

Abstract
Preclinical work suggests that the metabotropic glutamate receptor subtype 5 (mGlu5) may represent a novel target to treat neuropsychiatric disorders, including alcohol use disorder and obesity. The goal of this first-in-man study was to evaluate the safety, tolerability and pharmacokinetics (PK) of GET 73 (PubChem SID: 329974174), a novel mGluR5 negative allosteric modulator. This was a double-blind, placebo-controlled, ascending dose, phase I study conducted in healthy male volunteers in two experiments. GET 73 was administered as single ascending doses (N=48; Experiment 1; 10, 30, 100, 300, 450, 600-mg) or multiple ascending doses (N=32; Experiment 2; 100, 300, 450, 450-mg twice a day). Primary endpoints were the incidence of adverse events (AEs) among drug conditions and drug tolerability. The secondary endpoints were the PK parameters of GET 73 and its metabolite MET 2. Single GET 73 doses of up to 600-mg and repeated ascending doses of up to 450-mg twice/day were safe and well-tolerated. There were no serious or severe AEs. All AEs were mild or moderate in severity. Total GET 73 exposure increased with each increased GET 73 dose. A dose-related increase in mean maximum plasma drug concentration was observed after repeated dosing. Maximum plasma drug concentrations occurred between 0.5–2.05 hours after administration in all groups for both single and repeated doses. This first-in-human study indicates that GET 73, as single or multiple ascending doses, is safe and well-tolerated when administered to healthy male volunteers.
Keywords: glutamate receptor subtype 5 (mGlu5), allosteric modulator, GET 73, safety, tolerability
1. INTRODUCTION
Both alcohol use disorder (AUD) and obesity are associated with significant morbidity and mortality (Whitlock et al., 2009). However, effective treatments are limited for both AUD (Haass-Koffler et al., 2014) and obesity (Yanovski and Yanovski, 2014). There is a crucial need to develop novel effective treatments to help these patients. From a medications development standpoint, there is an important overlap between neurobiological pathways and mechanisms that are responsible for the development and maintenance of AUD and obesity (Volkow et al., 2013).
Recently, there has been an increased interest in the role of glutamate in neuropsychiatric disorders, and particularly in mediating the rewarding effects and other addictive properties related to food, alcohol and drugs of abuse. In fact, glutamate is a key neurotransmitter involved in the neurobiological mechanisms that lead to the excessive alcohol craving and drinking that characterizes AUD (Gilpin and Koob, 2008) as well as to the excessive food craving and intake that characterizes obesity (Delgado, 2013).
Glutamate is the main excitatory neurotransmitter and activates two classes of receptors: ionotropic glutamate receptors (iGluRs) that mediate fast excitatory glutamate transmission and metabotropic glutamate receptors (mGluRs), which are G-protein coupled receptors that mediate slow glutamate transmission. Blockade of glutamate transmission via both receptors reduces the rewarding effects of drugs of abuse, including alcohol (D’Souza, 2015; Duncan and Lawrence, 2012). Most iGluR antagonists show some efficacy in animal models of addiction, but exhibit serious side effects when tested in humans (Olive, 2009). By contrast, mGluR ligands have shown a safer profile when tested in clinical trials for various medical conditions, and experimental evidences indicate that mGluRs represent a promising target for developing new pharmacological treatments for AUD (Duncan and Lawrence, 2012; Meyers et al., 2015; Olive, 2009) and/or obesity (Ploj et al., 2010).
Pharmacological manipulations of mGluRs can be obtained through ligands acting at the orthosteric site or at allosteric sites located in the seven transmembrane portion of the receptor. Allosteric modulators of mGluRs represent a large field of opportunities for drug discovery, potentially leading to drugs characterized by more specific effects and few side effects respect to other drugs affecting glutamate transmission, such as N-methyl-D-aspartate (NMDA) antagonists and orthosteric ligands of mGluRs (Conn et al., 2009). Specifically, concerning the mGluRs of Group I, mGluR5 negative allosteric modulators (NAMs) reduce drug intake, reward, reinforcement, and relapse-related behaviors, mGluR5 positive modulators (PAMs) improve cognitive function, facilitate the extinction of drug-associated contextual memories, and reverse experimentally-induced deficits in cognitive function(Besheer et al., 2008; Mihov and Hasler, 2016; Olive, 2009, 2010).
GET 73 (N-[4-(trifluoromethyl)benzyl]-4-methoxybutyramide) (PubChem SID: 329974174) is a novel chemical entity that was selected among a series of γ–hydroxybutyric acid derivatives (Loche et al., 2012). GET 73 seems to partially act like a mGluR5 antagonist or negative allosteric modulator (Beggiato et al., 2013; Ferraro et al., 2011). GET 73 has been proposed as a potential novel pharmacotherapy for AUD and/or obesity. In Sardinian ethanol-preferring (sP) rats, a validated rodent model of AUD (Colombo et al., 2006), GET 73 reduces ethanol intake, and suppresses the alcohol deprivation effect and anxiety-related behavior (Loche et al., 2012). GET 73 shows a neuroprotective role against ethanol-induced neurotoxicity in primary cultures of rat hippocampal neurons (Tomasini et al., 2016). GET 73 also reduces preference for palatable food in Wistar rats (Ottani et al., 2007). In addition, previous in vitro and in vivo work conducted in different preclinical experimental models demonstrated the safety of GET 73 on the central nervous system (CNS), cardiovascular, respiratory and immune systems. Taken together, these results indicate that GET 73 may represent a safe and promising novel pharmacotherapy that should be investigated further. As such, the next step was to conduct the first-in-human evaluation of the safety and tolerability of oral GET 73 tested at different doses in healthy volunteers. The primary aims were to evaluate the safety and tolerability of a single GET 73 administration (Experiment 1) and repeated GET 73 administrations (Experiment 2) either once or twice daily over a 14-day period. Additional aims of these two clinical studies were to evaluate the pharmacokinetics (PK) profile of GET 73 and its main metabolite previously identified MET 2 (4-oxo-4-{[(4-trifluoromethyl)benzyl]amino}butanoic acid), during the same timeframe.
