

Abstract
Chlamydomonas reinhardtii is a unicellular chlorophyte alga that is widely-studied as a reference organism for understanding photosynthesis, sensory and motile cilia, and for development of an algal-based platform for producing biofuels and bio-products. Its highly repetitive, ~205 kbp circular chloroplast genome and ~15.8 kbp linear mitochondrial genome were sequenced prior to the advent of high-throughput sequencing technologies. Here, high coverage shotgun sequencing was used to assemble both organellar genomes de novo. These new genomes correct dozens of errors in the prior genome sequences and annotations. Genome sequencing coverage indicates that each cell contains on average 83 copies of the chloroplast genome and 130 copies of the mitochondrial genome. Using protocols and analyses optimized for organellar transcripts, RNA-Seq was used to quantify their relative abundances across 12 different growth conditions. 46% of total cellular mRNA is attributable to high expression from a few dozen chloroplast genes. RNA-Seq data were used to guide gene annotation, to demonstrate polycistronic gene expression, and to quantify splicing of psaA and psbA introns. In contrast to a conclusion from a recent study, we found that chloroplast transcripts are not edited. Unexpectedly, cytosine-rich polynucleotide tails were observed at the 3′ end of all mitochondrial transcripts. A comparative genomics analysis of 8 laboratory strains and 11 wild isolates of C. reinhardtii identified 2658 variants in the organellar genomes, which is one tenth as much genetic diversity as is found in the nucleus.
Keywords: Plastid, organelles, RNA-Seq, transcript editing, trans-splicing, rpoC1, rps2, Wendy transposon, ftsH, ycf1
Introduction
Chlamydomonas reinhardtii is a widely studied unicellular alga of the chlorophyte lineage. It has been a premiere model organism for studies of photosynthesis, nutrient homeostasis, and cilia structure and function (Harris, 2008). More recently, C. reinhardtii has shown great promise as a reference platform for the production of biofuels and bio-products (Rosales-Mendoza et al., 2012; Scranton et al., 2015). Endemic to soil and fresh water with a world-wide distribution, C. reinhardtii grows vegetatively as a haploid organism. Upon certain stress conditions, such as N deprivation, vegetatively growing cells become gametes, and matched pairs of “mt+” and “mt−” gametes can fuse to undergo sexual recombination. These features make C. reinhardtii a highly tractable species for genetic studies. Much of the research in C. reinhardtii is conducted on a few dozen standard laboratory strains whose lineage can be traced to a common ancestor isolated in 1945 (Gallaher et al., 2015). Additionally, a number of interfertile wild isolates of the species are available, and were found to have a sequence diversity of ~3% (Flowers et al., 2015). In 2007, the sequence of the ~112 Mbp nuclear genome of C. reinhardtii laboratory strain CC-503 was published (Merchant et al., 2007). This work facilitated a decade of important systems biology-scale study. However, the organellar genomes of the chloroplast and mitochondrion were not included in this work, and have been largely ignored by subsequent transcriptomics studies.
Each cell of C. reinhardtii has a single, large, cup-shaped chloroplast. This organelle is responsible for ~40% of the cell volume, and is the site of photosynthesis as well as considerable primary metabolism, including N and S assimilation. The chloroplast contains multiple copies of a 205 kbp circular genome, which is inherited uniparentally from the mt+ parent by daughter cells following sexual recombination. The sequence of that genome was first published by David Stern’s group in 2002 and was reported to contain 72 protein coding genes, a full complement of 29 tRNAs, and 5 rDNAs (Maul et al., 2002). This 2002 version of the genome was assembled from Sanger sequencing of cloned fragments from a number of laboratory strains, including CC-503. Notably, it was observed that the chloroplast genome contains over 20% repetitive DNA, mostly short dispersed repeats, and this feature is nearly unique to Chlamydomonas among chlorophyte species. The genome contains two ~21 kbp inverted repeats that are separated by ~80 kbp single copy regions. The 2002 sequence is referred to in this work by its GenBank accession number: BK000554.2.
An improved chloroplast genome was assembled some years later as part of a study on genetic drift (Smith and Lee, 2009). The 2009 version, referred to here as FJ423446.1, was assembled from public sequencing data of strain CC-503 made available from the Chlamydomonas nuclear genome sequencing project. The researchers identified 471 single nucleotide variants (SNVs) and 955 small insertions/deletions (InDels) relative to BK000554.2.
Aside from its role in photosynthesis, the chloroplast is an important target for the production of recombinant proteins in Chlamydomonas. Methods for the transformation of the nuclear genome have been developed, but expression from nuclear transgenes is typically anemic and unstable (Cerutti et al., 1997). In contrast, the chloroplast has been shown to readily incorporate transgenes by homologous recombination, and is capable of high level expression. This approach has been used successfully to express subunit vaccines, therapeutic antibodies, and nutraceuticals (Scranton et al., 2015).
Each Chlamydomonas cell contains multiple mitochondria, which together comprise ~1–3% of the cell volume (Harris, 2008). The complete ~15.8 kbp mitochondrial genome was submitted to GenBank in 1993 as accession U03843.1 (Vahrenholz et al., 1993). The mitochondrial genome is inherited uniparentally from the mt− parent by daughter cells following sexual recombination. The C. reinhardtii mitochondrial genome is much reduced relative to other plant species both in size and number of genes. Like animals, but to a higher degree than in plants, the Chlamydomonas mitochondrial genome is highly derived relative to its presumed free-living ancestor and ancestral mitochondrial genomes. It contains only 8 protein coding genes, 3 tRNA genes, and 15 rRNA genes. The mitochondrial genome is linear in structure, with 532 bp terminal inverted repeats followed by long single stranded 3′ extensions that may play a role in replication (Vahrenholz et al., 1993).
While dozens of RNA-Seq studies in C. reinhardtii have been published to date, the contribution of organellar transcripts has been largely neglected for two reasons. First, organellar transcripts are mostly excluded from RNA-Seq libraries. In the majority of studies, poly-adenylated transcripts are enriched from total RNA with oligo-d(T) beads as a means of diminishing the presence of rRNA. In contrast to most nuclear transcripts, organellar transcripts in C. reinhardtii are poly-adenylated to a much lesser degree, if at all, and poly-adenylation of organellar transcripts may function as a degradation signal (Komine et al., 2000). Therefore, most organellar mRNA is excluded from contributing to RNA-Seq libraries, and any transcripts that do leak through may not quantitatively reflect in vivo proportions. Second, the chloroplast and mitochondrial genomes and their annotations are not included with the commonly used reference sequence for C. reinhardtii (currently v5.5, Phytozome). Therefore, whatever organellar transcripts are present in RNA-Seq libraries are routinely discarded as unmapped reads. As of this writing (July 2017), none of the 29 published studies on RNA-Seq analysis in C. reinhardtii quantify transcription from chloroplast or mitochondrial genes.
Given the importance of the organelles for bioenergetic metabolism, we sought to systematically study the genomes and transcriptomes of the Chlamydomonas organelles. Could we improve on the existing genome sequences by leveraging the abundance of high-throughput data? How much variation is there in the chloroplast and mitochondrial genomes between different laboratory strains and wild isolates of C. reinhardtii, and how does that compare to variation in the nuclear genome? Lastly, what can RNA-Seq analysis reveal about transcript abundance and transcript editing in the chloroplast and mitochondria?
Results
De novo assembly of the organellar genomes
Previously, we re-sequenced the total cellular DNA of a number of laboratory strains of C. reinhardtii, and reported on the degree of variation among their nuclear genomes (Gallaher et al., 2015). As expected, a significant percentage (averaging ~25%) of those reads did not map to the nuclear genome. Assuming that many of these originated from the organellar genomes, we performed a de novo assembly on all DNA-Seq reads from strain CC-503 that did not map to the reference nuclear genome. Reads were re-mapped to the scaffolds to extend their ends until they could be joined manually. With each step in this iterative process, mapping of DNA-Seq reads was used to correct any discrepancies that were identified.
The final product of this process, referred to here as CPv4, is a circular genome of 205,535 bp (Figure 1 and Data S1). This is slightly larger than the versions presently available in GenBank: FJ423446.1 at 204,159 bp and BK000554.2 at 203,828 bp (Table 1). The average GC content is 34.6%, which is considerably lower than that of the nuclear genome: 64.1% (Figure S1). The final sequence was supported by DNA-Seq reads that align to the assembly at a 17,000× average coverage depth. Relative to FJ423446.1 (2009), the new sequence had one large 2.4 kbp inversion, 18 SNVs, and 22 InDels (Table S1).
Figure 1. Map of C. reinhardtii chloroplast genome, CPv4.
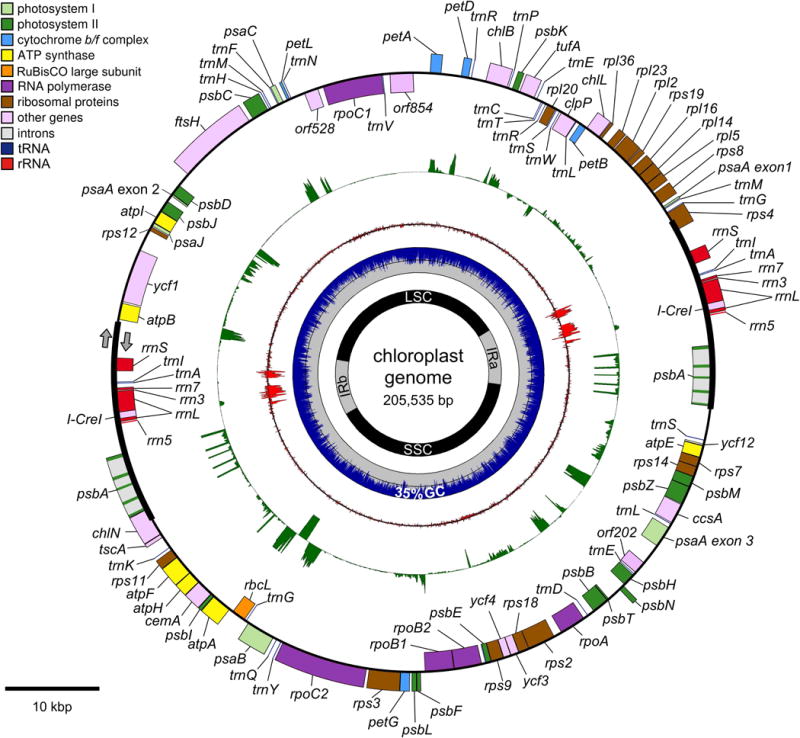
A map of the C. reinhardtii chloroplast genome, CPv4, is presented as a series of concentric rings. From outer to inner ring, they are as follows: Gene models are presented as a series of colored boxes. Genes transcribed in a clockwise fashion are placed outside of the line, and genes transcribed counter-clockwise are placed inside of the line. Categories of genes are color-coded according to the accompanying legend. Next, a green ring depicts RNA-Seq coverage track from a representative experiment on a linear scale with 1×105 reads per locus maximum. Strandedness is indicated as in the outer-most ring. Next, a red ring indicates the rRNA on a log10 scale. Next, a plot of the GC content in non-overlapping 25 bp windows with blue for %GC and gray for %AT. The 50% mark is indicated by a dashed white line, and the overall average GC content is indicated at the bottom of the ring. Next, a ring to indicate the position of the two inverted repeats (IRa and IRb), the long single copy region (LSC), and the short single copy region (SSC). A scale bar in the lower right indicates 10 kbp.
Table 1.
Comparison C. reinhardtii chloroplast genome versions
|
BK000554.2 (GB 2002) |
FJ423446.1 (GB 2009) |
cv11 (this work) |
CPv4 (this work) |
|
|---|---|---|---|---|
| size, bp | 203,828 | 204,159 | 205,713 | 205,535 |
| error frequency per 1 kbp | ||||
| mean ± stdev, % | 0.497 ± 0.525 | 0.168 ± 0.151 | 0.139 ± 0.069 | 0.136 ± 0.059 |
| maximum, % | 2.985 | 1.419 | 0.624 | 0.357 |
| minimum, % | 0.085 | 0.080 | 0.079 | 0.080 |
| coverage depth | ||||
| mean ± stdev, count | 16,648 ± 3,825 | 16,582 ± 3,758 | 16,711 ± 3,599 | 17,839 ± 1,993 |
| maximum, count | 48,588 | 60,589 | 34,989 | 29,049 |
| minimum, count | 0 | 0 | 16 | 1,345 |
| inferred insert size | ||||
| mean ± stdev, bp | 364 ± 77 | 365 ± 70 | 360 ± 32 | 360 ± 27 |
| > mean + 2 × stdev, % | 0.87 | 0.92 | 0.29 | 0.27 |
| < mean − 2 × stdev, % | 2.75 | 1.24 | 0.59 | 0.48 |
For the mitochondrion, de novo assembly produced a 15,789 bp linear genome called MTv4 (Figure 2 and Data S1). This is comparable in size to GenBank U03843.1, at 15,758 bp. The average GC content was 45.2% (Figure S1). The average coverage depth of DNA-Seq reads aligned to MTv4 was greater than 25,000-fold. We observed 11 SNVs and 12 InDels relative to U03843.1 (Table S1).
Figure 2. Map of the C. reinhardtii mitochondrial genome, MTv4.
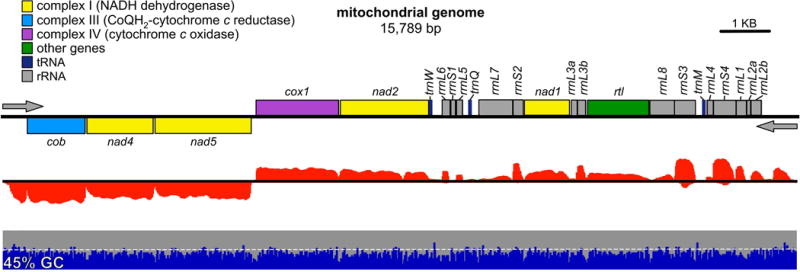
A map of the C. reinhardtii mitochondrial genome, MTv4, is presented as a series of tracks. Gene models are presented as colored boxes above and below the line to indicate strandedness. Categories of genes are color-coded according to the accompanying legend. The direction of transcription is indicated by gray arrows. Below that in red, an RNA-Seq coverage track from a representative experiment. Strandedness is indicated as above. Next, in blue and gray, a plot of the GC content in non-overlapping 25 bp windows, with blue for %GC and gray for %AT. The 50% mark is indicated by a dashed white line, and the overall average GC content is indicated at the far left. A scale bar in the upper right indicates 1 kbp.
