

Abstract
Amyloids have been identified as functional components of the extracellular matrix of bacterial biofilms. Streptococcus mutans is an established aetiologic agent of dental caries and a biofilm dweller. In addition to the previously identified amyloidogenic adhesin P1 (also known as AgI/II, PAc), we show that the naturally occurring antigen A derivative of S. mutans wall-associated protein A (WapA) and the secreted protein SMU_63c can also form amyloid fibrils. P1, WapA and SMU_63c were found to significantly influence biofilm development and architecture, and all three proteins were shown by immunogold electron microscopy to reside within the fibrillar extracellular matrix of the biofilms. We also showed that SMU_63c functions as a negative regulator of biofilm cell density and genetic competence. In addition, the naturally occurring C-terminal cleavage product of P1, C123 (also known as AgII), was shown to represent the amyloidogenic moiety of this protein. Thus, P1 and WapA both represent sortase substrates that are processed to amyloidogenic truncation derivatives. Our current results suggest a novel mechanism by which certain cell surface adhesins are processed and contribute to the amyloidogenic capability of S. mutans. We further demonstrate that the polyphenolic small molecules tannic acid and epigallocatechin-3-gallate, and the benzoquinone derivative AA-861, which all inhibit amyloid fibrillization of C123 and antigen A in vitro, also inhibit S. mutans biofilm formation via P1- and WapA-dependent mechanisms, indicating that these proteins serve as therapeutic targets of anti-amyloid compounds.
Keywords: amyloid, biofilm, Streptococcus mutans, amyloid inhibitors, biofilm inhibitors, genetic competence, bacterial cell density, sortase, P1, WapA, SMU_63c
Introduction
The concept of pathogenic amyloid was historically established in the context of neurodegenerative diseases [1]; however, functional amyloids have now been identified in organisms ranging from bacteria to humans [2, 3]. Amyloid represents a fibrous cross-β sheet quaternary structure of ordered peptide or protein aggregates that demonstrate common biophysical properties [4]. Functional amyloids exhibit the same biophysical characteristics as pathogenic amyloids, including binding of the amyloidophilic dyes thioflavin T (ThT) and Congo red (CR) and characteristic coloured birefringence when stained with CR and viewed under cross-polarized light [5]. When viewed by electron microscopy (EM), amyloid fibres are 5–13 nm in diameter and range in length from a few nanometers to several micrometers [6]. In their native environments, bacteria tend to exist in biofilms where they are encased in an extracellular matrix (ECM) of their own making. Over 40 % of biofilm organisms are estimated to produce amyloids [7]. The list of bacterial proteins that form functional amyloids is growing and includes the curli fibers of Escherichia coli and Salmonella sp. [8], the phenol-soluble modulins (PSMs) and the Bap protein from Staphylococcus aureus [9, 10], FapC from Pseudomonas aeruginosa [11] and TasA from Bacillus subtilis [12, 13].
Amyloidfibres are reported to have the tensile strength of steel and are resistant to degradation by proteases and detergents. It is believed that these amyloid characteristics contribute to biofilm architecture and integrity [3]. Identification of amyloid-forming proteins and molecules that inhibit their fibrillization therefore represents a promising avenue for effective therapeutic intervention against biofilm-dwelling organisms [8, 14]. Numerous small polyphenol molecules have been studied as drug candidates due to their capacity to inhibit fibril formation, particularly of neuropathogenic amyloid proteins [15, 16]. More recently, two additional small molecule amyloid inhibitors, AA-861, a derivative of benzoquinone and parthenolide, a sesquiterpene lactone, were shown to inhibit biofilm formation by B. subtilis, Bacillus cereus and E. coli and also inhibited fibrillization of their respective amyloid-forming proteins in vitro [14]. In addition, the polyphenolic compound, tannic acid (TA), has been shown to inhibit biofilm formation by S. aureus [17].
Streptococcus mutans, an established aetiologic agent of dental caries [18], is a quintessential biofilm dweller. We showed previously that amyloid is produced by laboratory and clinical strains of S. mutans, and is detectable in dental plaque [19]. Furthermore, a known inhibitor of amyloid fibril formation, epigallocatechin-3-gallate (EGCG), inhibits S. mutans biofilm. Our initial work identified adhesin P1 (Ag I/II, PAc) [20] as an amyloidogenic protein; however, S. mutans lacking P1 demonstrates residual amyloid-forming properties, indicating there are others. While secreted proteins present in the culture supernatant of a P1-deficient mutant can be triggered by mechanical agitation to form amyloid in vitro, a mutant lacking sortase, the transpeptidase that covalently links full-length P1 and several other proteins to the cell wall peptidoglycan, is defective in amyloid formation within biofilms [19]. These findings suggest a potential nucleation requirement at the surface of biofilm-grown cells, and that other amyloidogenic proteins may also be sortase substrates.
In the current study, we identified S. mutans wall-associated protein A (WapA) and an uncharacterized secreted protein, SMU_63c, as capable of amyloid fibrillization. We also determined that the naturally occurring C-terminal breakdown product of P1 (C123), known originally as antigen II (AgII) [21], represents the amyloidogenic moiety of P1. Immunogold electron microscopy (EM) experiments employing specific antibodies identified all three amyloidogenic proteins within the fibrous structure of the ECM of biofilms. We further demonstrate that amyloid inhibitors, including the polyphenolic small molecules TA and EGCG, as well as the benzoquinone derivative AA-861, inhibit amyloid fibrillization of C123 and antigen A (AgA) in vitro, as well as biofilm formation by S. mutans via P1-, WapA- and, to a lesser extent, SMU_63c-dependent mechanisms.
Methods
Bacterial strains and growth conditions
S. mutans serotype c strains NG8 and UA159 were used in this study. The P1-deficient mutant PC3370 [22] was used to identify non-P1 amyloid-forming proteins. Gene-deletion mutants were generated in both NG8 and UA159 backgrounds. The bacterial strains used in this study are listed in Table S1 (available in the online Supplementary Material). S. mutans was grown in either Todd–Hewitt yeast extract (THYE) (Beckton, Dickinson and Co., Sparks, MD), chemically defined medium (CDM-glucose) [23], biofilm medium (BM) [24] containing either glucose or sucrose or both, or Terleckyj-defined medium (TDM-glucose) [25] in 5 % CO2 at 37 °C. E. coli strains DH5α and Top10 (Invitrogen, Life Technologies) were used for plasmid preparation and purification. Strains BL21 Star (DE3) (Invitrogen, Life Technologies) and VS39 [26] were used for protein expression.
Fractionation of secreted proteins from PC3370
Cells from a stationary phase PC3370 culture grown in TDM (glucose) were removed by centrifugation and spent medium (containing 0.1 % NaN3) was filtered through a 0.2 µm Rapid-Flow Nalgene filter (Thermo Scientific), concentrated 100-fold using an Amicon stirred cell concentrator (EMD Millipore) with a 10 kDa cut-off membrane (Amicon Ultrafiltration Disc, Millipore Cat. no. 13642), followed by dialysis into 25 mM Tris buffer, pH 8.0. Proteins were fractionated by ion exchange chromatography on an ÄKTA Purifier system (GE Healthcare) using a 5 ml HiTrap DEAE FF column (GE Healthcare) with 25 mM Tris, pH 8 as the binding/wash buffer and 25 mM Tris, 1M NaCl, pH 8 as the elution buffer.
