrest 67 s | SR 91 bpm | — |
| 7 | 987692 | SR 71 bpm | Arrest 4 min | Exit block 49 s | — | — |
| 8 | 925852 | SR 65 bpm | Arrest 6 s | Arrest 12 s | SR 72 bpm | SR 71 bpm |
| 9 | 984478 | SR 76 bpm | Arrest 50 s | — | SR 60 bpm | SR 41 bpm |
| 10 | 474083 | SR 61 bpm | SR 46 bpm | — | — | — |
| 11 | 442404 | SR 55 bpm | Arrest 8 min | Arrest 33 min | SR 60 bpm | — |
Adenosine (100 μM) repeated after tertiapin.
Fig. 1. Functional, structural, and molecular features of the 3D human SAN pacemaker complex revealed by integrated ex vivo human heart mapping. (A).
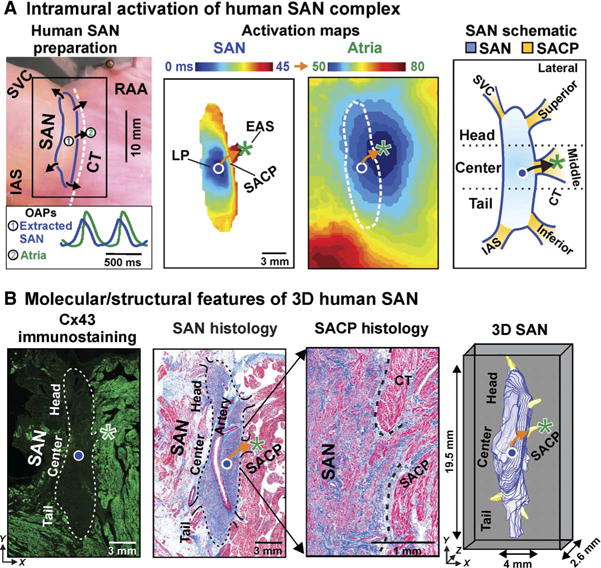
From left to right: Photograph of human SAN preparation of heart 118258 with SAN and atrial OAPs, SAN and atrial activation maps at baseline conditions, and schematic human SAN. Leading pacemaker (LP) is shown with circle, and EAS is shown with asterisk in activation maps and schematic. Arrows indicate the functionally and structurally defined SACP. (B) From left to right: Fluorescence image of Cx43 immunostaining of heart tissue with SAN outlined by dotted line, Masson’s trichrome staining of SAN pacemaker complex with fibrosis (blue) and cardiomyocytes (red), magnification of SAN histology showing the preferential SACP, and 3D reconstruction of the SAN (blue) with five SACPs (yellow). CT, crista terminalis; IAS, interatrial septum; RA, right atrium; SVC, superior vena cava.
On the basis of our previous canine and human functional-structural mapping (2, 7, 29), we defined the superior third of the SAN as the head, the middle third as the center, and the inferior third as the tail intranodal pacemaker regions (Fig. 1A). SACPs were functionally identified as the preferential conduction path between the SAN border and the EAS, which could only be defined using SAN and atrial activation maps. This and previous intramural mapping studies (2, 7, 29) revealed that the SAN is functionally insulated from the atria except at two to five discrete SACPs, which correspond to the discrete EASs (Fig. 1). A SAN intranodal leading pacemaker could activate one or more preferential SACPs simultaneously (3). To enable cross-heart analysis, we grouped all functionally identified SACPs based on their general anatomic locations (lateral superior/middle/inferior, superior vena cava, and septal pathways) (fig. S1). However, because of heart-specific variations, SACPs were not always revealed functionally in each region and all five may not be present in all human SAN complexes. The overall average number of functionally mapped SACPs per heart at all studied experimental conditions was 2.9 ± 1.0.
After optical mapping, SAN preparations (n = 5) were serially sectioned for Masson’s trichrome staining, immunostaining, and 3D reconstruction as previously described to confirm the structural basis of functionally observed SAN pacemakers and SACPs (2). Masson’s trichrome staining distinguished the SAN from neighboring atria as a dense fibrotic (45.8 ± 4.4% fibrosis) and cardiomyocyte compact region around the SAN artery, which was validated by negative immunostaining for the gap junction protein connexin 43 (Cx43) (Fig. 1B) (2, 19). The SAN was almost entirely structurally insulated from the surrounding atria by a border composed of fibrosis, fat, and discontinuous myofibers except in functionally defined SACP regions, where several intramural continuous branching myofiber tracts (100 to 400 μm thick and 2 to 3 mm long) of transitional cells were defined (Fig. 1B and figs. S2 and S3) (2).
SAN activation recorded by clinical catheter electrogram
To further support the translational application of our results, we integrated pseudo-atrial electrocardiogram (ECG) and clinically used bipolar catheters to record atrial and SAN activation simultaneously (17). Optical mapping in five human hearts validated that the slow upstroke component (~12 to 32 μV) preceding the sharp atrial deflection represented SAN activation when the bipolar catheter (7-French, 3.5-mm tip) was effectively placed: parallel to the elliptical human SAN structure and crista terminalis, with direct surface contact of both electrodes, and within at least 4.4 ± 1.8 mm from the leading pacemaker (Fig. 2A and fig. S4). When effectively placed, SAN conduction time (SACT) measurements by optical mapping and SAN electrogram were strongly correlated (P < 0.001) even when EAS shift was observed, or the morphology of the SAN electrogram changed because of the preferential intranodal conduction moving inferiorly versus superiorly (figs. S4 and S5). The accurate measurement of SACT by SAN electrogram represents the first step in diagnosing intranodal mechanisms underlying SAN function with clinically available techniques.
Fig. 2. Adenosine-induced SAN dysfunction recorded by optical mapping and SAN electrograms in ex vivo human hearts.
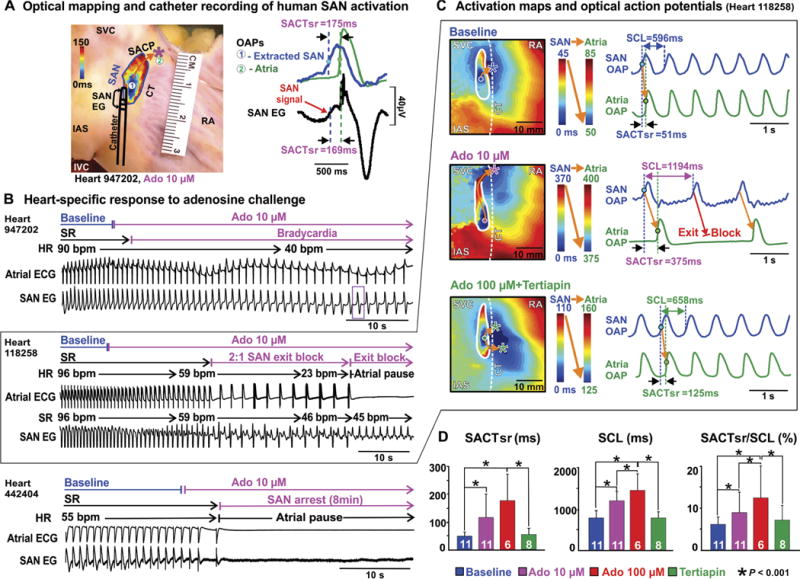
(A) Left: Heart 947202 preparation showing the location of the SAN and bipolar catheter. Circles #1 and #2 indicate SAN leading pacemaker and EAS (asterisk), respectively. The orange arrow indicates the preferential SACP. Right: OAPs of extracted SAN (blue) and atria (green) with SAN electrogram (EG) (black) during adenosine (10 μM) perfusion. (B) Atrial ECG and SAN electrogram recordings of adenosine (10 μM)–induced bradycardia in heart 947202, 2:1 exit block followed by complete exit block in heart 118258, and SAN arrest in heart 442404. Purple box in (B) shows SAN beat presented in (A). (C) SAN and atria activation maps and OAPs during baseline, adenosine (10 μM), and adenosine (100 μM) + tertiapin in heart 118258, respectively. (D) Quantification of adenosine-induced SAN conduction and automaticity inhibition restored by tertiapin. Bar graphs summarize the average drug effect; white numbers show the number of hearts used. Data are means ± SD. P values were determined by analysis of variance (ANOVA) after Tukey’s adjustment. Ado, adenosine; HR, heart rate; IVC, inferior vena cava; SACTsr, sinoatrial conduction time during sinus rhythm; SR, sinus rhythm.
