

Abstract
A small ring phosphacycle (1,2,2,3,4,4-hexamethylphosphetane) is found to catalyze deoxygenative N-N bond-forming Cadogan heterocyclization of o-nitrobenzaldimines, o-nitroazobenzenes and related substrates in the presence of hydrosilane terminal reductant. The reaction provides a chemoselective catalytic synthesis of 2H-indazoles, 2H-benzotriazoles, and related fused heterocyclic systems with good functional group compatibility. On the basis of both stoichiometric and catalytic mechanistic experiments, the reaction is proposed to proceed via catalytic PIII/PV=O cycling, where DFT modelling suggests a turnover limiting (3+1) cheletropic addition between the phosphetane catalyst and nitroarene substrate. Strain/distortion analysis of the (3+1) transition structure highlights the controlling role of frontier orbital effects underpinning the catalytic performance of the phosphetane.
Graphical Abstract
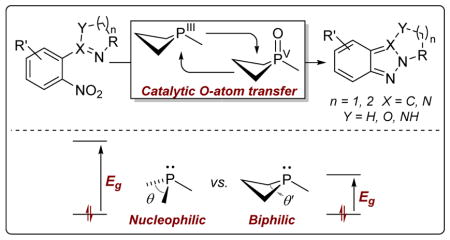
Tricoordinate phosphorus reagents are versatile O-atom acceptors,1 and the conversion PIII→PV=O drives numerous valuable synthetic transformations.2–4 Despite great utility, the inefficient generation of stoichiometric phosphine oxide waste inherent to these methods is regarded as a key limitation. In recent years, though, several new catalytic methods involving cycling through phosphine oxide intermediates have emerged,5 both in redox-neutral6–7, and redox-driven modes.8–10 As part of a program aimed at developing designer main group compounds as broadly useful biphilic11 catalysts in organic synthesis, 12 we recently reported the use of phosphetanes as O-atom transfer catalysts operating via PIII/PV=O redox cycling.12c In this Communication, we advance this biphilic catalysis concept by describing a catalytic N-N bond forming heterocyclization enabled by phosphetane-catalyzed reductive O-atom transfer. Beyond broadening the repertoire of phosphine oxide redox-catalyzed organic transformations, this study documents a dominant electronic basis for the superior performance of phosphetane catalysts that provides a framework for future targeted design of geometrically deformed main group compounds as biphilic catalysts.
Cadogan13 and Sundberg14 have shown that phosphine-mediated exhaustive deoxygenation of o-functionalized nitrobenzene derivatives drives phenylnitrene-like azacyclizations. Most commonly, these transformations employ superstoichiometric amounts of phosphorus-based reagents at elevated temperatures; the standard Cadogan protocol calls for reaction in neat refluxing triethylphosphite (bp 156 °C), although milder conditions have been reported in some cases.15 We questioned whether inherently nontrigonal biphilic phosphines, which colocalize donor and acceptor behavior at P, might facilitate O-atom transfer from the nitro substrate under milder conditions and in a manner conducive to PIII/PV=O redox cycling (Figure 1).
Figure 1.