ThT fluorescence assays
The ThT fluorescence assays were performed as described previously [19] with slight modifications. Aliquots (250 µl) from either concentrated dialyzed S. mutans spent medium, ion-exchange fractions derived from spent medium, or purified recombinant proteins (~1 mg ml−1) were stirred at 4 °C in a 1.5 ml Eppendorf tube using a 10×3 mm micro stir bar (Fisher Scientific) for 3–5 days on a Fisher Scientific Isotemp stir plate at the highest setting. For evaluation of amyloid inhibition by TA, AA-861, EGCG or parthenolide, the purified proteins were stirred in the presence of the inhibitor at the stated inhibitor concentration. AA-861 was dissolved in dimethyl sulfoxide (DMSO) as a 10 mM stock, EGCG was dissolved in PBS (potassium phosphate buffer, 20 mM, pH 7.2, sodium chloride, 0.9 %, w/v) as a 10 mM stock, TA was dissolved in 95 % ethanol as a 10 mM stock, and parthenolide was dissolved in DMSO as a 10 mM stock. Single-use aliquots of AA-861, EGCG and parthenolide were stored at −20 °C. TA was made fresh for each use, and the tubes were wrapped in aluminum foil to protect the TA from light. For ThT measurements, ThT was added to 200 µl of stirred or unstirred protein samples for a final ThT concentration of 2 µM, vortexed for a few seconds and pipetted onto 96-well all-black flat-bottom Microfluor plates (Thermo Scientific Nunc Cat. # 7605) and incubated in the dark for 15–30 min. Fluorescence intensities were measured using a BioTek Synergy 2 spectrophotometer. Excitation was at 440 nm, and fluorescence emission intensity was measured at 485 nm. All experiments were done in triplicate or quadruplicate, and the background fluorescence intensity of 2 µM ThT solution alone was subtracted from that of protein samples.
CR birefringence assay
CR birefringence was evaluated as described previously [19]. Protein samples were stirred as for the ThT assay, and 50–100 µl of the aggregated samples were pelleted in a microcentrifuge at maximum speed. Pellets were resuspended in 10 µl of CR solution and incubated for 30–60 min at room temperature. Visualization was done using a Zeiss Scope A1 equipped with a computer-controlled ProgRes C5 Jenoptik inverted camera equipped with cross-polarized filters.
Mass spectrometric identification of potential amyloid-forming proteins
Ion-exchange fractions were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and the major bands were excised and sent to the proteomics facility of the Interdisciplinary Center for Biotechnology Research of the University of Florida for liquid chromatography–mass spectrometry (LC-MS) analysis and protein identification. S. mutans proteins with a predicted secretion signal sequence were considered for further evaluation.
Cloning, expression and purification of recombinant proteins
The SignalP 4.1 prediction tool [27] was used to predict the signal sequence cleavage sites of GbpA, GbpC, WapA, GtfC, SMU_63c and SMU_2147c. Genes encoding each protein were PCR-amplified, omitting the signal sequences, using S. mutans UA159 genomic DNA as template. PCR primers and their corresponding restriction sites are listed in Table S2. Details of the cloning experiments, expression and purification of recombinant proteins are found in the supplementary information.
Generation of anti-SMU_63c and anti-AgA polyclonal antibodies
Polyclonal rabbit antisera were raised against purified recombinant AgA and SMU_63c using the services of Lampire Biological Laboratories, Everett, PA. Specificity was confirmed by ELISA and Western blot analysis.
Biofilm production and inhibition
Biofilm production was analysed using a 96-well plate assay employing crystal violet (CV) staining and spectrophotometry as described previously [19, 28]. The BM [24] contained 0.8 % sucrose, or 0.8 % glucose, with the indicated inhibitors or solvent-only controls.
EM
Transmission electron microscopy (TEM) analysis of aggregated recombinant proteins was done as previously described [19] using a 400-mesh formvar-carbon coated grid, FCF400-Cu-UB (Electron Microscopy Sciences, Hatfield, PA) stained with 1 % uranyl acetate for 30 s. Imaging of P1-A3VP1, GbpA, SMU_63c and SMU_2147c was done on a Hitachi H-7000 transmission electron microscope (Hitachi High Technologies America, Schaumburg, IL) with digital images acquired using a Veleta 2k×2k camera and iTEM software (Olympus Soft-Imaging Solutions, Lakewood, CO). TEM imaging of P1-C123, GtfC and AgA was done on a Tecnai G2 Spirit Twin (FEI, Hillsboro, Oregon) with digital images acquired using a Gatan UltraScan 2k×2k camera and Digital Micrograph software (Pleasanton, CA).
Analysis of S. mutans biofilms was done by field emission-scanning EM (FE-SEM) as previously described [29, 30]. The BM contained 16 mM glucose and 4 mM sucrose, with or without 10 µM TA or 50 µM AA-861, or the corresponding ethanol or DMSO solvent controls. Biofilms were grown on vertically deposited hydroxylapatite (HA) disks in 12-well culture plates (Corning, New York) for 24 h [31], and analysed using a Hitachi 4800 field emission-scanning electron microscope (Hitachi, Tokyo, Japan) under 5 kV accelerate voltage.
RESULTS
Screening for non-P1 amyloid-forming proteins
To identify non-P1 amyloid-forming proteins, extracellular proteins from a P1-deficient S. mutans mutant were harvested and fractionated by ion-exchange chromatography. Five distinct peaks were observed (not shown). Aliquots from pooled fractions corresponding to each peak, as well as the flow-through containing unbound proteins, were mechanically agitated (stirred) to facilitate nucleation and amyloid aggregation and tested for ThT binding. Pooled fractions corresponding to three of the five peaks demonstrated a significant increase in ThT fluorescence upon stirring (Fig. 1a). Stirred samples that tested positive for ThT-binding also exhibited CR-induced birefringence (not shown). Corresponding unstirred ion-exchange fraction samples were analysed by SDS-PAGE (Fig. 1b). Major bands from the samples with and without a substantial increase in ThT fluorescence after stirring (Fig. 1a, b) were excised for LC-MS analysis and protein identification. The proteins identified were: glucan binding protein A (GbpA), GbpB, WapA, β-d-fructosyl transferase (β-DFT), glucosyltransferase C (GtfC), SMU_2147c and SMU_63c (Table S3). All are secreted proteins with predicted signal sequences. β-DFT was present in almost all fractions, but was most abundant in fractions with little or no ThT binding activity (A8/A9). Of the proteins identified, WapA was the only additional sortase substrate.
Fig. 1.

Identification and characterization of amyloid-forming proteins. Spent medium (TDM-glucose) from a P1-deficient S. mutans mutant was concentrated and fractionated by ion-exchange chromatography. (a) ThT fluorescence assay showing ThT uptake by unstirred and stirred (aggregated) samples of pooled fractions corresponding to ion exchange peaks, as well as load and flow-through (FT) fractions. (b) SDS-PAGE of unstirred samples shown in (a), as well as the starting material (load), FT fractions and starting material prior to filtration. (c) SDS-PAGE of purified recombinant protein candidates. (d) ThT fluorescence assay of unstirred and stirred recombinant proteins. ThT uptake was measured at pH 4 and pH 8. Error bars represent the mean±sem of four independent replicates.