Robustness of SAN automaticity and conduction challenged by adenosine
As an important endogenous heart rhythm regulator and pharmacological tool, adenosine perfusion led to a range of heart-specific SAN automaticity and conduction inhibitions, from mild bradycardia to SAN exit block, atrial pauses, and arrest (Fig. 2B and fig. S6), which provided a methodological framework to study the mechanism of human SAN robustness. Contrary to our previous canine study (29), some of the human SAN preparations experienced complete SAN arrest. Adenosine-induced SAN exit block or SAN arrest led to atrial pauses during sinus rhythm in 5 of 11 hearts at 10 μM (6 s to 8 min) and in 6 of 7 hearts at 100 μM (12 s to 33 min) (Table 1). Figure 2 shows examples of SAN and atrial activation maps at intrinsic baseline rhythm and during adenosine-induced disturbance of SAN automaticity and conduction. These disturbances were demonstrated by leading pacemaker shift away from the central SAN region, preferential SACP shifts, and exit conduction blocks (Fig. 2C). In all hearts, adenosine significantly prolonged sinus CL (SCL) (P = 0.002 at 10 μM adenosine, P < 0.001 at 100 μM adenosine), SACT at sinus rhythm (SACTsr) (P = 0.038 at 10 μM adenosine, P < 0.001 at 100 μM adenosine), and SACTsr/ SCL (P < 0.001) (Fig. 2D and table S2). Figure 3 shows that adenosine-induced dynamic changes in intranodal conduction and SACP shift were seen not only by intramural optical mapping but also by the SAN electrogram. Using our integrated mapping approach and adenosine challenge, we revealed that the robust protection of SAN rhythm to adenosine challenge is heart-specific: Some hearts maintained rhythm, whereas others experienced failure, manifesting as SAN arrest and exit block.
Fig. 3. Exit and intranodal block revealed by ex vivo intramural optical mapping and SAN electrogram during adenosine treatment (10 mM) in heart 118258. (A).
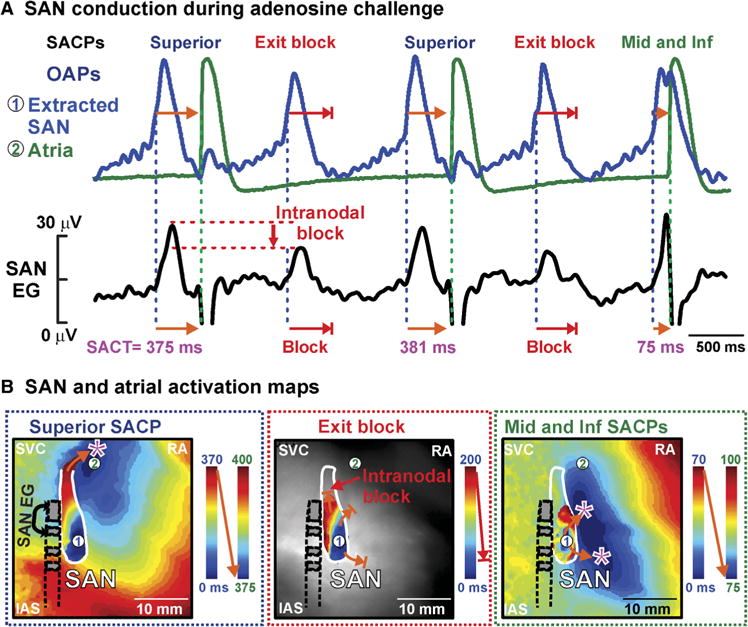
OAPs and SAN electrogram show exit block from SAN to atria. Intranodal block seen as a reduction in amplitude on SAN electrogram. (B) From left to right: Activation maps for complete activation of the SAN that activated atria through the superior SACP, partial SAN activation and exit block, and partial SAN activation that activated atria through the inferior (inf) and middle (mid) lateral SACPs. SACT, sinoatrial conduction time.
To further confirm that the observed negative chronotropic effects were due to potassium GIRK channel activation, tertiapin (10 to 100 nM), a selective GIRK channel blocker (36), was added after adenosine perfusion (n = 8). Tertiapin restored stable sinus rhythm and conduction (Fig. 2, fig. S6, and Table 1) to near-baseline values. In three preparations, perfusion of adenosine (100 μM) was repeated after tertiapin, as shown in fig. S6, which prevented the depressant effects of adenosine seen when administered before tertiapin. Reversing and preventing adenosine-induced SAN depression by tertiapin confirmed that adenosine’s negative effects on intrinsic SAN function primarily stem from activation of the potassium GIRK channels, and thus blocking or down-regulating GIRK channels could be a potential strategy for treatment of SAN dysfunction related to elevated adenosine (23, 24).
Robust sinus rhythm maintained by shifts between redundant and diverse pacemakers and conduction pathways
By combining optical mapping with serial histology images of the 3D human SAN (2), we demonstrated the consequences of leading pacemaker and preferential SACP shifts in human hearts after exposure to adenosine (Fig. 4). Adenosine caused the leading SAN pacemaker to shift in either superior or inferior directions up to 3.9 ± 2.4 mm at 10 μM (n = 11) and 7.1 ± 3.5 mm at 100 μM (n = 6). When adenosine provoked atrial pauses, the maximum distance of the leading pacemaker shift was greater after these pauses than during adenosine-induced bradycardia episodes (6.3 ± 3.2 mm versus 3.2 ± 2.0 mm, P = 0.024). Moreover, the magnitude of the leading pacemaker shift strongly correlated with prolongation of SACT (fig. S7).
Fig. 4. Multiple SAN intranodal leading pacemakers and SACPs revealed by adenosine challenge.
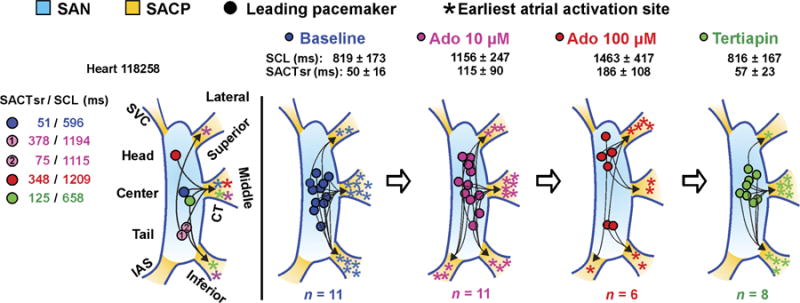
From left to right: SAN leading pacemaker and SACP shift during baseline (blue), 10 μM adenosine (pink), 100 μM adenosine (red), and tertiapin (green) for heart 118258 and (right) for all hearts during sinus rhythm.
Our intramural mapping revealed that conduction through the inferior septal SACP was observed in four hearts during adenosine-mediated bradycardia, but not during baseline in any hearts (Fig. 4), which suggests that the sole purpose of the septal SACP may be to maintain conduction when all lateral SACPs are suppressed. However, although SAN leading pacemaker shift was often observed, the EAS shifted in only 6 of 11 (10 μM) and 4 of 6 (100 μM) adenosine-treated hearts when there was a change in preferential SACP (Fig. 4). The occurrence and direction of EAS shift were not correlated with the direction of leading pacemaker shift (Fig. 4) or SACT and SCL prolongation, suggesting two independent fail-safe mechanisms for automaticity and conduction. Furthermore, in all eight hearts perfused with tertiapin, the leading pacemaker sites returned to within 2.2 ± 0.7 mm of baseline sites and, in seven of eight hearts, restored conduction through baseline preferential SACPs. Our results suggest that negative chronotropic regulation of human SAN rhythm relies on the presence of multiple possible intranodal pacemakers and SACPs with heterogeneous sensitivity to adenosine regulation.
