

Abstract
Small single-stranded nucleic acid phages effect lysis by expressing a single protein, the amurin, lacking muralytic enzymatic activity. Three amurins have been shown to act like ‘protein antibiotics’ by inhibiting cell-wall biosynthesis. However, the L lysis protein of the canonical ssRNA phage MS2, a 75 aa polypeptide, causes lysis by an unknown mechanism without affecting net peptidoglycan synthesis. To identify residues important for lytic function, randomly mutagenized alleles of L were generated, cloned into an inducible plasmid and the transformants were selected on agar containing the inducer. From a total of 396 clones, 67 were unique single base-pair changes that rendered L non-functional, of which 44 were missense mutants and 23 were nonsense mutants. Most of the non-functional missense alleles that accumulated in levels comparable to the wild-type allele are localized in the C-terminal half of L, clustered in and around an LS dipeptide sequence. The LS motif was used to align L genes from ssRNA phages lacking any sequence similarity to MS2 or to each other. This alignment revealed a conserved domain structure, in terms of charge, hydrophobic character and predicted helical content. None of the missense mutants affected membrane-association of L. Several of the L mutations in the central domains were highly conservative and recessive, suggesting a defect in a heterotypic protein–protein interaction, rather than in direct disruption of the bilayer structure, as had been previously proposed for L.
Keywords: bacteriophage, lysis, Escherichia coli
Introduction
Small lytic phages with single-stranded nucleic acid genomes achieve lysis and release of the progeny virions by the expression of a single gene [1]. These phages can be broadly classified into ssDNA and ssRNA phages. The ssDNA phage φX174 is the prototype of the ubiquitous family Microviridae, with a 5.4 kb genome and 10 genes. In the φX174 genome, the lysis gene E is entirely embedded within the +1 reading frame of the essential assembly gene D [2]. The ssRNA phages defined the family Leviviridae and were traditionally further subdivided into two genera, Allolevivirus and Levivirus, represented by the prototype male-specific coliphages Qβ and MS2 (Fig. 1a). These phages are the simplest viruses, with 3.5–4.3 kb genomes and three core genes encoding an RNA-dependent RNA polymerase or replicase (Rep), a major capsid protein (Coat) and Mat, the maturation or attachment protein (one molecule per virion, named as A in MS2, A2 in Qβ). The two genera differ in the structure and role of a fourth essential gene in each case, with the Alloleviviruses having the A1 gene, a translational read-through extension of coat, and the Leviviruses having the L lysis gene, with a reading frame overlapping and out-of-frame with the end of coat and the beginning of rep. Moreover, the Mat protein of Qβ (A2) was found to be necessary and sufficient for lysis [3].
Fig. 1.

Cysteine scanning and alignment of L homologues identifies the importance of the conserved LS motif. (a) Genome maps of four ssRNA phages representing the four known genetic architectures of lysis genes. The lysis genes are highlighted in grey. (b) Lysis profiles of various cysteine scanning mutants. ▲, L; ⊞, LC29S; □, LS9C; ◊, LS15C; ♦, LS35C; Δ, LS49C; X, LS58C; ○, LL73C. (c) L-like lysis proteins from different leviviruses are aligned with respect to the conserved LS motif (yellow), preceded by a stretch of hydrophobic residues (underlined) and highly basic N-termini. Basic and acidic residues are highlighted in red and blue, respectively. GenBank accession numbers for the lysis proteins shown in (c) are as follows: MS2 (CAA23990.1), M12 (AAF19634.1), fr (CAA33137.1), GA (CAA27498.1), JP34 (AAA72211.1), KU1 (AAF67675.1), Hgal1 (YP_007237174.1), C1 (YP_007237128.1), AP205 (NP_085469.1), PP7 (NP_042306.1), PRR1 (YP_717670.1).
The genetic simplicity of the ssDNA and ssRNA phages made it straightforward to define the cistron required for lysis. The term amurin has been proposed for these lysis proteins, which lack muralytic activity but somehow subvert peptidoglycan (PG) integrity [1]. The mechanism by which amurins effect lysis remained controversial for decades. One model supported by multiple reports was that φX174 E formed a ‘membrane tunnel’ that traversed the entire envelope and allowed release of the cytosolic contents, including the assembled virions [4, 5]. Ultimately a genetic approach based on selection of host mutants resistant to the induction of the amurin genes cloned under a plasmid-borne inducible promoter was successful [6]. Remarkably, both the φX174 E and Qβ A2 amurins were found to be specific inhibitors of enzymes of the conserved pathway for murein precursor biosynthesis: A2 inhibits MurA, the first committed step of the pathway, whereas E inhibits MraY, which catalyses the formation of the first lipid-linked precursor [7–9]. More recently, a Levivirus specific for the conjugational pilus of an IncM R-factor was shown to have a lysis cistron, lysM, that evolved in a genomic location different from the position of L astride the coat–rep interface in MS2 [10]. By use of a similar approach, it was shown that LysM is a specific inhibitor of MurJ, the Lipid II flippase of Escherichia coli (K. R. Chamakura, L. T. Sham, R. M. Davis, L. Min, H. Cho, N. Ruiz, T. G. Bernhardt, R. F. Young, unpublished results). Thus, for all three of these small phages, the expression of these proteins, either from plasmid-cloned cistrons or in the context of phage infection, caused blockage of the flow of murein precursors, generally leading to lysis as a result of failed septation events. The obvious similarity to murein-specific chemotherapeutic agents has led to these lysis proteins being designated as ‘protein antibiotics’ [1].