Purification and in vitro characterization of potential amyloid-forming proteins
Genes encoding GbpA, GtfC, SMU_63c, SMU_2147c and WapA [specifically the natural truncation polypeptide corresponding to antigen A (aa 30–323)] [32, 33], were cloned and expressed in E. coli and the recombinant proteins were purified and confirmed by SDS-PAGE (Fig. 1c). Because an S. mutans sortase-deficient mutant is defective in amyloid formation [19], an additional sortase substrate, GbpC, which was not identified in our initial screen but demonstrated high amyloid prediction scores by computational analyses [34, 35], was also evaluated in these experiments. Unlike the proteins listed above, for which little structural information is available at present, the complete tertiary structure of P1 has been modelled based on crystal structures of several partial polypeptides [36–38]. Importantly, it is now recognized that the C-terminal region of P1 exists in two separate forms: firstly, in the context of the full-length molecule where it is positioned in proximity to the cell wall, and secondly, as a naturally processed and isolated C123 polypeptide that associates with covalently attached full-length P1 on the cell surface [39]. Two previously described recombinant polypeptides, A3VP1 and C123, each of which contains one of P1′s two β-rich globular regions, were evaluated in the current study for independent formation of amyloid fibrils.
We found previously that A3VP1 and C123 both exhibit measurable increases in ThT uptake after stirring at neutral pH [19], hence these polypeptides were included as controls. Similar to P1-A3VP1 and C123, GtfC and AgA exhibited a notable increase in ThT fluorescence when stirred at pH 8, whereas the other proteins tested, including GbpA, GbpC, SMU_63c and SMU_2147c, did not (Fig. 1d). Considering that S. mutans produces large quantities of organic acids (acidogenicity), and can survive and thrive at low pH (aciduricity) [40], we also evaluated induction of aggregation at pH 4. A3VP1, C123, GtfC and AgA also demonstrated ThT uptake following stirring at pH 4, albeit less than when stirred at pH 8. GbpC did not exhibit ThT uptake at either pH, while GbpA showed a slight uptake of ThT at both pH 4 and 8. SMU_63c demonstrated obvious ThT uptake at pH 4, but not at pH 8, and also demonstrated uptake of ThT at pH 4 even when the protein was not stirred.
To confirm whether aggregated proteins that tested positive in the ThT uptake assay were amyloid-like in nature, the samples were also analysed using a more specific indicator, CR-induced birefringence. When stirred at pH 8, and to a lesser extent when stirred at pH 4, aggregated C123 and AgA exhibited orange or bright yellow-green birefringence when observed under cross-polarized filters (Fig. 2). SMU_63c also exhibited bright green birefringence, but only when tested at pH 4. In contrast, A3VP1 did not exhibit birefringence when tested at either pH. The stirred GbpA, GtfC and SMU_2147c samples were also negative for CR-induced birefringence (Fig. S1). GbpC also did not demonstrate detectable CR-induced birefringence upon stirring (not shown).
Fig. 2.
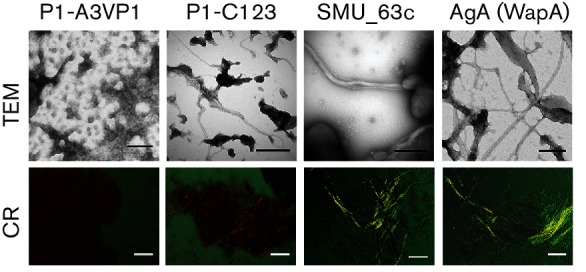
CR-induced birefringence and visualization of amyloid fibrils by TEM. Aggregated recombinant proteins (stirred at pH 8 for A3VP1, C123 and AgA, and pH 4 for SMU_63c) were evaluated for the presence of amyloid by CR-induced birefringence (bottom panel) and TEM (top panel). Scale bars represent 50 µm (bottom panel), and 200 nm (top panel). Orange or yellow-green birefringence, and amyloid fibrils, were observed for the P1-C123, SMU_63c and WapA-AgA, but not for the P1-A3VP.
Visualization of amyloid fibrils by TEM
To visualize the amyloid fibrils, or lack thereof, samples were also analysed by TEM (Fig. 2 and S1). We showed previously by TEM that full-length P1 forms amyloid fibrils [19]. In the current study, we also investigated whether either A3VP1 or C123 could form fibrils independently. Confirmative of the CR-induced birefringence results, we observed characteristic amyloid fibrils, ranging from ~7 to 12 nm wide, in the aggregated C123, AgA and SMU_63c samples (Fig. 2). The fibrils ranged in length from ~15 nm to several micrometers and appeared most abundantly in the AgA sample. The C123, SMU_63c and AgA fibrils also appeared to assemble into higher-order structures ~90–150 nm wide. Also consistent with the CR birefringence results, the aggregates formed by A3VP1 (Fig. 2), GbpA, GtfC and 2147c (Fig. S1) were amorphous and non-fibrillar. Although positive in the ThT uptake assay, neither A3VP1 (Fig. 2) nor GtfC (Fig. S1) displayed detectable fibrils by TEM or demonstrated CR-induced birefringence, underscoring the importance of confirming amyloid formation by means other than dye-uptake screening assays. TEM analyses also suggested the presence of non-fibrillar aggregates in the stirred AgA, SMU_63c and C123 samples. Amyloid fibrils are insoluble in SDS, therefore stirred samples of C123 and AgA were boiled in 2 % SDS and analysed by SDS-PAGE to estimate the proportion of amyloid present (not shown). By densitometry comparison to unstirred samples, ~40–50 % of the stirred C123 and AgA proteins were solubilized by boiling in SDS and able to migrate into the gel. Therefore, the insoluble amyloid fraction represented ~50–60 % of the total protein in the stirred samples.
To further evaluate the monomeric-to-amyloid transition in the stirred samples, far-UV circular dichroism (CD) was used to assess potential conformational changes upon fibrillization (Fig. S2). The non-amyloidogenic polypeptide A3VP1 displayed a CD spectrum indicative of both β-sheet and α-helical content at pH 8. This did not change upon stirring. At the non-amyloidogenic pH of 8, both unstirred and stirred SMU_63c samples displayed a CD spectrum indicative of α-helical content. However, at the amyloid-promoting pH of 4 there was a pronounced change in the CD spectrum of SMU_63c, indicating a conformational change to a predominantly β-sheet structure. When stirred at the pH shown in the other experiments described above to support amyloid formation, each S. mutans amyloid-forming protein demonstrated a change in its CD spectra.
Effect of gene deletions on biofilm formation and architecture
Given the clear ability of P1, AgA and SMU_63c to form amyloid in vitro, the genes encoding them were deleted individually and in combination to generate a panel of single, double and triple mutants. Gene deletions were confirmed by DNA sequence analysis and Western blot analysis of SDS extracts of S. mutans whole cells (Fig. S3a). P1 and SMU_63c migrated at the appropriate sizes (185 and 75 kDa, respectively) in those strains still containing the corresponding gene. SMU_63c has a theoretical molecular mass of 59 kDa, although SDS-PAGE (Fig. 1c) and Western blot analysis (Fig. S3a) showed that both recombinant and native proteins migrate as a ~75 kDa band. As stated above, WapA exists in both an unprocessed form that migrates at 75 kDa, and as a truncated amino-terminal AgA fragment (32 kDa) [32] that corresponds to the recombinant polypeptide used in the current study. Deletion of one, two or all three genes encoding the amyloid-forming proteins did not significantly affect planktonic growth of S. mutans in the presence (not shown) or absence (Fig. S3b) of oxygen.