2. METHODS AND MATERIALS
2.1 Study design and randomization
GET 73 was evaluated in two phase I clinical studies. Both studies were randomized, double-blind, placebo-controlled with dose escalations, that were designed to characterize the safety, tolerability and preliminary PK of GET 73 and its metabolite MET 2 in healthy male volunteers. Only the Pharmacy staff was un-blinded for the purposes of final drug preparation. The Pharmacy staff was provided with randomization lists for dose preparation and each dose was in compliance. The randomization schedule ensured that there was only one placebo in the first four volunteers dosed. The research staff was responsible for enrolling participants and assigniong interventions.
Experiment 1
Experiment 1 employed a single ascending dose designed to assess the safety, tolerability and PK of single oral doses of GET 73. Eighty-eight participants were screened and 48 healthy male volunteers (N = 48) were randomized into six groups of eight individuals each, six to receive GET 73 and two to receive placebo (Fig 1A). Groups were dosed sequentially with escalation to the next dose following review of emerging safety and tolerability data. Participants were admitted to a clinical research unit on the evening of Day-1, dosing occurred on the morning of Day 1, and they were discharged ~24 hours after dosing on Day 2. Participants returned to the clinical research unit on Day 3 for additional study assessments. A follow-up visit was conducted on Day 15. For a complete outline of the study schedule, see Table 1 in Data in Brief. For the first dose level (group 1) the group was divided into four subgroups to allow staggered dosing as an additional safety precaution. Group 1A and 1B contained 1 individual each, group 1C contained 2 individuals and group 1D contained 4 individuals. The randomization schedule was modified to ensure that there was only one placebo participant in the first four individuals dosed. A minimum of 24 hours elapsed between dosing of the first participant in each subgroup. After review of safety and tolerability data, subsequent dose levels were administered as complete groups.
Figure 1. Study flowcharts related to the Experiment 1 (A) and Experiment 2 (B).
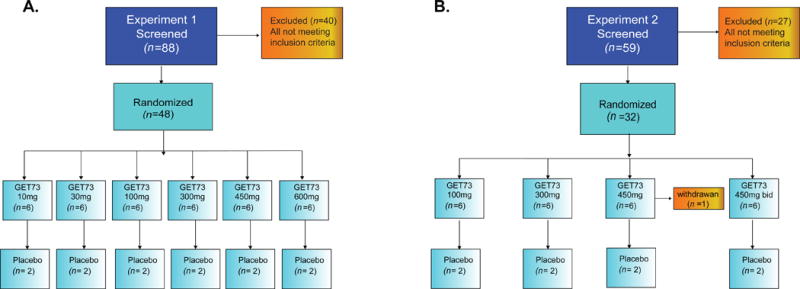
A) Experiment 1, Eighty-eight participants were screened and 48 healthy male volunteers (N = 48) were randomized into six groups of eight participants, six to receive GET 73 and two to receive placebo. B) Experiment 2, Fifty-nine participants were screened and 32 healthy male volunteers (N = 32) were randomized into four groups of eight participants, six to receive GET 73 and two to receive placebo. Individuals in groups 1 - 3 received 14 doses according to the randomization schedule, while individuals in group 4 received 28 doses. All groups were dosed sequentially with escalation to the next dose following review of emerging safety and tolerability data.
Experiment 2
Experiment 2 employed a multiple ascending dose design to evaluate the safety, tolerability and PK of multiple oral doses of GET 73. Fifty-nine participants were screened and 32 healthy male volunteers (N = 32) were randomized into four groups of eight individuals each, six to receive GET 73, and two to receive placebo. Individuals in groups 1 - 3 received 14 doses according to the randomization schedule, while individuals in group 4 received 28 doses (Fig 1B). Groups were dosed sequentially with escalation to the next dose following review of emerging safety and tolerability data, as specified in section 2.6. Individuals in groups 1 - 3 received once daily oral doses of GET 73 or placebo for 14 sequential days; individuals in group 4 received twice daily oral doses of GET 73 or placebo for 14 sequential days, dosing at 12 hour intervals. Participants were admitted to the clinical research unit on the evening of Day –1 and had the first dose administered on the morning of Day 1. Participants remained hospitalized until the morning of Day 3. Once discharged, participants returned to the clinical research unit on the mornings of Days 5, 7, 9, 11 and 13 for dosing and study procedures. On Days 4, 6, 8, 10 and 12, individuals took the specified dose(s) at home. On the evening of Day 14, individuals returned to the clinical research unit and remained hospitalized until the morning of Day 16. A follow-up visit was conducted on Day 28. For a complete outline of the study schedule, see Table 2 in Data in Brief.
2.2 Dose justification
In accordance with the guidelines provided by the European Medicines Agency (EMA), Guideline on strategies to identify and mitigate risks for first-in human clinical trials with investigational medical products (CHMP/SWP/28367/07), the maximum recommended safe starting dose (MRSD) for GET 73 was calculated step by step, starting from the no-observed-adverse-effect-level (NOAEL) values obtained in 28-day oral toxicity studies conducted in rat and dog. NOAEL values were converted to human equivalent dose (HED) based on surface body area and HED values of 16.1 mg/kg and 69.4 mg/kg were obtained from rat and dog NOAEL values, respectively.
The more conservative 16.1 mg/kg NOAEL value obtained from the toxicity rat study was selected and divided by a safety factor of 100, leading to 0.161 mg/kg. The starting dose calculated for a subject of 70 kg was thus 11.3 mg. Finally, a starting dose of 10 mg was considered appropriate for the first in human administration of GET 73, taking into account both the preclinical toxicology results, and the pharmacological active dose (PAD) determined in rats (Loche et al., 2012).