Despite this success with three single gene lysis systems, the function of the original amurin, MS2 L, remains unclear. L is a 75-amino acid protein that has been reported to be present in the membrane fractions [11]. Work from the van Duin group using plasmid-borne L genes showed that the highly basic N-terminal half of the protein is dispensable for lytic activity, whereas C-terminal truncations were found to be non-functional [12]. An experiment with a synthetic peptide comprising the C-terminal 25 amino acids of L was reported to dissipate proton motive force (pmf) of E. coli inverted membrane vesicles and cause fluorescent dye leakage in reconstituted liposomes [13]. Surprisingly, immuno-electron microscopy studies using an antibody raised against a hybrid L protein, suggested that L was enriched in zones of adhesion between inner and outer membranes, also known as Bayer’s bridges [14]. Moreover, biochemical analysis of the murein in cells lysed by L was reported to have a decreased average chain length of glycan strands and altered cross-linking [15]. These disparate observations were not easily reconciled into a clear conceptual framework. However, it was proposed that L causes pmf-depleting lesions in the inner membrane, thereby somehow activating host autolytic enzymes such as lytic transglycoslyases and D-D endopeptidases [15]. Although this ‘autolysis’ remains as an attractive general model for L function, neither an operational schema nor molecular details for such a pathway have been forthcoming.
Recently a genetics-based approach, analogous to the approach used for E and A2, was employed to address the host target of the L amurin. Although a cellular target was not identified, it was demonstrated that the lytic function of L requires an interaction with the host chaperone DnaJ, and that this requirement could be by passed by deletion of the basic N-terminal domain [16]. Here we report a comprehensive genetic analysis and identify residues and regions of L that are critical for lysis. Results are discussed in terms of a model for L function.
Methods
Bacterial strains, culture growth, plasmids and reagents
The bacterial strains used in this study are described in Table 1 and the primers are listed in Table S1 (available in the online Supplementary Material). LB broth and LB agar were used as growth medium and were supplemented with appropriate antibiotics and inducers. When indicated, ampicillin (Amp), chloramphenicol (Cam) and arabinose were added to the growth media at concentrations of 100 µg ml−1, 10 µg ml−1 and 0.2–0.4 % (w/v), respectively. Bacterial growth and lysis were monitored as previously described [17]. The his6-Lsyn gene from pKC12 (bla araC Para :: his6-Lsyn) was amplified with primers KC19 and KC31 using Phusion DNA polymerase (New England Biolabs). The PCR product was gel-purified and digested with enzymes KpnI and HindIII and ligated into pBAD33 digested with the same enzymes to generate pKC17 (cat araC Para :: his6-Lsyn). Plasmids pKC18, pKC19, pKC20 and pKC21 were constructed by site-directed mutagenesis of the plasmid pBAD24 L with primers KC407, KC408, KC409 and KC410, respectively. Rabbit polyclonal serum raised against MS2 L peptide ‘TPASTNRRRPFKHEDC’ and goat anti-rabbit-HRP (ThermoFisher Scientific) were used as primary and secondary antibodies, respectively. Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich.
Table 1. Strains and plasmids used in this study.
| Strain/plasmid | Relevant genotype or description | Reference |
|---|---|---|
| Strain | ||
| XL1-Blue |
recA endA1 gyrA96 thi hsdR17 supE44 relA1 lac [F′ :: Tn10 proA+B+lacIq
Δ(lacZ)M15] |
Stratagene |
| MG1655 | ilvG- rfb-50 rph-1 | [34] |
| TB28 | MG1655 lacIZYA <>frt | [35] |
| Plasmid | ||
| pBAD24 | bla araC Para | [22] |
| pBAD33 | cat araC Para | [22] |
| pQ | [36] | |
| pRE-L | L gene from MS2 clone under the lambda late promoter pR' | [36] |
| pRE-LC29S | pRE-L with C29S mutation | [36] |
| pRE-LC29S, S9C | pRE-LC29S with S9C mutation | [36] |
| pRE-LC29S, S15C | pRE-LC29S with S15C mutation | [36] |
| pRE-LC29S, S35C | pRE-LC29S with S35C mutation | [36] |
| pRE-LC29S, S49C | pRE-LC29S with S49C mutation | [36] |
| pRE-LC29S, S58C | pRE-LC29S with S58C mutation | [36] |
| pRE-LC29S, L73C | pRE-LC29S with L73C mutation | [36] |
| pRE-LA45E | pRE-L with A45E mutation | [36] |
| pKC12 | cat araC Para :: his6-Lsyn | [16] |
| pKC17 | cat araC Para :: his6-Lsyn | This study |
| pBAD24 L | bla araC Para :: L | This study |
| pKC18 | bla araC Para :: LL44I | This study |
| pKC19 | bla araC Para :: LA45V | This study |
| pKC20 | bla araC Para :: LL48I | This study |
| pKC21 | bla araC Para :: LL48V | This study |
Selection of non-functional mutants
The L gene was randomly mutagenized using GeneMorph II Random Mutagenesis kit (Agilent Technologies) as per the instructions provided with the kit and cloned into pBAD24 [16]. The ligated plasmids were transformed into XL1-Blue and plated on LB agar plates supplemented with Amp and arabinose (0.2 %, w/v). Colonies that survived on the inducer plates were isolated, grown in liquid culture, and the plasmid DNA was extracted using a Qiagen mini-prep kit, and sequenced at Eton Biosciences (San Diego) with primers KC30 and KC31. The plasmids with a single-missense change in the L gene were transformed into TB28 and the lysis profiles of the mutants were compared with that of the wild-type allele. Mutants that did not cause lysis in liquid culture were scored as non-functional alleles of L.
Monitoring accumulation of L allele products
To quantify the expression of mutant alleles of L, a 1 ml sample was collected at 40 min post-induction and mixed with 111 µl cold 100 % TCA as previously described [18]. The TCA precipitates were collected by centrifugation at 13 000 r.p.m. for 10 min in a micro centrifuge. The pellets were washed three times with 1 ml cold acetone and air-dried. The dried pellets were resuspended in 2× sample loading buffer with β-mercaptoethanol and boiled for 10 min. Normalized amounts of protein (~0.4 OD550 units) were analysed by SDS-PAGE and Western blotting as previously described [18]. The antibodies against MS2 L and goat anti-rabbit-HRP were used at a 1 : 3000 dilution.