S. mutans biofilm formation is mediated by both sucrose-dependent and independent mechanisms [18, 41]. Biofilm development of the WT and mutant strains was quantified by CV staining and spectrophotometry [28] (Fig. 3a). When S. mutans was grown in a BM containing glucose as the sole carbon source, the single spaP and wapA deletion mutants produced significantly less biofilm compared to the WT strain. The effect of elimination of these amyloid-forming proteins was even more pronounced in the ∆spaP/∆wapA double-deletion strain and the ∆spaP/∆smu_63 c/∆wapA triple-deletion strain. In contrast to deletion of spaP or wapA, deletion of smu_63 c resulted in a slight increase in biofilm production compared to the WT strain. In addition, deletion of smu_63 c in combination with spaP or wapA rescued the spaP and wapA deletion phenotypes, resulting in biofilm production comparable to WT levels. As mentioned above, P1 and WapA are both sortase substrates. An S. mutans mutant lacking srtA demonstrated diminished biofilm formation comparable to mutant strains lacking both P1 and WapA. In contrast, when the cells were grown in a BM containing sucrose as the sole carbon source, the effects of elimination of the amyloid-forming proteins were masked by glucan production, resulting in no significant differences in biofilm accumulation among the different strains, except when glucan production was disrupted by deletion of the genes encoding the glucosyl transfer enzymes glucosyltransferase B, C and D (∆gtfBCD) (Fig. 3b).
Fig. 3.

Evaluation of biofilms produced by S. mutans WT and single-, double- and triple-deletion mutants by CV staining and FE-SEM. (a and b) CV assays of cells grown in BM containing glucose (a) or sucrose (b) as the carbon source. (c) Visualization of biofilms grown on 16 mM glucose and 4 mM sucrose by FE-SEM. Scale bars represent 2 µm. *P<0.05; ***P<0.001; ****P<0.0001.
FE-SEM was used to evaluate the effects of the various gene deletions on biofilm formation and structure on hydroxyapatite discs. As expected, biofilms of the single spaP and wapA deletion mutants appeared to have fewer cells, when compared to the WT (Fig. 3c). Such decreases of cellular accumulation were more evident in the ∆spaP/∆wapA and ∆spaP/∆smu_63 c/∆wapA mutants, which further corroborated the results of the CV assays. We also observed a pronounced and unexplained increase in the non-cellular content of the ∆wapA mutants. It had been reported previously that deletion of wapA causes impaired biofilm formation [42]. Our current observations reiterate the importance of both P1 and WapA to biofilm production and architecture.
In contrast to the WT strain, deletion of smu_63 c, singly or in combination with spaP or wapA, resulted in biofilms exhibiting a dense mass of cells piled on top of each other. There was no observable effect of smu_63 c deletion on growth rate or final cell yield when S. mutans was grown in broth culture (Fig. S3b). When the Δsmu_63 c mutant was complemented by reintroduction of this gene into the chromosome, the biofilm cell density reverted to the WT phenotype (Fig. S4). These results suggest that SMU_63c plays a role in the regulation of S. mutans cell density within biofilms where cell–cell or cell–surface interactions would occur. Interestingly, the hyper-density phenotype was only overcome when both spaP and wapA were also deleted in combination with smu_63 c (triple mutant), which resulted in a biofilm with sparsely distributed cells.
Further evaluation of SMU_63c
SMU_63c has not yet been characterized. A protein blast analysis of its predicted amino acid sequence showed SMU_63c to be limited to a small subset of streptococcal bacteria. It is highly conserved, with 99–100 % identity in all S. mutans strains sequenced to date [43]. Additionally, SMU_63c-like proteins, ranging from 45 % to 76 % identity, were identified among S. macacae, S. criceti, S. sobrinus, S. gallolyticus, S. oligofermentas and S. salivarius. It is predicted by SignalP 4.1 to have a long N-terminal signal sequence of 49 amino acids. Topology analysis using TMPred (www.ch.embnet.org/software/TMPRED_form.html) predicts two to four transmembrane segments, but none were found using the TMHMM prediction tool [44]. Our experimental analysis showed that a significant amount of SMU_63c is released into the medium during planktonic growth, although some remains associated with the cell surface where it is extractable with SDS (Fig. S3a).
Given that SMU_63c appears to regulate cell density within biofilms, we examined the context of smu_63 c within the S. mutans genome. The gene is located directly downstream of the ComRS operon [45], albeit in the opposite orientation (Fig. 4a). It has been shown that deletion of comR or comS severely reduces transformation efficiency in S. mutans [45]. Considering the proximity of smu_63 c to these competence-related genes, we analysed the expression of smu_63 c in response to the two peptide pheromones that trigger genetic transformation in S. mutans: synthetic competence-stimulating peptide (CSP) [46] or ComX-iducing peptide (XIP) [45]. RT-PCR analyses revealed that smu_63 c expression is up-regulated more than fourfold in THYE planktonic cultures treated with 0.4 µM CSP (Fig. 4b), and approximately five- and 10-fold in CDM (1 % glucose) planktonic cultures treated with 1 or 5 µM synthetic XIP, respectively (Fig. 4c). Additionally, Western blot analysis showed that cells treated with 0.1 or 0.4 µM CSP had higher levels of SMU_63c in SDS extracts of whole cells compared to samples without CSP (Fig. 4d). As expected, treatment with CSP did not lead to increased levels of WapA (Fig. 4f) or P1 (not shown). Western blot analysis also showed that concentrated spent medium from cells treated with 5 µM XIP had a threefold increase in SMU_63c compared to that from cells treated with 1 % DMSO solvent control or 1 µM XIP (Fig. 4e).
Fig. 4.

Characterization of SMU_63c. (a) Schematic representation of the genetic locus containing smu_63 c. (b and c) Quantitative real-time PCR analysis of smu_63 c expression with and without addition of CSP in THYE medium (b) or XIP in CDM-glucose (c). (d) Western blot densitometry analysis of SMU_63c present in cell surface SDS extracts of S. mutans in THYE, with and without addition of CSP. (e) Western blot densitometry analysis of SMU_63c in concentrated spent medium from CDM-glucose cultures grown with or without XIP. (f) Western blot analysis of WapA in the same samples as (d). (g) Evaluation of the genetic competence of S. mutans WT, ∆smu_63 c mutant, complemented mutant and pBGK integration vector control strain. Error bars represent the mean±sem of three or four independent replicates. Statistical significance was evaluated by Student's t-test or one-way ANOVA.
We also measured transformation efficiency following addition of 0.8 µM CSP and found that the ∆smu_63 c mutant was approximately threefold more competent than the WT strain (Fig. 4g). This hyper-transformable phenotype was reversed when smu_63 c (plus kan) was reintroduced into the ∆smu_63 c mutant strain (Fig. 4g). The transformation efficiency of the ∆smu_63 c mutant containing the integration vector pBGK [47] (kan resistance) was not significantly different from that of the ∆smu_63 c mutant with no kan resistance vector (Fig. 4g).
Amyloid-forming proteins P1, WapA and SMU_63c are part of the biofilm matrices
Immunogold-labelling TEM with a cocktail of three different anti-P1 monoclonal antibodies and polyclonal rabbit antisera against AgA and SMU_63c was used to characterize the amyloid proteins in the biofilms. When probed with anti-P1 and anti-AgA in a double-labelling assay, binding of both the anti-P1 and anti-AgA antibodies was clearly evident on the cell surface of the WT strain and within the aggregates of extracellular fibrous matrices (Fig. S5a). However, little to no signal was observed in the biofilms of the ΔspaP/ΔwapA double-mutant negative control, which was treated in exactly the same way as the WT strain (Fig. S5b). A high level of reactivity of the anti-SMU_63c rabbit antiserum was also observed in the extracellular fibrous matrix of the WT biofilms (Fig. S5c). Little or no signal was observed with the Δsmu_63 c mutant strain (Fig. S5d).