2.3 Study setting and approval
This study was conducted at LCG Bioscience, Cambridge, UK, from 03 February 2010 to 03 November 2010. The study was reviewed and approved by the Medicine and Healthcare products Regulatory Agency (MHRA). A written informed consent was obtained for each participant prior to conducting any procedure. The study was conducted in accordance with the European Good Clinical Practice (GCP) guideline as issued by the European Community (EU Directive 2001/20/EC), the Medicines for Human Use regulations (SI 2004/1031), the Committee of Proprietary Medicinal Products (CPMP), International Conference on Harmonisation (ICH) 1997 guideline on GCP (CPMP/ICH/135/95) and the principles enunciated in the Declaration of Helsinki. The trial was registered in the EudraCT (N. 2009-015377-12), now EU Clinical Trial Register from January 2016. The authors confirm that all ongoing and related trials for this drug/intervention are registered. The analytical component of this study was conducted in accordance with the medicines for Human Use (Clinical Trials) Regulations (Statutory Instrument 2004 No. 1031) in laboratories that operate in compliance with the UK Good Laboratory Practice Regulations (Statutory Instrument No. 3106), OECD Principles of good Laboratory Practice (Paris 1998) and the EC Commission Directive 2004/10/EC (February 2004).
2.4 Medication stopping criteria
In accordance with the guidelines provided by the European Medicines Agency (EMA), Guideline on strategies to identify and mitigate risks for first-in human clinical trials with investigational medical products CHMP/SWP/28367/07), the study protocol defined stopping rules.
GET 73 dose would not be escalate further if one of the following circumstances occurred in individuals on active study treatment within the same dose group: 1) severe GET 73-related adverse events (AEs) of the same character in two individuals on active drug, 2) GET 73-related laboratory abnormalities of the same character in two individuals on active drug, 3) clinically significant GET 73-related changes in vital signs or electrocardiograms (ECGs) of the same character in two individuals on active drug, 4) three overall occurrences of any one of the above circumstances, or 5) dosing could have been stopped at any time if the tolerability and safety of a dose was unacceptable to participants or investigators. Laboratory abnormalities outside the specified reference range were flagged and identified as high or low in the Individual Subject Data Listings.
2.5 Selection of study population
It was anticipated that the study design and sample size would provide sufficient data to assess the safety, tolerability and PK in the chosen study populations. To be eligible for inclusion into this study, each participant fulfilled the following criteria at the screening visit: 1) male, 2) 18 - 65 years old inclusive, 3) body mass index ≤ 30 kg/m2, 4) registered with a UK general practitioner (GP), 5) able and willing to give written consent, and 6) willing to use a double barrier form of contraception through the study and for a period of three months afterward.
Exclusion criteria included: 1) any condition or disease detected during the medical/physical examination that would render the participant unsuitable for the study, or place the participant at undue risk or interfere with the ability to complete the study, 2) confirmed significant allergic reaction against any drug, or history of multiple allergies, 3) use of any prescribed medication, over the counter medication (excluding paracetamol) or herbal product within 1 week prior to day 1 of the study, 4) likely to need concomitant medication during the study period, 5) alcohol consumption averaging > 3 drinks/day, 6) regular smoker (≥ 5 cigarettes/day, or smokers of pipes or cigars), or unwilling to abstain from smoking from 72 hours prior to drug administration until follow-up (nicotine replacement products may be used up to the equivalent of 5 cigarettes/day), 7) loss/donation ≥ 500-mL of blood in the 3 months prior to day 1 of the study, 8) participation in an investigational drug/device study within 3 months prior to day 1 of the study, 9) clinically significant deviation from normal blood pressure (BP) at screening, 10) clinically significant findings on ECGs taken at screening, 11) clinically relevant laboratory abnormalities identified at screening and, 12) positive drug screen or alcohol breath test.
At the time when this first-in-human study was conducted, GET 73 has been subjected to three genotoxicity studies: Ames, Chromosome aberration and Micronucleus. Embryo-Fetal Toxicity studies in rats and rabbits are ongoing, so at the time this Phase 1 study was conducted, only males were included due to safety precautions.
2.6 Study medication, schedule and dose escalation
The study medications, GET 73 (10-mg batch#: TRF09207 and 100-mg batch#:TRF0920) and matching placebo (batch#: TFR09205 and TFR09205) were prepared as oral capsules by Doppel (Rozzano, Italy). Study medication was stored in the pharmacy, at 15 - 25 °C. Participants were instructed to abstain from grapefruit, grapefruit juice, caffeine or xanthine-containing products (chocolate, tea, coffee, cola, etc.) for 72 hours before the first dose until after the follow-up visit. They were also required to abstain from alcohol for 24 hours prior to screening. Participants fasted for at least 10 hours before dosing on Day1 (Experiment 1 and Experiment 2), Day 14 (Experiment 2), and on all days when clinical laboratory samples were taken, according with the planned assessments and procedures specified in Table 3 and 4 in Data in Brief. On Day 1 (Experiment 1 and Experiment 2) and Day 14 (Experiment 2) participants did not eat until after the 4 hour post-dose time-point, after which they received a standardized light lunch. A light snack was provided approximately 13 hours post-dose. Participants in Experiment 2, group 4 (twice daily dosing) were required to fast for 1 hour prior to the evening dose, and for 1 hour post dose on Day 1 and Day 14.
The starting dose for this study was calculated, as specified in section 2.2, from the NOAEL obtained in the rat in a previous toxicological study (Study N ° 20060018TRB by CERB) and was determined to be 100 mg/kg. The PK of GET73 following oral repeated administration in the rat was explored in a subsequent independent study (unpublished data) in which the NOAEL level of exposure in this species, measured as the maximum plasma concentration (Cmax) and the area under the concentration-time curve up to the last measurable concentration (AUC0-t) were determined. At the NOAEL Cmax was 6700 ng/mL in male rats and 7840 ng/ml in female rats and AUC0-t was 12500 ng*h/mL for males and 14900 ng*h/mL for females. For safety reasons, the more conservative values of exposure were used for this first human study, such that Cmax of GET 73 was not to exceed 6700 ng/ml, and AUC0-t was not to exceed 12500 ng*h/mL, reducing the exposure of male volunteers. The decision to escalate was made based on an interim safety report, summarizing the safety and PK data.