Evaluation of four different chloroplast genome versions
In addition to the final de novo chloroplast assembly presented with this work, CPv4, and the two separate accessions that are currently part of GenBank, BK000554.2 (2002) and FJ423446.1 (2009), a fourth version of the chloroplast genome was produced by us in parallel using a different approach. DNA-Seq reads from strain CC-503 were aligned to FJ423446.1 (2009), and used to identify likely variants. The GenBank sequence was then manually edited to reflect these variants, and the edits were verified by re-aligning the DNA-Seq reads to the resulting edited genome in an iterative process. The final 205,713 bp version of this variant-edited reconstruction is referred to here as cv11 (Table 1 and Data S2).
With four different versions of ostensibly the same ~200 kbp molecule prepared by different methodologies, we evaluated the relative quality of each approach. To that end, a set of 99 million paired-end DNA-Seq reads were aligned in parallel to each genome version, and the resulting alignments were analyzed using a variety of different metrics. Given that the same pool of DNA-Seq reads was aligned to each genome version, any differences in alignment quality metrics should be the result of errors in the underlying genome version.
First, the per-base error frequency was examined; defined here as the percentage of base calls that differ from the reference sequence, including InDels, for each position in the genome. It was assumed that there is some basal error frequency that is inherent to the technology, but that differences between the genome versions above the basal frequency would indicate inaccuracies in the reference sequence. When averaged across the entire chloroplast genome, BK000554.2 (2002) had a significantly higher mean error frequency of 0.497% than the other genome versions (Table 1). FJ423446.1, cv11, and CPv4 had mean error frequencies of 0.168%, 0.139%, and 0.136%, respectively. To determine if the errors were uniformly or heterogeneously distributed, the per locus error frequency was averaged across non-overlapping 1 kbp windows, and plotted for each genome version (Figure 3). In BK000554.2 (2002) and FJ423446.1 (2009), there were distinct regions with high error frequencies, reaching maximums of 2.99% and 1.42%, respectively. By contrast, the errors in cv11 and CPv4 were more evenly distributed, and reached maximums of 0.624% and 0.357%, respectively. The distribution of error frequencies in 1 kbp windows is presented as a violin plot in Figure 4A.
Figure 3. Error frequency heterogeneity of chloroplast genome versions.
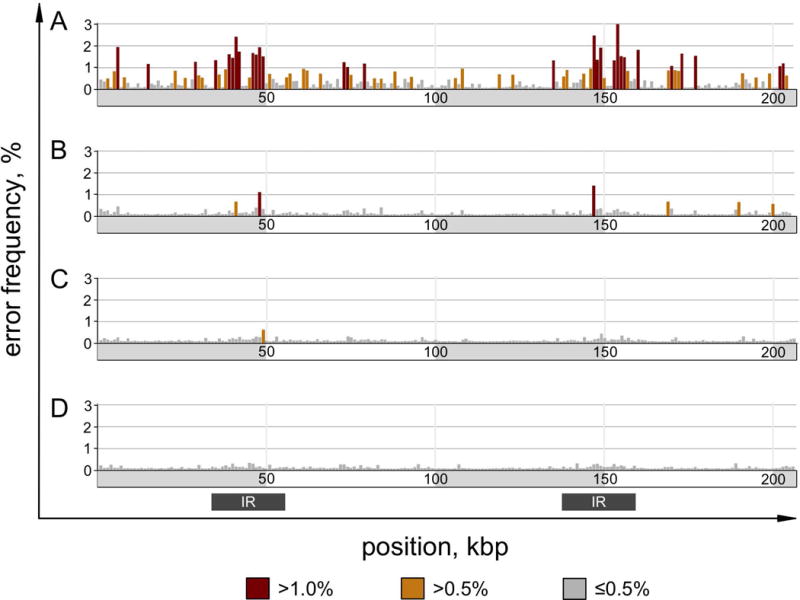
DNA-Seq reads were aligned to each of four genome versions in parallel using the same parameters. An error rate was determined for each locus as the percentage of mismatched base calls or InDels relative to the total. The per-locus error rate was averaged across non-overlapping 1 kbp windows over the length of each genome version and plotted. Bars are color coded as indicated by the legend. A gray bar below indicates the position of the inverted repeats. The four genome versions are as follows: (A) BK000554.2 from NCBI GenBank submitted in 2002, (B) FJ423446.1 from NCBI GenBank submitted in 2009, (C) cv11, which is a variant-corrected reconstruction of FJ423446.1, and (D) CPv4, which is a de novo assembly.
Figure 4. Evaluating genome versions.
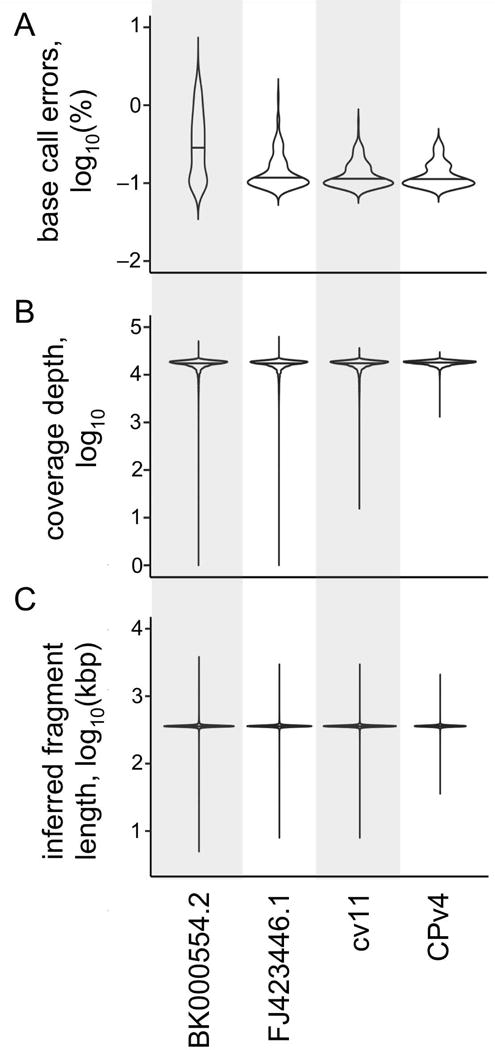
As in Figure 3, four different versions of the C. reinhardtii chloroplast genome were compared using a variety of metrics. A set of 1×108 paired-end DNA-Seq reads (100 +100 nt) were aligned to each genome version in parallel using the same parameters. (A) The error rate was determined as percentage of mismatched base calls relative to total base calls for each locus in each genome version. This error rate was averaged across all loci in non-overlapping 1 kbp windows, log10-transformed, and then plotted as a violin plot. A base call was considered errant if it differed from the reference, or if it represented an insertion or deletion. (B) The depth of coverage at each locus was determined, and log10-transformed. The distribution of coverage is presented as a violin plot for each genome version. (C) The inferred size of the DNA fragments in the DNA-Seq library were determined by the relative position of their alignments in each genome version. The average of the inferred fragment sizes was calculated for each locus, log10-transformed, and then plotted as a violin plot. In each panel, the median of the distribution is indicated by a horizontal line.
Next, the per-locus depth of coverage was evaluated. While some variation in depth of coverage is expected (Benjamini and Speed, 2012), extremely high or extremely low coverage suggest the presence of inaccuracies in the reference sequence. The mean depth of coverage was nearly uniform at ~17,000 reads per locus across all four genome versions (Table 1). However, as with the error frequency, we observed heterogeneity in the coverage depth. To examine this, the coverage depth was averaged across non-overlapping 1 kbp windows. For each window, the percentage of loci that were more than 1.5 times the mean or less than 0.5 times the mean were plotted for each genome version (Figure S2). The distribution of coverage depth for all ~200,000 loci in each genome version is presented in Figure 4B. While the median coverage for all four genomes is nearly identical, cv11 and CPv4 have fewer extremes than the two GenBank versions. Only cv11 and CPv4 have no loci where the coverage drops to zero, with minimum coverage depths of 16 and 1,345 reads/locus, respectively.
Lastly, we examined the inferred fragment sizes of the paired end DNA-Seq libraries. This determination is based on the relative positions of the alignments of the left-end read and the right-end read for each sequenced fragment. While some distribution in fragment sizes is expected, extremes of fragment size suggest missing or extraneous sequence in the reference. As expected, the mean inferred fragment size was nearly identical at ~360 bp for each of the four genome versions (Table 1). However, the inferred insert size was not uniform across each genome version. The inferred fragment size was averaged across non-overlapping 1 kbp windows, and the percentage of loci with insert sizes greater than 1.5 times the mean or less than 0.5 times the mean were plotted for each genome version (Figure S3). In this analysis, hot spots of high variation are observable in the two GenBank versions, and to a lesser extent in cv11 and CPv4. The distribution of inferred fragment sizes is shown in Figure 4C. BK000554.2 (2002) has the widest distribution with 3.6% of loci having inferred fragment sizes outside of the mean ± 2 standard deviations versus 0.74% for CPv4.
Relative copy number of organellar genomes
In the DNA-Seq libraries of strain CC-503, 19.4% of reads aligned to the chloroplast genome and 2.1% aligned to the mitochondrial genome (Figure 5A). From this and the known sizes of the three genomes, we wished to estimate the relative copy number of the organellar genomes per cell. Raw read counts do not correspond in a one-to-one fashion with template copy number as a result of differences in GC content, which can skew PCR amplification, as well as differences in the mappability of short sequencing reads (Benjamini and Speed, 2012). In the case of C. reinhardtii, the GC content of the three genomes are significantly different (Figure S1). To account for this, the coverage at each locus was corrected for GC content and mappability using Hidden Markov Model approach (Ha et al., 2012). The resulting distribution of coverage is presented in Figure 5D. Based on the median coverage for each genome, there are an estimated 83 copies of the chloroplast genome for every one copy of the nuclear genome. This is in excellent agreement with a previous study that estimated 80 – 90 copies of the chloroplast genome per cell by fluorescence microscopy of DAPI-stained chloroplasts (Misumi et al., 1999). We estimate that there are 130 copies of the mitochondrial genome per cell (Figure 5D). An earlier study of the mitochondrial genome estimated that its per cell copy number was similar to that of the chloroplast genome (46 copies for the mitochondria versus 52 copies for the chloroplast), which is lower than our estimate (Ryan et al., 1978).
Figure 5. DNA-Seq and RNA-Seq coverage.
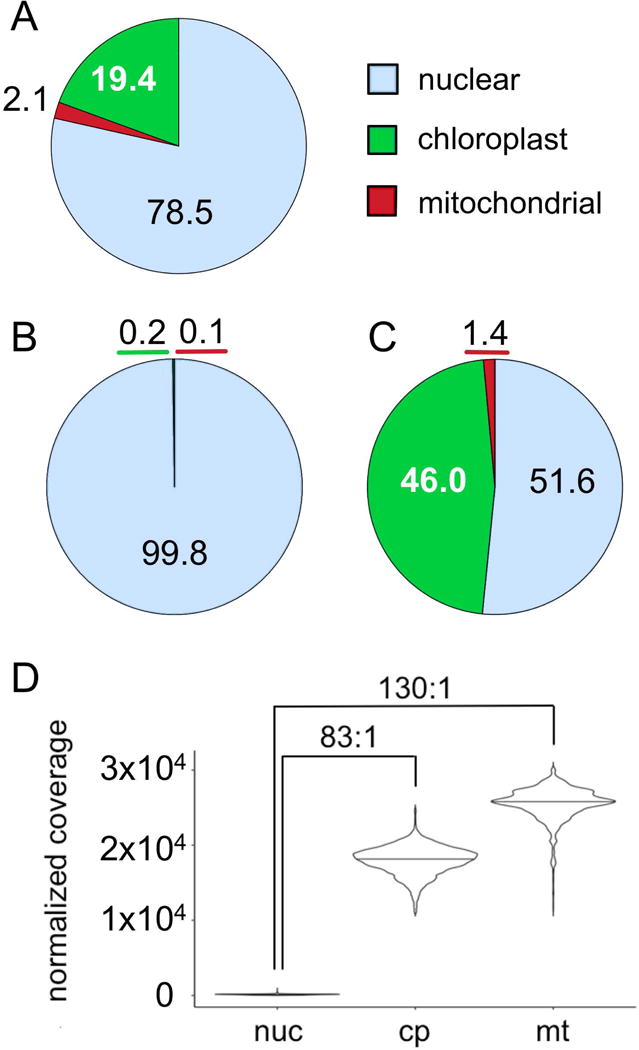
(A) A set of 1×108 paired-end DNA-Seq reads were aligned to the nuclear, chloroplast, and mitochondrial genomes of strain CC-503. The percentage of reads aligned to each genome is presented as a pie chart with the color scheme as indicated. (B and C) The percentage of 3×107 reads generated from RNA-Seq libraries generated with a poly(A) based protocol (B) or an rRNA-depletion protocol (C) from a representative experiment are presented as pie charts. (D) The depth of DNA-Seq coverage at each locus was adjusted by HMMcopy to account for variation in GC-content and sequence complexity. The distribution of corrected depth of coverage was plotted for each genome as a violin plot. A horizontal bar in each violin indicates the median of coverage for that genome. Values in excess of the mean plus or minus three times the standard deviation were trimmed. Brackets above the violin plot indicate the ratio of the median for the chloroplast and mitochondrial genomes relative to the nuclear genome.
RNA-Seq-guided gene annotations
To annotate the chloroplast de novo assembly, CPv4, putative protein coding ORFs were identified computationally in the nucleotide sequence. Then, RNA-Seq libraries prepared with an rRNA-depletion protocol on samples of RNA from a variety of conditions were sequenced and aligned to CPv4. The resulting coverage was then used to validate or reject the computationally identified ORFs (Data S3). When the resulting genes were compared to the annotations in FJ423446.1 (2009), 68 protein coding ORFs were identical between the GenBank annotations and those of CPv4. However, there were some exceptions.