SACP conduction filtering during overdrive atrial pacing and atrial fibrillation protection of robust SAN pacemaker function
Atrial pacing at a rate above sinus rhythm is used to overdrive suppress the SAN automaticity and evaluate SAN function clinically (17). Specifically, direct or indirect SAN recovery times (SNRTd/SNRTi) corrected by sinus rhythm before pacing (cSNRTd/cSNRTi) measure the time it takes for the intranodal pacemaker or atrial activity, respectively, to recover from overdrive suppression by atrial pacing (17). Here, we used atrial overdrive pacing (CL = 500 ms) in parallel with adenosine to further challenge SAN robustness (Fig. 5). Five hundred milliseconds was chosen as the CL for comparison, because this CL limited the occurrence of SAN entrance block. SAN entrance block or filtering of overdrive pacing by SACPs (29) was observed when hearts were paced at CLs faster than 500 ms and decreased the reliability of SNRT measurements (fig. S8). At baseline, 10 of 11 hearts exhibited normal clinical values of cSNRTi (<525 ms) (27) because of the absence of both SAN automaticity and conduction depression by atrial pacing (Fig. 5A and table S2). Only aged heart 442404 (69-year-old male) had an abnormal baseline cSNRTi (1630 ms) because of post-pacing exit block.
Fig. 5. SAN intranodal automaticity and conduction strongly affected by combination of atrial pacing and adenosine in ex vivo human hearts. (A).
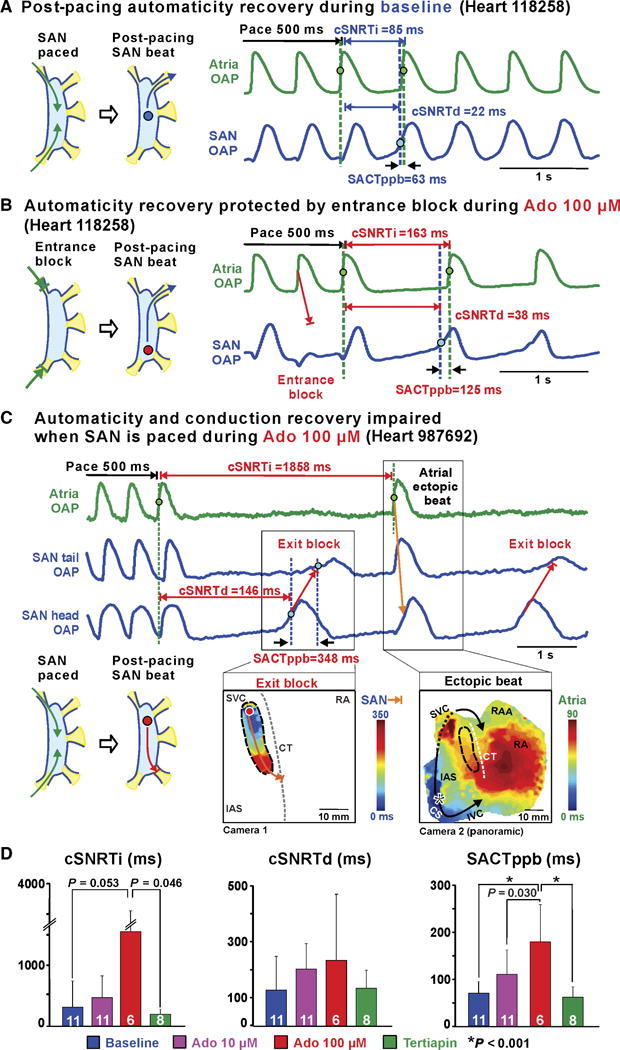
Baseline SAN activation and conduction pattern during and after atrial pacing. (B) Adenosine impaired SAN automaticity and conduction; conversely, entrance block, caused by conduction depression, prevented overdrive suppression of SAN automaticity. (C) Top: Atrial and SAN OAPs during 500-ms CL pacing and post-pacing with 100 μM adenosine. Bottom: SAN exit block (left) and atrial ectopic beat (right) activation maps during 100 μM adenosine. (D) Bar graphs summarizing average drug effect on cSNRTi (left), cSNRTd (middle), and SACTppb (right). White numbers show the number of hearts used. Data are means ± SD.P values were determined by ANOVA after Tukey’s adjustment. CS, coronary sinus; RAA, right atrial appendage; SACTppb, sinoatrial conduction time of first post-pacing SAN beat; SNRTi/d, indirect/direct SAN recovery time; cSNRTi/d, corrected SNRTi/d.
Adenosine (100 μM) significantly prolonged SACT of the post-pacing beat (SACTppb) (P < 0.001) in hearts with normal baseline values (Fig. 5). Figure 5B shows that adenosine-induced conduction impairment led to SAN entrance block, which prevented overdrive suppression of SAN automaticity even at 500 ms pacing. Conversely, if entrance block was absent, overdrive pacing exaggerated the negative chronotropic effects of adenosine and caused SAN exit block and post-pacing atrial pause (Fig. 5C). Adenosine-induced entrance block, seen in 8 of 11 hearts, prevented significant prolongation of cSNRTd (P = 0.52; Fig. 5 and fig. S9). With adenosine, a greater dispersion of leading pacemakers and all possible SACPs were observed after pacing that were not seen at baseline (fig. S10). Moreover, in accordance with our recent study (18), fast pacing provoked atrial fibrillation in 7 of 11 preparations under adenosine (fig. S11). However, just as with atrial pacing, SAN intranodal pacemakers were partially protected from fast atrial fibrillation rhythm by entrance block in SACPs, similar to our recent observations in canine SAN (29). Spontaneous termination of fibrillation led to only one tachycardia-bradycardia arrhythmia episode due to SAN exit block in heart 987692 (fig. S11). Tertiapin returned cSNRTi, cSNRTd, and SACTppb (Fig. 5D, fig. S9, and table S2) to near-baseline values, confirming that GIRK channel activation affects SAN conduction and automaticity.
Heterogeneous A1R and GIRK protein expression profile in the human SAN underlying the robust regulation of sinus rhythm
As our optical mapping consistently demonstrated a high sensitivity of the central SAN pacemaker to adenosine’s bradycardic effect, we tested the hypothesis that expression of A1R and GIRK1/4, the main cardiac proteins responsible for adenosine’s negative chronotropic effects, was higher in the human SAN center than the adjacent atria (19, 29). Our methods to extract pure SAN protein made it possible to compare the protein profile among the three (head, center, and tail) intranodal pacemaker regions (Fig. 6A) (19). Immunoblotting showed higher A1R (3.0 ± 1.2–fold, P = 0.003) and GIRK4 (2.5 ± 1.2–fold, P = 0.028) expression in the SAN center than the adjacent right atrium (Fig. 6B). GIRK1 expression was not significantly higher in the SAN center versus the right atrium (1.5 ± 1.1–fold, P = 0.24). Furthermore, there were heart-specific differences in which SAN pacemaker region (head, center, or tail) had the highest protein expression relative to the right atrium (Fig. 6C). A1R and GIRK protein expression is heterogeneously distributed in the human SAN and atria, which may provide the molecular basis for the regionally diverse response of SAN pacemakers and SACPs to adenosine challenge underlying robust protection.
Fig. 6. Heterogeneous A1R and GIRK1/4 protein expression in human SAN and atria revealed by ex vivo molecular mapping.