The proposed dose escalation sequence for Experiment 1 of the study was 30-, 100-, 300-, 600- and 1000-mg. The interim safety report for the group dosed with 600 mg indicated that AUC0-t level was just under the planned maximum of 12500 ng*h/mL, so the highest planned dose 1000-mg was not administered, and a lower dose 450-mg, intermediate between 300-mg and 600-mg, was introduced. Doses for Experiment 2 were selected based upon the results of Experiment 1, such that the maximum dose for Experiment 2 was not to exceed the maximum tolerated dose from Experiment 1. Based on the results obtained on Experiment 1, the GET 73 dose in Experiment 2 was not to exceed 600-mg (once a day) and 450-mg (twice a day). The dose escalation sequence for Experiment 2 was 100, 300, 450 (once a day), and 450 (twice a day).
A minimum of six participants had to complete the dosing schedule in order to make a decision regarding escalation to a higher dose level. This ensured that the data included at least four individuals on active treatment. A minimum interval of seven days from the last dose was required between dosing of sequential groups when the dose was escalated.
2.7 Treatment compliance
For doses given at the clinical research unit the dosing procedure were checked independently by the research staff (Experiment 1 and Experiment 2). For doses taken home (Experiment 2), compliance was monitored by completion of a study diary by the participants, telephonic communication with the research staff to confirm dosing, and return of the containers.
2.8 Adverse events
The Medical Dictionary for Regulatory Affairs (MedDRA) was used to code the adverse events. An adverse event (AE) was defined as: “any untoward medical occurrence in a participant administered a study medication or matching placebo and which did not necessarily have a causal relationship with the treatment”. An AE could have been any unfavorable and unintended sign (including an abnormal laboratory finding), symptom or disease temporally associated with use of the study medication or matching placebo, whether or not related to the investigational product. The condition of each participant was monitored throughout the study. Participants were instructed to spontaneously report AEs occurring at any time during the study. The nature, time of onset, duration, and severity were documented, together with the relationship to drug administration (unrelated, unlikely, possible or probable). The AE severity was recorded as mild, moderate or severe.
2.9 Clinical laboratory, physical examination, vital signs and 12-lead ECG
The schedule of clinical laboratory, chemistry, hematology, urinalysis, physical examination, vital signs, 12-lead ECG, PK assessments through the studies are presented in Table 3 in Data in Brief for Experiment 1 and Table 4 in Data in Brief for Experiment 2. The list of Clinical laboratory assessments is reported on Table 5 in Data in Brief.
2.10 PK assessments
The purpose of including PK analytical data was to measure GET 73 and the main metabolite MET 2 (also known as ACGET 20) concentrations in human plasma samples. The method was validated at Quotient Bioresearch Ltd. (Studies QBR110528QB02 and QBR103278/4). The procedures for the development and the validation of the method were based on the United States (US) Food and Drug Administration (FDA) recommendations Guidance for Industry Bioanalytical Method Validation - U.S. Department of Health and Human Services, FDA Center for Drug Evaluation and Research, Center for Veterinary Medicine, May 2001, BP.
The PK sampling in humans was decided based on data derived from a prior study on rats (unpublished data). The rapid absorption and elimination of GET 73 justified the short sampling (every 30 min) in the first part of the curve and more distanced drawings later. Blood samples for PK evaluation in plasma were collected into tubes containing lithium heparin prior to dosing and at various time points after dosing up to 48 h for Experiment 1 and up to 360 h for Experiment 2. The plasma samples were obtained from individuals from six single ascending dose groups in Experiment 1 (10-mg, 30-mg, 100-mg, 300-mg, 450-mg and 600-mg) and four multiple ascending dose groups in Experiment 2 (100-mg 300-mg, 450-mg and 450-mg twice a day).
Concentrations of GET 73 in human plasma samples were measured by LC-MS/MS after SLE+ (supported liquid extraction) over the calibration range of 2-1000 ng/ml according to the validated method and the relevant SOPs. Concentrations of MET 2 in human plasma samples were measured by LC-MS/MS after protein precipitation and dilution over the calibration range of 20 - 10,000 ng/ml. All instrument control, data collection, peak area integration and storage were performed using Analyst (versions 1.4.2 and 1.5.1, Applied Biosystems Inc., OA, US). Peak areas were then imported into Watson LIMS (version 7.2 Thermo Fisher Scientific Inc., MA, US) for regression and quantification. The mass spectrometer response (peak area ratio of analyte to internal standard) of each calibration standard was calculated by Watson LIMS and plotted against the nominal (prepared) concentration. A weighted linear regression analysis was used to calculate an equation of the calibration line. Concentrations of GET 73 and MET 2 in the plasma samples were back calculated from the calibration lines.
Dataset from all participants for whom a PK sample was obtained and analyzed was considered evaluable for PK concentration analysis. PK analysis of GET 73 and MET 2 concentration time data were conducted in accordance with Data Magik (Laburnum House, East Grimstead, Salisbury, Wiltshire, SP5 3RT, UK) and Quotient procedures. Values for the following PK parameters were estimated: 1) maximum plasma concentration (Cmax), 2) the time to reach maximum plasma concentration (tmax) and 3) the area under the plasma concentration-time curve AUC(0-t). Pharmacokinetic parameters were derived by non-compartmental analysis using WinNonlin (Version 4.1b, Pharshight Corporation, Mountain View, CA, US).
2.11 Statistical analysis
Reporting of the study results was of a descriptive nature and presented using summary statistics: number of subjects (n), mean (M), standard deviation (SD) or standard error of the mean (SEM), co-efficient of variation (CV), median, minimum, maximum or frequency distributions (n, %). Statistical analysis was performed after all participants had their follow-up visit and the study database had been locked. Data management and statistical analysis was performed by Data Magik Ltd. All the statistical procedures described below were performed by the package SAS® (v.9.13., Cary, NC, US).
3. RESULTS
3.1 Study population
Experiment 1
The total number of individuals screened was 88, among which 48 were enrolled into the study (Table 1). All enrolled participants completed the study and were included in the analyses for safety and PK.
TABLE 1.