Phosphine biphilicity and N–N bond forming Cadogan heterocyclization via PIII/PV=O-catalyzed reductive O-atom transfer.
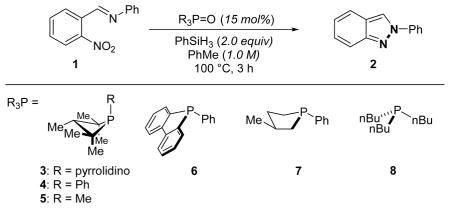
Starting from our reported conditions for deoxygenative carbonyl functionalization as a point of departure, treatment of 1 with substiochiometric quantities of aminophosphetane P-oxide 3·[O] (15 mol%) and phenylsilane (2 equiv.) in PhMe at 100 °C was indeed found to afford indazole 2, albeit in modest yield (Table 1, entry 1). Further optimization studies converged on the simple 1,2,2,3,4,4-hexamethylphosphetane P-oxide 5·[O] as the optimal catalyst for this transformation (see SI for details); in the event, phosphetane P-oxide 5·[O] is found to catalyze deoxygenative heterocyclization delivering product 2 in 83% GC yield within 3 h at 100 °C (entry 3). Neither phospholane-based (6·[O] and 7·[O], entries 4, 5) nor acyclic (8·[O], entry 6) phosphorus precatalysts exhibit similar catalytic reactivity under otherwise identical conditions. Control experiments (entries 7 and 8) confirm that no reaction is observed in the absence of either 5·[O] or terminal reductant PhSiH3; the transformation is demonstrably under catalyst control. Consistent with the notion of PIII/PV=O redox cycling, the use of tricoordinate (σ3-P) precatalyst 1,2,2,3,4,4-hexamethylphosphetane 5 affords indazole 2 with qualitatively similar results as 5·[O] (entry 9). That said, the ease with which phosphine oxide 5·[O] –an air stable solid– is both handled on laboratory scale and reduced in situ to 516 recommended its use as precatalyst in subsequent studies.
Table 1.
Phosphacycles as catalysts for deoxygenative heterocyclization of o-nitrobenzaldimine 1.a
| ||||
|---|---|---|---|---|
|
| ||||
| Entry | R3P=O | Silane | Conversion (%) | Yield (%) |
| 1 | 3·[O] | PhSiH3 | 49 | 20 |
| 2 | 4·[O] | PhSiH3 | 45 | 31 |
| 3 | 5·[O] | PhSiH3 | 99 | 83 |
| 4 | 6·[O] | PhSiH3 | 21 | 1 |
| 5 | 7·[O] | PhSiH3 | 96 | 24 |
| 6 | 8·[O] | PhSiH3 | 21 | 6 |
| 7 | none | PhSiH3 | 14 | 0 |
| 8 | 5·[O] | none | 12 | 0 |
| 9 | 5 | PhSiH3 | 99 | 93 |
Conversion and yield determined via GC vs. internal standard.
Results of our studies into the scope of this catalytic transformation are collected in Table 2. With respect to N-substitution, aliphatic substituents (Table 2A) are well tolerated including 1° (9), 2° (10), and 3° (11) moieties, and basic nitrogen functionality may be incorporated without incident (compare 12 and 13). Likewise, aromatic substrates of diverse substitution (Table 2B) are suitable substrates; halogenated (17–19), electron-rich (20, 21), electron-poor (23, 25), unsaturated (24) and sterically demanding (26) N-aryl substituted substrates all undergo smooth catalytic indazole formation. Polyheterocyclic products (30, 31) may be similarly prepared. Free hydroxyl moieties do not inhibit deoxygenative heterocyclization (22, 32), although such substrates may undergo in situ silylation by the PhSiH3 reductant; desilylative workup ensures recovery of the free OH group. Challenging substrates for the current method include, unsurprisingly, those with multiple nitro moieties (Table 2C, 33); evidently the catalyst does not selectively recognize the o-imino nitro moiety. However, a range of other reducible functionality including nitriles (23), amides (29), and esters (36) are all carried through the deoxygenative cyclization reaction without incident.
Table 2.
Examples of catalytic Cadogan synthesis.a

|
Yields reported for isolated products.
Treatment with TBAF prior to silica gel chromatography.
Single stereoisomer by HPLC.
The reaction is similarly amenable to non-benzaldimine substrates, permitting the synthesis of diverse heterocyclic structures through catalytic N-N bond formation (Table 2D,E). For instance, deoxygenative heterocyclization of 2-(2-pyridyl)nitrobenzene delivers the fused polyheterocyclic pyrido[1,2-b]indazole (39) in near quantitative yield under standard catalytic conditions within 4 h. Relatedly, indazolodihydroimidazoles (40), -tetrahydropyrimidines (41), and -dihydrooxazoles (42) are accessible under standard conditions. Stereochemistry adjacent to the reaction centers is retained upon cyclization (42). Beyond indazole synthesis, the preparation of N-aryl 2H-benzotriazoles (43–45) is achieved by deoxygenative heterocyclization of the corresponding substituted o-nitroazobenzene.
In situ spectral monitoring of catalytic reactions provides information regarding both the mechanistic course of the transformation and speciation of active phosphorus compounds during catalysis. 1H NMR spectra (400 MHz, toluene-d8, 100 °C) of a standard catalytic transformation (1 equiv. of 1, 15 mol% of 5·[O], 2 equiv. of PhSiH3, 1M in C7D8) show the appearance of product indazole 2 over the course of ca. 1.5 h at the expense of starting imine substrate 1; no long-lived intermediates are observed by 1H NMR spectroscopy. Under identical conditions, 31P NMR spectra show the rapid conversion of 5·[O] (δ 53.2 ppm) to an epimeric mixture of the corresponding phosphetane 5 (δ 32.4 ppm (major, anti); δ 18.9 ppm (minor, syn)), which persists in solution until the reaction is complete (see SI). From these data, we infer that 5 represents the catalytic resting state, with the initial deoxygenation of substrate 1 as the turnover limiting step.17 Regrettably, direct kinetic observation of reaction steps following initial deoxygenation is therefore precluded; presumably, though, the nitroso derivative formed by deoxygenation of 1 is an obligate intermediate that proceeds on to observed product in a series of fast following steps.18
Under stoichiometric pseudo-first order conditions with excess phosphine reagent, we find that consumption of substrate 1 is markedly faster with phosphetane 5 than nBu3P 8 (krel ≈ 8, see SI Figure S2); phosphetane 5 is evidently superior to acyclic 8 for Cadogan cyclization. In an effort to understand the origin of this rate difference, we undertook DFT modelling studies (Figure 2). Direct O-atom transfer from nitro-methane to phosphetane 5′ and trimethylphosphine 8′ (via TS15′ and TS18′, respectively) is found to be high in energy. Instead, a stepwise mechanism proceeding through pentacoordinate azadioxaphosphetanes I5′ and I8′ is found at lower energies. The rate-controlling transition structure along this pathway (i.e. TS25′ and TS28′) involves a (3+1) cheletropic addition19,20 of MeNO2 to 5′ and 8′, respectively. Subsequent decomposition of I5′ and I8′ via retro-(2+2) fragmentation (TS25′ and TS28′) then follows with low barrier. This (3+1)/retro-(2+2) pathway is mechanistically analogous and orbitally equivalent to known reactivity of phosphines(-ites) with O3.20
Figure 2.