In the first publication of the chloroplast genome, rpoC1, the gene encoding the β′ subunit of the plastid-encoded RNA polymerase (PEP), was split into two genes labeled rpoC1a and rpoC1b (Maul et al., 2002). This was due to an inability to identify a single ORF spanning the two regions, or to identify a full-length transcript by RT-PCR. The annotation of rpoC1 as two separate genes was propagated in the subsequent genome version (Smith and Lee, 2009). Curiously, we found that the intergenic region between rpoC1a and rpoC1b almost perfectly overlaps with the 2.4 kbp inversion that distinguishes the GenBank versions from CPv4 (see de novo assembly section above). In contrast to the GenBank versions, CPv4 includes a single, continuous 5799 bp ORF that spans rpoC1a, rpoC1b and the “intergenic” region between them. We observed high coverage of RNA-Seq reads across the full-length CPv4 rpoC1 gene, which suggests that the updated gene model is correct (Figure 6A). To further validate this, total soluble protein from Chlamydomonas cultures was subjected to a proteomics analysis by mass spectrometry. We identified 24 distinct peptides uniquely attributable to the 1932 aa rpoC1 protein predicted in CPv4 (Data S4). These were distributed throughout the protein, including 8 peptides from the portion of the gene that is intergenic in the prior annotations (Figure 6A, purple boxes). Based on these results, it seems likely that the choice to split rpoC1 into two separate genes in the earlier versions was made erroneously, albeit justifiably, due to an error in the underlying assembly.
Figure 6. Corrected rpoC1 and rps2 gene models.
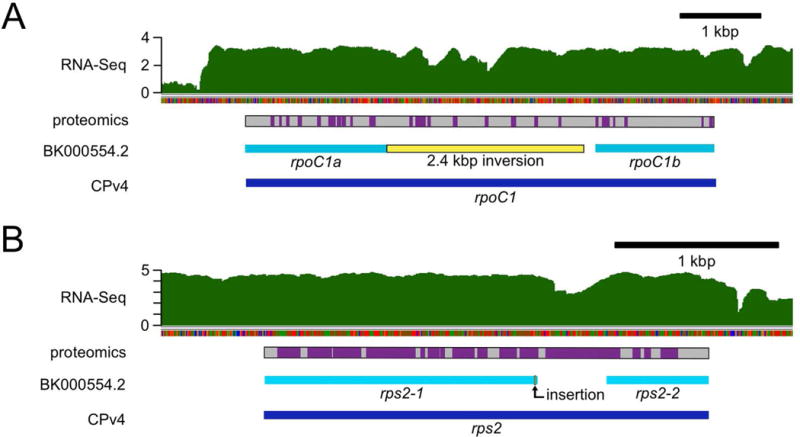
In the course of this work, we identified many errors in the genetic sequence of the previously available chloroplast genomes, such as GenBank BK000554.2. Some of these errors likely resulted in mis-annotated chloroplast genes. Here, RNA-Seq coverage and proteomics data were used to validate the improved CPv4 gene models. (A) In BK000554.2, the gene encoding the β′ subunit of the plastid-encoded RNA polymerase, rpoC1, was annotated as two separate genes: rpoC1a and rpoC1b (cyan bars). In contrast, CPv4 is annotated with a single ORF (dark blue bar) encoding a 1932 aa protein that spans both of the previous models. Relative to CPv4, there is a 2.4 kbp inversion (yellow bar) in BK000554.2 in the region between rpoC1a and rpoC1b. RNA-Seq coverage from a representative sample is presented on a log10 scale in green. A gray bar indicates the protein sequence encoded by CPv4 rpoC1, with purple boxes to indicate peptides that were identified by mass spectrometry. (B) Similarly, the gene encoding the S2 ribosomal protein was annotated as two genes in BK000554.2: rps2-1 and rps2-2 (cyan bars). A single nucleotide insertion (bent arrow) in that assembly causes a frame shift that leads to a premature stop codon. The CPv4 rps2 ORF (dark blue bar) spans both genes and encodes a 910 aa protein.
In the 2002 BK000554.1 assembly and the corresponding manuscript, two adjacent genes, ORF570 and ORF208, were proposed to contribute to the S2 ribosomal small subunit protein (Maul et al., 2002). These were labeled rps2-1 and rps2-2, respectively, and this labeling persisted in work by other groups (Smith and Lee, 2009). Relative to CPv4, there is a single nucleotide insertion in BK000554.1 near the 3′ end of the rps2-1 gene. The resulting frame shift leads to a stop codon a few bases further downstream. In CPv4, a single ORF spans rps2-1, rps2-2, and the intervening sequence. High coverage of RNA-Seq reads throughout the combined rps2 gene suggests that the CPv4 version of the gene is expressed (Figure 6B). Further, peptides were identified by mass spectrometry for most of the rps2 protein as annotated in CPv4 (Data S4 and Figure 6B).
A few ORFs, such as orf2971, were identified in the GenBank chloroplast genome versions, but were never assigned a gene name or function. Based on the RNA-Seq analyses described below, we observed that this gene is expressed at moderate levels under most conditions (Data S5), and the protein product of this gene was identified by 11 distinct peptides by mass spectrometry (Data S4). When orf2971 was compared to the nr database with BLASTP, there were 22 hits with E-values less than 1×10−100 in species other than C. reinhardtii (Data S6). All of the 22 hits were to genes found in chloroplasts of various chlorophyte species, and all but two of them were annotated as ftsH, with the remaining two receiving generic descriptors.
The ftsH gene was originally identified as cell division gene in Escherichia coli (Ogura et al., 1991). To examine the relationship between C. reinhardtii orf2971 and other ftsH family members, a protein similarity network was constructed that included ftsH proteins from 41 different species including E. coli (Figure S4A and Table S2). C. reinhardtii orf2971 clustered with the ftsH family members from the chloroplast genomes of 21 other chlorophyte species. This group was distinct from, but still connected to, a second group of ftsH orthologs in cyanobacteria, streptophytes, and fungi. Interestingly, this second cluster included several nucleus-encoded ftsH-family genes from the chlorophyte lineage, such as FTSH4 and FTSH11 from C. reinhardtii. Sequence similarity with the other ftsH-family proteins was largely localized to a conserved AAA domain (PFAM00004: ATPase family associated with various cellular activities). The 22 chloroplast-encoded chlorophyte genes were all predicted to encode for large proteins, with a median size of 3480 aa (Table S2). This is significantly larger than the other proteins in this analysis, which had a median size of 706 aa, or the 644 aa E. coli ftsH. Based on sequence similarity and the conserved domains, this analysis suggests that C. reinhardtii orf2971 is an ortholog of the other chloroplast-encoded ftsH genes from the chlorophyte lineage, and we have labelled it as such. However, the much larger size of these chlorophyte lineage proteins suggest that they may have diverged significantly from other members of the ftsH family.
A second chloroplast gene, orf1995, was identified in both GenBank versions and in CPv4, but did not receive a gene name. RNA-Seq analysis indicates that the gene is expressed at high levels in vegetatively growing cells (Data S5), and 46 distinct peptides were identified by mass spectrometry that could be attributed to the orf1995 gene product (Data S4). Previously, the orf1995 gene was found to encode a large transmembrane protein that is essential for cell survival (Boudreau et al., 1997). This work speculated that the gene might be a ycf1 ortholog, despite significant sequence divergence from the ycf1 genes of land plants. When compared to the nr database with BLASTP, we found 42 hits with E-values less than 1×10−100 in species other than C. reinhardtii (Data S6). 41 out of 42 of these hits were annotated as ycf1, with the remaining one given a generic gene name, orf2032. A protein similarity network reveals that C. reinhardtii orf1995 is tightly clustered with genes annotated as ycf1 in 40 other chlorophyte species (Figure S4B and Table S2). A second, unconnected cluster grouped ycf1 genes from 12 streptophytes species. This analysis agrees with the work of de Vries and colleagues, in which they found that the ycf1 gene sequence had diverged significantly between chlorophytes and streptophytes (de Vries et al., 2015).
The C. reinhardtii chloroplast genome carries two copies of the Wendy Transposon (Fan et al., 1995). Wendy II, which lies between psaA exon 3 and psbH, is not annotated in FJ423446.1. There was low, but clearly observable expression at that locus of a transcript that would encode a 202 aa polypeptide (Figure S5A). This is labeled orf202 in CPv4. Wendy I, which lies between rpoC1 and petA, expresses a much larger transcript that would encode an 854 aa polypeptide labeled orf854 (Figure S5B). Interestingly, this transcript extends beyond the boundary of Wendy I by an additional 508 nt. There is 80% identity between the first 200 aa of orf202 and orf854, which suggests significant sequence divergence. Previously, Wendy I was reported to contain two ORFs; one of 140 aa and another of 271 aa (Fan et al., 1995). However, the nucleotide sequence reported in that work has a 1 nt insertion relative to the sequence of CPv4. This InDel creates a stop codon that divides what would otherwise be a single large ORF into two smaller ones.
Previous versions of the C. reinhardtii chloroplast genome annotated only the CDS portion of the genes. However, coverage of RNA-Seq reads to the chloroplast suggest that all genes have 5′ and 3′ UTRs of variable lengths. Further, the RNA-Seq coverage confirmed that several chloroplast genes are co-transcribed in clusters as polycistronic transcripts (Figure S6). Some of these, such as rpl23–rpl2–rps19 (Figure S6B), psbB–psbT (Figure S6F), atpA–psbI–cemA–atpH–atpF (Figure S6K), and chlN–tscA (Figure S6L), have been reported previously (Rymarquis et al., 2006; Drapier et al., 1998; Hahn et al., 1998). We identified at least 16 clusters of co-transcribed genes (Figure S6).
In addition to the protein-coding genes, tscA, which aides in the trans-splicing of psaA, 29 tRNA genes, and 5 rRNA genes were annotated in CPv4 (Goldschmidt-Clermont et al., 1991).
The new mitochondrial assembly, MTv4, was annotated as described above for the chloroplast (Data S3). Consistent with previous studies (Gray and Boer, 1988), all genes appear to be expressed as two polycistronic messages from a bi-directional transcription start site between nad5 and cox1 (Figure 2).
Quantification of organellar transcripts by RNA-Seq
The majority or RNA-Seq work in C. reinhardtii has been performed using libraries that are prepared from poly(A)-enriched RNA. Since chloroplast and mitochondrion-encoded transcripts are not poly-adenylated to the same degree as most nucleus-encoded transcripts, it is expected that organellar transcripts are underrepresented in most RNA-Seq work. To test this directly, libraries were prepared from a common sample of total RNA using either a poly(A)-enrichment protocol or rRNA-depletion protocol, and then aligned to the nuclear, chloroplast, and mitochondrial genomes (Figures 5B and C). Surprisingly, nearly half of the RNA-Seq reads (46.0%) were chloroplast-encoded when the rRNA-depletion strategy was used. In contrast, only 0.2% of reads originated from the chloroplast when poly(A)-enriched libraries were analyzed. This amounts to a greater than 400-fold enrichment. Similarly, mitochondrial transcripts increased from 0.1 to 1.4% of total reads when the two protocols were compared, which equates to a 27-fold enrichment.
In order to examine RNA metabolism from both the chloroplast and mitochondrion, RNA-Seq libraries were prepared from a wide variety of conditions including: diurnally grown cultures sampled in the dark and in the light, and cultures grown in medium with and without Fe or Cu. A previous RNA-Seq study of nuclear transcription in C. reinhardtii during the sexual cycle was performed using an rRNA-depletion protocol (Lopez et al., 2015). That data was reanalyzed here to examine organellar transcript abundance. The expression of each chloroplast gene was quantified in terms of fragments per kbp of gene per million mapped reads (FPKMs) for each of these experiments, and is presented on a log10 scale on the left portion of Figure 7. Fold-change comparisons between pairs of matched samples are presented on the right. The transcript abundances of nucleus-encoded subunits of photosystem I, photosystem II, and cytochrome b6f are also included for comparison. Across all conditions examined, transcript abundance from chloroplast genes is high: ranging from 100 – 100,000 FPKMs. Differences in transcript abundance between conditions are relatively minor; an exception being gametes compared to vegetatively growing cells. The eight protein coding genes of the mitochondrion are presented in a similar fashion in Figure 8. As a validation of this analysis, the abundance of nucleus-encoded transcripts from sentinel genes known to be up or down-regulated under these conditions were examined using the same pipeline, and found to have the expected expression patterns (Figure S7).
Figure 7. RNA-Seq analysis of chloroplast genes and related nuclear genes.
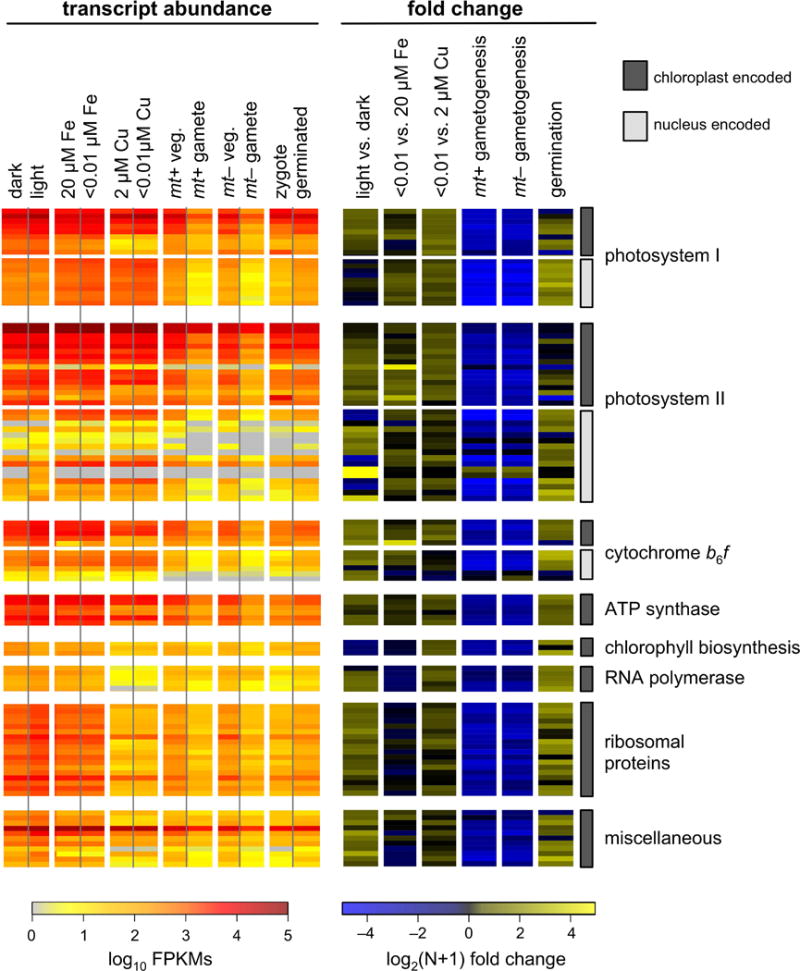
Transcript abundances for all chloroplast-encoded genes were determined by RNA-Seq from a series of experiments. The resulting transcript abundance determinations were calculated in terms of log10 transformed FPKMs, and are presented as a heat map in the panel on the left. Nucleus-encoded, chloroplast-targeted transcripts for subunits of photosystem I, photosystem II and cytochrome b6f are included for comparison. Chloroplast-encoded genes and nucleus-encoded genes are distinguished by the presence of a dark gray or light gray bar, respectively, to the right of the figure. On the right, pairs of samples were compared in terms of log2(N+1) transformed fold change.