Demographic and baseline characteristics of the subjects enrolled in Experiment 1 of the clinical study.
| Placebo 0-mg (n=12) |
GET 73 10-mg (n=6) |
GET 73 30-mg (n=6) |
GET 73 100-mg (n=6) |
|
|---|---|---|---|---|
| Age (years) | 35.8 (11.7) | 25.3 (6.3) | 41.2 (11.8) | 29.8 (10.4) |
| Weight (kg) | 77.8 (10.7) | 78.8 (12.7) | 80.0 (10.1) | 83.9 (10.2) |
| Height (cm) | 179.5 (4.0) | 179.5 (6.4) | 178.7 (3.9) | 177.8 (5.8) |
| BMI (kg/m2) | 24.2 (3.5) | 24.4 (3.1) | 25 (2.6) | 26.4 (1.7) |
|
| ||||
| Placebo 0-mg (n=12) |
GET 73 300-mg (n=6) |
GET 73 450-mg (n=6) |
GET 73 600-mg (n=6) |
|
|
| ||||
| Age (years) | 35.8 (11.7) | 32.3 (14.3) | 36 (8.5) | 42.2 (9.0) |
| Weight (kg) | 77.8 (10.7) | 80.3(6.2) | 84.6 (7.3) | 87.8 (3.8) |
| Height (cm) | 179.5 (4.0) | 180.3 (4.3) | 179.3 (8.3) | 180.8 (1.6) |
| BMI (kg/m2) | 24.2 (3.5) | 24.8 (2.5) | 26.4 (2.5) | 26.9 (1.3) |
Results are reported as M and (SD); BMI: Body Mass Index
Experiment 2
The total number of individuals screened was 59, of these, 32 were enrolled into the study (Table 2). All participants in groups 1 - 3 received 14 doses, while all individuals in group 4 received 28 doses. All individuals completed the study except one who withdrew after Day 15, the only assessments not completed were those related to Day 16. The volunteer did not report any AE and we included this individual in all data analysis sets.
TABLE 2.
Demographic and baseline characteristics of the subjects enrolled in Experiment 2 of the clinical study.
| Placebo 0-mg (n=8) |
GET 73 100-mg (n=6) |
GET 73 300-mg (n=6) |
GET 73 450-mg (n=6) |
GET 73 450-mg twice/day (n=6) |
|
|---|---|---|---|---|---|
| Age (years) | 34.5(10.7) | 38.2(13.9) | 47.5(16.1) | 42.7(9.6) | 44.2(9.0) |
| Weight (kg) | 79.6 (8.91) | 78.1(8.65) | 75.4(5.88) | 80.2(5.27) | 83.8(11.78) |
| Height (cm) | 182.8(8.3) | 172.2(1.8) | 173.8(3.9) | 175.2(5.3) | 178.7(6.2) |
| BMI (kg/m2) | 23.9(2.60) | 26.3(2.68) | 25.0(2.34) | 26.2(2.09) | 26.1(1.92) |
Results are reported as M and (SD); BMI: Body Mass Index
3.2 Adverse Events
Experiment 1
Adverse events were observed in all treatment groups, with the highest incidence occurring in the 10-mg, the 600-mg and the 450-mg group each with 2 participants and the placebo group with 4 participants experiencing an adverse event. Except for the 300-mg group, Treatment Emergent Adverse Events (TEAEs) headache and nausea.
Experiment 2
Adverse events were observed in all treatment groups, with the highest incidence occurring in the 450-mg group. TEAEs were seen in all treatment groups. The most commonly occurring TEAE was headache. The only other TEAEs occurring in more than one treatment group were: nasopharyngitis, a catheter site related reaction and vessel puncture hematoma.
3.3 Medication stopping criteria
There were no clinical events for which the stopping criteria had to be applied either in Experiment 1 or Experiment 2 of the study.
3.4 Pharmacokinetic analysis
Experiment 1
The derived PK parameters of GET 73 are displayed in Fig 2 and summarized in Table 6 in Data in Brief. A GET 73 dose-related increase in M Cmax plasma drug concentration was observed, ranging from 48.35 (95% CI: 27.47 - 69.23) ng/mL in the 10-mg group, to 5015.00 (95% CI: 3061.38 – 6968.62) ng/mL in the 600-mg group, with tmax plasma GET 73 concentration, displayed as the absolute median, occurring between 0.5 - 0.75 hours in all groups. Total exposure also increased with increase in GET 73 dose, values of AUC(0-t) ranging from 60.04 (95% CI: 25.97 – 94.11) ng*h/mL in the 10-mg group to 13338.25 (95% CI: 7495.02 - 19181.48) ng*h/mL in the 600-mg group.
Figure 2. Experiment 1, GET 73 pharmacokinetics plasma concentration.
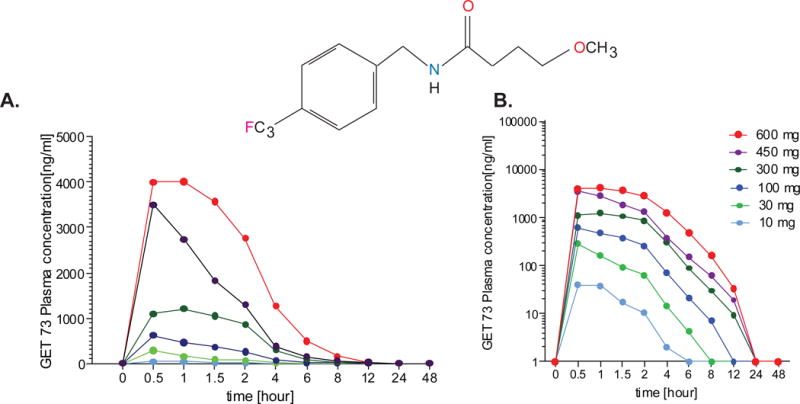
A GET 73 dose-related increase in mean maximum plasma drug concentration was observed, ranging from 48.35 (95% CI: 27.47 - 69.23) ng/mL in the 10-mg group, to 5015.00 (95% CI: 3061.38 – 6968.62) ng/mL in the 600-mg group, with median maximum plasma GET 73 concentration between 0.5 - 0.75 hours in all groups. Total exposure also increased with increase in GET 73 dose, ranging from 60.04 (95% CI: 25.97 – 94.11) ng/mL*h in the 10-mg group to 13338.25 (95% CI: 7495.02 - 19181.48) ng/mL*h in the 600-mg group. Results are reported as M ± SEM.