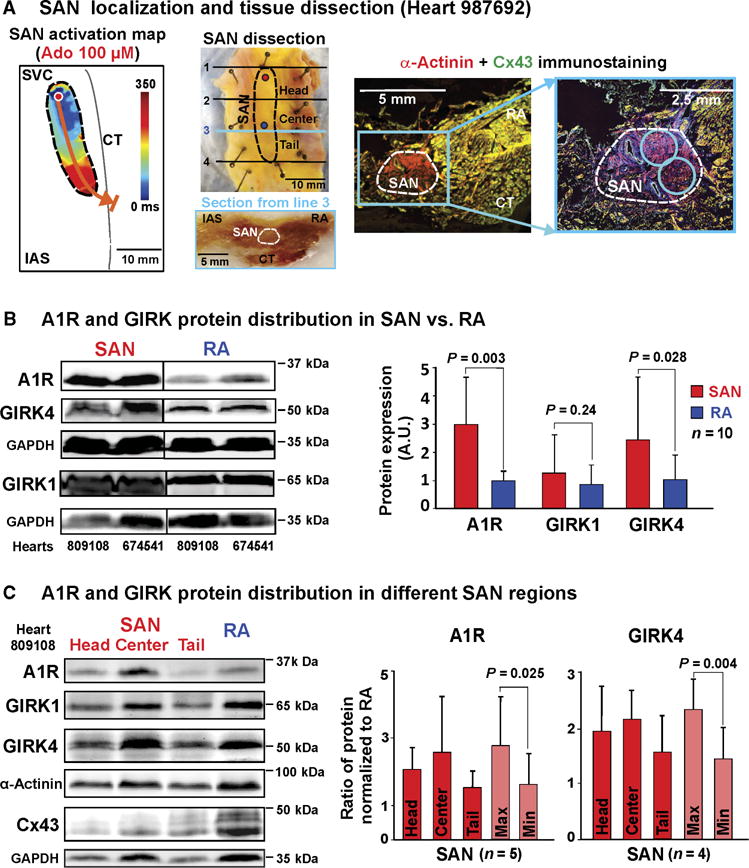
(A) SAN localization and tissue dissection. Left: SAN activation map of heart 987692. SAN leading pacemaker is shown with red circle. Middle: Outline of functionally defined SAN overlaid on photograph of dissected SAN preparation. Blue and red circles indicate the leading pacemaker locations during baseline and 100 μM adenosine, respectively. Lines 1 to 4 indicate the dissection lines for the SAN head, center, and tail regions. Beneath is shown a photograph of transmural section of frozen SAN preparation. Right: Immunostaining showing α-actinin (red)–positive and Cx43 (green)–negative regions of the human SAN indicated by the white dotted line. Blue circles show the locations where tissue was collected for SAN protein isolation. (B) Left: Representative immunoblot from hearts 809108 and 674541. Right: Bar graphs summarizing A1R and GIRK4 protein expression in the SAN central region and the right atria (RA) (n = 10, samples from unmapped human hearts). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)–normalized band density is shown as means ± SD; n, number of human hearts; comparison of protein expression in SAN versus RA was done using mixed models in package lme4. P value was determined by pairwise comparisons with Tukey’s adjustment. A.U., arbitrary unit. (C) Left: Representative immunoblot from each SAN region and RA for heart 809108. Right: SAN-to-RA ratio of GAPDH-normalized band density shown as means ± SD; n, number of human hearts (four unmapped and one mapped heart); values of SAN/RA ratios were compared to 1 using two-sided one-sample test; for each protein Max versus Min, P values were determined by two-sided paired t test.
DISCUSSION
We have developed our integrated approach that includes near-infrared optical mapping, histological analysis, and molecular mapping to reveal the functional, structural, and molecular characteristics underlying robustness of the SAN pacemaker and conduction complex in the human heart. Near-infrared optical mapping is the only currently available method to resolve intramural activation and conduction of the human SAN pacemaker complex (2, 7) and thus can provide mechanistic insights into the function of different SAN pacemakers and SACPs. Our results show that the robust protection of SAN rhythm regulation relies on the integrative collaboration of all SAN pacemakers and conduction pathways. Consequently, to treat SAN arrhythmias, we suggest that targeted repair of specific intranodal pacemakers or SACPs may restore SAN robustness.
Intrinsic mechanisms of SAN function and dysfunctions, such as fibrosis and molecular remodeling, have been extensively studied in animal models, which established a foundation of theories on heart rate regulation and possible mechanisms of SAN arrhythmias (13, 29, 37, 38). However, these animal model studies have also highlighted interspecies variations that affect SAN function, including how the SAN excites the atrium, dynamic changes in the leading pacemaker site (39), fibrotic content and architecture (29), and protein expression profiles (1). These interspecies differences prevent the direct translation of animal model findings to clinical application. Furthermore, clinical studies of human SAN function have been limited by surface electrode mapping approaches (5, 14–17) that are unable to elaborate the detailed activity of the 3D SAN within the atrial wall. Therefore, there is a critical need for an alternative approach to obtain in-depth knowledge of the mechanisms underlying the SAN robustness directly from the unique human SAN complex (2, 19).
The redundant molecular/functional features of the human SAN complex at both the cellular and tissue level may be essential for the robust protection of heart rhythm. Single-cell studies of ion channel function (12, 40) revealed redundancies that provide robust pacemaking at the cellular level. Our study reveals that the human SAN complex has functional redundancies at a tissue level by integrating backup intranodal pacemakers and SACPs to robustly maintain heart rhythm. We found that during adenosine challenge, the pacemaker cells of the central SAN were more depressed than other regions of the SAN complex because of its high sensitivity to adenosine. Meanwhile, the SAN head or tail pacemakers, with lower sensitivity to adenosine, could maintain a slower sinus rhythm and prevent complete arrest (Fig. 2). Thus, the heterogeneous sensitivity of the multiple intranodal SAN pacemakers represents redundancies that act as a fail-safe to prevent complete SAN automaticity inhibition (38). The presence of redundant pacemakers within the human SAN complex is further supported by adenosine-mediated shifts in the preferential SACPs, which were independent from the intranodal leading pacemaker shift and ensured that SAN activation had a viable path to the atria if conduction was blocked in the preferential SACP. If the SAN would lose the backup redundancy of intranodal pacemakers and SACPs, such as due to disease or age, it would be prone to SAN exit block and arrest. Under the experimental conditions studied, we functionally mapped 2.9 ± 1.0 SACPs per heart based on the discrete anatomical locations of EAS. It could have been possible that two distinct SACPs were grouped in the same anatomic region, and there could be more SACPs not active under the conditions studied. The same SACP may serve for both exit and entrance conduction or preferentially serve as an exit SACP (lateral pathways) or an entrance SACP (superior vena cava and septal pathways) (3, 41). Moreover, we demonstrated in the human heart that SACPs also play a role in protecting intranodal pacemakers from overdrive suppression by entrance block during atrial pacing or atrial fibrillation (Fig. 5B and figs. S9 and S11). However, the fast activation of SACPs could depress their excitability and lead to exit block after termination of atrial fibrillation (fig. S11), which could be one of the mechanisms of clinical tachycardia-bradycardia arrhythmias (3, 16, 28).
Our current findings provide a framework for understanding a potential molecular basis underlying the robust regulation of human SAN pacemaker function. We revealed that the heterogeneous expression of A1R and GIRK4 proteins among different human SAN pacemaker regions may predispose the variation and severity of adenosine-induced SAN automaticity and intranodal conduction disturbances. A1R stimulation activates a G protein–coupled outward potassium current (IK,Ado) (31, 32) to produce membrane hyperpolarization, which may create a source-sink mismatch, or a failure of conduction because of inadequate electrical loading, between the intranodal pacemakers or SACPs and surrounding atrial myocardium (28, 29, 42). This hyperpolarization could suppress both conduction and automaticity in the SAN pacemaker complex, leading to pacemaker shifts, intranodal blocks, exit block, or complete automaticity arrest (Fig. 2). Our current and recent (18) ex vivo human heart studies together show that the SAN has the highest expression of A1R and GIRK4 proteins within the human atria and suggest that the SAN may be more sensitive to adenosine than the surrounding atria. The difference in A1R and GIRK4 protein expression between the SAN and atria in humans is greater than previously observed in canine hearts (29). This relatively high gradient may explain the incidence of adenosine-induced SAN arrest in humans, which is not observed in canine hearts (29). Heart-specific variations in A1R and GIRK4 protein expression among different regions of the SAN pacemaker complex (Fig. 6) may be responsible for the heart-specific directions of intranodal leading pacemaker shifts (Fig. 4) and incidence of SAN exit block versus SAN arrest during adenosine perfusion (Fig. 2). Studies in human and canine SAN have highlighted the importance of structural remodeling in SND (2, 29). These studies suggest that region-specific SAN disease remodeling, such as fibrotic infiltration (13, 37), could contribute to the intrinsic region-specific SAN conduction abnormalities and arrhythmias. Disease-induced structural remodeling may exacerbate region-specific conduction abnormalities induced by adenosine and incapacitate the fail-safe mechanisms of robust heart rhythm protection, and predispose to SND.