Membrane fractionation
Cultures of TB28 (500 ml) harbouring plasmids with L alleles were grown to an OD550 ~0.2, induced with arabinose, and the cultures were collected at 40 min post-induction by centrifugation at 10 000 g (Sorvall LYNX 6000 Superspeed Centrifuge) for 10 min. Pelleted cells were resuspended in ~3 ml PBS (pH 7.2) supplemented with Protease Inhibitor Cocktail (Sigma; 1 µl/35 OD550 units of original culture) and lysed by passing three times through an Aminco French Pressure cell at 16 000 psi. After intact cells were cleared from the lysate by centrifuging at 10 000 g, a 500 µl volume of the supernatant was saved as the total fraction. The rest of the supernatant (~2.5 ml) was centrifuged at 100 000 g in a TLA100.3 rotor (Beckman TL100 centrifuge) for 1 h and the supernatant and membrane pellet were collected as the soluble fractions and membrane, respectively. The three fractions were normalized to ~0.4 OD550 units, mixed with 2× sample loading buffer, boiled at 100 °C for 10 min, resolved on SDS-PAGE gels and analysed by Western blotting as described above.
Results
Discovery of a serine residue essential for function allows alignment of diverse L-like amurins
In order to facilitate both cysteine-accessibility studies and also experiments with hetero-bifunctional thiol-specific cross-linkers to identify binding partners, we conducted site-directed mutagenesis on gene L to create a variety of single-Cys alleles, including five Ser-Cys substitutions in the background LC29S where the single natural Cys codon was replaced by Ser. Although most of the substitutions had no effect on L lytic function, a single change, S49C, conferred an absolute lysis defect, both in the LC29S (not shown) and the parental L contexts (Fig. 1b), without affecting L accumulation (not shown). We noticed that this essential Ser residue could be used as part of a Leu-Ser dipeptide motif (LS motif) for aligning the lysis protein sequences, not only from the closely related F-specific Leviviruses but also from Leviviruses that use different conjugational or Type IV pili for infection (E. coli phages Hgal1 and C1; Pseudomonas phages PP7 and PRR1) and from the Acinetobacter baumannii phage AP205, where the lysis gene evolved in a distinct genomic location (Fig. 1a, c). These lysis genes have been shown to function in E. coli [19–21]. In this alignment, although sequence similarity could not be generally detected, organizing the sequences using the LS motif allowed a putative domain structure for L-like amurins to be assigned: (1) a positively charged N-terminus (4–36 aa, 3–6 net positive charges); (2) a hydrophobic sequence rich in aromatic and large beta-branched aliphatic residues (10–17 aa); (3) the LS dipeptide; and (4) a phage-specific C-terminal domain of variable length (4 to 31 residues) but either neutral or containing one net charge. Based on the previous truncation analysis of L and our results indicating the N-terminal domain confers a dependency on the host DnaJ chaperone, Domain 1 would be dispensable [16].
Near-saturating mutational analysis for L lytic function
To interrogate the new L domain structure for functional significance, a mutational analysis involving selection for non-lethal L alleles was implemented. The general approach was to create a mutagenized library of PCR-mutagenized L alleles in a plasmid in which the lysis gene was cloned under a tightly regulated arabinose-inducible pBAD promoter [22]. The transformants carrying this library were plated for surviving cells under inducing conditions. The plasmid DNAs from 396 survivors were extracted and sequenced. Several false positives also survived on the inducer plates, probably due to the lower concentration of arabinose (0.2 %, w/v) in the inducer plates. Nineteen of these alleles were wild-type and three had only silent mutations. Of the remaining 374 alleles, only 139 had a single base change, with the rest having frameshifts or multiple mutations. Of these single base change alleles, 36 mutant alleles had no lysis defect in liquid culture and were scored as false positives, leaving 103 lysis-defective, single base change alleles (Table S2). A summary of this analysis based on a total of 67 unique non-functional single mutation alleles obtained from the lysis-defect selection, as well as a mutant allele obtained by site-directed mutagenesis, is depicted in Fig. 2 and Table S2. The mutant hunt was close to saturation, as could be judged from the frequency of repeat alleles that were identified late in the procedure. Moreover, of 29 possible nonsense mutations that could be accessed by single base changes in the L gene, 23 were obtained, distributed throughout the cistron. Generally, the mutational analysis supports the domain concept proposed above. Since Domain 1 has been previously reported to be non-essential, missense alleles in this region conferring a lysis defect were not expected. Accordingly, three of the missense alleles we did obtain in Domain 1 were changes to the start codon (M1I, M1T), which ablate L production entirely, and T3I, T3S, P6L, Q8L and C29R, which exhibited a severe accumulation defect (Table S2). Since the C29S allele is fully functional, we suspect that C29R is also lysis-defective due to proteolytic instability, but because this position is within the epitope used for raising the antibody, this could not be confirmed. Only one allele, Q33H, was lysis-defective and exhibited normal accumulation (Table S2 and Fig. 3). The simplest interpretation is that even in the dispensable N-terminal Domain 1, missense changes can occur that block function; e.g. by preventing DnaJ from activating L through interaction with Domain 1. In any case, by far the highest incidence of missense changes with lysis-defective phenotypes was obtained in the LS dipeptide motif, and in the regions of Domains 2 and 4 surrounding the LS sequence, supporting the importance of the motif and its context. Moreover, a significant proportion of the inactivating missense changes were conservative, especially in terms of hydrophobic and polar character, e.g. L44V, F47L, F47Y, S49T (similar to S49C), F51L and L56F. The inactivating effect of such substitutions, coupled with their undiminished levels of protein accumulation (Fig. 3), suggests that L makes a specific protein–protein contact that depends on these mutationally sensitive residues, rather than acting as a membrane-disrupting peptide as previously suggested [13]. To further test the sensitivity of these positions to other conservative changes, we generated four additional alleles (L44I, A45V, L48I and L48V) by site-directed mutagenesis and followed their lysis profiles in liquid cultures (Fig. 4a). Interestingly, none of these changes blocked the lytic function of L, even L44I, despite the absolute lysis defect associated with L44V. The delayed onset of lysis did not uniformly correlate with the intracellular levels of L (Fig. 4b). Most of the other mutations in Domains 2 and 4 more distal to the key LS motif represent more drastic changes in side-chain character. In addition, all of the alleles tested were recessive to wild-type L, even when the mutant allele was expressed from a higher-copy plasmid (Fig. 5), suggesting that L lytic function does not require homo-oligomerization but instead requires a heterotypic interaction with a host protein. Finally, all of the defective alleles tested showed unperturbed association with the membrane fraction (Fig. 6). Taken together, the results of the mutational analysis indicate that L has a host membrane protein target and interacts with it through the LS motif and the nearby residues in essential Domains 2 and 4.