Experiment 2
The PK plasma concentration for the GET 73 single and repeated dose administration is depicted on Fig 3 and the derived GET 73 PK parameters by dose/treatment and day are displayed in Table 7 in Data in Brief for Cmax, on Table 8 in Data in Brief for tmax, on Table 9 in Data in Brief for AUC0-t and on Table 10 in Data in Brief for Cmin.
Figure 3. Experiment 2, GET 73 repeated dose administration.
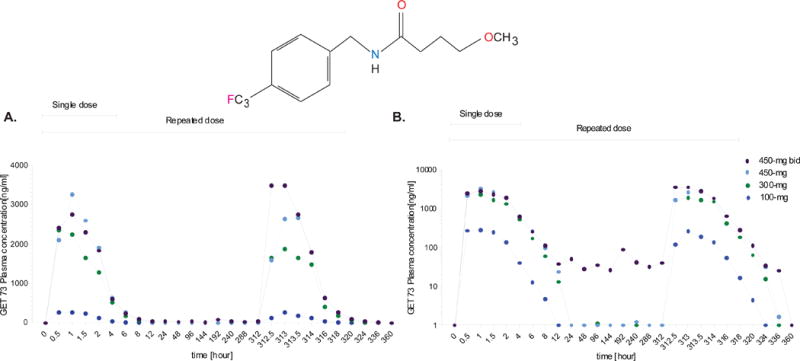
A dose-related increase in mean maximum plasma drug concentration was observed on Day 14 (336 hours) and the median maximum plasma drug concentration occurred between 0.5 – 2.05 hours in all groups on Day 14 (336 hours). Total exposure also increased with an increase in GET 73 dose. Results are reported as M ± SEM.
A dose-related increase in mean Cmax and total exposure (AUC(0-t)) increased with an increase in GET 73 dose on both Day 1 (single dose) and Day 14 (repeat dose). tmax, displayed as the absolute median occurred between 0.5 – 2.05 hours in all groups for both Day 1 and Day 14. There was no notable difference in the mean (± 95 CIs) values of Cmax, tmax,AUC(0-t) within dose groups between Day 1 and Day 14.
3.5 Accumulation of GET 73
There was no evidence of GET 73 accumulation over 14 days of repeated administration. Cmin values for each dose group on Days 1 and 14 suggests there was only a small accumulation of the parent drug, GET 73: 100-mg group samples contained no drug on either Days 1 or 14; a mean value of 0.36 (±95 CIs: −0.35 to 1.07) ng/mL was observed for both the 300-mg and 450-mg groups after dosing on Day 1, but no drug was present for either of these groups on Day 14. As might be expected, some drug was present in the 450-mg twice daily (bid) group after dosing on Day 1and after dosing on Day 14.
3.6 Metabolite MET 2
The results for MET 2 were very consistent with those of the parent drug (Fig 4). The derived MET 2 PK parameters by dose/treatment and day are displayed in Table 11 in Data in Brief for Cmax,, on Table 12 in Data in Brief for tmax, on Table 13 in Data in Brief for AUC0-t and on Table 14 in Data in Brief for Cmin.
Figure 4. Experiment 2, MET 2 repeated dose administration.
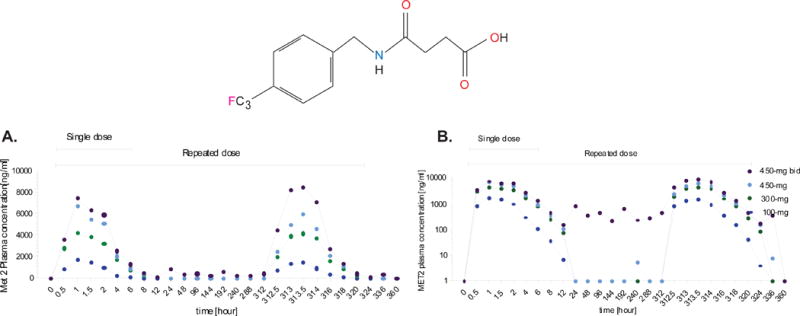
A dose-related increase in mean maximum plasma MET 2 concentration was observed on Day 1 and 14. The maximum plasma MET 2 concentration occurred between 0.5 – 2.05 hours in all groups both on day 1 and on Day 14. Similarly, MET 2 total exposure also increased with an increase in GET 73 dose. Results are reported as M ± SEM.
A dose-related increase in mean Cmax plasma MET 2 concentration and MET 2 total exposure increased with an increase in GET 73 on both Day 1 and Day 14. The tmax of MET 2, displayed as the absolute median, occurred between 0.5 – 2.05 hours in all groups.
There was no notable difference in the mean (± 95 CIs) values of Cmax, tmax,AUC(0-t) within dose groups between Day 1 and Day 14.
3.7 Accumulation of MET 2
Over the course of the 14 days repeated administration of GET 73, there was no evidence of MET 2 accumulation. Exposure (as measured by Cmax and AUC0-t) was comparable on Day 14 of dosing to that seen on Day 1 of dosing, at all dose levels. The MET 2 Cmin values for each dose group on Days 1 and 14 shows that there was no accumulation of the main metabolite in the 100-mg, 300-mg or 450-mg dose groups. However, as might be expected, some metabolite was present in the 450-mg twice day group after dosing on Day 1; and after dosing on Day 14. Plasma MET 2 concentrations had returned to 0 by 24 hours in the 100-mg, 300-mg and 450-mg groups after a single dose and all subsequent mean trough values for these dose group were also reported as 0 at pre-dose on Days 3 – 14, except for the 450-mg group at Day 9.The 450-mg twice day dose group had MET 2 present at all trough time-points, at 24 hours after two doses and when compared to the GET 73 results, on Day 7 on Day 9 and on Day 14.