Greater knowledge of intranodal conduction in the human 3D SAN pacemaker complex is critically needed for successful targeted ablation of SAN tachyarrhythmias, as was shown in the clinical study by Jacobson et al. (43). However, diagnosis of region-specific SAN impairments is challenging in clinics because leading pacemaker and preferential SACP shifts are heart-specific. Data from animal models suggest that pacemaker locations can be predicted from heart rate, with fast activity from the superior SAN and slow activity from the inferior SAN (41, 44, 45). It was thought that this paradigm applied to humans, because clinical surface electrode mapping observed an inferior shift of EAS during bradycardia (46). We also demonstrated that the EAS could shift inferiorly during adenosine-mediated bradycardia in three hearts because of conduction through septal SACPs, which was not seen in any hearts at baseline (Fig. 4). However, the direction and occurrence of SACP and EAS shifts were independent of changes in leading pacemaker location, making analysis of intranodal pacemaker and conduction functions solely from atrial activation patterns challenging. Here, we circumvent clinical barriers by using optically validated analysis of clinical bipolar catheter recordings from the SAN (Fig. 2 and figs. S4 and S5). The small, preceding SAN component was more distinguishable from faster, larger atrial components when SACT was longer (Fig. 3A and fig. S5). However, further studies are needed to optimize catheter orientation, electrode size and spacing, and contact force to determine whether this type of recording is feasible in vivo. Such validated human SAN electrogram recordings (17) may help clinicians differentiate disorders of SAN conduction from disorders of impulse generation and target the specific SAN regions involved in SND and SAN arrhythmias.
The development of a “biological pacemaker” (47) based on gene or cell therapy (48) will greatly benefit from in-depth knowledge of the mechanisms involved in human heart rhythm regulation and could provide alternative treatments for SND. Currently, the only treatments available for SND are implantable artificial pacemakers that act as a crutch to support but not heal the heart because they can maintain the heart beat but have a limited response to autonomic regulation and increased physical activity. On the basis of our study, we suggest that a successful biological pacemaker should incorporate the robust, fail-safe nature of the human SAN complex. For example, instead of a single pacemaker, an integrated complex of interconnected pacemakers should be designed, each with heterogeneous sensitivities to known neuromodulators of SAN function that would allow for dynamic regulation and improved protection from rhythm failure. Targeting SACPs with gene therapy to reduce local GIRK4 expression could treat patients suffering from SAN exit block during conditions with elevated endogenous adenosine without affecting other cardiac functions.
In conclusion, robustness—through functional, structural, and molecular redundancies—is an important property of the human SAN, which allows it to efficiently maintain its automaticity and conduction even during external and internal perturbations. The use of an integrated 3D mapping approach to study human hearts is necessary to uncover the mechanisms underlying the dynamic regulation of automaticity, conduction, and the robustness of the 3D SAN complex. More detailed knowledge of the human SAN complex during normal and pathologic conditions is fundamental for the development of novel treatments for SAN arrhythmias.
MATERIALS AND METHODS
Study design
The objective of our study was to elucidate the functional, structural, and molecular mechanisms responsible for human SAN pacemaker complex robustness. We developed an integrated approach that combined optical mapping, histological analysis, and molecular mapping to study the explanted human heart in controlled laboratory experiments. Sample sizes were chosen on the basis of power calculations after previous animal and human studies (18, 29) to estimate population means and SD.
Human hearts (n = 21) were obtained from The Ohio State University Cardiac Transplant Team and Lifeline of Ohio Organ Procurement Organization in accordance with the Ohio State University Institutional Review Board. All donor hearts were randomized for functional or molecular experiments, whereas the transplanted hearts were only used for molecular experiments due to possible SAN artery disruption. The characteristics of the hearts used for functional and molecular experiments are shown in tables S1 and S3. Most of the mapped hearts did not have known cardiac diseases, but the majority had chronic hypertension or diabetes. All optical mapping experiments were conducted within 6 to 8 hours of perfusion to avoid the effects of washout and photobleaching of the dye, and edema (7). Baseline conditions of the same heart served as the control group to study the effects of subsequent adenosine or tertiapin perfusion on SAN automaticity and conduction. Hearts that did not recover from SAN depression during treatment with 10 μM adenosine were not further exposed to 100 μM adenosine. Pacing protocols were replicated at least twice for each condition in each heart. Molecular experiments were run in duplicate. Hearts were coded with six-digit case numbers that blinded investigators to the clinical etiology of the hearts until data analysis was complete.
Optical mapping experiment setup and protocol
Explanted human hearts were coronary-perfused and optically mapped, as previously described (2, 7, 18). In nine atrial preparations, we performed optical mapping of the whole atria from the epicardium (Fig. 1A) (18). In another two right atrial preparations, simultaneous dual-sided optical mapping of the SAN region was used to ensure that the intramural leading pacemaker sites were reliably mapped from the epicardium by near-infrared imaging (fig. S12) (18, 29, 49). Imaging was simultaneously conducted with two to four MiCAM Ultima-L CMOS cameras (SciMedia Ltd) with a 3.3 × 3.3 cm2 (330 μm2 resolution, 100 × 100 pixels) optical field of view sampled at 1000 frames/s. Two customized bipolar pacing electrodes were placed on the atrial surface of the heart. In addition, a far-field pseudo-atrial ECG was recorded by two silver-silver chloride plaque electrodes (9 mm diameter, World Precision Instruments) (Fig. 2).
The SAN preparations were immobilized with 10 μM blebbistatin (Tocris Bioscience) and stained with near-infrared dye di-4-ANBDQBS (10 to 40 μM; University of Connecticut Health Center) (7). Then, SAN preparations were imaged during sequential perfusion of regular Tyrode’s solution (baseline, n = 11), 10 μM (n = 11) and 100 μM (n = 7) adenosine (Sigma-Aldrich), and 10 to 100 nM tertiapin (Tocris Bioscience) (n = 8) (36). In three of the preparations, 100 μM adenosine was repeated after tertiapin perfusion (Table 1). The time interval between drug applications was 15 to 30 min. After sinus rhythm recording, all preparations were paced at a CL of 500 ms to evaluate the SNRTd/SNRTi (28) and paced incrementally until a functional refractory period was reached or atrial fibrillation was induced (49). This protocol was repeated after drug application. SACT during SAN exit block was measured as the full SACT, as described previously (29).
Optical mapping data analysis
Near-infrared optical mapping data were analyzed through a custom-made MATLAB program, as recently described (29, 49). Analysis of SAN OAP morphology and activation patterns allowed for the identification of the leading pacemaker (area of earliest SAN depolarization) and EAS, where SAN activation exited through SACPs and excited the atria. The signals recorded from cardiac tissue optically mapped with near-infrared dye carry weighted information from at least 4 mm deep, and the human SAN typically lies intramurally beneath 1 to 2 mm of atrial and connective tissue. Therefore, raw OAPs from the SAN region had double upstroke components, slow SAN and fast atrial. Thus, intramural SAN optical signals were extracted from the overlaying atrial signals, as previously described (28). SACT was measured during sinus rhythm (SACTsr; Fig. 2) and the first post-pacing SAN beat (SACTppb; Fig. 5). SNRTi was calculated as the interval from the last paced atrial beat to the first post-pacing atrial beat, which is the traditional method for SNRT measurement in the clinical setting. SNRTd was calculated as the interval from the last paced atrial beat to the first spontaneous SAN activation measured by optical mapping and SAN electrogram (50). cSNRTi/cSNRTd was calculated by subtracting the preceding SCL from SNRTi or SNRTd.
Clinical catheter recording of SAN activity
We complimented our optical SAN study with a clinical bipolar catheter to record SAN activity (17) and offer clinical translation of our ex vivo observations. Electrical activity of the SAN was recorded in five hearts with a 7-French bipolar sensing catheter (3.5 mm tip, 2 mm inter-electrode distance; Biosense Webster) placed on the atrial surface and filtered between 0.1 and 100 Hz. SAN activation was defined by SAN electrogram recordings as a slow deflection of 12 to 32 μV preceding the fast atrial deflection (Fig. 2 and figs. S4 and S5) (17). Catheter position was adjusted until a preceding deflection of >10 μV amplitude was observed, and the catheter distance was measured from the tip and base of the distal electrode and proximal electrode to the leading pacemaker and EAS (fig. S4).