Fig. 2.

Mutational analysis of MS2 L. The L primary structure is represented as in Fig. 1(c). The four domains of L are represented by numbered boxes. Missense alleles with lysis defects but without a defect in protein accumulation are indicated above the L sequence. Missense changes that do not affect lytic function are indicated below the L sequence. Green asterisks indicate all possible codon positions where a nonsense mutation could be accessed by a single nucleotide change; underlined asterisks indicate positions where no nonsense mutants were obtained in the mutagenesis. Basic and acidic residues are highlighted in red and blue, respectively.
Fig. 3.
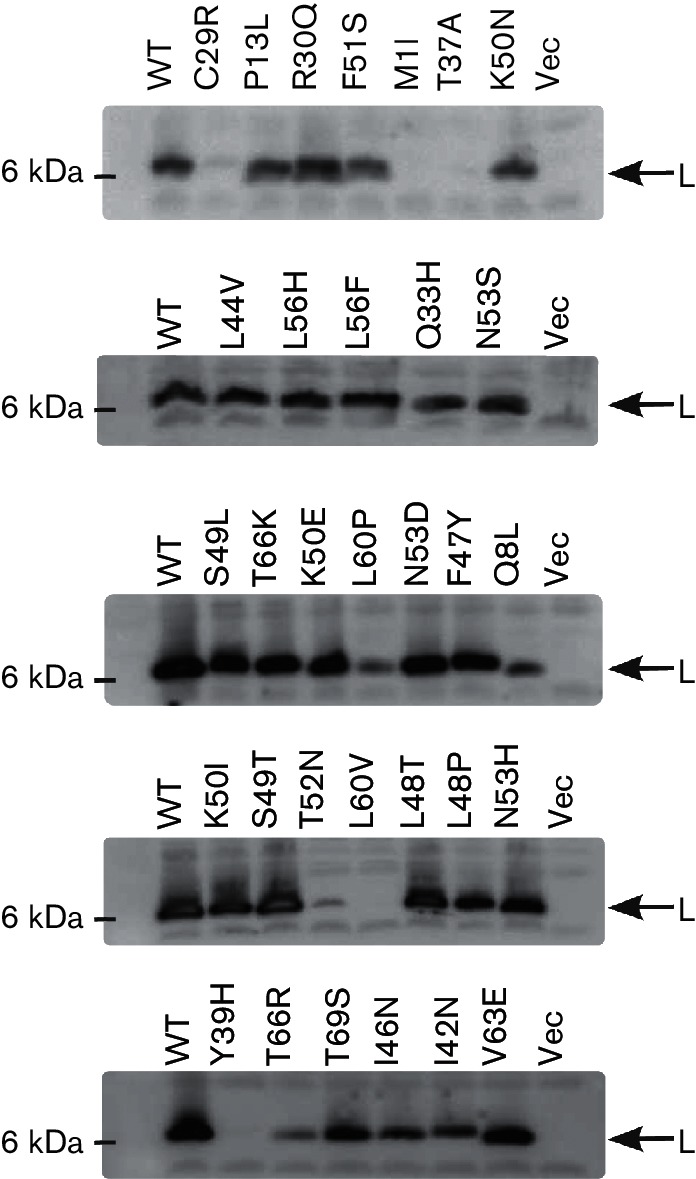
Immunoblots of mutant alleles of L. Protein samples from various L mutants were collected 5 min prior to the onset of lysis, normalized to OD550 ~0.4 units and immunoblotted with anti-L antibody. The wild-type (WT), empty vector (Vec) and the mutant alleles are indicated directly above each blot. The molecular mass standard in kDa is represented on the left. The bands corresponding to L are indicated by an arrow on the right.
Fig. 4.