4. DISCUSSION
The primary objectives of this first-in-man trial were to evaluate the safety and tolerability of single and repeated doses of oral GET 73. In this study, the new chemical entity GET 73 demonstrated an acceptable safety and tolerability in health male volunteers. Single doses of 10-mg to 600-mg were administered to participants in Experiment 1; while in Experiment 2 doses of 100-mg to 450-mg were administered once daily for a period of 14 days, and a dose of 450-mg was administered twice per day for the same period. The main limitation of this trial was the small sample size, however the planned enrollment and the completers proved sufficient to establish safety, tolerability and preliminary PK information of the GET 73 compared to placebo.
The use of sub-groups for the first dose level (Experiment 1) was in line with the EMA recommendations for safety, rights, and well-being of research volunteers. A systematic review of safety and tolerability data did not indicate safety concerns, so that no sub-groups strategy was adopted for other dose level groups (Experiment 2). The occurrence severity and relationship of TEAEs reported in this study were similar across all treatment groups. All TEAEs were mild or moderate in severity. Importantly, no serious or severe adverse events occurred in any of the two experiments.
A possible, non-significant, decrease in heart rate of 6 - 8 beats per minute was noted in the 450-mg dose group of Experiment 1. As this was not evident in any other dose group or in participants receiving repeated doses of GET 73, it is considered likely to be a chance finding due to the small number of participants exposed at each dose level. No morphological changes were noted on the 12-lead ECGs, and although there were occasional out-of-range findings among the ECG parameters of individual participants, there did not appear to be any effect of the GET 73 upon mean values for the treatment groups. GET 73 was well-tolerated at the maximum tolerated dose (MTD) tested here with no significant adverse events.
After both single and repeated doses a dose-related increase in mean Cmax and total exposure drug concentration was observed in the 10-mg and in the 600-mg group. Plasma drug concentrations had returned to 0 in all groups.
There was not a clear relationship between dose and exposure, and a greater than proportional increase was observed between 10-mg and 30-mg (Experiment 1; SAD), 100 and 300 mg (Experiment 2; MAD).
The exposure to MET 2 was approximately proportional to GET 73 dose. Caution should be exercised when reviewing these results due to the small number of subjects (n = 6, within each dose group), however the reason of no proportionality in the exposure to GET 73 may be indicated in different biotransformation competence of participants. Also, the small number of subjects (n = 6 per dose level), while adequate to ensure safety in healthy volunteers, it was insufficient to draw reliable PK-related conclusions. For the PK, we limited our analysis to descriptive statistics. Consequently, data on proportionality and accumulation would not be a reliable conclusion in this study and were not conducted. Based on the accumulation index, however, AUC ratio was close to 1 both or the GET 73 and its metabolite MET 2 indicating that there is no accumulation that can raise safety concerns. Notably, the biotransformation of GET 73 and the exposure to the main metabolite did not give any concerns in term of safety, at least in healthy male subjects over the dose range studied.
There are only a few medications approved for AUD and obesity, therefore, identifying new mechanisms of action and developing novel treatment strategies is necessary. GET 73 provides a novel mechanism of action. Taken together with preclinical data, these Phase 1 results support further investigation of GET 73 in clinical studies to evaluate the efficacy, safety and tolerability in patients suffering from AUD and/or obesity.
5. CONCLUSION
In this first in human study, GET 73 is well tolerated in healthy male volunteers at single doses of up to 600-mg and multiple doses of up to 450-mg twice/day with no dose-limiting toxicity or serious events observed. GET 73 is rapidly absorbed with rate and extent of exposure increasing with increasing dose and t1/2 compatible with multiple dose administration. The safety, tolerability and preliminary PK profile of GET 73 demonstrated in this Phase 1 study support its further development.
Supplementary Material
S1 Table. Experiment 1 single ascending dose schedule.
S2 Table. Experiment 2, multiple ascending dose schedule.
S3 Table. Experiment 1 assessment and procedures.
S4 Table. Experiment 2 assessment and procedures.
S5 Table - Clinical Laboratory AssessmentsS6 Table. Experiment 1 GET 73 PK parameters: Cmax (ng/mL), AUC0-t (ng*h/mL) and tmax (h) by dose/treatment and day.
S7 Table. Experiment 2 GET 73 PK parameters: Cmax (ng/mL) by dose/treatment and day.
S8 Table. Experiment 2 GET 73 PK parameters: tmax (h) by dose/treatment and day.
S9 Table. Experiment 2 GET 73 PK parameters: AUC0-t (ng*h/mL) by dose/treatment and day.
S10. Table. Experiment 2 GET 73 PK parameters: Cmin (ng/mL) by dose/treatment and day.
S11 Table. Experiment 2 MET 2 parameters: Cmax (ng/mL) by dose/treatment and day.
S12 Table. Experiment 2 MET 2 parameters: tmax (h) by dose/treatment and day.
S13 Table. Experiment 2 MET 2 parameters: AUC0-t (ng*h/mL) by dose/treatment and day.
S14 Table. Experiment 2 PK parameters: Cmin (ng/mL) by dose/treatment and day.
Acknowledgments
The authors would like to thank Dr. Anthony Priestley, Dr. Disala Fernando and the research staff at LCG Bioscience, Cambridge, UK for the logistic support of the trial. This study was funded by CT Laboratories, San Remo, Italy. RC and AL are employees of CT Laboratories. RMS has received consultant fees from CT Laboratories and travel and honoraria from D&A Pharma and Lundbeck.
Footnotes
Referred to by: Haass-Koffler, C.L., Goodyear, K., Long, V.M., Tran, H.H., Loche, A., Cacciaglia, R., Swift, R.M., Leggio, L., Dataset for Phase I clinical trial for safety and tolerability of GET 73 in single and repeated doses including preliminary PK parameters. Data in Brief, submitted
The other authors report no biomedical financial interests or potential conflicts of interest.