Figure 8. RNA-Seq analysis of mitochondrion-encoded genes and related nuclear genes.
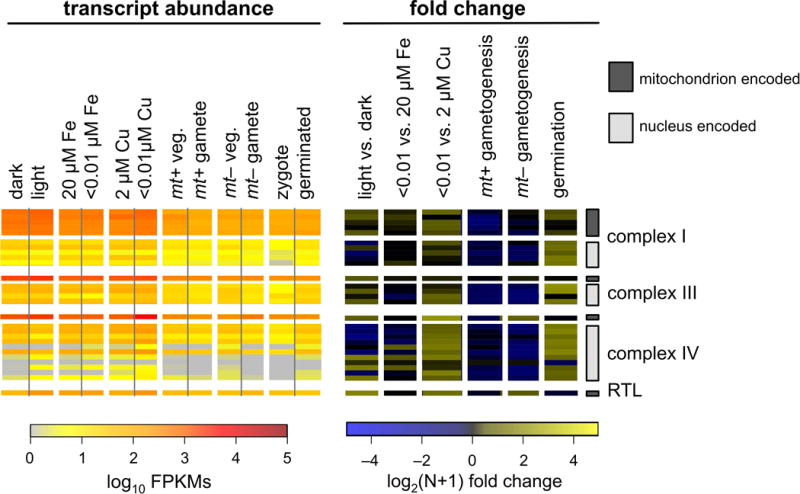
Transcript abundances for all mitochondrion-encoded genes were determined by RNA-Seq from a series of experiments. The resulting transcript abundance determinations were calculated in terms of log10 transformed FPKMs, and are presented as a heat map in the panel on the left. Nucleus-encoded, mitochondrion-targeted transcripts for subunits of complex I, complex III and complex IV are included for comparison. Mitochondrion-encoded genes and nucleus-encoded genes are distinguished by the presence of a dark gray or light gray bar, respectively, to the far right of the figure. On the right, pairs of samples were compared in terms of log2(N+1) transformed fold change.
Next, we wished to identify the nuclear genes with expression patterns most similar to that of the chloroplast genes. The RNA-Seq expression estimates of all nuclear, chloroplast, and mitochondrial genes were calculated as the fold change between pairs of conditions, as described above for Figures 7 and 8, and the complete dataset was subjected to a k-means clustering analysis. With 10 centers, the majority of chloroplast genes, 56 out of 75 (75%), co-clustered with 498 nuclear genes (Data S7). As expected, this cluster contained many nucleus-encoded, chloroplast-targeted members of the photosynthetic apparatus, such as PSAD, PSBO and PETM. The GreenCut is a list of ~600 genes that are conserved in plants and green algae but absent from non-photosynthetic organisms (Karpowicz et al., 2011). 108 out of 498 (22%) of the nuclear genes that co-clustered with the chloroplast genes are members of the GreenCut, which represents a significant enrichment (p = 2.6×10−55). Next, we queried the nuclear genes in this cluster for enrichment of gene ontology (GO) terms. There was statistically significant enrichment (p < 0.05) of 12 GO terms for biological processes, which included photosynthesis, chlorophyll metabolic processes, and carbon fixation. The full list of genes in this cluster, their annotations, membership in the GreenCut, and GO term enrichment are in Supplemental Data S7.
Given that 75 protein coding genes in the chloroplast are responsible for nearly half of RNA-Seq reads (Figure 5C), it would be expected that the majority of the most highly expressed genes in the cell are in the chloroplast. All transcripts across 12 different experimental conditions were quantified in terms of FPKMs and ranked. The 100 most abundant transcripts for each experiment are included in Data S8. The top 20 were classified as originating in either the nucleus, chloroplast, or mitochondrion, and are presented graphically in Figure 9. As many as 19 of the top 20 genes are chloroplast derived, depending on condition. The only conditions where chloroplast transcript abundance is significantly reduced are the ones in which gametogenesis is induced. A single mitochondrial transcript, cox1, is in the top 20 for one sample; the mt− gamete. For the most abundant nucleus-encoded transcripts, many, including LHCBM1, RBCS2 and PCY1, encode proteins that are chloroplast targeted.
Figure 9. Twenty most abundant cellular transcripts.
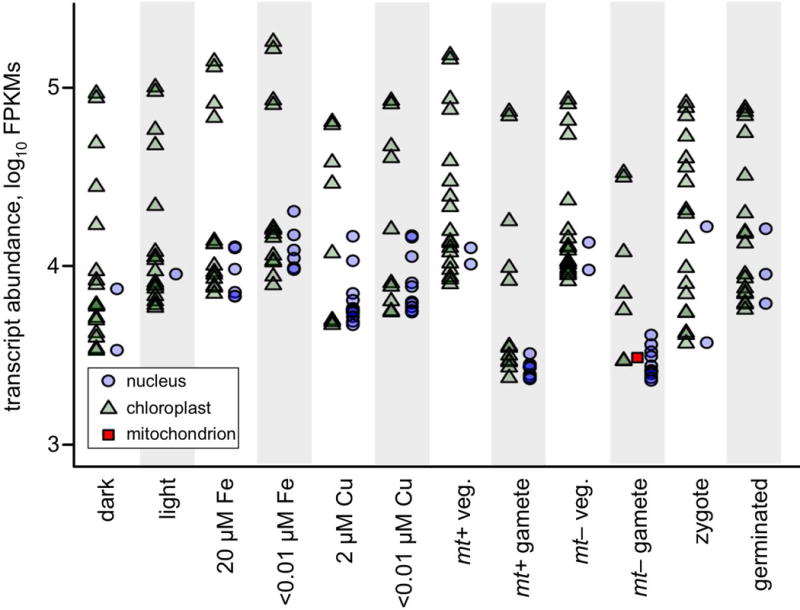
Transcript abundances were quantified in terms of FPKMs by RNA-Seq from 12 different cultures of C. reinhardtii grown under various conditions as indicated in the figure (see text for details). The 20 most abundant transcripts in each sample were plotted on a log10 scale with following symbols: nucleus-encoded transcripts as blue circles, chloroplast-encoded transcripts as green triangles, and mitochondrion-encoded transcripts as red squares. Additional information on these genes, including gene names and FPKMs, can be found in Supplemental Data S8.
Comparison of RNA-Seq preparation methods
Despite the fact that most organellar RNA is filtered out during the preparation of RNA-Seq libraries by the poly(A)-enrichment approach, a small number of transcripts do remain (Figure 5B). We wished to determine what effect the choice of library preparation method has on quantification of chloroplast and mitochondrial transcripts. RNA-Seq libraries were prepared from a common set of samples of total RNA using either poly(A)-enrichment or rRNA-depletion, and then sequenced and aligned to the nuclear, mitochondrial, and chloroplast genomes.
The result is presented as a series of pair-wise comparisons in Figure 10. While the majority of nucleus-encoded transcripts are minimally affected by the choice of library preparation protocol, a few transcripts, such as those expressed from most histone genes, are detected at significantly higher levels in rRNA-depletion libraries. As expected, the chloroplast and mitochondrial transcripts are quantified at levels many orders of magnitude higher when the rRNA-depletion method is used. The effect is much more pronounced for chloroplast transcripts than it is for mitochondrial transcripts. Interestingly, the degree to which chloroplast transcripts are underestimated by the poly(A) enrichment method is not a linear function. Instead it increases with increasing transcript abundance. This is evident by the slope of linear regressions fit to the comparison of transcript abundances by poly(A)-enrichment versus rRNA-depletion (Figures 10C and D). For nucleus encoded genes, the line is close the diagonal, with slopes of 0.96 and 0.94 for the +Fe and −Fe samples, respectively. For the chloroplast genes, the corresponding slopes are 0.44 and 0.39.
Figure 10. Comparison of RNA-Seq library preparation methods.
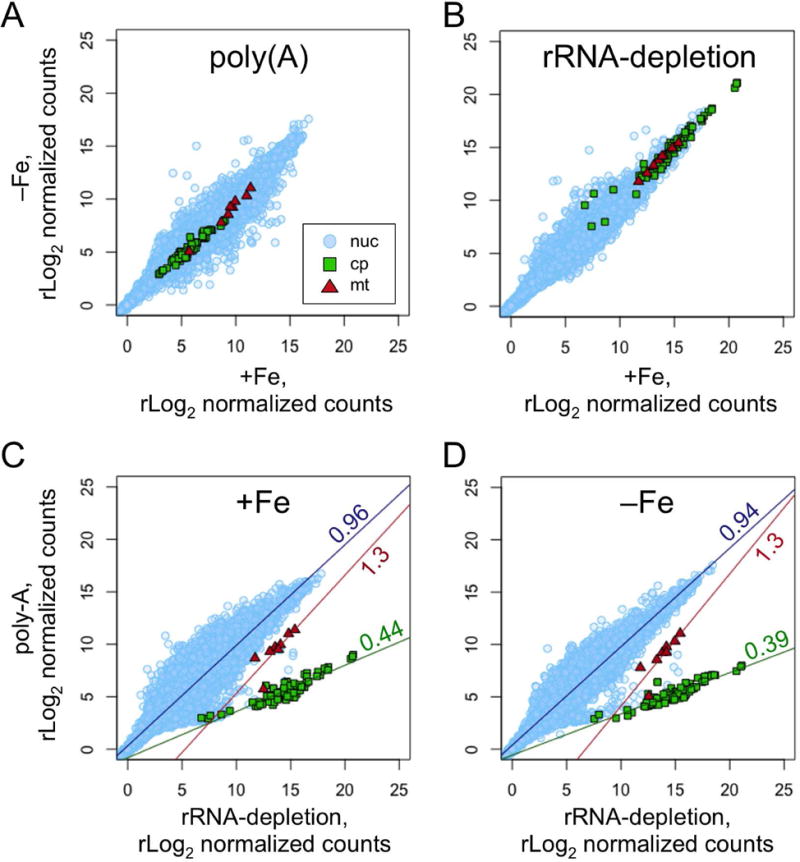
Total RNA was collected from cultures grown in media with 20 μM Fe (+Fe) or 4 h after transfer to media with <0.01 μM Fe (−Fe). The purified RNA was then used to construct RNA-Seq libraries using either a poly(A)-enrichment protocol or an rRNA-depletion protocol. Sequencing reads from each library were aligned to the nuclear, chloroplast and mitochondrial genomes in parallel. The number of counts per gene were determined and rLog2 normalized by DESeq2. The resulting counts per gene were plotted as pair-wise scatter plots with nuclear genes as light blue circles, chloroplast genes as green squares, and mitochondrial genes as red triangles. Comparisons of the −Fe sample versus the +Fe sample for libraries prepared by (A) the poly(A) protocol, or by (B) the rRNA-depletion protocol are shown. Comparisons of the poly(A) protocol versus the rRNA-depletion protocol for (C) the +Fe sample, or for (D) the −Fe sample are shown. For (C) and (D), a linear regression was fit to each set of genes and plotted. The slope of the line is indicated.
Transcript splicing
The C. reinhardtii chloroplast genome contains a few genes whose transcripts require splicing to generate mature mRNA. One of these is psaA, which encodes photosystem I chlorophyll a binding apoprotein, A1. The psaA gene is split into three independently transcribed genes at distant loci on the chloroplast genome (Kück et al., 1987). Two group II introns assemble from portions of these three transcripts plus one additional non-coding RNA, tscA, and are then spliced out with the aid of a number of nucleus-encoded proteins (Goldschmidt-Clermont et al., 1991). In order to quantify the degree of splicing of the psaA transcript, a pseudo-assembly was constructed to contain both the spliced and unspliced versions of the gene, and reads from a number of RNA-Seq studies were aligned to it. The depth of coverage at the splice sites was used to estimate the relative abundance of the spliced and unspliced forms. In each case for psaA, the large majority of transcripts, 71.7% to 96.7%, were in the spliced form (Figure 11A). Differences between conditions, such as light versus dark or plus versus minus Fe, were minor and generally not statistically significant.
Figure 11. Splicing of psaA and psbA.