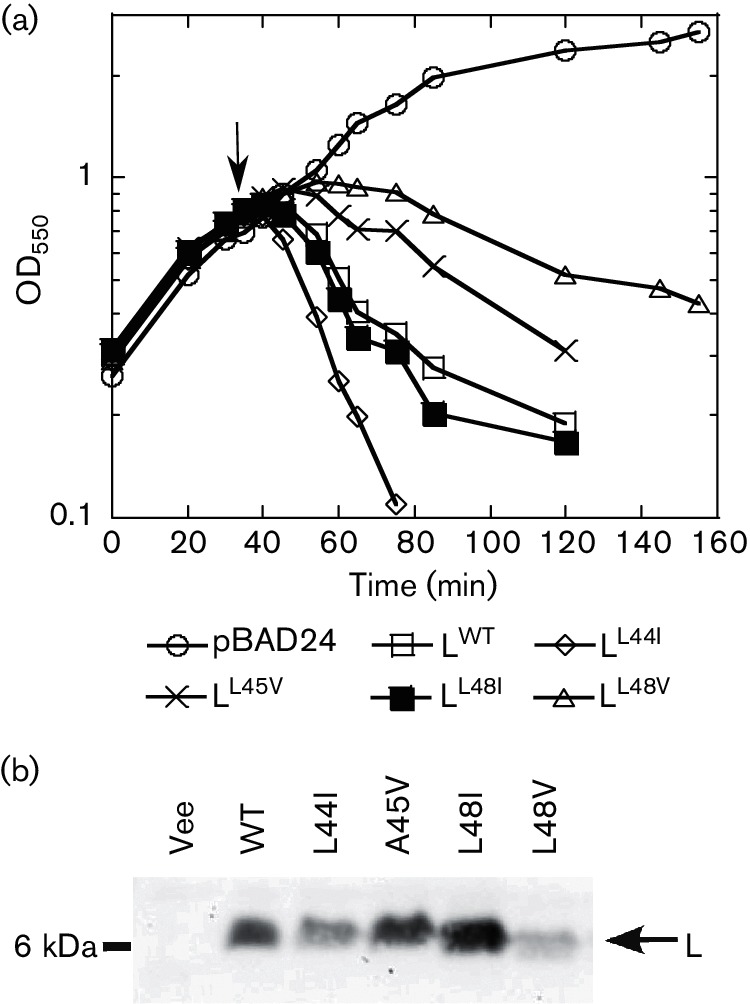
Conservative changes in domains 2 and 3 result in varied lysis profiles. (a) Lysis profiles of isoleucine and valine substitution mutants of residues at positions 44, 45 and 48 of L. ○, pBAD24 (empty vector); □, L; ◊, LL44I; X, LA45V; ■, LL48I; Δ, LL48V. (b) Protein samples were collected at the time point indicated by an arrow in (a) and immunoblotted with anti-L antibody. The molecular mass standard (in kDa) and the bands corresponding to L are indicated on the left and right sides of the blot, respectively. The various substitution mutants, wild-type (WT) and empty vector control (Vec) are indicated directly above the blot.
Fig. 5.
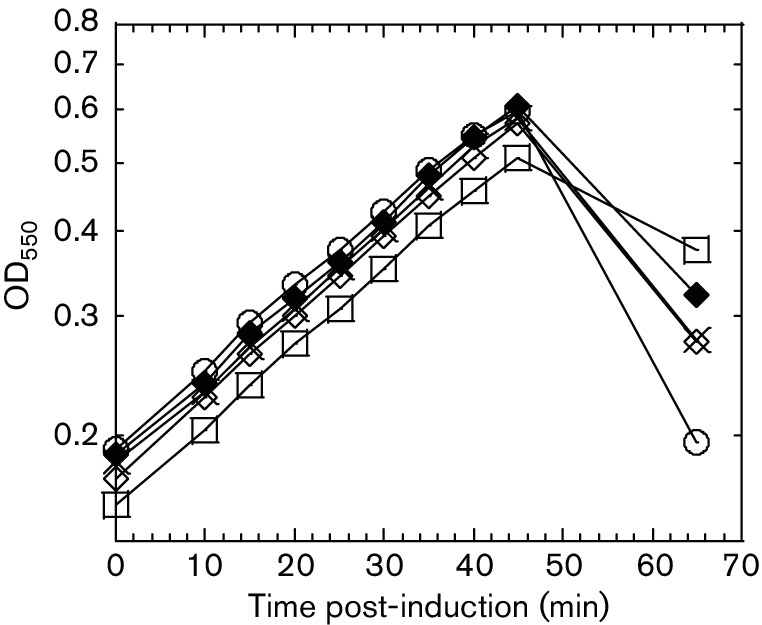
L mutant alleles are recessive. The lysis profiles of pBAD24 L alleles and pKC17 (his6-L) co-transformants in the TB28 background. Culture growth was monitored by measuring OD550; cultures were induced at time 0, and OD550 was measured at indicated time points. ○, pBAD24 L and pKC17; □, pBAD24 LK50E and pKC17; ◊, pBAD24 LL44V and pKC17; X, pBAD24 LF47Y and pKC17; ♦, pBAD24 LS49T and pKC17.
Fig. 6.
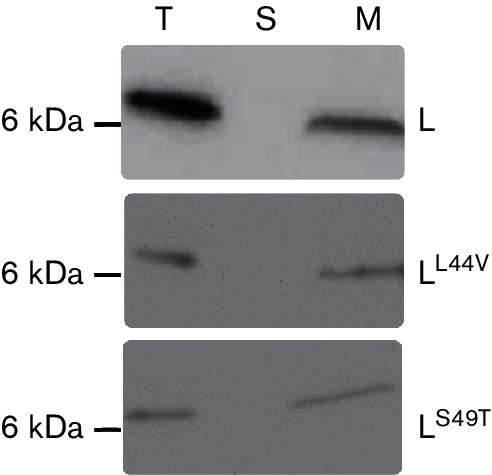
Both wild-type and mutant alleles of L are membrane-associated. Cell lysates from wild-type and mutant alleles (L44V and S49T) were fractionated into soluble and membrane fractions by high-speed ultra-centrifugation. The normalized fractions were analysed by immunoblotting with anti-L antibody. The molecular mass standard in kDa is represented on the left. Total (T), soluble (S) and membrane (M) fractions are indicated above the blot.