Histology, immunostaining, and immunoblotting
To confirm the anatomical SAN and SACP locations of optically mapped preparations, SAN tissues (n = 5) were formalin-fixed, paraffin-embedded, and serially sectioned at 5 μm thickness from epicardium to endocardium, as described previously (2, 19, 29). Tissue sections at an average 20 μm interval were stained with Masson’s trichrome (Sigma-Aldrich), and sister sections were immunolabeled with α-actinin and Cx43 (Sigma-Aldrich). High-resolution (×20) histology images of human SAN pacemaker complex (Fig. 1B) were sequentially stacked, and artificial deformation across the z axis was minimized using a 3D image alignment approach by applying a global elastic constraint via an ImageJ plug-in (TrakEM2), as we described in our recent study (2).
To molecularly map the A1R and GIRK1/4 protein expression in the human SAN complex, tissues from 1 optically mapped and 10 unmapped hearts were collected and flash-frozen in liquid nitrogen. We extracted protein from pure SAN pacemaker tissue (19) and performed immunoblotting, as recently described (19, 29). On the basis of anatomic and functional data (2, 7), frozen SAN tissues (cryo blocks) were cut perpendicular to the epicardium into head, center, and tail blocks (~4 to 6 mm long). In keeping with previous studies (3, 7, 19), we describe the most superior third of the SAN as the head, the middle third as the center, and the inferior third as the tail (Figs. 1A and 6A). Cryosections were collected from both ends of the cryo blocks at 20 μm thickness. Masson’s trichrome staining and Cx43/α-actinin double immunolabeling were performed on cryosections, which were used to guide the SAN tissue collection from cryo block by 16-gauge (1.3 mm internal diameter) biopsy needles. SAN protein was confirmed by both negative Cx43 and positive α-actinin immunoblotting. Primary antibodies against A1R, GIRK1, GIRK4, Cx43, and α-actinin were used to quantify corresponding proteins in atrial tissue homogenates (table S4) (29). Cy5- and Cy3-conjugated goat anti-rabbit antibodies (1:2000; Jackson ImmunoResearch Laboratories) were used as secondary antibodies. Specific bands were detected on a Typhoon 9410 imager (GE Healthcare) and quantified by densitometry analysis (ImageQuant, GE Healthcare). Protein expression was normalized to GAPDH. Immunostaining showed the SAN and atrial cardiomyocyte–specific staining of A1R and GIRK1/4 antibodies used (fig. S13).
Statistical analysis
Data are presented as means ± SD. All analyses were done in R 3.3.0 (R Foundation for Statistical Computing). Comparison of treatments (baseline, 10 μM adenosine, 100 μM adenosine, and tertiapin) for SCL, SACTsr, SACTsr/SCL ratio, SNRTi, cSNRTi, SACTppb, as well as leading pacemaker shift distance and protein distributions was done using mixed models in package lme4. Treatments were considered as fixed effect, and hearts were considered as random effect. Pairwise comparisons were done with Tukey’s adjustment. Proportions of atrial pause events between different treatments were compared using a pairwise proportion test with continuity adjustment and Holm correction. Comparison of the leading pacemaker shift distance in two groups used two-sample two-tailed Welch t test. Normality assumption was verified using an Anderson-Darling test and visual inspection. Correlation between SACT prolongation (%) and SCL with leading pacemaker shift distance was evaluated using General Linear Model in Minitab 17, where SACT and SCL were treated as covariates and heart as a random effect. P values of <0.05 were considered significant. Individual subject-level data are reported in table S5.
Supplementary Material
Fig. S1. Discrete SACP locations seen functionally by ex vivo optical mapping in individual human hearts distributed into five anatomical regions.
Fig. S2. High-resolution histology sections through five SACPs in heart 118258.
Fig. S3. 3D reconstruction of the preferential lateral middle SACPs in heart 118258.
Fig. S4. SAN catheter electrode positions and distance measurements to SAN leading pacemakers.
Fig. S5. SAN catheter electrode recordings during SAN conduction through different SACPs in heart 685884.
Fig. S6. Atrial ECGs of adenosine-induced SAN arrhythmias restored and prevented by tertiapin in heart 925852.
Fig. S7. Correlations between leading pacemaker shift and the percent of SCL prolongation or the percent of SACT prolongation from baseline conditions.
Fig. S8. SNRT measurements affected by entrance block.
Fig. S9. Effects of adenosine and tertiapin on post-pacing SAN automaticity and conduction in heart 118258.
Fig. S10. Mapping of multiple SAN intranodal leading pacemakers and SACPs revealed by overdrive atrial pacing challenge.
Fig. S11. Long pause after atrial fibrillation termination due to SAN exit block in heart 987692.
Fig. S12. Simultaneous sub-epicardial and sub-endocardial SAN optical mapping during adenosine exposure (10 μM) in heart 925852.
Fig. S13. Immunostaining showing cell specificity of used antibodies in heart 474083.
Table S1. Human heart information for optical mapping experiments.
Table S2. Effects of adenosine and tertiapin on the human SAN function.
Table S3. Human heart information for molecular mapping experiments.
Table S4. Antibodies used for immunostaining and immunoblotting.
Table S5. Individual subject-level data and exact P values.
One-sentence summary.
Multiple redundant and diverse intranodal pacemakers and conduction pathways allow the human sinoatrial node to robustly maintain cardiac rhythm.
Editor’s Summary.
The human heart beats more than 100,000 times per day. Arrhythmia, or irregular heartbeat, can occur due to heart disease, changes in diet or hormones, electrolyte imbalances, or for other reasons—but these inconsistencies only infrequently lead to total loss of heart function. Li et al. uncovered how the heart is hardwired to maintain consistency. Optical and molecular mapping of human hearts ex vivo coupled with electrocardiograms and histology revealed that the sinoatrial node is home to multiple pacemakers, specialized cardiomyocytes that generate electrical heartbeat-inducing impulses. This means that multiple conduction pathways can deliver the electrical impulses required for rhythm control, so total cardiac arrest occurs only when all pacemakers and conduction pathways fail. Understanding inherent cardiac robustness may help develop treatments for arrhythmias.
Acknowledgments
We thank the Lifeline of Ohio Organ Procurement Organization and the Division of Cardiac Surgery at The Ohio State University Wexner Medical Center for providing the explanted hearts. The human heart repository program is supported by the Dorothy M. Davis Heart and Lung Research Institute. We thank P. Ssekayombya for help with the manuscript revision.
Funding: This work was supported by the NIH (HL115580 and HL135109 to V.V.F.; HL113084 to P.M.L.J.; HL084583, HL083422, and HL114383 to P.J.M.; and HL114940 to B.J.B.), American Heart Association Grant-in-Aid #16GRNT31010036, CR Webb Fund in Cardiovascular Research, and The Ohio State University Heart and Vascular Center TriFit Challenge Discovery Fund to V.V.F.