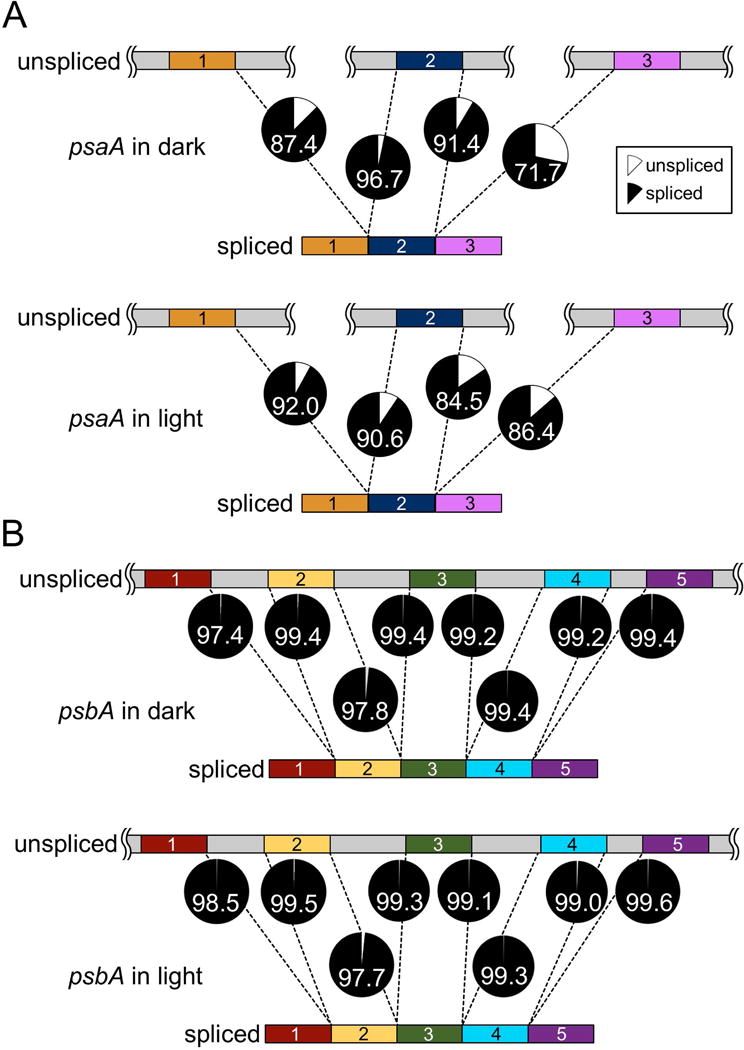
Mature psaA mRNA is the result of trans-splicing between three independently transcribed RNA fragments. Maturation of psbA mRNA requires the excision of four type I introns. To quantify the efficiency of these splicing reactions, a pseudo-assembly containing both the spliced and unspliced forms of the psaA and psbA transcripts was constructed in silico, and RNA-Seq reads from the light versus dark experiment were aligned to it. The mean depth of coverage was determined for the 20 NTs upstream and downstream of each splice site in both the spliced and unspliced forms. The ratio of depth of coverage from either the spliced form (black) or the unspliced form (gray) relative to the total is presented as a pie chart for each splice site for (A) psaA and (B) psbA. The number in each pie indicates the percentage that is spliced for that locus. RNA-Seq libraries were prepared from rRNA-depleted RNA from samples collected at the end of the dark phase, or one hour into the light phase, as indicated.
The gene encoding photosystem II D1 protein, psbA, is present in two copies within the inverted repeat regions. The gene is divided into five exons by the presence of four large group-I introns that must be spliced out to form the mature psbA transcript (Erickson et al., 1984; Holloway et al., 1999). As with psaA, RNA-Seq data was used to quantify the percentage of spliced versus unspliced transcripts. In contrast to a prior study of psbA transcript splicing in response to light, we observed nearly complete splicing under all conditions examined (Deshpande et al., 1997). This ranged from 97.7 to 99.7% in all samples, including those from diurnally grown cultures sampled in the light and dark phases (Figure 11B).
Lastly, there are two copies of 23S rRNA gene, rrnL, within the inverted repeats that are split by a group-I intron called I-CreI (Dürrenberger and Rochaix, 1991). The fact that an rRNA-depletion protocol was used to generate the RNA-Seq data precludes quantitative analysis of rrnL splicing. Qualitatively, however, it appears that the significant majority of rrnL is in the spliced form (Figure 1).
Chloroplast transcripts are not edited
Editing of chloroplast transcripts – primarily C to U deamination – is widespread in land plants, but is generally understood to be absent in the chlorophyte lineage (Stern et al., 2010). In contrast to this, Shi and colleagues recently reported that they had identified 68 examples of edited loci in C. reinhardtii chloroplast transcripts (Shi et al., 2016). For their analysis, they aligned RNA-Seq reads to BK000554.2 (2002), and identified non-reference base calls. In contrast to that study, we did not observe evidence for editing in the alignments of our RNA-Seq data when using the same criteria (frequency of non-reference base call ≥50%, base call quality score ≥ 20, 10 counts minimum).
To resolve this discrepancy, we identified two procedural differences between the Shi study and this work. First, Shi and colleagues used PASS for RNA-Seq read alignment, while STAR was used in this work (Campagna et al., 2009; Dobin et al., 2013). To determine if the different analysis pipelines could be the cause of our discordant results, we aligned RNA-Seq data with both alignment tools, PASS and STAR, in parallel, and compared the results. Second, the previous study aligned reads to BK000554.2 (2002), which has hundreds of inaccuracies relative to CPv4 (see section above). To examine this effect, reads were aligned to BK000554.2 and CPv4 in parallel, and compared.
Each of the 68 loci identified by Shi were then evaluated to compare the effects of using BK000554.2 versus CPv4 as a reference sequence, and of using PASS versus STAR for the alignment. The results are detailed in Table S3. 40 of the 68 loci were attributable to single nucleotide errors in BK000554.2. An additional 10 loci were due to 1 – 3 nt InDels at or within a few nts of the putative edit site in BK000554.2 that lead to misalignment of the RNA-Seq reads. Of the putative editing sites, 10 were at intron-exon boundaries. In contrast to STAR, the PASS aligner was developed for DNA-Seq reads and does not properly account for gaps in the alignment due to mRNA splicing. The remaining 8 sites are all within highly repetitive sequences. PASS aligner, but not STAR, was found to align reads too promiscuously between different degenerate repeat sequences, which creates a number of non-reference base calls. Taken together, it seems that both the choice of alignment tool, and the choice of a reference sequence contributed to erroneously identifying 68 edited loci in the chloroplast transcripts in the previous study.
Heterogeneous polynucleotide 3′ tails on mitochondrial transcripts
Upon inspection of RNA-Seq read alignments to the mitochondrial genes, we observed evidence for heterogeneous polynucleotide tails at the 3′ end of all eight protein coding transcripts (Zimmer et al., 2009). Examples of this for each of the mitochondrial genes are presented in Figure S8. These tails varied in their start site by up to 6 nt, and varied a great deal in terms of nucleotide composition. While all four nucleotides were observed, C was over represented at 65%, and G was under represented at 1%. A and U were observed at 14% and 21%, respectively. Given the degenerate nature of these sequences, their length was difficult to determine. The polynucleotide 3′ tails appeared in all samples examined, and regardless of which alignment tool was used (STAR, PASS, BWA-MEM). We did not observe this phenomenon in the chloroplast-encoded transcripts.
Comparative genomics analysis
The chloroplast and mitochondrial genomes presented here were generated from the DNA of the same strain as the reference nuclear genome, strain CC-503. However, dozens of other strains are commonly used in laboratory research. In order to examine the degree of genetic divergence between strains for the organellar genomes, we performed a comparative genomics analysis on seven additional standard laboratory strains and 11 wild isolates. As a control, we re-analyzed strain CC-503 sequence data from two independent sources. A VCF-formatted table of all variants is included in Data S9.
The eight standard laboratory strains that were included in this analysis are all descended from a single zygospore isolated in 1945 (Gallaher et al., 2015). For these strains, only one SNV and two InDels were identified in the chloroplast genome (Table 2). This low number of variants is consistent with the uniparental inheritance of the organelle genomes from a recent common ancestor. Likewise, the mitochondrial genome had five SNVs and one InDel. The reference strain, CC-503, was sequenced both by this group and by a group from NYU (Flowers et al., 2015). Curiously, four of the five mitochondrial SNVs detected were unique to strain CC-503 as sequenced by the NYU group.
Table 2.
Summary of variants
| CP SNVs | CP InDels | MT SNVs | MT InDels | |
|---|---|---|---|---|
| lab strains | 1 | 2 | 5 | 1 |
| wild isolates | 1754 | 492 | 130 | 13 |
| combined | 1755 | 494 | 135 | 13 |
For the 11 wild isolates, we detected 1,754 SNVs and 492 InDels in the chloroplast genome and 130 SNVs and 13 InDels in the mitochondrial genome (Table 2). For any one strain, this ranged between 447 and 944 total organellar variants. Given the size of the organellar genomes, this corresponds to a variant frequency of between 0.20% and 0.43%, which is 10-fold lower than the ~3% variant frequency that was previously reported for the nuclear genomes of these same strains (Flowers et al., 2015).
Next, we wished to determine what effects the 2,397 total combined variants might have chloroplast and mitochondrial genes. In the chloroplast, the majority of variants,1465 out of 2249 (65.1%), were found outside of protein coding sequences (Table 3). A similar distribution, 97 out of 148 (65.5%), was found outside of protein coding sequences in the mitochondrial genome. Within the chloroplast protein coding genes, 724 (32.2%) were SNVs that change one codon to another. The altered codons were split almost equally between synonymous and non-synonymous codons. There were 50 codon-altering SNVs in the mitochondrial genes, but 46 of those were synonymous changes. There were 60 InDels within the coding sequences of chloroplast genes, but 58 of those maintain the gene’s reading frame (i.e. occur in multiples of three). Only two frameshifting InDels were found in the chloroplast; one in strain CC-2342 and the other in strains CC-2936 and CC-2937. Both of these variants disrupt the orf854 gene of the Wendy I transposon, which is likely unnecessary for normal chloroplast function. The only intragenic InDel in the mitochondrion removes two codons in the nad1 gene of strain CC-1373.
Table 3.
Predicted effects of variants
| CP Count | CP Percent | MT Count | MT Percent | |
|---|---|---|---|---|
| frameshift InDel | 2 | 0.1% | 0 | 0.0% |
| frameshift-preserving InDel | 58 | 2.6% | 1 | 0.7% |
| non-synonymous codon | 363 | 16.1% | 4 | 2.7% |
| synonymous codon | 361 | 16.1% | 46 | 31.1% |
| UTR | 855 | 38.0% | 27 | 18.2% |
| intronic | 11 | 0.5% | 0 | 0.0 |
| intergenic | 599 | 26.6% | 70 | 47.3% |
| total | 2249 | 100% | 148 | 100% |
The C. reinhardtii chloroplast genome harbors two copies of the Wendy transposon. It is believed that these integrated into the chloroplast genome sometime since the divergence of C. reinhardtii and C. moewusii approximately 500 MYA (Fan et al., 1995; Munakata et al., 2016). We examined the DNA-Seq reads extending outward from the terminal inverted repeats of Wendy for each copy in each of the 11 wild isolates to see if we could find evidence for transposition. In each case, the DNA-Seq coverage indicated that the Wendy transposons have been stably located in the chloroplast genome for at least as long as the divergence of the North American isolates of C. reinhardtii included in this analysis.
In the previous study, the distribution of variants within the nuclear genomes of the wild isolates of C. reinhardtii corresponded to the geographic distribution of those strains (Flowers et al., 2015). For comparison, we performed a similar analysis on the organellar variants in the same strains. As expected, there was a nearly perfect overlap for all of the laboratory strains (Figure 12). There was also a near overlap of the two strains from Minnesota, USA: CC-1952 and CC-2290. However, for the other wild isolates, there was no clear correlation between the principal components and the geographic origin as had been observed for the nuclear genomes (Flowers et al., 2015).
Figure 12. Comparative genomics analysis of organelles.
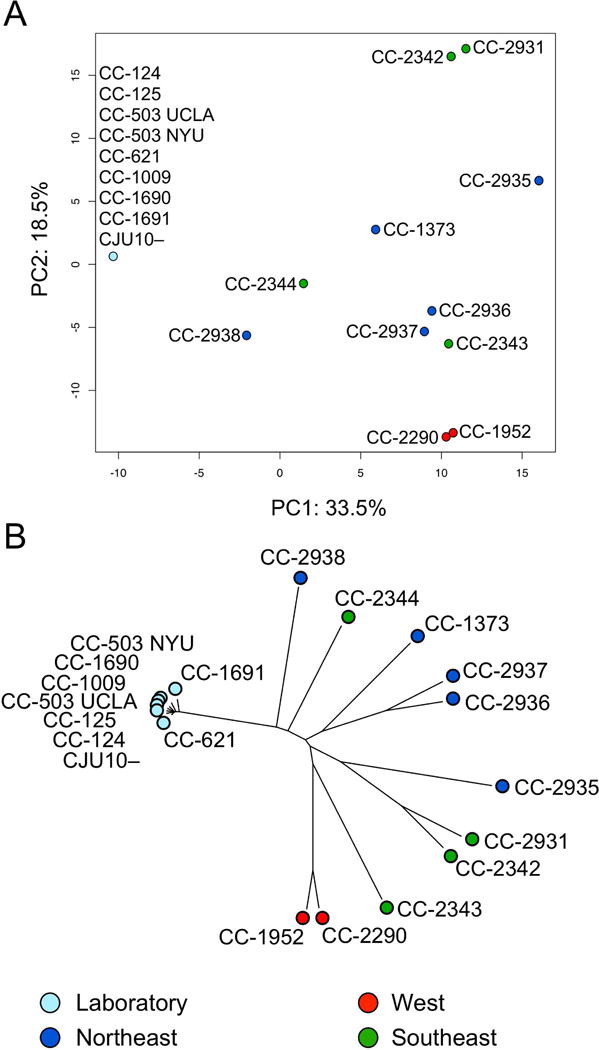
DNA-Seq reads from 11 wild isolates and 8 different laboratory strains of C. reinhardtii were aligned to CPv4 and MTv4 and used to identify variants in the organellar genomes. One strain, CC-503, was sequenced independently by this group (UCLA) and another (NYU), and both samples were analyzed independently. (A) The full set of 2058 SNVs and 606 InDels was used to perform a principle component analysis. Strains are color coded by region as in (Flowers et al., 2015). (B) A neighbor-joining phylogenetic analysis of the same strains. The strains in both panels are color coded by geographic origin as in Flowers et al.
Discussion
Due in large part to the high degree (~20%) of repetitive sequence, the C. reinhardtii chloroplast genome was a singular challenge to assemble. Different assembly tools (Ray and SPA) were tried and only minimally successful on their own, despite having 17,000-fold coverage of high quality 100+100 paired end DNA-Seq data. In this work, we developed a number of tools to leverage this abundant sequencing data to identify and resolve problematic loci in the assembly based on heterogeneity of base-call error frequency, coverage depth, and inferred DNA-Seq fragment size. Even armed with these tools, significant manual intervention was necessary to solve such a repetitive sequence.
Given these difficulties, it is not surprising that many errors were identified in the previous classical sequencing-based assemblies. The hundreds of SNVs and small InDels identified in BK000554.2 (2002) could be attributed to the fact that that assembly was based on data from a few different laboratory strains. However, our comparative genomics data suggests that this is unlikely. We found a total of only six variants in eight different strains, despite the fact that these strains have all been maintained as separate cultures for many decades. The majority of the InDel variants we identified in FJ423446.1 (2009) relative to CPv4, were due to repetitive sequence. Many of the single nucleotide errors identified in FJ423446.1 relative to CPv4 were found to flank larger insertions and deletions. This suggests that they may be a second-order effect of trying to assemble sequencing data across large gaps.