Identification of amyloid inhibitors that impair S. mutans biofilm formation
We used the WT and the triple-deletion mutant to screen for inhibition of S. mutans biofilm formation by a panel of polyphenolic compounds known to inhibit fibrillization of pathogenic amyloids in vitro: curcumin, resveratrol, quercetin, epicatechin gallate (EGC), EGCG and TA [15], as well as two additional molecules, AA-861 and parthenolide, that inhibit biofilm and amyloid formation by B. subtilis [14].
Of all the compounds tested TA, EGCG and AA-861 had the most pronounced effects on S. mutans biofilm formation, with TA being the most potent S. mutans biofilm inhibitor identified so far. When grown in BM-glucose, biofilm production by the WT and ∆smu_63 c single-deletion mutant strains was significantly inhibited by 5 µM TA compared to growth in the absence of inhibitor (Fig. 5a). However, mutants lacking P1 (∆spaP), WapA (∆wapA) or both (ΔspaP/ΔwapA or ΔspaP/Δsmu_63c/ΔwapA), did not differ significantly in biofilm production when grown in the presence of 5 µM TA or absence of TA (Fig. 5a). As shown earlier in Fig. 3(a), when S. mutans is grown in BM-glucose, mutants lacking either P1 or WapA are significantly impaired in biofilm formation compared to the WT. In this study, we further found that in contrast to the WT and Δsmu_63 c strains, the diminished biofilm produced by mutants lacking spaP and/or wapA is no longer susceptible to inhibition by 5 µM TA, suggesting that P1 and WapA serve as targets of this compound. Similar effects were observed with EGCG and AA-861 (Fig. S6a, b, respectively).
Fig. 5.

Inhibition of biofilm formation by TA, EGCG or AA-861. (a) CV assays of S. mutans WT and deletion-mutant biofilms grown in BM-glucose and in the presence of the indicated concentrations of TA. b–d: CV assays of the same biofilms grown in BM-sucrose in the presence of indicated concentrations of TA (b); EGCG (c); and AA-861 (d). Error bars represent the mean±sem of six independent replicates. Statistical significance was evaluated by two-way ANOVA using Tukey’s multiple comparisons. Asterisks indicate the statistical significance of the comparison of the mean±sem value of each deletion mutant to that of the WT under the same inhibitor concentration: *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. (e) FE-SEM of S. mutans WT and mutant biofilms grown in BM-glucose and sucrose with 10 µM TA (bottom panel) or the 0.1 % ethanol diluent control (top panel). Scale bars represent 2 µm.
S. mutans possesses at least three Gtf enzymes that produce adhesive glucans with sucrose as substrate. This sucrose-dependent pathway is known to play a central role in S. mutans biofilm formation and cariogenicity [48, 49]. When biofilms were grown in BM-sucrose, the Gtf-mediated inter-cellular adhesion and accumulation masked the effects of deletion of genes encoding the amyloid-forming proteins, resulting in no significant differences in total biofilm amount among the strains. Therefore, we next tested whether biofilms grown in sucrose were also susceptible to inhibition by TA and compared the effects of this compound on strains with and without amyloid-forming proteins. Five micromolar TA inhibited biofilm formation by the WT strain, and to a lesser extent the Δsmu_63 c and ΔwapA mutants, during growth in BM-sucrose (Fig. 5b). However, when spaP was deleted singly or in combination with wapA or smu_63c, or both, biofilm development of the mutant strains was significantly less sensitive to inhibition by 5 or 10 µM TA (Fig. 5b). These results reiterate that P1 and WapA serve as targets of TA inhibition of S. mutans biofilm formation.
Biofilm production of the WT, Δsmu_63 c and ΔwapA strains was almost completely inhibited by 50 µM EGCG, while inhibition of the ∆spaP and ∆spaP/∆smu_63 c mutants was significantly diminished (Fig. 5c). The loss of sensitivity to 50 µM EGCG was further enhanced in mutants lacking both spaP and wapA (ΔspaP/ΔwapA and ΔspaP/Δsmu_63c/ΔwapA). In contrast to the WT strain, mutants lacking both P1 and WapA produced biofilms even at the higher EGCG concentration of 100 µM, and the EGCG insensitivity was further amplified when smu_63 c was deleted as well (Fig. 5c). Taken together, these results suggest that P1 is a primary target of biofilm inhibition by EGCG, and that in the absence of P1, WapA (AgA) and SMU_63c appear to serve as secondary targets.
Biofilm formation by the WT strain and Δsmu_63 c mutant was also significantly inhibited by 50 µM of AA-861 (Fig. 5d). In contrast, inhibition of biofilm formation by this concentration of AA-861 was significantly diminished in mutants lacking spaP or wapA, either singly or in combination. These results suggest that, similar to the results described above for TA, P1 and WapA can also serve as targets of biofilm inhibition by AA-861. However, unlike for TA, there was no additive effect in loss of sensitivity to AA-861 upon additional deletion of smu_63c; therefore, its protein product appears completely unaffected by this molecule. Lastly, despite parthenolide's anti-biofilm effect on B. subtilis and anti-fibrillization effect on TasA [14], this molecule had no effect on S. mutans biofilm formation, even at concentrations as high as 600 µM (Fig. S6c). All the amyloid inhibitors used above were tested to ensure that their mechanism of action did not involve a negative impact on cell growth (Fig. S7).
FE-SEM analysis reveals effects of TA on biofilm architecture
FE-SEM was used to visualize biofilms of the WT, ΔspaP, ΔwapA, ΔspaP/ΔwapA and ΔspaP/Δsmu_63c/ΔwapA strains grown in the presence and absence of 10 µM TA. WT cells treated with the 0.1 % ethanol diluent control produced biofilms containing cells that clumped together in a dense mass (Fig. 5e), akin to those observed for the Δsmu_63 c mutants (Fig. 3c). It is not clear how ethanol causes an alteration of biofilm architecture, but it was recently reported that alcohols (ethanol and isopropanol) cause a similar increase in biofilm production by S. aureus [50]. Similar to the CV assay (Fig. 5a, b), S. mutans WT biofilms grown in 10 µM TA demonstrated a clear reduction in cell density compared to the diluent control cells (Fig. 5e). As shown earlier for biofilms grown without ethanol (Fig. 3c), biofilms of the ΔspaP, ΔwapA, ΔspaP/ΔwapA and ΔspaP/Δsmu_63c/ΔwapA mutants grown in the presence of 0.1 % ethanol were substantially less dense than the WT (Fig. 5e). Importantly, there were no differences observed for these mutants when biofilms were grown with or without 10 µM TA (Fig. 5e), again indicating that P1 and WapA serve as TA targets since TA sensitivity is lost when genes encoding these amyloid-forming proteins are deleted.
Amyloid inhibitors affecting biofilm development also inhibit fibrillization of C123 and AgA in vitro
ThT uptake was significantly reduced when C123 was stirred in the presence of 10 or 100 µM TA, 50 or 200 µM EGCG, or 50 or 200 µM AA-861, compared to samples stirred in the absence of inhibitor (Fig. 6a). ThT uptake was also significantly reduced when AgA was stirred in the presence of the same inhibitors compared to samples stirred without them (Fig. 6b). Stirring C123 or AgA in the presence of parthenolide, which did not affect S. mutans biofilm production, did not affect ThT uptake by these proteins (Fig. 6a, b). TA, EGCG, AA-861 or parthenolide alone did not affect ThT fluorescence in the absence of added protein (Fig. 6c). The influence of the inhibitors of biofilm formation on amyloid fibrillization was also confirmed by TEM. When either purified C123 (Fig. 6d) or AgA (Fig. 6e) was stirred in the presence of parthenolide, there was no detectable effect on fibril formation. However, when these proteins were stirred in the presence of TA, we observed the accumulation of amorphous material instead of fibrillar amyloid aggregates. Inhibition of amyloid fibrillization by SMU_63c could not be tested in these assays because its amyloid formation occurs at low pH and in the absence of mechanical stirring.