Discussion
New perspective on L mode of action
Despite its early identification as the lysis protein in one of the most intensively studied phages of all time, MS2 L has long eluded operational or mechanistic characterization at the molecular level. Until recently, the extant literature on L focused on a consensus in which its essential C-terminal domain acted as a membrane-disrupting polypeptide that subverted the energization of the bi-layer and somehow induced an autolytic response involving unknown host proteins [13]. We have interrogated L function by genetic analysis based on analysis of survivors of L expression from a plasmid vector. First, this approach revealed that the host DnaJ chaperone was required for L function, interacting with the N-terminal, highly basic domain of L [16]. Both this domain and thus the DnaJ interaction were shown to be dispensable, and a model was proposed in which the N-terminus acted as a regulatory domain that inhibited the interaction of L with a target protein. In this scenario, then, DnaJ acts not as the target but instead binds the regulatory domain and displaces it from its inhibitory position. Here, in the next phase of our genetic approach, we have probed the full length of the L protein by mutational analysis, revealing a distinct domain structure with differential mutational sensitivity and highlighting a dipeptide motif, Leu48-Ser49, that is conserved in heterologous amurins. In addition, many of the missense changes found to abolish lytic function without affecting accumulation or membrane-localization of L are very conservative and indicative of a protein–protein interface (e.g. L44V is defective but L44I is fully functional). Taken together, these data support a model in which the LS motif and its neighbouring subdomains form the core of an essential heterotypic protein–protein interaction domain. Moreover, the fact that all the inactivating mutations are recessive suggests that L does not act as an oligomeric lytic factor, as would be the case for models in which L is proposed to form membrane-permeating lesions.
Evolution of L-like amurins
The study of ssRNA phages began with the F-pilus-specific RNA phages of E. coli, and indeed the original classification of these Leviviridae into two genera, the Leviviruses and Alloleviviruses. This was largely dependent on the fact that the former had an independent lysis gene L, overlapping the coat and rep genes, whereas the latter did not, instead effecting lysis with a secondary function of the Mat protein (A2 in Qβ) [7, 23]. Many Leviviridae, either MS2-like or Qβ-like based on the arrangement of genes, were sequenced, and in all the MS2-like phages, the L sequences were related (Fig. 1c). However, more recently the genome sequences of seven Leviviridae specific for other retractable pili have become available, four (PRR1, Hgal1, M and C-1) for conjugational pili of R-factor plasmids [10, 19, 20], two for polar pili (PP7 for Pseudomonas; phiCb5 for Caulobacter crescentus) [24] and one for a Type IV pilus (AP205 for A. baumannii) [21]. Although there is no significant sequence similarity, the arrangements of the genes in PRR1, Hgal1, C-1 and PP7 all resemble that of MS2, with a small ORF overlapping the coat-rep gene junction. In each case, the small gene was identified as the lysis gene by testing the induction of a plasmid-based clone. However, in the other three phages, AP205, M and phiCb5, there was no ORF at the coat-rep junction [10, 21, 24]. In AP205 and M, the lysis gene was identified by cloning and testing for inducible lysis, and in both cases, its position was novel: in AP205, it was a separate gene at the 5′ end of the genome, whereas in M, it was a gene that is embedded out of frame at a different position within rep. Very recently, the lysis protein of the latter, LysM, was shown to be an inhibitor of MurJ, the Lipid II flippase of E. coli, and thus, operationally, resembles the A2 (Mat) protein of Qβ, a specific inhibitor of MurA, the first enzyme in the peptidoglycan biosynthesis pathway; in both cases, lysis is effected by inhibition of cell-wall synthesis. The work we have reported here, with the identification of the LS motif and the domain structure of L, clearly groups all the lysis proteins found encoded at the coat-rep junction and the AP205 lysis protein with MS2 L. Moreover, the implication is that the L-mediated pathway to lysis, which operates without affecting net cell-wall biosynthesis, interacts with a conserved target common to these diverse Gram-negative genera. In addition, since the L gene in AP205 is located in a completely different place in the genome compared to the L genes straddling the coat-rep boundary, it seems clear that the conservation of the LS motif and the domain structure derives from convergent evolution that occurred after speciation of a progenitor Leviviridae to the respective retractable pili. This may also be true for the L proteins in PRR1, Hgal1, C-1 and PP7, which, although occupying the same coat-rep junction region of their respective genomes, share no sequence homology. In support of this notion, other work has shown that for MS2, the region of rep within which the key hydrophobic and LS elements are encoded in the out-of-frame L cistron in MS2 is not conserved, so the emergence of L-like genes in this region may simply reflect the limited availability of genomic space for sequence variation.
Comparative analysis of the domain structure of L proteins
Based on our mutational analysis of MS2 L, we have postulated four domains as significant for lytic function of L-like amurins, including the dipeptide sequence LS as a completely conserved motif (Fig. 2). Assuming the LS sequence has to function from the same molecular context in all of the L-like proteins, then a comparative analysis of the putative domain structure in unrelated L-like amurins may make it possible to infer properties of subcellular localization, membrane topology and potential target proteins. We have chosen, as a comparison, a set of six L sequences from F-specific Leviviridae, which we designate as the homologous set, and the five L-like proteins from Leviviridae specific for other retractable pili, here designated as the heterologous set (Fig. 1c). Confidence for the meaningfulness of this comparison stems from the fact that the L sequences from four of the five heterologous phages (Hgal1, C1, AP205 and PRR1) have been shown to be necessary and sufficient for lysis if induced from a plasmid clone in E. coli. Preliminary results from this laboratory have demonstrated similar lytic capacity for the cloned L gene of PP7 [16].
The simplest conclusions from the alignment pertain to the non-essential Domain 1, where there are clearly common characteristics for all the L proteins. In all cases, Domain 1 is marked by a high content of charged residues, biased towards basic residues. Despite a wide variation in size, from seven residues in AP205 to 36 in MS2 L, Domain 1 in all L proteins has a net predicted charge of +3 to +6. The simplest notion is that, although L is firmly associated with the membrane, Domain 1 is disposed in the cytosol, consistent with its ability to bind DnaJ. Unstructured basic domains have been identified in other amurins [25, 26] and may serve in initial localization to the surface of the membrane.