The research presented here demonstrates the importance of having an accurate reference sequence. For example, Shi and colleagues published a report in which they identified 68 examples of editing in chloroplast transcripts. We demonstrate that 50 of the 68 editing examples are directly due to SNV and small InDel errors in the BK000554.2 (2002) chloroplast genome that they used as their reference sequence (Table S3). As another example, it has been understood since 2002 that the β′ subunit of the chloroplast PEP was encoded by two genes (Maul et al., 2002). Here we show that this erroneous belief was due to a 2.4 kbp inversion in previous versions of the chloroplast genome that falls within the rpoC1 gene.
We had to optimize both the library preparation protocols, and our analysis pipeline to perform RNA-Seq studies on the chloroplast and mitochondrial transcriptomes of C. reinhardtii. For the RNA-Seq library preparation, choosing an rRNA-depletion approach instead of a poly(A) selection resulted in a 400-fold increase in the contribution of chloroplast transcripts to the library. The reduced contribution of chloroplast mRNA in libraries prepared by the poly(A) method reduced the transcript abundance estimates by several orders of magnitude. More alarmingly, this reduction was non-linear; high abundance transcripts were underestimated to a greater degree than were low abundance transcripts. This suggests that attempts to examine chloroplast gene expression with poly(A)-enrichment libraries should be met with skepticism.
We found the rRNA-depletion kit to be highly effective in removing nuclear and chloroplast rRNA, but not mitochondrial rRNA, from our libraries. However, rRNA is the dominant form of RNA in the cell, and a significant portion remained. Additionally, some of the oligo probes from the kit remained as contaminants in the RNA-Seq library preparation and were detectable in the sequencing data. For the most accurate quantification of total cellular protein coding transcripts, we found that it was necessary to filter out any remaining rRNA in silico. Additionally, as a consequence of using rRNA-depletion instead of a poly(A) enrichment to exclude rRNA, other types of non-coding RNA were detected in the data and affected the quantification of protein-coding transcripts. We identified numerous examples of non-coding snoRNA genes in the intronic and UTR portions of protein coding genes that artificially inflated the expression estimates of the adjacent protein coding genes. For example, we found that the U3 snoRNA gene is located in the 3′ UTR of Cre07.g350976 (Antal et al., 2000). Unless the snoRNA sequence is filtered out in silico, Cre07.g350976 artificially appears among the most highly abundant transcripts in the cell. To facilitate removal of these non-coding sequences, we compiled the nuclear, chloroplast and mitochondrial rRNA sequences and over 300 snoRNA sequences (Chen et al., 2008) into a multi-fasta file that can be used to filter non-coding reads from RNA-Seq data prepared by rRNA-depletion (Data S10).
In plants, editing of the chloroplast transcripts is wide-spread (Stern et al., 2010). Based on the extremely low mismatch frequency that we observed between the RNA-Seq data and the CPv4 chloroplast genome, we found no evidence for transcript editing in C. reinhardtii. The unexpected observation of Shi et al. of wide-spread editing of chloroplast transcripts in Chlamydomonas appears to be wrong (Shi et al., 2016). Our analysis suggests that all examples of transcript editing in Chlamydomonas are better explained by inaccuracies in the reference chloroplast genome sequence that they used, their failure to account for intron splicing, and abundance of highly repetitive DNA in the chloroplast genome.
A particularly unexpected result in this work was the observation of C-rich polynucleotide tails at the 3′ ends of all eight mitochondrial protein coding transcripts. This observation was recapitulated across each of the different RNA-Seq conditions examined, and regardless of which alignment program was used (STAR, PASS, BWA-MEM). Previous studies have reported on 3′ poly(A) tails, as well as poly(U) tails, on mitochondrial transcripts that are thought to be a signal for degradation (Zimmer et al., 2009). In our data, runs of poly(A) and poly(U) were present, albeit at a lower frequency than poly(C). Interestingly, mitochondrial transcripts were less reduced than chloroplast transcripts by the use of the poly(A)-enrichment library preparation method as compared to the rRNA-depletion method (Figure 10). This could be due to stretches of poly(A) being common in the polynucleotide tails of the mitochondrial transcripts.
The degenerate nature of these polynucleotide tails, and their placement at the 3′ ends of the transcripts suggest that ribonucleotides are added in a template-independent manner, possibly by a polynucleotide phosphorylase (PNPase) or nucleotidyltransferase (NTR) (Schuster and Stern, 2009). The nucleus-encoded PAP4 protein (Cre14.g625950) has NTR activity, and there is evidence from a GFP-fusion that it can translocate to mitochondria, thus making it a good candidate for this activity (Zimmer et al., 2009). Another possibility is the nucleus-encoded PNP1 protein (Cre04.g214501). This protein is known to poly-adenylate transcripts in the chloroplast, but may also be targeted to the mitochondria (Zimmer et al., 2009).
One striking result of this study is that there is approximately one tenth as much genetic variation between strains in the organellar genomes as had been reported for the nuclear genome (Flowers et al., 2015). This is despite the fact that the abundance of reactive oxygen species in both the chloroplast and the mitochondrion make those compartments somewhat inhospitable to DNA. One factor that is likely to be important for the relative paucity of variants in the chloroplast and mitochondrion is the high copy number of the organellar genomes relative to the nuclear one. Since C. reinhardtii grows vegetatively as a haploid organism, each cell contains just one copy of each chromosome in its nucleus. Any spontaneous mutations, unless lethal, will be propagated to all daughter cells during vegetative growth. In contrast, we determined that there are over 80 copies of the chloroplast genome, and approximately 130 copies of the mitochondrial genome per cell (Figure 5D). Any spontaneous mutations that occur in the organellar genomes will be in competition with many other wild type alleles. Double strand breaks in DNA are mainly repaired by one of two pathways, homologous recombination (HR) or non-homologous end joining (NHEJ). It would be expected that DNA repair by HR, which uses a homologous DNA strand as a template for error-free repair, would be facilitated by the high genome copy number observed in the organelles. In support of this, DNA repair by HR has been demonstrated in the C. reinhardtii chloroplast (Cerutti et al., 1995). In contrast, only the more error-prone NHEJ pathway is available to repair double strand breaks in the haploid nuclear genome of vegetatively growing cells. In cases where a copy of an organelle genome is damaged and not repaired, the new mutation will be present at a very low allele frequency, which makes it susceptible to loss from genetic drift (Kimura and Ohta, 1968). Lastly, the organellar genomes are inherited uni-parentally during meiosis: the mitochondrial genome from the mt− parent and the chloroplast genome from the mt+ parent (Harris, 2008). Any variants that had accumulated in the organelle genomes of the opposite sex parent would therefore be lost following sexual recombination.
Experimental Procedures
De novo assembly of organellar genomes
DNA-Seq data for strain CC-503 was published previously (Gallaher et al., 2015), and is available from NCBI’s Sequence Read Archive (SRA) at accession SRR1797945. The raw sequencing data, consisting of 99,579,111 100+100 nt paired end reads, were aligned to the C. reinhardtii reference nuclear genome (v5) from Phytozome using bwa-mem (v.0.7.7) with default settings. The resulting sam-formatted alignment file was filtered using samtools view (v.1.3) to retain only reads that did not map to the nuclear genome. The resulting unmapped reads were converted back to fastq format with Picard tools samtofastq (v1.77). This yielded 42,733,538 total reads. Next, the reads were used for a de novo assembly using Ray (v.2.3.1) with default parameters. The scaffolds produced by Ray were compared to the newest existing mitochondrial and chloroplast genomes (GenBank accessions U03843.1 and FJ423446.1, respectively) using a local instance of the blastn tool (v.2.2.26). Those scaffolds with significant hits to the chloroplast and mitochondrial genomes were used as a base to manually assemble complete genomes. To fill in the missing sequence, the sequencing reads described above were re-aligned to the assembly and evaluated by manual examination with Integrative Genomics Viewer (IGV v2.3.94) from the Broad Institute. This process was used to extend and join the contigs in an iterative process until both organelle genomes were complete. The final versions of each de novo assembly are identified here as CPv4 and MTv4 for the chloroplast and mitochondrial genomes, respectively (Data S1).
Like the previous versions of the C. reinhardtii chloroplast genome, CPv4 contains two ~22 kbp inverted repeats, separated by two single copy regions. Reads spanning the transitions between the inverted repeats and the single copy regions validate that the inverted repeats are in the correct position and orientation. The continuity in the depth of DNA-Seq coverage between the inverted repeats and the single copy regions suggest that each region is present in CPv4 in the correct ratio. However, the relative orientations of the two single copy regions relative to each other cannot be inferred from relatively short Illumina sequencing data. The choice of orientation for these two regions is arbitrary, but was chosen to be consistent with GenBank U03843.1 and FJ423446.1.
Variant-corrected chloroplast genome reconstruction
In parallel to the de novo chloroplast genome assembly described above, a version of the chloroplast genome was produced by editing GenBank FJ423446.1 (2009). The same DNA-Seq reads used above were aligned to the GenBank assembly with bwa-mem. Variants were identified with the Genome Analysis Toolkit (GATK) from the Broad Institute as described previously (Gallaher et al., 2015). GenBank FJ423446.1 was manually edited to reflect the high confidence variants identified by GATK. Changes were evaluated by re-mapping the DNA-Seq reads, followed by manual review of the edits on IGV. Changes were made as needed in an iterative process. The final result of this approach is referred to here as “cv11” and is included in fasta format as Data S2.
Evaluating assemblies
The DNA-Seq reads described above were aligned to four different chloroplast genome versions in parallel by bwa-mem using default parameters. This included BK000554.2 and FJ423446.1 from GenBank, and cv11 and CPv4 described above. The resulting alignment files were analyzed using in-house PERL scripts as follows.
Base call errors were determined by sam2errorFreq.pl (v1.3), which compares each base call to the assembly. Base calls that are soft-clipped, as indicated by the cigar string, are ignored. Base calls flagged as being a deletion or insertion by the cigar string were treated as errors and charged to the adjacent locus. The percentage of errant base calls relative to the total was calculated for each locus, and for non-overlapping 1000 nt windows. The resulting data were then plotted using ggplots2 in R.
Coverage depth was calculated for each set of alignments with bedtools genomeCov (v2.25.0) with the -d and -split parameters. The resulting data were further analyzed by bam2covDep.pl (v1.0), which determines the overall mean coverage for the assembly. Next, the script determines the percentage of loci in non-overlapping 1000 nt windows that fall within various bins relative to the overall mean.
Inferred fragment length analysis was performed by sam2length.pl (v1.3), which takes fragment length data from the alignments of paired-end DNA-Seq data, and determines the mean inferred length for all fragments aligned to the assembly. Next, the script determines the percentage of loci in non-overlapping 1000 nt windows that fall within various bins relative to the overall mean.
The PERL scripts used in these analyses (sam2errorFreq.pl, bam2covDep.pl, and sam2length.pl) are available as the evaluatingAssemblies suite via BitBucket at https://bitbucket.org/gallaher/evaluatingassemblies.
Variants between the different chloroplast and mitochondrial genome versions were identified by multiple sequence alignment with MUSCLE v3.8.31 (Edgar, 2004).
RNA-Seq strains and culture conditions
This analysis incorporated RNAseq data from twelve different conditions from four different studies as follows:
Dark versus Light dataset
Strain CC-4351 (Matagne 325, cw15 arg7–8) was transformed with pCB412, an ARG7-expressing plasmid. Cultures of this strain were grown in a photobioreactor in high salt medium (HSM) supplemented with Kropat’s trace elements (Kropat et al., 2011) Cells were entrained on a 12 h/12 h light-dark cycle. Total RNA was collected from triplicate samples collected at the end of the dark phase (“dark”) and 1 h into the light phase (“light”) (Strenkert et al., manuscript in preparation). Total RNA was collected and purified as described previously (Strenkert et al., 2011). The RNA was depleted of rRNA by means of the RiboZero plant leaf kit, and the remaining RNA was used to generate RNA-Seq libraries by means of the KAPA Stranded RNA-Seq kit. Sequencing was performed on an Illumina HiSeq 2000 with 50 nt single end reads.
Fe dataset
Strain CC-4532 was grown in flasks on a shaking platform in tris-acetate-phosphate (TAP) medium supplemented with Hutner’s trace elements including 20 μM Fe (Urzica et al., 2012). At t=0, a sample of RNA was collected (“20 μM Fe”). The remaining samples were washed, and grown for an additional 4h in Fe free medium and then collected (“<0.01 μM Fe”). Total RNA was collected and purified as described previously (Urzica et al., 2012). RNA-Seq libraries were constructed and sequenced using two approaches. In the first, libraries were made by means of the Illumina Stranded Total RNA Library kit, which enriches for poly-adenylated mRNA by binding to oligo(T) magnetic beads. The resulting libraries were sequenced as 100 nt single end reads on an Illumina HiSeq 2000. In the second case, the same samples of purified RNA were subjected to rRNA-depletion by means of the RiboZero plant leaf kit, followed by a modified Illumina Stranded Total RNA Library kit protocol that omitted the oligo-d(T) bead binding step. The resulting libraries were sequenced as 100+100 nt paired end reads on an Illumina HiSeq 2000. For head to head comparisons between the poly-A libraries and the rRNA-depletion libraries, only the first read of the rRNA-depletion libraries were used.
Cu dataset
A strain resulting from a cross between CC-124 and CC-4425, was grown in flasks in TAP medium supplemented with Kropat’s trace elements including 2 μM Cu (“2 μM Cu”) or without Cu (“<0.01 μM Cu”) for three consecutive rounds. Cells were collected at a concentration between 6 – 8×106. RNA was collected and purified as described previously (Urzica et al., 2012). The RNA was depleted of rRNA by means of the RiboZero plant leaf kit, and libraries were constructed from the remaining RNA by means of the KAPA Stranded RNA-Seq kit. Sequencing was performed on an Illumina HiSeq 2000 with 50 nt single end reads.