Fig. 6.

Anti-biofilm compounds inhibit fibrillization of P1-C123 and WapA-AgA in vitro. (a and b) ThT uptake of C123 (a) or AgA (b) stirred in the absence or presence of the indicated inhibitors. (c) ThT fluorescence in the absence of added proteins. (d and e) Transmission electron micrographs of C123 (d) or AgA (e) stirred without inhibitor, or in the presence of 200 µM TA or 200 µM parthenolide. Scale bars represent 200 nm. Error bars represent mean±sem of three independent replicates. Statistical significance was evaluated by one-way ANOVA using Dunnett’s multiple comparisons. Asterisks indicate statistically significant differences between protein stirred in the presence of inhibitor compared to the absence of inhibitor: *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
Discussion
Our previous studies have shown that the S. mutans P1 adhesin is an amyloid-forming protein [19]. Here, we expand those findings to show that the C123 fragment of P1 (antigen II) represents the amyloid-forming moiety. We also identify two additional amyloid-forming proteins, WapA and the heretofore-uncharacterized secreted protein SMU_63c. WapA, like P1, is a sortase substrate and is processed to generate AgA. Also similar to P1, this naturally occurring cleavage product is capable of amyloid fibrillization.
SMU_63c was previously annotated as an unknown hypothetical protein. In this study, we have generated evidence that SMU_63c forms amyloid at low pH and acts as an apparent negative regulator of genetic competence and biofilm cell density. These functions need to be examined further to fully understand the mechanisms by which SMU_63c regulates competence and biofilm cell density.
In bacteria that produce functional amyloids, there is often one principal amyloid-forming protein, with other accessory proteins that aid in the production, secretion or nucleation of fibrillization [8]. S. mutans is different in that at least three unrelated proteins from the same organism have now been shown to produce amyloid, to be associated with fibrous ECM structures and to affect biofilm formation. It is not yet known whether these proteins interact with each other in vivo, whether they produce heterogeneous amyloids, or whether they produce amyloids in a sequential manner. The fact that WapA and P1, which function as adhesins during the initial stages of biofilm development, produce fragments that form amyloid better under neutral pH, while SMU_63c forms amyloid under acidic conditions, suggests that the prevailing environmental pH may act as an in vivo regulator of fibrillization of the different S. mutans proteins. Amyloid formation by the Bap protein of S. aureus has been reported to occur in response to acidic pH and low concentrations of calcium [10]. It was also shown recently that in S. aureus, eDNA triggers the conversion of PSMs to functional amyloids [51]. S. mutans also produces eDNA within biofilms [52] but its relationship to amyloid is not yet known in this organism. We have found both eDNA and cell-surface proteins, including P1, to be contained within S. mutans extracellular vesicles, whose content but not production is negatively impacted by the absence of sortase [52]. Thus another plausible mechanism, in addition to a potential nucleation event at the cell surface, exists to explain the observed negative impact of sortase deficiency on amyloid production within S. mutans biofilms [19, 52].
The tertiary structure and cell surface architecture of P1 has been studied extensively [20, 36–39]. Interestingly, C123, which we demonstrate herein to represent the amyloid-forming portion of P1, also associates with covalently attached full-length P1 on the cell surface [39]. The crystal structure of WapA (also known as AIII) [53] is not yet known. It is known to bind collagen [54], affect chain length and biofilm architecture [42], promote dendritic cell maturation [55] and like P1 has been evaluated as a vaccine candidate [56]. In this study, we showed that a recombinant polypeptide corresponding to the naturally occurring processed form of WapA, AgA, [32, 33] is amyloidogenic. To our knowledge, it has not yet been established whether or how AgA may interact with covalently attached full-length WapA or other proteins on the cell surface. Our results show that both AgA and unprocessed WapA can be extracted from whole bacterial cells with SDS (Fig. S3a). It is intriguing that both WapA and P1 are covalently attached by sortase to the cell wall in their full-length forms, and are also processed to generate truncated amyloidogenic fragments. It seems likely, therefore, that the truncated fragments of both P1 and WapA are responsible for the cell-associated amyloid observed in S. mutans, and that covalent linkage of the full-length proteins to the cell wall may be necessary to initiate nucleation of fibrillization. Such a mechanism may be more widespread than previously recognized. The Bap protein of S. aureus, which also contains a sortase recognition motif at its C-terminus, is also cleaved by an unknown mechanism, thus enabling its ability to form amyloid aggregates [10]. Hence our data support an emerging model in streptococcal and staphylococcal species of dual-function cell surface adhesins that, when cleaved, participate in the formation of an extracellular amyloid scaffolding matrix that is likely environmentally regulated [10, 57].
Taken together, our results reveal a clear correlation between the inhibition of amyloid fibril formation and the inhibition of biofilm formation by S. mutans. TA, EGCG and AA-861 were effective at inhibiting S. mutans biofilm formation in vivo, as well as amyloid fibrillization of C123 and AgA in vitro. As expected, deletion mutants lacking the sucrose-independent adhesins P1 and/or WapA were impaired in biofilm formation during growth in glucose. Furthermore, these mutants demonstrated significantly diminished sensitivity to amyloid inhibitors when these compounds were used to impede biofilm development, while the same amyloid inhibitors prevented biofilm production in strains producing both P1 and WapA, suggesting that their adhesive function may be related to their capacity to form amyloid. Amyloid inhibitors were also effective when S. mutans biofilms were grown in sucrose, suggesting a functional cooperation of the amyloid forms of P1 and WapA with the adhesive extracellular glucans formed under these growth conditions. This is consistent with the emerging recognition of bacterial amyloid functioning in the context of a multi-component exopolysaccharide and eDNA-containing matrix [58]. S. mutans mutants lacking amyloidogenic proteins still formed biofilms when grown in BM-sucrose, although these strains were significantly less sensitive to inhibition by the small-molecule amyloid inhibitors, compared to WT cells. It is likely then that in the presence of sucrose, but in the absence of P1 and WapA, extracellular glucans mediate cell–cell and cell–substratum interactions to produce biofilms. When P1 and WapA are also present, and in their amyloid form, they can interact with those glucans to further stabilize and support robust biofilm production. When P1 and WapA are produced, but are prevented from fibrillization by amyloid inhibitors, only monomeric or small oliogomeric forms of the proteins would be present. These apparently do not participate in cooperative interactions with the glucans, and may even interfere with glucan function. This results in diminished biofilm accumulation of sucrose-grown cells treated with amyloid inhibitors. When P1 and/or WapA are completely absent due to gene deletion, amyloid inhibitors have a diminished impact on biofilm formation compared to untreated cells because their targets of activity are gone, and the glucans can mediate cell–cell and cell–substratum interactions unhindered by monomeric or oliogomeric P1 and WapA. It has been reported that EGCG disrupts the ability of P. aeruginosa Fap to form amyloid fibrils by stabilizing protein oligomers [59]. In addition, these authors found that treatment of P. aeruginosa biofilms with EGCG decreased the minimal bactericidal concentration of tobramycin, an effect that was amplified when fap genes were overexpressed. TA, a relatively non-toxic component of beverages such as tea, red wine and beer, has also been shown to inhibit biofilm formation by S. aureus via an increase in expression of the transglycosylase IsaA [17], but it is not yet known whether it might also inhibit fibrillization of PSMs or Bap or other yet-to-be-identified amyloidogenic proteins in this organism. AA-861 and parthenolide were shown by Kolter and coworkers to be quite effective at inhibiting biofilm formation by B. subtilis, B. cereus and E. coli, as well as inhibiting fibrillization of B. subtilis TasA [14]. While AA-861 was an effective biofilm and amyloid inhibitor in S. mutans, parthenolide was not. This suggests some degree of specificity of bacterial amyloid formation that may enable directed targeting of certain bacteria. The identification of multiple amyloidogenic proteins that contribute to S. mutans biofilm formation makes this a highly suitable organism for the continued study and screening of anti-amyloid and anti-biofilm compounds, and for further characterization of bacterial amyloids to elucidate their individual and collective functional activities and the factors that regulate their formation.