The simplest approach to a comparative analysis requires the assumption that Domain 1 will prove to be non-essential for the heterologous L proteins. On this basis, the alignment of L with the five heterologous L sequences was used for seeding secondary structure analysis of L using the Jpred algorithm [27], resulting in a high-confidence prediction for alpha-helical structure from Leu37 to the C-terminus of the molecule (Fig. S1). This predicted alpha-helical domain coincides exactly with Domains 2–4, the essential domains for L function. Moreover, the TMHMM algorithm, widely used for transmembrane domain (TMD) prediction, predicts with uniformly high confidence that L and its five functional homologues are integral membrane proteins, which agrees with the behaviour of L in subcellular fractionation experiments, even when its N-terminal domain is in complex with DnaJ [16]. In each case, the predicted membrane-embedded domain begins at the start of Domain 2 and extends through the LS motif and into Domain 4. In addition, a by-product of the L mutagenesis was the isolation of two Pro substitution alleles, L44P and A45P, with normal lysis function (Fig. 2). Although proline residues are severe helix-breakers in solvent-exposed structures, they are often tolerated at the ends of TMDs [28], suggesting that this domain is embedded in the bi-layer. Taken together, these considerations suggest a structural model in which all of the essential residues in L are in a helical structure that spans the bilayer. However, the existence of a TMD is not easily reconciled with the detailed primary structure of the L proteins from the F-specific homologous set. In each of these cases, there is a charged residue (Lys50 in L) adjacent to the LS motif. Unbalanced charged residues are rarely found in TMDs in bacterial integral membrane proteins, except in some transport proteins where there is solvent exposure deep in the membrane-embedded helices [29].
Thus, although MS2 L, and, by extension, the other L proteins are undoubtedly membrane proteins, canonical requirements for a simple TMD cannot be met. Moreover, even for the heterologous L sequences, where it is not necessary to embed a charged residue to accommodate a TMD, the predicted TMD would also place the LS motif within the bi-layer, where specific interactions presumably including the H-bonding capacity of the serine hydroxyl group would be hard to envision unless the membrane is unusually distorted. However, recent findings suggest that membrane distortion might be integral to normal cell-wall growth. To account for homeostatic growth of the PG sacculus, models for the recruitment of the PG biosynthetic complexes to sites of negative membrane curvature have been proposed and supported by in vivo and in silico experiments [30, 31]. Disruption of such a system by L interacting with a membrane-embedded target protein would account for the ability of L proteins to effect lysis without inhibiting net incorporation of PG into the existing wall, as has been reported for MS2 L [32, 33]. Thus interaction of L with its target, perhaps via attraction to distorted membrane, would lead to a lytic catastrophe. To address this general hypothesis for L lysis, experiments are underway to assess the distribution of L molecules relative to the sites of PG biosynthesis.
Funding information
This work was supported by Public Health Service grant GM27099 and by the Center for Phage Technology at Texas A&M University, jointly sponsored by Texas A&M AgriLife.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgements
The constructs, preliminary results and analysis pertaining to the cysteine alleles of L have been previously reported in the dissertation of Dr Brenley McIntosh. The clerical assistance of Daisy Wilbert at Center for Phage Technology is highly appreciated.
Supplementary Data
Footnotes
A supplementary figure and two supplementary tables are available with the online Supplementary Material.
Abbreviations: PG, peptidoglycan; pmf, proton motive force; TMD, transmembrane domain.
Edited by: S. P. Diggle and M. Whiteley
References
- 1.Bernhardt TG, Wang IN, Struck DK, Young R. Breaking free: "protein antibiotics" and phage lysis. Res Microbiol. 2002;153:493–501. doi: 10.1016/S0923-2508(02)01330-X. [DOI] [PubMed] [Google Scholar]
- 2.Sanger F, Coulson AR, Friedmann T, Air GM, Barrell BG, et al. The nucleotide sequence of bacteriophage φX174. J Mol Biol. 1978;125:225–246. doi: 10.1016/0022-2836(78)90346-7. [DOI] [PubMed] [Google Scholar]
- 3.Winter RB, Gold L. Overproduction of bacteriophage Qβ maturation (A2) protein leads to cell lysis. Cell. 1983;33:877–885. doi: 10.1016/0092-8674(83)90030-2. [DOI] [PubMed] [Google Scholar]
- 4.Lubitz W, Halfmann G, Plapp R. Lysis of Escherichia coli after infection with φX174 depends on the regulation of the cellular autolytic system. J Gen Microbiol. 1984;130:1079–1087. doi: 10.1099/00221287-130-5-1079. [DOI] [PubMed] [Google Scholar]
- 5.Witte A, Wanner G, Bläsi U, Halfmann G, Szostak M, et al. Endogenous transmembrane tunnel formation mediated by φX174 lysis protein E. J Bacteriol. 1990;172:4109–4114. doi: 10.1128/jb.172.7.4109-4114.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernhardt TG, Roof WD, Young R. Genetic evidence that the bacteriophage φX174 lysis protein inhibits cell wall synthesis. Proc Natl Acad Sci USA. 2000;97:4297–4302. doi: 10.1073/pnas.97.8.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernhardt TG, Wang IN, Struck DK, Young R. A protein antibiotic in the phage Qβ virion: diversity in lysis targets. Science. 2001;292:2326–2329. doi: 10.1126/science.1058289. [DOI] [PubMed] [Google Scholar]
- 8.Zheng Y, Struck DK, Bernhardt TG, Young R. Genetic analysis of MraY inhibition by the φX174 protein E. Genetics. 2008;180:1459–1466. doi: 10.1534/genetics.108.093443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng Y, Struck DK, Young R. Purification and functional characterization of φX174 lysis protein E. Biochemistry. 2009;48:4999–5006. doi: 10.1021/bi900469g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rumnieks J, Tars K. Diversity of pili-specific bacteriophages: genome sequence of IncM plasmid-dependent RNA phage M. BMC Microbiol. 2012;12:277. doi: 10.1186/1471-2180-12-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beremand MN, Blumenthal T. Overlapping genes in RNA phage: a new protein implicated in lysis. Cell. 1979;18:257–266. doi: 10.1016/0092-8674(79)90045-X. [DOI] [PubMed] [Google Scholar]