Sexual cycle dataset
An mt+ strain, CC-620, and an mt− strain, CJU−, were used for study of the Chlamydomonas sexual cycle described previously (Lopez et al., 2015). Cultures of both the mt+ and the mt− strain were grown in HSM, and samples were collected as “mt+ vegetative” and “mt− vegetative”, respectively. Cells were induced to undergo gametogenesis by transferring to HSM minus N for 15 h, after which samples were collected as “mt+ gametes” and “mt− gametes”. The gametes of both mating types were combined, and a sample was collected 1 d later as “zygote”. Zygotes were transferred to TAP medium and incubated in the light for 24 h, after which a sample was collected as “germinated”. Libraries were prepared using the RiboZero Plant Leaf kit and KAPA Stranded RNA-Seq kit, and sequenced as 50 nt single end reads on an Illumina HiSeq 2000.
RNA-Seq data analysis
Sequencing data from the experiments described above were first aligned with RNA STAR (v.2.5.1a –alignIntronMax 5000 –outReadsUnmapped Fastx) to a pseudo-assembly containing nuclear, chloroplast and mitochondrial rRNA and snoRNA loci (Data S10) to exclude these from downstream analysis. The remaining unmapped reads were then aligned to the nuclear (Phytozome v5), chloroplast (CPv4, this work), and mitochondrial (MTv4, this work) genomes. Default parameters were used except for maximum intron size, which was limited to 5 kbp. Counts of sequencing reads per gene and FPKMs were determined by cuffdiff v2.0.2 (Trapnell et al., 2013). Weighted rLog2 transformation of counts and differential expression significance testing were performed with DESeq2 (Love et al., 2014). Figures were produced in R.
The mRNA transcripts from all 12 conditions were ranked in abundance, and the top 20 for each condition were plotted in R using ggplots2.
k-means Clustering Analysis
A table was generated of FPKMs from each sample described above for each nuclear, chloroplast, and mitochondrial gene. Low expressing genes were filtered if their maximum FPKM was less than 1 for all experiments. Next, the fold changes in FPKMs between pairs of related samples (light vs. dark, Fe− vs. Fe+, Cu− vs. Cu+, mt+ gamete vs. vegetative, mt− gamete vs. vegetative, and germinated vs. zygote) were calculated. Genes were sorted into clusters with the Kmeans tool in the amap package in R with the following settings: centers=10, iter.max=500, nstart=50, method= “euclidean”. The cluster with the highest percentage of chloroplast genes was selected for further analysis. Enrichment of GO terms was performed with the Algal Functional Annotation Tool (Lopez et al., 2011). Enrichment of GreenCut genes was calculated by the hypergeometric distribution in R.
Depth of coverage analysis
The DNA-Seq reads described above were simultaneously aligned to the nuclear genome (Phytozome v5), and to CPv4 and MTv4 (this work) using bwa-mem. The resulting sam-formatted alignment files were compressed and sorted with samtools (v1.3). The percentage of reads assigned to each of the three genomes was calculated using in-house scripts and plotted as a pie chart using Microsoft Excel.
RNA-Seq libraries from the study of C. reinhardtii grown in medium with and without Fe (described above) were prepared using either a poly-A or an rRNA-depletion protocol. The resulting reads were aligned to the three genomes using STAR (v.2.5.1a) with default parameters and –alignIntronMax 5000 (Dobin et al., 2013). The percentage of reads assigned to each genome was calculated using in-house scripts. Values presented in the figure are the mean of two samples prepared by each protocol.
For the genome copy number analysis, DNA-Seq coverage was corrected for mappability and GC-content with HMMcopy (v0.1.0) following the manufacturers standard protocol (Ha et al., 2012). First, wiggle tracks of coverage, GC-content, and mappability were prepared using readCounter, gcCounter, and mapCounter, respectively. The wiggle files were imported into R and used to determine the corrected coverage at each locus with correctReadcount. The mean, median, and standard deviation were determined for each genome in R. Loci with coverage in excess of three standard deviations of the mean were filtered to remove outliers. This filter excluded 0.18% of the data. Coverage data was plotted as a violin plot with the ggplots2 package in R.
Annotations
First, ORFs were identified by IGV. These were then validated or rejected based on manual examination of RNA-Seq alignments. Gene names were informed by the previous annotations available from GenBank. The tRNA genes were predicted in silico with tRNAscan-SE (v2.0) (Lowe and Eddy, 1997) and cross-referenced with the PlantRNA database (http://plantrna.ibmp.cnrs.fr/plantrna/, accessed on Feb. 17, 2017). The rRNA genes were mapped forward from GenBank FJ423446.1 and U03843.1 to CPv4 and MTv4 by sequence homology. The chloroplast rRNA genes were qualitatively validated by means of an RNA-Seq library that was prepared as described below, except that the rRNA-depletion protocol was performed improperly resulting in incomplete removal of the rRNA. The protocol was repeated a second time on the same samples of RNA using the proper procedure. Triplicate samples of both library preparation batches were aligned simultaneously to CPv4, MTv4, the C. reinhardtii nuclear genome (Phytozome v5), and the A. thaliana chloroplast rRNA locus (GenBank KX551970.1 bases 130,619 - 137,676). The last of these was included to capture contaminating probes from the rRNA-depletion kit. Secondary alignments were filtered out with samtools. Next, the coverage was determined by bedtools genomecov (v2.25.0) using -ibam -d -split -strand flags. Coverage from each library was normalized per 1×108 mapped reads. The ratio of coverage between the incomplete versus complete rRNA-depletion libraries was calculated for each replicate, then averaged over the three replicates, and log10 transformed. This ratio was then plotted using circos.
Protein similarity networks
Protein sequences were selected from the NCBI database and this work, and compiled into a multifasta file. Similarity was calculated via BLASTP with the following settings: -evalue 1e-10 -outfmt 6. The similarity network was constructed from the resulting file with the BLAST2SimilarityGraph plugin in Cytoscape (v2.7.0).
Organelle genome maps
Gene maps of the chloroplast and mitochondrial genomes were produced with OrganellarGenomeDRAW (v1.1.1) (Lohse et al., 2013). GC-coverage was calculated in non-overlapping 25 bp windows with in-house scripts. Total mRNA coverage and rRNA coverage were determined with bedtools genomecov (v.2.25.0). Concentric circular tracks of mRNA coverage, rRNA coverage, GC content, and structure were generated for the chloroplast genome map with circos (v0.69). Final compositing and labeling was performed with Pixelmator (v.3.6).
Splicing of the psaA and psbA genes
Splice junctions for the psaA and psbA genes of the chloroplast were identified by the position of split RNA-Seq reads aligned to chloroplast genome. A pseudo-assembly was generated to contain both the fully spliced and fully unspliced versions of both genes. RNA-Seq reads prepared by the rRNA-depletion protocol from each of the samples described above were aligned to the pseudo-assembly using bwa-mem (v0.7.7). The depth of coverage at each locus was determined with bedtools genomecov (v.2.25.0). Next, the depth of coverage was averaged over a window of 50 bp adjacent to each splice site in both the spliced and unspliced versions. Finally, the ratio of spliced depth of coverage relative to the combined depth of coverage was calculated and plotted as a pie chart in Microsoft Excel.
Proteomics
Cells from diurnally grown cultures of C. reinhardtii were collected by centrifugation at 1450×g, 4°C. The cell pellet was washed once with 1 ml of 10 mM phosphate buffer (pH 7), and then resuspended in fresh phosphate buffer. Cells were broken by two cycles of slowly freezing to −80°C and thawing to room temperature. Soluble proteins were digested with sequencing-grade modified porcine trypsin. The resulting polypeptides were loaded on a Q-Exactive Plus Orbitrap mass spectrometer (Thermo Electron, Waltham, MA) coupled to Waters NanoAcquity or Next-Gen 3 high performance liquid chromatography systems (Waters Corporation, Milford, MA) through 75 um × 70 cm columns packed with Phenomenex Jupiter C-18 derivatized 3 um silica beads (Phenomenex, Torrance, CA). Samples were loaded onto columns with 0.05% formic acid in water and eluted with 0.05% formic acid in Acetonitrile over 100 minutes. Twelve high resolution (17.5K nominal resolution) data-dependent MS/MS scans were recorded for each survey MS scan (70K nominal resolution) using normalized collision energy of 30, isolation width of 2.0 m/z, and rolling exclusion window lasting 30 seconds before previously fragmented signals are eligible for re-analysis. Unassigned charge and singly charge precursor ions were ignored. The resulting MS/MS spectra were converted to ASCII text (.dta format) using MSConvert (http://proteowizard.sourceforge.net/tools/msconvert.html) which precisely assigns the charge and parent mass values to an MS/MS spectrum as well as converting them to centroid. The data files were then interrogated via target-decoy approach using MSGFPlus with a +/- 20 ppm parent mass tolerance, partial tryptic enzyme settings, and a variable posttranslational modification of oxidized Methionine. MS/MS search results were then collated into tab separated ASCII text files listing the best scoring identification for each spectrum. Results were filtered to 1% FDR using an MSGF+ supplied Q-Value that assesses reversed sequence decoy identifications for a given MSGF score across each dataset. Filter passing results were reported in an Excel file (Data S4). Using the protein references as a grouping term, unique peptides belonging to each protein were counted, as were all PSMs belonging to all peptides for that protein (i.e. a protein level observation count value).
Organelle transcript editing
RNA-Seq reads from each sample were filtered to remove rRNA and snoRNA reads, and then aligned to the nuclear, chloroplast, and mitochondrial genomes as described above. Alignment was performed in parallel using two different alignment programs. The first, STAR (v2.4.0j), was run with default parameters except for –alignIntronMax 1000. The second, PASS (v2.31), was run using -flc 1 -fid 90 -fle 30 -sam -trim 5 20, to closely approximate the analysis conducted by Shi and colleagues (Shi et al., 2016). SNP calls were made using PASS -program genotype -f 0.5 -q 20 -c 10 2000 as in Shi et al. Putative edit sites were manually reviewed in IGV.
Evaluation of mitochondrial polynucleotide 3′ tails
Polynucleotide tails were observed as soft-clipped bases at the 3′ ends of all eight mitochondrial genes when RNA-Seq reads were manually examined with IGV. To quantify this phenomenon, 100 nt RNA-Seq reads from the +/− Fe experiment (described above) were trimmed by scythe (v. 0.981 https://github.com/vsbuffalo/scythe) to remove contaminating Illumina adaptor sequence. Reads were further processed by sickle (v 1.210 https://github.com/najoshi/sickle) to trim base calls with quality scores less than 30 from the edges of the reads. Next, the 3′ terminal 25 nucleotides of each mitochondrial gene were used as search terms for grep, a pattern matching algorithm, to isolate the relevant RNA-Seq reads. The number of A, C, G, and U nucleotides downstream of the 3′ terminal 25 nucleotides were counted for each gene in each sample, and calculated as a percentage of the total.
Comparative genomics of organellar genomes
DNA sequencing data was published previously (Gallaher et al., 2015; Flowers et al., 2015). Accession numbers and strain names are described in Table S4. Reads were mapped to CPv4 and MTv4 using bwa-mem with default settings. The resulting sam-formatted alignment files were compressed and sorted with samtools (v1.3). Duplicate reads were marked, and read groups were added with Picard Tools (v1.77) from the Broad Institute. Further analysis was performed with the GATK suite of tools (v3.6) from the Broad Institute. Variants were called on each bam-formatted alignment file using GATK HaplotypeCaller with the following settings: –genotypeing_mode DISCOVERY, -ploidy 1, -stand_emit_conf 10, -stand_call_conf 30, –emitRefConfidence GVCF. The resulting g.vcf files were combined into a raw.vcf file with GATK GenotypeGVCFs. GATK BaseRecalibrator was run twice in series on the bam-formatted alignment files with the raw.vcf file used for -knownSites. The base qualities of the reads were then recalibrated with GATK PrintReads, and GATK HaplotypeCaller and GenotypeGVCFs were re-run as before. Finally, reads were filtered with GATK VariantFiltration using “QD < 2.0 || FS > 60.0 || MQ < 25.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0” for SNPs and “QD < 2.0 || FS > 200.0 || ReadPosRankSum < −20.0” for InDels. The final variant set was extensively reviewed by visualization of HaplotypeCaller bamout files on IGV. The effect of variants on chloroplast and mitochondrial genes was predicted with snpEff (v4.3r) using default settings (Cingolani et al., 2012). High and moderate impact variants (i.e. those that fall within coding sequences) were reviewed by visualization on IGV, and variant classifications were revised as needed.
A data table of strains and their variants was used to construct a principle component analysis in R. An unrooted phylogenetic tree was generated with the ape package in R, as described previously (Saitou and Nei, 1987).
Supplementary Material
Data S1. FASTA sequences of CPv4 and MTv4.
Data S2. Variant-edited chloroplast genome version, cv11.
Data S3. Annotations of CPv4 and MTv4.
Data S4. Proteomics analysis.
Data S5. RNA-Seq Quantitation Data.
Data S6. Sequence homology analysis of select chloroplast genes.
Data S7. k-means Clustering Analysis.
Data S8. Top 100 most abundant cellular transcripts across all conditions examined by RNA-Seq.
Data S9. VCF-formatted table of variants across strains.
Data S10. FASTA file of decoy rRNA and snoRNA sequences.
Figure S1. GC content of nuclear, chloroplast, and mitochondrial genomes.
Figure S2. Coverage heterogeneity of chloroplast genome versions.
Figure S3. Inferred insert size heterogeneity of chloroplast genome versions.
Figure S4. Protein similarity networks for ftsH and ycf1.
Figure S5. Expression from Wendy transposons.
Figure S6. Examples of polycistronic expression in the chloroplast.
Figure S7. Validation of RNA-Seq analysis with nucleus-expressed sentinel genes.
Figure S8. Evidence for cytosine-rich polynucleotide 3′ tails in mitochondrial genes.
Table S1. Variants between GenBank and de novo assemblies of chloroplast and mitochondrial genomes.
Table S2. Protein Similarity Network Accessions.
Table S3. Evaluation of chloroplast transcript editing.
Table S4. Strains and accession numbers used for comparative genomics analysis of C. reinhardtii organelles.
Acknowledgments
Funding was provided by the U.S. National Institutes of Health R24 grant (GM092473) and by the U.S. Department of Energy (DE-FC03-02ER63421). The proteomics analysis was performed using the Environmental Molecular Sciences Laboratory, a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research (proposal ID 49262). Thanks to Weihong Yan for her assistance with the UCSC Genome Browser website.
Footnotes
DR SEAN D GALLAHER (Orcid ID : 0000-0002-9773-6051)
The authors declare no conflict of interest.