Funding information
This work was supported by NIH R01DE021789 to L. J. B., R01 DE019452 to Z.T . W., R21 DE025348 to L. J. B. and Z. T. W., and a T90 DE021990 post-doctoral fellowship to R. N. B.
Acknowledgements
We thank the Brady laboratory members, especially Paula Crowley, for helpful discussions; Ann Hochschild and Viknesh Sivanathan of Harvard Medical School for the pVS72 plasmid and E. coli VS39 expression strains; Karen Kelly of the UF Interdisciplinary Center for Biotechnology Research Electron Microscopy Core facility for help with TEM analyses; Wandy L. Beatty at Washington University Department of Microbiology, St. Louis, MO for technical assistance on immunogold labeling and TEM analysis; and Patricia L. Clark and Micayla Bowman of the University of Notre Dame, Department of Chemistry and Biochemistry for their help with the CD analyses.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
Footnotes
Abbreviations: CD, circular dichroism; CDM, chemically defined medium; CR, Congo red; CSP, competence-stimulating peptide; ECM, extra cellular matrix; EGC, epicatechingallate; EGCG, epigallocatechin-3-gallate; EM, electron microscopy; FE-SEM, field emission scanning EM; MAbs, monoclonal antibody; PBS, potassium phosphate buffer; PSM, phenol-soluble modulin; TA, tannic acid; TDM, Terleckyj-defined medium; TEM, transmission EM; ThT, thioflavin T; THYE, Todd–Hewitt yeast extract; WapA, wall-associated protein A.
Seven supplementary figures and three supplementary tables are available with the online Supplementary Material.
Edited by: P. Zuber and T. Msadek
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Fowler DM, Kelly JW. Functional amyloidogenesis and cytotoxicity—insights into biology and pathology. PLoS Biol. 2012;10:e1001459. doi: 10.1371/journal.pbio.1001459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid—from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Nilsson MR. Techniques to study amyloid fibril formation in vitro. Methods. 2004;34:151–160. doi: 10.1016/j.ymeth.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Howie AJ, Brewer DB. Optical properties of amyloid stained by Congo red: history and mechanisms. Micron. 2009;40:285–301. doi: 10.1016/j.micron.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Naiki H, Gejyo F. Kinetic analysis of amyloid fibril formation. Methods Enzymol. 1999;309:305–318. doi: 10.1016/s0076-6879(99)09022-9. [DOI] [PubMed] [Google Scholar]
- 7.Otzen D, Nielsen PH. We find them here, we find them there: functional bacterial amyloid. Cell Mol Life Sci. 2008;65:910–927. doi: 10.1007/s00018-007-7404-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR. Diversity, biogenesis and function of microbial amyloids. Trends Microbiol. 2012;20:66–73. doi: 10.1016/j.tim.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 2012;8:e1002744. doi: 10.1371/journal.ppat.1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taglialegna A, Navarro S, Ventura S, Garnett JA, Matthews S, et al. Staphylococcal bap proteins build amyloid scaffold biofilm matrices in response to environmental signals. PLoS Pathog. 2016;12:e1005711. doi: 10.1371/journal.ppat.1005711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dueholm MS, Petersen SV, Sønderkær M, Larsen P, Christiansen G, et al. Functional amyloid in P seudomonas . Mol Microbiol. 2010;77:1009–1020. doi: 10.1111/j.1365-2958.2010.07269.x. [DOI] [PubMed] [Google Scholar]
- 12.Chai L, Romero D, Kayatekin C, Akabayov B, Vlamakis H, et al. Isolation, characterization, and aggregation of a structured bacterial matrix precursor. J Biol Chem. 2013;288:17559–17568. doi: 10.1074/jbc.M113.453605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romero D, Aguilar C, Losick R, Kolter R. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc Natl Acad Sci USA. 2010;107:2230–2234. doi: 10.1073/pnas.0910560107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romero D, Sanabria-Valentín E, Vlamakis H, Kolter R. Biofilm inhibitors that target amyloid proteins. Chem Biol. 2013;20:102–110. doi: 10.1016/j.chembiol.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 16.Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, et al. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J Virol. 2003;77:10288–10294. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payne DE, Martin NR, Parzych KR, Rickard AH, Underwood A, et al. Tannic acid inhibits Staphylococcus aureus surface colonization in an IsaA-dependent manner. Infect Immun. 2013;81:496–504. doi: 10.1128/IAI.00877-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamada S, Slade HD. Biology, immunology, and cariogenicity of Streptococcus mutans. Microbiol Rev. 1980;44:331–384. doi: 10.1128/mr.44.2.331-384.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oli MW, Otoo HN, Crowley PJ, Heim KP, Nascimento MM, et al. Functional amyloid formation by Streptococcus mutans. Microbiology. 2012;158:2903–2916. doi: 10.1099/mic.0.060855-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brady LJ, Maddocks SE, Larson MR, Forsgren N, Persson K, et al. The changing faces of Streptococcus antigen I/II polypeptide family adhesins. Mol Microbiol. 2010;77:276–286. doi: 10.1111/j.1365-2958.2010.07212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell MW, Bergmeier LA, Zanders ED, Lehner T. Protein antigens of Streptococcus mutans: purification and properties of a double antigen and its protease-resistant component. Infect Immun. 1980;28:486–493. doi: 10.1128/iai.28.2.486-493.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crowley PJ, Brady LJ, Michalek SM, Bleiweis AS. Virulence of a spaP mutant of Streptococcus mutans in a gnotobiotic rat model. Infect Immun. 1999;67:1201–1206. doi: 10.1128/iai.67.3.1201-1206.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wenderska IB, Lukenda N, Cordova M, Magarvey N, Cvitkovitch DG, et al. A novel function for the competence inducing peptide, XIP, as a cell death effector of Streptococcus mutans. FEMS Microbiol Lett. 2012;336:104–112. doi: 10.1111/j.1574-6968.2012.02660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouvet A, Van De Rijn I, Mccarty M. Nutritionally variant streptococci from patients with endocarditis: growth parameters in a semisynthetic medium and demonstration of a chromophore. J Bacteriol. 1981;146:1075–1082. doi: 10.1128/jb.146.3.1075-1082.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terleckyj B, Willett NP, Shockman GD. Growth of several cariogenic strains of oral streptococci in a chemically defined medium. Infect Immun. 1975;11:649–655. doi: 10.1128/iai.11.4.649-655.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sivanathan V, Hochschild A. Generating extracellular amyloid aggregates using E. coli cells. Genes Dev. 2012;26:2659–2667. doi: 10.1101/gad.205310.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen TN, Brunak S, Von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 28.O'Toole GA, Kolter R. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol. 1998;28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 29.Wen ZT, Burne RA. LuxS-mediated signaling in Streptococcus mutans is involved in regulation of acid and oxidative stress tolerance and biofilm formation. J Bacteriol. 2004;186:2682–2691. doi: 10.1128/JB.186.9.2682-2691.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bitoun JP, Nguyen AH, Fan Y, Burne RA, Wen ZT. Transcriptional repressor Rex is involved in regulation of oxidative stress response and biofilm formation by Streptococcus mutans. FEMS Microbiol Lett. 2011;320:110–117. doi: 10.1111/j.1574-6968.2011.02293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koo H, Nino de Guzman P, Schobel BD, Vacca Smith AV, Bowen WH. Influence of cranberry juice on glucan-mediated processes involved in Streptococcus mutans biofilm development. Caries Res. 2006;40:20–27. doi: 10.1159/000088901. [DOI] [PubMed] [Google Scholar]