References
- Beggiato S, O’Connor WT, Tomasini MC, Antonelli T, Loche A, Tanganelli S, Cacciaglia R, Ferraro L. GET73 increases rat extracellular hippocampal CA1 GABA levels through a possible involvement of local mGlu5 receptor. Synapse. 2013;67:678–691. doi: 10.1002/syn.21672. [DOI] [PubMed] [Google Scholar]
- Besheer J, Faccidomo S, Grondin JJ, Hodge CW. Regulation of motivation to self-administer ethanol by mGluR5 in alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008;32:209–221. doi: 10.1111/j.1530-0277.2007.00570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G, Lobina C, Carai MA, Gessa GL. Phenotypic characterization of genetically selected Sardinian alcohol-preferring (sP) and -non-preferring (sNP) rats. Addict Biol. 2006;11:324–338. doi: 10.1111/j.1369-1600.2006.00031.x. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza MS. Glutamatergic transmission in drug reward: implications for drug addiction. Front Neurosci. 2015;9:404. doi: 10.3389/fnins.2015.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado TC. Glutamate and GABA in Appetite Regulation. Front Endocrinol (Lausanne) 2013;4:103. doi: 10.3389/fendo.2013.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JR, Lawrence AJ. The role of metabotropic glutamate receptors in addiction: evidence from preclinical models. Pharmacol Biochem Behav. 2012;100:811–824. doi: 10.1016/j.pbb.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Ferraro L, Beggiato S, Tomasini MC, Antonelli T, Loche A, Tanganelli S. GET73 modulates rat hippocampal glutamate transmission: evidence for a functional interaction with mGluR5. Pharmacol Rep. 2011;63:1359–1371. doi: 10.1016/s1734-1140(11)70700-9. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Koob GF. Neurobiology of alcohol dependence: focus on motivational mechanisms. Alcohol Res Health. 2008;31:185–195. [PMC free article] [PubMed] [Google Scholar]
- Haass-Koffler CL, Leggio L, Kenna GA. Pharmacological approaches to reducing craving in patients with alcohol use disorders. CNS Drugs. 2014;28:343–360. doi: 10.1007/s40263-014-0149-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loche A, Simonetti F, Lobina C, Carai MA, Colombo G, Castelli MP, Barone D, Cacciaglia R. Anti-Alcohol and Anxiolytic Properties of a New Chemical Entity, GET73. Front Psychiatry. 2012;3:8. doi: 10.3389/fpsyt.2012.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JL, Salling MC, Almli LM, Ratanatharathorn A, Uddin M, Galea S, Wildman DE, Aiello AE, Bradley B, Ressler K, Koenen KC. Frequency of alcohol consumption in humans; the role of metabotropic glutamate receptors and downstream signaling pathways. Transl Psychiatry. 2015;5:e586. doi: 10.1038/tp.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihov Y, Hasler G. Negative Allosteric Modulators of Metabotropic Glutamate Receptors Subtype 5 in Addiction: a Therapeutic Window. Int J Neuropsychopharmacol. 2016;19 doi: 10.1093/ijnp/pyw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF. Metabotropic glutamate receptor ligands as potential therapeutics for addiction. Curr Drug Abuse Rev. 2009;2:83–98. doi: 10.2174/1874473710902010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF. Cognitive effects of Group I metabotropic glutamate receptor ligands in the context of drug addiction. Eur J Pharmacol. 2010;639:47–58. doi: 10.1016/j.ejphar.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottani A, Leone S, Vergara FB, Tacchi R, Loche A, Bertolini A. Preference for palatable food is reduced by the gamma-hydroxybutyrate analogue GET73, in rats. Pharmacol Res. 2007;55:271–279. doi: 10.1016/j.phrs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Ploj K, Albery-Larsdotter S, Arlbrandt S, Kjaer MB, Skantze PM, Storlien LH. The metabotropic glutamate mGluR5 receptor agonist CHPG stimulates food intake. Neuroreport. 2010;21:704–708. doi: 10.1097/WNR.0b013e32833b4fe7. [DOI] [PubMed] [Google Scholar]
- Tomasini MC, Borelli AC, Beggiato S, Tanganelli S, Loche A, Cacciaglia R, Ferraro L, Antonelli T. GET73 Prevents Ethanol-Induced Neurotoxicity in Primary Cultures of Rat Hippocampal Neurons. Alcohol Alcohol. 2016;51:128–135. doi: 10.1093/alcalc/agv094. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Tomasi D, Baler RD. The addictive dimensionality of obesity. Biol Psychiatry. 2013;73:811–818. doi: 10.1016/j.biopsych.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, Halsey J, Qizilbash N, Collins R, Peto R. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373:1083–1096. doi: 10.1016/S0140-6736(09)60318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanovski SZ, Yanovski JA. Long-term drug treatment for obesity: a systematic and clinical review. JAMA. 2014;311:74–86. doi: 10.1001/jama.2013.281361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Table. Experiment 1 single ascending dose schedule.
S2 Table. Experiment 2, multiple ascending dose schedule.
S3 Table. Experiment 1 assessment and procedures.
S4 Table. Experiment 2 assessment and procedures.
S5 Table - Clinical Laboratory AssessmentsS6 Table. Experiment 1 GET 73 PK parameters: Cmax (ng/mL), AUC0-t (ng*h/mL) and tmax (h) by dose/treatment and day.
S7 Table. Experiment 2 GET 73 PK parameters: Cmax (ng/mL) by dose/treatment and day.
S8 Table. Experiment 2 GET 73 PK parameters: tmax (h) by dose/treatment and day.
S9 Table. Experiment 2 GET 73 PK parameters: AUC0-t (ng*h/mL) by dose/treatment and day.
S10. Table. Experiment 2 GET 73 PK parameters: Cmin (ng/mL) by dose/treatment and day.
S11 Table. Experiment 2 MET 2 parameters: Cmax (ng/mL) by dose/treatment and day.
S12 Table. Experiment 2 MET 2 parameters: tmax (h) by dose/treatment and day.
S13 Table. Experiment 2 MET 2 parameters: AUC0-t (ng*h/mL) by dose/treatment and day.
S14 Table. Experiment 2 PK parameters: Cmin (ng/mL) by dose/treatment and day.