Footnotes
Author contributions: V.V.F. designed the experiments. N.L., B.J.H., A.J.I., L.V.S., T.A.C., and V.V.F. performed human heart optical mapping experiments and analyzed data. N.L., T.A.C., and J.Z. performed molecular and structural experiments and data analysis. N.L., A.J.I., and S.O.Z. performed statistical analysis. N.L., B.J.H., and V.V.F. wrote the manuscript. A. Kilic and P.M.L.J. were involved in processing human atrial tissue samples. A. Kalyanasundaram, J.P.D., B.J.B., A. Kilic, P.J.M., R.W., and J.D.H. provided critical comments and significantly improved the manuscript. All authors approved the final version.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data and materials are available in this article.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/400/eaam5607/DC1
Materials and Methods
REFERENCES AND NOTES
- 1.Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, DiFrancesco D, Baruscotti M, Longhi R, Anderson RH, Billeter R, Sharma V, Sigg DC, Boyett MR, Dobrzynski H. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation. 2009;119:1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369. [DOI] [PubMed] [Google Scholar]
- 2.Csepe TA, Zhao J, Hansen BJ, Li N, Sul LV, Lim P, Wang Y, Simonetti OP, Kilic A, Mohler PJ, Janssen PML, Fedorov VV. Human sinoatrial node structure: 3D microanatomy of sinoatrial conduction pathways. Prog Biophys Mol Biol. 2016;120:164–178. doi: 10.1016/j.pbiomolbio.2015.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fedorov VV, Glukhov AV, Chang R. Conduction barriers and pathways of the sinoatrial pacemaker complex: Their role in normal rhythm and atrial arrhythmias. Am J Physiol Heart Circ Physiol. 2012;302:H1773–H1783. doi: 10.1152/ajpheart.00892.2011. [DOI] [PubMed] [Google Scholar]
- 4.Gomes JA, Winters SL. The origins of the sinus node pacemaker complex in man: Demonstration of dominant and subsidiary foci. J Am Coll Cardiol. 1987;9:45–52. doi: 10.1016/s0735-1097(87)80081-5. [DOI] [PubMed] [Google Scholar]
- 5.Boineau JP, Canavan TE, Schuessler RB, Cain ME, Corr PB, Cox JL. Demonstration of a widely distributed atrial pacemaker complex in the human heart. Circulation. 1988;77:1221–1237. doi: 10.1161/01.cir.77.6.1221. [DOI] [PubMed] [Google Scholar]
- 6.Keith A, Flack M. The form and nature of the muscular connections between the primary divisions of the vertebrate heart. J Anat Physiol. 1907;41:172–189. [PMC free article] [PubMed] [Google Scholar]
- 7.Fedorov VV, Glukhov AV, Chang R, Kostecki G, Aferol H, Hucker WJ, Wuskell JP, Loew LM, Schuessler RB, Moazami N, Efimov IR. Optical mapping of the isolated coronary-perfused human sinus node. J Am Coll Cardiol. 2010;56:1386–1394. doi: 10.1016/j.jacc.2010.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitano H. Biological robustness. Nat Rev Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 9.Alonso A, Jensen PN, Lopez FL, Chen LY, Psaty BM, Folsom AR, Heckbert SR. Association of sick sinus syndrome with incident cardiovascular disease and mortality: The atherosclerosis risk in communities study and cardiovascular health study. PLOS ONE. 2014;9:e109662. doi: 10.1371/journal.pone.0109662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.John RM, Kumar S. Sinus node and atrial arrhythmias. Circulation. 2016;133:1892–1900. doi: 10.1161/CIRCULATIONAHA.116.018011. [DOI] [PubMed] [Google Scholar]
- 11.Verkerk AO, Wilders R. Pacemaker activity of the human sinoatrial node: An update on the effects of mutations in HCN4 on the hyperpolarization-activated current. Int J Mol Sci. 2015;16:3071–3094. doi: 10.3390/ijms16023071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verkerk AO, Wilders R, van Borren MMGJ, Peters RJG, Broekhuis E, Lam K, Coronel R, de Bakker JMT, Tan HL. Pacemaker current (If) in the human sinoatrial node. Eur Heart J. 2007;28:2472–2478. doi: 10.1093/eurheartj/ehm339. [DOI] [PubMed] [Google Scholar]
- 13.Morris GM, Kalman JM. Fibrosis, electrics and genetics. Perspectives in sinoatrial node disease. Circ J. 2014;78:1272–1282. doi: 10.1253/circj.cj-14-0419. [DOI] [PubMed] [Google Scholar]
- 14.Stiles MK, Brooks AG, Roberts-Thomson KC, Kuklik P, John B, Young GD, Kalman JM, Sanders P. High-density mapping of the sinus node in humans: Role of preferential pathways and the effect of remodeling. J Cardiovasc Electrophysiol. 2010;21:532–539. doi: 10.1111/j.1540-8167.2009.01644.x. [DOI] [PubMed] [Google Scholar]
- 15.Joung B, Hwang HJ, Pak HN, Lee MH, Shen C, Lin SF, Chen PS. Abnormal response of superior sinoatrial node to sympathetic stimulation is a characteristic finding in patients with atrial fibrillation and symptomatic bradycardia. Circ Arrhythm Electrophysiol. 2011;4:799–807. doi: 10.1161/CIRCEP.111.965897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higa S, Tai CT, Lin YJ, Liu TY, Lee PC, Huang JL, Hsieh MH, Yuniadi Y, Huang BH, Lee SH, Ueng KC, Ding YA, Chen SA. Focal atrial tachycardia: New insight from noncontact mapping and catheter ablation. Circulation. 2004;109:84–91. doi: 10.1161/01.CIR.0000109481.73788.2E. [DOI] [PubMed] [Google Scholar]
- 17.Gomes JA, Kang PS, Sherif N El. The sinus node electrogram in patients with and without sick sinus syndrome: Techniques and correlation between directly measured and indirectly estimated sinoatrial conduction time. Circulation. 1982;66:864–873. doi: 10.1161/01.cir.66.4.864. [DOI] [PubMed] [Google Scholar]
- 18.Li N, Csepe TA, Hansen BJ, Sul LV, Kalyanasundaram A, Zakharkin SO, Zhao J, Guha A, Van Wagoner DR, Kilic A, Mohler PJ, Janssen PML, Biesiadecki BJ, Hummel JD, Weiss R, Fedorov VV. Adenosine-induced atrial fibrillation: Localized reentrant drivers in lateral right atria due to heterogeneous expression of adenosine A1 receptors and GIRK4 subunits in the human heart. Circulation. 2016;134:486–498. doi: 10.1161/CIRCULATIONAHA.115.021165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N, Csepe TA, Hansen BJ, Dobrzynski H, Higgins RSD, Kilic A, Mohler PJ, Janssen PML, Rosen MR, Biesiadecki BJ, Fedorov VV. Molecular mapping of sinoatrial node HCN channel expression in the human heart. Circ Arrhythm Electrophysiol. 2015;8:1219–1227. doi: 10.1161/CIRCEP.115.003070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Headrick JP, Peart JN, Reichelt ME, Haseler LJ. Adenosine and its receptors in the heart: Regulation, retaliation and adaptation. Biochim Biophys Acta. 2011;1808:1413–1428. doi: 10.1016/j.bbamem.2010.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Watt AH. Sick sinus syndrome: An adenosine-mediated disease. Lancet. 1985;325:786–788. doi: 10.1016/s0140-6736(85)91449-7. [DOI] [PubMed] [Google Scholar]
- 22.Funaya H, Kitakaze M, Node K, Minamino T, Komamura K, Hori M. Plasma adenosine levels increase in patients with chronic heart failure. Circulation. 1997;95:1363–1365. doi: 10.1161/01.cir.95.6.1363. [DOI] [PubMed] [Google Scholar]
- 23.Alboni P, Ratto B, Cappato R, Rossi P, Gatto E, Antonioli GE. Clinical effects of oral theophylline in sick sinus syndrome. Am Heart J. 1991;122:1361–1367. doi: 10.1016/0002-8703(91)90578-6. [DOI] [PubMed] [Google Scholar]
- 24.Alboni P, Menozzi C, Brignole M, Paparella N, Gaggioli G, Lolli G, Cappato R. Effects of permanent pacemaker and oral theophylline in sick sinus syndrome the THEOPACE study: A randomized controlled trial. Circulation. 1997;96:260–266. doi: 10.1161/01.cir.96.1.260. [DOI] [PubMed] [Google Scholar]
- 25.Burnett D, Abi-Samra F, Vacek JL. Use of intravenous adenosine as a noninvasive diagnostic test for sick sinus syndrome. Am Heart J. 1999;137:435–438. doi: 10.1016/s0002-8703(99)70488-6. [DOI] [PubMed] [Google Scholar]
- 26.Resh W, Feuer J, Wesley RC., Jr Intravenous adenosine: A noninvasive diagnostic test for sick sinus syndrome. Pacing Clin Electrophysiol. 1992;15:2068–2073. doi: 10.1111/j.1540-8159.1992.tb03023.x. [DOI] [PubMed] [Google Scholar]