Accession Numbers and Data Availability
The chloroplast and mitochondrial assemblies, gene annotations, variants from 20 strains, and RNA-Seq coverage data can all be viewed on the UCSC Genome Browser hosted at http://genomes.mcdb.ucla.edu/CreOrganelles/ The DNA-Seq data from strain CC-503 that was used to generate de novo assemblies CPv4 and MTv4 and to produce cv11 is available from the NCBI Short Read Archive at accession SRR1797945. All RNA-Seq data including raw sequencing reads, assemblies, annotations, and processed mRNA abundance tables are available from the NCBI Gene Expression Omnibus at accession GSE101944.
Previously available DNA-Seq data from additional strains was used for the comparative genomics analysis. These strains and the related accession numbers are detailed in Table S4.
References
- Antal M, Mougin A, Kis M, Boros E, Steger G, Jakab G, Solymosy F, Branlant C. Molecular characterization at the RNA and gene levels of U3 snoRNA from a unicellular green alga, Chlamydomonas reinhardtii. Nucleic Acids Res. 2000;28:2959–68. doi: 10.1093/nar/28.15.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Speed TP. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40:e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau E, Turmel M, Goldschmidt-Clermont M, Rochaix JD, Sivan S, Michaels A, Leu S. A large open reading frame (orf1995 ) in the chloroplast DNA of Chlamydomonas reinhardtii encodes an essential protein. Mol Gen Genet MGG. 1997;253:649–653. doi: 10.1007/s004380050368. [DOI] [PubMed] [Google Scholar]
- Campagna D, Albiero A, Bilardi A, Caniato E, Forcato C, Manavski S, Vitulo N, Valle G. PASS: a program to align short sequences. Bioinformatics. 2009;25:967–968. doi: 10.1093/bioinformatics/btp087. [DOI] [PubMed] [Google Scholar]
- Cerutti H, Johnson AM, Boynton JE, Gillham NW. Inhibition of chloroplast DNA recombination and repair by dominant negative mutants of Escherichia coli RecA. Mol Cell Biol. 1995;15:3003–11. doi: 10.1128/mcb.15.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti H, Johnson AM, Gillham NW, Boynton JE. Epigenetic Silencing of a Foreign Gene in Nuclear Transformants of Chlamydomonas. PLANT CELL ONLINE. 1997;9:925–945. doi: 10.1105/tpc.9.6.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Chen CJ, Vallon O, Huang ZP, Zhou H, Qu LH. Genomewide Analysis of Box C/D and Box H/ACA snoRNAs in Chlamydomonas reinhardtii Reveals an Extensive Organization Into Intronic Gene Clusters. Genetics. 2008;179 doi: 10.1534/genetics.107.086025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w 1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande NN, Bao Y, Herrin DL. Evidence for light/redox-regulated splicing of psbA pre-RNAs in Chlamydomonas chloroplasts. RNA. 1997;3:37–48. [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapier D, Suzuki H, Levy H, Rimbault B, Kindle KL, Stern DB, Wollman FA. The chloroplast atpA gene cluster in Chlamydomonas reinhardtii. Functional analysis of a polycistronic transcription unit. Plant Physiol. 1998;117:629–41. doi: 10.1104/pp.117.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürrenberger F, Rochaix JD. Chloroplast ribosomal intron of Chlamydomonas reinhardtii: in vitro self-splicing, DNA endonuclease activity and in vivo mobility. EMBO J. 1991;10:3495–501. doi: 10.1002/j.1460-2075.1991.tb04913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JM, Rahire M, Rochaix JD. Chlamydomonas reinhardii gene for the 32 000 mol. wt. protein of photosystem II contains four large introns and is located entirely within the chloroplast inverted repeat. EMBO J. 1984;3:2753–62. doi: 10.1002/j.1460-2075.1984.tb02206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan WH, Woelfle MA, Mosig G. Two copies of a DNA element, “Wendy”, in the chloroplast chromosome of Chlamydomonas reinhardtii between rearranged gene clusters. Plant Mol Biol. 1995;29:63–80. doi: 10.1007/BF00019119. [DOI] [PubMed] [Google Scholar]
- Flowers JM, Hazzouri KM, Pham GM, et al. Whole-Genome Resequencing Reveals Extensive Natural Variation in the Model Green Alga Chlamydomonas reinhardtii. Plant Cell. 2015;27:2353–69. doi: 10.1105/tpc.15.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallaher SD, Fitz-Gibbon ST, Glaesener AG, Pellegrini M, Merchant SS. Chlamydomonas Genome Resource for Laboratory Strains Reveals a Mosaic of Sequence Variation, Identifies True Strain Histories, and Enables Strain-Specific Studies. Plant Cell. 2015;27:2335–52. doi: 10.1105/tpc.15.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt-Clermont M, Choquet Y, Girard-Bascou J, Michel F, Schirmer-Rahire M, Rochaix JD. A small chloroplast RNA may be required for trans-splicing in Chlamydomonas reinhardtii. Cell. 1991;65:135–43. doi: 10.1016/0092-8674(91)90415-u. [DOI] [PubMed] [Google Scholar]
- Gray MW, Boer PH. Organization and Expression of Algal (Chlamydomonas reinhardtii) Mitochondrial DNA. Philos Trans R Soc London B Biol Sci. 1988;319 doi: 10.1098/rstb.1988.0038. [DOI] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012;22:1995–2007. doi: 10.1101/gr.137570.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn D, Rg Nickelsen J, Hackert A, Kü U. A single nuclear locus is involved in both chloroplast RNA trans-splicing and 3Ј end processing. Plant J. 1998;15:575–581. [Google Scholar]
- Harris E. The Chlamydomonas Sourcebook: Introduction into Chlamydomonas and its laboratory use. Elsevier Academic Press; 2008. [Google Scholar]
- Holloway SP, Deshpande NN, Herrin DL. The catalytic group-I introns of the psbA gene of Chlamydomonas reinhardtii : core structures, ORFs and evolutionary implications. Curr Genet. 1999;36:69–78. doi: 10.1007/s002940050474. [DOI] [PubMed] [Google Scholar]
- Karpowicz SJ, Prochnik SE, Grossman AR, Merchant SS. The GreenCut2 resource, a phylogenomically derived inventory of proteins specific to the plant lineage. J Biol Chem. 2011;286:21427–21439. doi: 10.1074/jbc.M111.233734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Ohta T. THE AVERAGE NUMBER OF GENERATIONS UNTIL FIXATION OF A MUTANT GENE IN A FINITE POPULATION’. Genetics. 1968;61:763–771. doi: 10.1093/genetics/61.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komine Y, Kwong L, Anguera MC, Schuster G, Stern DB. Polyadenylation of three classes of chloroplast RNA in Chlamydomonas reinhardtii. RNA. 2000;6:598–607. doi: 10.1017/s1355838200992252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropat J, Hong-Hermesdorf A, Casero D, Ent P, Castruita M, Pellegrini M, Merchant SS, Malasarn D. A revised mineral nutrient supplement increases biomass and growth rate in Chlamydomonas reinhardtii. Plant J. 2011;66:770–780. doi: 10.1111/j.1365-313X.2011.04537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kück U, Choquet Y, Schneider M, Dron M, Bennoun P, Rochaix JD. Structural and transcription analysis of two homologous genes for the P700 chlorophyll a-apoproteins in Chlamydomonas reinhardii: evidence for in vivo trans-splicing. EMBO J. 1987;6:2185–2195. doi: 10.1002/j.1460-2075.1987.tb02489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW–a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41:W575–W581. doi: 10.1093/nar/gkt289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez DA, Hamaji T, Kropat J, et al. Dynamic changes in the transcriptome and methylome of Chlamydomonas reinhardtii throughout its life cycle. Plant Physiol. 2015;169:00861. doi: 10.1104/pp.15.00861. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez D, Casero D, Cokus SJ, Merchant SS, Pellegrini M. Algal Functional Annotation Tool: a web-based analysis suite to functionally interpret large gene lists using integrated annotation and expression data. BMC Bioinformatics. 2011;12:282. doi: 10.1186/1471-2105-12-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul JE, Lilly JW, Cui L, dePamphilis CW, Miller W, Harris EH, Stern DB. The Chlamydomonas reinhardtii plastid chromosome: islands of genes in a sea of repeats. Plant Cell. 2002;14:2659–79. doi: 10.1105/tpc.006155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant SS, Prochnik SE, Vallon O, et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science (80-) 2007;318:245–250. doi: 10.1126/science.1143609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misumi O, Suzuki L, Nishimura Y, Sakai A, Kawano S, Kuroiwa H, Kuroiwa T. Isolation and phenotypic characterization of Chlamydomonas reinhardtii mutants defective in chloroplast DNA segregation. Protoplasma. 1999;209:273–282. [Google Scholar]
- Munakata H, Nakada T, Nakahigashi K, Nozaki H, Tomita M. Phylogenetic Position and Molecular Chronology of a Colonial Green Flagellate, Stephanosphaera pluvialis (Volvocales, Chlorophyceae), among Unicellular Algae. J Eukaryot Microbiol. 2016;63:340–8. doi: 10.1111/jeu.12283. [DOI] [PubMed] [Google Scholar]
- Ogura T, Tomoyasu T, Yuki T, Morimura S, Begg KJ, Donachie WD, Mori H, Niki H, Hiraga S. Structure and function of the ftsH gene in Escherichia coli. Res Microbiol. 1991;142:279–82. doi: 10.1016/0923-2508(91)90041-8. [DOI] [PubMed] [Google Scholar]
- Rosales-Mendoza S, Paz-Maldonado LMT, Soria-Guerra RE. Chlamydomonas reinhardtii as a viable platform for the production of recombinant proteins: current status and perspectives. Plant Cell Rep. 2012;31:479–494. doi: 10.1007/s00299-011-1186-8. [DOI] [PubMed] [Google Scholar]
- Ryan R, Grant D, Chiang KS, Swift H. Isolation and characterization of mitochondrial DNA from Chlamydomonas reinhardtii. Proc Natl Acad Sci USA. 1978;75:3268–72. doi: 10.1073/pnas.75.7.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymarquis LA, Higgs DC, Stern DB. Nuclear suppressors define three factors that participate in both 5′ and 3′ end processing of mRNAs in Chlamydomonas chloroplasts. Plant J. 2006;46:448–461. doi: 10.1111/j.1365-313X.2006.02711.x. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schuster G, Stern D. Chapter 10 RNA Polyadenylation and Decay in Mitochondria and Chloroplasts. Prog Mol Biol Transl Sci. 2009;85:393–422. doi: 10.1016/S0079-6603(08)00810-6. [DOI] [PubMed] [Google Scholar]
- Scranton MA, Ostrand JT, Fields FJ, Mayfield SP. Chlamydomonas as a model for biofuels and bio-products production. Plant J. 2015;82:523–531. doi: 10.1111/tpj.12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Wang S, Xia EH, Jiang JJ, Zeng FC, Gao LZ. Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci Rep. 2016;6:30135. doi: 10.1038/srep30135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Lee RW. Nucleotide diversity of the Chlamydomonas reinhardtii plastid genome: addressing the mutational-hazard hypothesis. BMC Evol Biol. 2009;9:120. doi: 10.1186/1471-2148-9-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DB, Goldschmidt-Clermont M, Hanson MR. Chloroplast RNA Metabolism. Annu Rev Plant Biol. 2010;61:125–155. doi: 10.1146/annurev-arplant-042809-112242. [DOI] [PubMed] [Google Scholar]
- Strenkert D, Schmollinger S, Sommer F, Schulz-Raffelt M, Schroda M. Transcription factor-dependent chromatin remodeling at heat shock and copper-responsive promoters in Chlamydomonas reinhardtii. Plant Cell. 2011;23:2285–301. doi: 10.1105/tpc.111.085266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urzica EI, Casero D, Yamasaki H, et al. Systems and trans-system level analysis identifies conserved iron deficiency responses in the plant lineage. Plant Cell. 2012;24:3921–3948. doi: 10.1105/tpc.112.102491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahrenholz C, Riemen G, Pratje E, Dujon B, Michaelis G. Mitochondrial DNA of Chlamydomonas reinhardtii: the structure of the ends of the linear 15.8-kb genome suggests mechanisms for DNA replication. Curr Genet. 1993;24:241–247. doi: 10.1007/BF00351798. [DOI] [PubMed] [Google Scholar]
- Vries J, de Sousa FL, Bölter B, Soll J, Gould SB. YCF1: A Green TIC? Plant Cell. 2015;27:1827–33. doi: 10.1105/tpc.114.135541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer SL, Schein A, Zipor G, Stern DB, Schuster G. Polyadenylation in Arabidopsis and Chlamydomonas organelles: the input of nucleotidyltransferases, poly(A) polymerases and polynucleotide phosphorylase. Plant J. 2009;59:88–99. doi: 10.1111/j.1365-313X.2009.03853.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. FASTA sequences of CPv4 and MTv4.
Data S2. Variant-edited chloroplast genome version, cv11.
Data S3. Annotations of CPv4 and MTv4.
Data S4. Proteomics analysis.
Data S5. RNA-Seq Quantitation Data.
Data S6. Sequence homology analysis of select chloroplast genes.
Data S7. k-means Clustering Analysis.
Data S8. Top 100 most abundant cellular transcripts across all conditions examined by RNA-Seq.
Data S9. VCF-formatted table of variants across strains.
Data S10. FASTA file of decoy rRNA and snoRNA sequences.
Figure S1. GC content of nuclear, chloroplast, and mitochondrial genomes.
Figure S2. Coverage heterogeneity of chloroplast genome versions.
Figure S3. Inferred insert size heterogeneity of chloroplast genome versions.
Figure S4. Protein similarity networks for ftsH and ycf1.
Figure S5. Expression from Wendy transposons.
Figure S6. Examples of polycistronic expression in the chloroplast.
Figure S7. Validation of RNA-Seq analysis with nucleus-expressed sentinel genes.
Figure S8. Evidence for cytosine-rich polynucleotide 3′ tails in mitochondrial genes.
Table S1. Variants between GenBank and de novo assemblies of chloroplast and mitochondrial genomes.
Table S2. Protein Similarity Network Accessions.
Table S3. Evaluation of chloroplast transcript editing.
Table S4. Strains and accession numbers used for comparative genomics analysis of C. reinhardtii organelles.