- 32.Yoder S, Cao C, Ugen KE, Dao ML. High-level expression of a truncated wall-associated protein A from the dental cariogenic Streptococcus mutans. DNA Cell Biol. 2000;19:401–408. doi: 10.1089/10445490050085898. [DOI] [PubMed] [Google Scholar]
- 33.Russell RR. Wall-associated protein antigens of Streptococcus mutans. J Gen Microbiol. 1979;114:109–115. doi: 10.1099/00221287-114-1-109. [DOI] [PubMed] [Google Scholar]
- 34.Tsolis AC, Papandreou NC, Iconomidou VA, Hamodrakas SJ. A consensus method for the prediction of 'aggregation-prone' peptides in globular proteins. PLoS One. 2013;8:e54175. doi: 10.1371/journal.pone.0054175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh I, Seno F, Tosatto SC, Trovato A. PASTA 2.0: an improved server for protein aggregation prediction. Nucleic Acids Res. 2014;42:W301–W307. doi: 10.1093/nar/gku399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heim KP, Crowley PJ, Long JR, Kailasan S, Mckenna R, et al. An intramolecular lock facilitates folding and stabilizes the tertiary structure of Streptococcus mutans adhesin P1. Proc Natl Acad Sci USA. 2014;111:15746–15751. doi: 10.1073/pnas.1413018111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larson MR, Rajashankar KR, Crowley PJ, Kelly C, Mitchell TJ, et al. Crystal structure of the C-terminal region of Streptococcus mutans antigen I/II and characterization of salivary agglutinin adherence domains. J Biol Chem. 2011;286:21657–21666. doi: 10.1074/jbc.M111.231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larson MR, Rajashankar KR, Patel MH, Robinette RA, Crowley PJ, et al. Elongated fibrillar structure of a streptococcal adhesin assembled by the high-affinity association of alpha- and PPII-helices. Proc Natl Acad Sci USA. 2010;107:5983–5988. doi: 10.1073/pnas.0912293107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heim KP, Sullan RM, Crowley PJ, El-Kirat-Chatel S, Beaussart A, et al. Identification of a supramolecular functional architecture of Streptococcus mutans adhesin P1 on the bacterial cell surface. J Biol Chem. 2015;290:9002–9019. doi: 10.1074/jbc.M114.626663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemos JA, Quivey RG, Koo H, Abranches J. Streptococcus mutans: a new Gram-positive paradigm? Microbiology. 2013;159:436–445. doi: 10.1099/mic.0.066134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu L, Kreth J, Cross SE, Gimzewski JK, Shi W, et al. Functional characterization of cell-wall-associated protein WapA in Streptococcus mutans. Microbiology. 2006;152:2395–2404. doi: 10.1099/mic.0.28883-0. [DOI] [PubMed] [Google Scholar]
- 42.Lévesque CM, Voronejskaia E, Huang YC, Mair RW, Ellen RP, et al. Involvement of sortase anchoring of cell wall proteins in biofilm formation by Streptococcus mutans. Infect Immun. 2005;73:3773–3777. doi: 10.1128/IAI.73.6.3773-3777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, et al. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krogh A, Larsson B, Von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 45.Mashburn-Warren L, Morrison DA, Federle MJ. A novel double-tryptophan peptide pheromone controls competence in Streptococcus spp. via an Rgg regulator. Mol Microbiol. 2010;78:589–606. doi: 10.1111/j.1365-2958.2010.07361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Senadheera D, Cvitkovitch DG. Quorum sensing and biofilm formation by Streptococcus mutans. Adv Exp Med Biol. 2008;631:178–188. doi: 10.1007/978-0-387-78885-2_12. [DOI] [PubMed] [Google Scholar]
- 47.Wen ZT, Burne RA. Construction of a new integration vector for use in Streptococcus mutans. Plasmid. 2001;45:31–36. doi: 10.1006/plas.2000.1498. [DOI] [PubMed] [Google Scholar]
- 48.Kuramitsu HK. Virulence factors of mutans streptococci: role of molecular genetics. Crit Rev Oral Biol Med. 1993;4:159–176. doi: 10.1177/10454411930040020201. [DOI] [PubMed] [Google Scholar]
- 49.Bowen WH, Koo H. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res. 2011;45:69–86. doi: 10.1159/000324598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luther MK, Bilida S, Mermel LA, Laplante KL. Ethanol and isopropyl alcohol exposure increases biofilm formation in Staphylococcus aureus and Staphylococcus epidermidis. Infect Dis Ther. 2015;4:219–226. doi: 10.1007/s40121-015-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwartz K, Ganesan M, Payne DE, Solomon MJ, Boles BR. Extracellular DNA facilitates the formation of functional amyloids in Staphylococcus aureus biofilms. Mol Microbiol. 2016;99:123–134. doi: 10.1111/mmi.13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao S, Klein MI, Heim KP, Fan Y, Bitoun JP, et al. Streptococcus mutans extracellular DNA is upregulated during growth in biofilms, actively released via membrane vesicles, and influenced by components of the protein secretion machinery. J Bacteriol. 2014;196:2355–2366. doi: 10.1128/JB.01493-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Russell MW, Harrington DJ, Russell RR. Identity of Streptococcus mutans surface protein antigen III and wall-associated protein antigen A. Infect Immun. 1995;63:733–735. doi: 10.1128/iai.63.2.733-735.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han TK, Zhang C, Dao ML. Identification and characterization of collagen-binding activity in Streptococcus mutans wall-associated protein: a possible implication in dental root caries and endocarditis. Biochem Biophys Res Commun. 2006;343:787–792. doi: 10.1016/j.bbrc.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 55.Li H, Wang D. Streptococcus mutans wall-associated protein A promotes TLR4-induced dendritic cell maturation. Scand J Immunol. 2014;80:121–126. doi: 10.1111/sji.12194. [DOI] [PubMed] [Google Scholar]
- 56.Han TK, Dao ML. Enhancement of salivary IgA response to a DNA vaccine against Streptococcus mutans wall-associated protein A in mice by plasmid-based adjuvants. J Med Microbiol. 2007;56:675–680. doi: 10.1099/jmm.0.47020-0. [DOI] [PubMed] [Google Scholar]
- 57.Di Martino P. Bap: a new type of functional amyloid. Trends Microbiol. 2016;24:682–684. doi: 10.1016/j.tim.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 58.Taglialegna A, Lasa I, Valle J. Amyloid structures as biofilm matrix scaffolds. J Bacteriol. 2016;198:2579–2588. doi: 10.1128/JB.00122-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stenvang M, Dueholm MS, Vad BS, Seviour T, Zeng G, et al. Epigallocatechin gallate remodels overexpressed functional amyloids in Pseudomonas aeruginosa and increases biofilm susceptibility to antibiotic treatment. J Biol Chem. 2016;291:26540–26553. doi: 10.1074/jbc.M116.739953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.