- 27.Fragakis N, Iliadis I, Sidopoulos E, Lambrou A, Tsaritsaniotis E, Katsaris G. The value of adenosine test in the diagnosis of sick sinus syndrome: Susceptibility of sinus and atrioventricular node to adenosine in patients with sick sinus syndrome and unexplained syncope. Europace. 2007;9:559–562. doi: 10.1093/europace/eum099. [DOI] [PubMed] [Google Scholar]
- 28.Lou Q, Glukhov AV, Hansen B, Hage L, Vargos-Pinto P, Billman GE, Carnes CA, Fedorov VV. Tachy-brady arrhythmias: The critical role of adenosine-induced sino-atrial conduction block in post-tachycardia pauses. Heart Rhythm. 2013;10:110–118. doi: 10.1016/j.hrthm.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lou Q, Hansen BJ, Fedorenko O, Csepe TA, Kalyanasundaram A, Li N, Hage LT, Glukhov AV, Billman GE, Weiss R, Mohler PJ, Györke S, Biesiadecki BJ, Carnes CA, Fedorov VV. Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual-sided intramural optical mapping. Circulation. 2014;130:315–324. doi: 10.1161/CIRCULATIONAHA.113.007086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krapivinsky G, Gordon EA, Wickman K, Velimirović B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 31.West GA, Belardinelli L. Correlation of sinus slowing and hyperpolarization caused by adenosine in sinus node. Pflugers Arch. 1985;403:75–81. doi: 10.1007/BF00583285. [DOI] [PubMed] [Google Scholar]
- 32.Belardinelli L, Giles WR, West A. Ionic mechanisms of adenosine actions in pacemaker cells from rabbit heart. J Physiol. 1988;405:615–633. doi: 10.1113/jphysiol.1988.sp017352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Csepe TA, Zhao J, Sul LV, Wang Y, Hansen BJ, Li N, Ignozzi AJ, Bratasz A, Powell KA, Kilic A, Mohler PJ, Janssen PML, Hummel JD, Simonetti OP, Fedorov VV. Novel application of 3D contrast enhanced CMR to define fibrotic structure of the human sinoatrial node in-vivo. Eur Heart J Cardiovasc Imaging. 2017;18:862–869. doi: 10.1093/ehjci/jew304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jose AD, Taylor RR. Autonomic blockade by propranolol and atropine to study intrinsic myocardial function in man. J Clin Invest. 1969;48:2019–2031. doi: 10.1172/JCI106167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jose AD, Collison D. The normal range and determinants of the intrinsic heart rate in man. Cardiovasc Res. 1970;4:160–167. doi: 10.1093/cvr/4.2.160. [DOI] [PubMed] [Google Scholar]
- 36.Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U. The G protein–gated potassium current IK,ACh is constitutively active in patients with chronic atrial fibrillation. Circulation. 2005;112:3697–3706. doi: 10.1161/CIRCULATIONAHA.105.575332. [DOI] [PubMed] [Google Scholar]
- 37.Csepe TA, Kalyanasundaram A, Hansen BJ, Zhao J, Fedorov VV. Fibrosis: A structural modulator of sinoatrial node physiology and dysfunction. Front Physiol. 2015;6:37. doi: 10.3389/fphys.2015.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res. 2000;47:658–687. doi: 10.1016/s0008-6363(00)00135-8. [DOI] [PubMed] [Google Scholar]
- 39.Shibata N, Inada S, Mitsui K, Honjo H, Yamamoto M, Niwa R, Boyett MR, Kodama I. Pacemaker shift in the rabbit sinoatrial node in response to vagal nerve stimulation. Exp Physiol. 2001;86:177–184. doi: 10.1113/eph8602100. [DOI] [PubMed] [Google Scholar]
- 40.Maltsev VA, Lakatta EG. The funny current in the context of the coupled-clock pacemaker cell system. Heart Rhythm. 2012;9:302–307. doi: 10.1016/j.hrthm.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fedorov VV, Chang R, Glukhov AV, Kostecki G, Janks D, Schuessler RB, Efimov IR. Complex interactions between the sinoatrial node and atrium during reentrant arrhythmias in the canine heart. Circulation. 2010;122:782–789. doi: 10.1161/CIRCULATIONAHA.109.935288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedorov VV, Hucker WJ, Dobrzynski H, Rosenshtraukh LV, Efimov IR. Postganglionic nerve stimulation induces temporal inhibition of excitability in the rabbit sinoatrial node. Am J Physiol Heart Circ Physiol. 2006;291:H612–H623. doi: 10.1152/ajpheart.00022.2006. [DOI] [PubMed] [Google Scholar]
- 43.Jacobson JT, Kraus A, Lee R, Goldberger JJ. Epicardial/endocardial sinus node ablation after failed endocardial ablation for the treatment of inappropriate sinus tachycardia. J Cardiovasc Electrophysiol. 2013;25:236–241. doi: 10.1111/jce.12318. [DOI] [PubMed] [Google Scholar]
- 44.Boineau JP, Schuessler RB, Roeske WR, Autry LJ, Miller CB, Wylds AC. Quantitative relation between sites of atrial impulse origin and cycle length. Am J Physiol. 1983;245:H781–H789. doi: 10.1152/ajpheart.1983.245.5.H781. [DOI] [PubMed] [Google Scholar]
- 45.Beau SL, Hand DE, Schuessler RB, Bromberg BI, Kwon B, Boineau JP, Saffitz JE. Relative densities of muscarinic cholinergic and β-adrenergic receptors in the canine sinoatrial node and their relation to sites of pacemaker activity. Circ Res. 1995;77:957–963. doi: 10.1161/01.res.77.5.957. [DOI] [PubMed] [Google Scholar]
- 46.Sanders P, Morton JB, Kistler PM, Spence SJ, Davidson NC, Hussin A, Vohra JK, Sparks PB, Kalman JM. Electrophysiological and electroanatomic characterization of the atria in sinus node disease: Evidence of diffuse atrial remodeling. Circulation. 2004;109:1514–1522. doi: 10.1161/01.CIR.0000121734.47409.AA. [DOI] [PubMed] [Google Scholar]
- 47.Rosen MR. Gene therapy and biological pacing. N Engl J Med. 2014;371:1158–1159. doi: 10.1056/NEJMcibr1408897. [DOI] [PubMed] [Google Scholar]
- 48.Hu YF, Dawkins JF, Cho HC, Marban E, Cingolani E. Biological pacemaker created by minimally invasive somatic reprogramming in pigs with complete heart block. Sci Transl Med. 2014;6:245ra94. doi: 10.1126/scitranslmed.3008681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hansen BJ, Zhao J, Csepe TA, Moore BT, Li N, Jayne LA, Kalyanasundaram A, Lim P, Bratasz A, Powell KA, Simonetti OP, Higgins RSD, Kilic A, Mohler PJ, Janssen PML, Weiss R, Hummel JD, Fedorov VV. Atrial fibrillation driven by micro-anatomic intramural re-entry revealed by simultaneous sub-epicardial and sub-endocardial optical mapping in explanted human hearts. Eur Heart J. 2015;36:2390–2401. doi: 10.1093/eurheartj/ehv233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomes JA, Hariman RI, Chowdry IA. New application of direct sinus node recordings in man: Assessment of sinus node recovery time. Circulation. 1984;70:663–671. doi: 10.1161/01.cir.70.4.663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Discrete SACP locations seen functionally by ex vivo optical mapping in individual human hearts distributed into five anatomical regions.
Fig. S2. High-resolution histology sections through five SACPs in heart 118258.
Fig. S3. 3D reconstruction of the preferential lateral middle SACPs in heart 118258.
Fig. S4. SAN catheter electrode positions and distance measurements to SAN leading pacemakers.
Fig. S5. SAN catheter electrode recordings during SAN conduction through different SACPs in heart 685884.
Fig. S6. Atrial ECGs of adenosine-induced SAN arrhythmias restored and prevented by tertiapin in heart 925852.
Fig. S7. Correlations between leading pacemaker shift and the percent of SCL prolongation or the percent of SACT prolongation from baseline conditions.
Fig. S8. SNRT measurements affected by entrance block.
Fig. S9. Effects of adenosine and tertiapin on post-pacing SAN automaticity and conduction in heart 118258.
Fig. S10. Mapping of multiple SAN intranodal leading pacemakers and SACPs revealed by overdrive atrial pacing challenge.
Fig. S11. Long pause after atrial fibrillation termination due to SAN exit block in heart 987692.
Fig. S12. Simultaneous sub-epicardial and sub-endocardial SAN optical mapping during adenosine exposure (10 μM) in heart 925852.
Fig. S13. Immunostaining showing cell specificity of used antibodies in heart 474083.
Table S1. Human heart information for optical mapping experiments.
Table S2. Effects of adenosine and tertiapin on the human SAN function.
Table S3. Human heart information for molecular mapping experiments.
Table S4. Antibodies used for immunostaining and immunoblotting.
Table S5. Individual subject-level data and exact P values.