- 12.Berkhout B, de Smit MH, Spanjaard RA, Blom T, van Duin J. The amino terminal half of the MS2-coded lysis protein is dispensable for function: implications for our understanding of coding region overlaps. EMBO J. 1985;4:3315–3320. doi: 10.1002/j.1460-2075.1985.tb04082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goessens WH, Driessen AJ, Wilschut J, van Duin J. A synthetic peptide corresponding to the C-terminal 25 residues of phage MS2 coded lysis protein dissipates the protonmotive force in Escherichia coli membrane vesicles by generating hydrophilic pores. Embo J. 1988;7:867–873. doi: 10.1002/j.1460-2075.1988.tb02886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walderich B, Höltje JV. Specific localization of the lysis protein of bacteriophage MS2 in membrane adhesion sites of Escherichia coli. J Bacteriol. 1989;171:3331–3336. doi: 10.1128/jb.171.6.3331-3336.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walderich B, Ursinus-Wössner A, van Duin J, Höltje JV. Induction of the autolytic system of Escherichia coli by specific insertion of bacteriophage MS2 lysis protein into the bacterial cell envelope. J Bacteriol. 1988;170:5027–5033. doi: 10.1128/jb.170.11.5027-5033.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chamakura KR, Tran JS, Young R. MS2 lysis of Escherichia coli depends on host chaperone DnaJ. J Bacteriol. 2017;199:e00058-17. doi: 10.1128/JB.00058-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith DL, Chang CY, Young R. The λ holin accumulates beyond the lethal triggering concentration under hyperexpression conditions. Gene Expr. 1998;7:39–52. [PMC free article] [PubMed] [Google Scholar]
- 18.Moussa SH, Kuznetsov V, Tran TA, Sacchettini JC, Young R. Protein determinants of phage T4 lysis inhibition. Protein Sci. 2012;21:571–582. doi: 10.1002/pro.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ruokoranta TM, Grahn AM, Ravantti JJ, Poranen MM, Bamford DH. Complete genome sequence of the broad host range single-stranded RNA phage PRR1 places it in the Levivirus genus with characteristics shared with Alloleviviruses. J Virol. 2006;80:9326–9330. doi: 10.1128/JVI.01005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kannoly S, Shao Y, Wang IN. Rethinking the evolution of single-stranded RNA (ssRNA) bacteriophages based on genomic sequences and characterizations of two R-plasmid-dependent ssRNA phages, C-1 and Hgal1. J Bacteriol. 2012;194:5073–5079. doi: 10.1128/JB.00929-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klovins J, Overbeek GP, van den Worm SH, Ackermann HW, van Duin J. Nucleotide sequence of a ssRNA phage from Acinetobacter: kinship to coliphages. J Gen Virol. 2002;83:1523–1533. doi: 10.1099/0022-1317-83-6-1523. [DOI] [PubMed] [Google Scholar]
- 22.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murphy FA, Fauquet CM, Bishop DHL, Ghabrial SA, Jarvis AW, et al. Virus Taxonomy: Classification and Nomenclature of Viruses: Sixth Report of the International Committee on Taxonomy of Viruses Vienna. Vienna: Springer-Verlag; 1995. [Google Scholar]
- 24.Kazaks A, Voronkova T, Rumnieks J, Dishlers A, Tars K. Genome structure of caulobacter phage phiCb5. J Virol. 2011;85:4628–4631. doi: 10.1128/JVI.02256-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernhardt TG, Roof WD, Young R. The Escherichia coli FKBP-type PPIase SlyD is required for the stabilization of the E lysis protein of bacteriophage φX174. Mol Microbiol. 2002;45:99–108. doi: 10.1046/j.1365-2958.2002.02984.x. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka S, Clemons WM., Jr Minimal requirements for inhibition of MraY by lysis protein E from bacteriophage φx174. Mol Microbiol. 2012;85:975–985. doi: 10.1111/j.1365-2958.2012.08153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drozdetskiy A, Cole C, Procter J, Barton GJ. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43:W389–W394. doi: 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yohannan S, Yang D, Faham S, Boulting G, Whitelegge J, et al. Proline substitutions are not easily accommodated in a membrane protein. J Mol Biol. 2004;341:1–6. doi: 10.1016/j.jmb.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 29.Kuk AC, Mashalidis EH, Lee SY. Crystal structure of the MOP flippase MurJ in an inward-facing conformation. Nat Struct Mol Biol. 2017;24:171–176. doi: 10.1038/nsmb.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ursell TS, Nguyen J, Monds RD, Colavin A, Billings G, et al. Rod-like bacterial shape is maintained by feedback between cell curvature and cytoskeletal localization. Proc Natl Acad Sci USA. 2014;111:E1025. doi: 10.1073/pnas.1317174111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgenstein RM, Bratton BP, Nguyen JP, Ouzounov N, Shaevitz JW, et al. RodZ links MreB to cell wall synthesis to mediate MreB rotation and robust morphogenesis. Proc Natl Acad Sci USA. 2015;112:12510–12515. doi: 10.1073/pnas.1509610112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holtje JV, van Duin J. MS2 phage induced lysis of E. coli depends upon the activity of the bacterial autolysins. In: Nombela C, editor. Microbial Cell Wall Synthesis and Autolysis. New York, NY: Elsevier Science Publishers; 1984. pp. 195–199. (editor) [Google Scholar]
- 33.Bernhardt TG. Ph.D thesis. College Station, TX: Texas A&M University; 2001. Breaking free: small phages inhibit murein synthesis to lyse their host. [Google Scholar]
- 34.Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a sex-factor-affinity site in E. coli as γδ. Cold Spring Harb Symp Quant Biol. 1981;45:135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- 35.Bernhardt TG, de Boer PA. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McIntosh BK. Ph.D thesis. College Station, TX: Texas A&M University; 2008. Bacteriophage MS2 L protein: genetic and biochemical charecterization. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.