DFT mechanisms of nitro deoxygenation (M06-2X/6-311++g(d,p)). Relative electronic energies in kcal/mol with unscaled zero-point correction.
To understand further the superiority of the phosphetane with respect to deoxygenation, the (3+1) transition structures TS25′ and TS28′ were analyzed within the distortion/interaction model21 (Figure 3). Despite the presence of the small ring in 5′, the destabilizing distortion energy (ΔEd‡) within transition structures TS25′ and TS28′ is nearly identical (39.2 vs. 38.8 kcal/mol, respectively), being driven to an over-whelming extent by pyramidalization of the nitro substrate, not by geometric reorganization about phosphorus. By consequence, the lower overall energy of TS25′ arises from a significantly larger stabilizing interaction energy (ΔEi‡) for phosphetane 5′ (−17.5 kcal/mol) than Me3P 8′ (−9.7 kcal/mol). The inference from this analysis is that the differential performance of the small-ring phosphetane and trimethylphosphine – both of which compositionally are simple trialkylphosphines – is driven primarily by electronic (orbital) as opposed to enthalpic (ring strain) effects.
Figure 3.

Transition structures and distortion/interaction analyses for (3+1)-transition states (M06-2X/6-311++g(d,p)). (A) Phosphetane TS25′ (B) Me3P TS28′. Phosphine distortion energy (ΔEdP‡) in green, nitro-methane distortion energy (ΔEdN‡) in blue, fragment interaction energy (ΔEi‡) in red, activation energy (ΔE‡) in black. All energies in kcal/mol without zero-point correction.
The difference in interaction energies for the (3+1) addition transition structures can be rationalized by frontier orbital inspection. Figure 4 depicts the Kohn-Sham HOMO and LUMO of each reactant distorted to the transition state structure. Whereas both phosphetane and trimethylphosphine exhibit HOMOs (nonbonding lone pair) of nearly identical energy (−6.95 eV), LUMO(5′) resides ca. 0.8 eV lower in energy than LUMO(8′). In effect, the geometric constraint enforced by the 4-membered ring of the phosphetane 5′ results in a marked lowering of the LUMO that preferentially permits synergistic interactions of HOMO(5′) → LUMO(MeNO2) and HOMO(MeNO2) → LUMO(5′) in a [π4s+ω2s] fashion.22 We note that the low frontier orbital energy gap of phosphetanes has previously been invoked by Chesnut and Quin to rationalize nonmonotonic chemical shift anisotropy trends in phosphacycloalkanes.23 Relatedly, Hudson24 and Westheimer25 have noted the extent to which ring constraint increases electrophilic character at phosphorus. The importance of orbital effects in reactions of strained ring systems has been noted by Hoz.26
Figure 4.
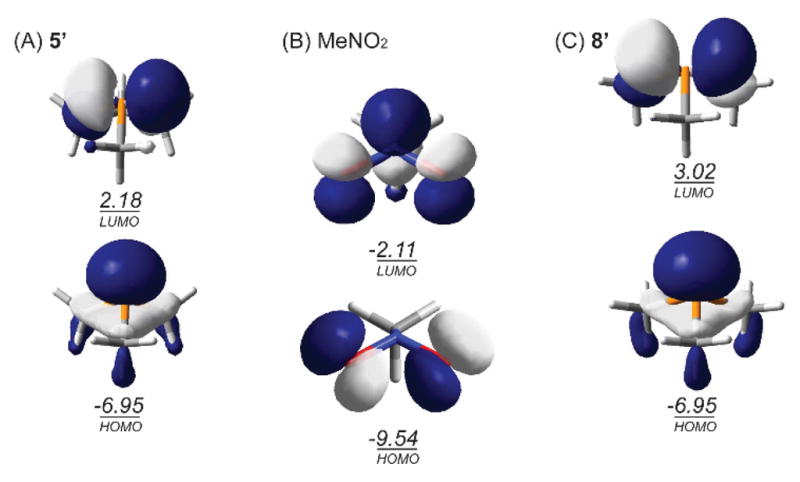
Frontier Kohn-Sham orbital plots and orbital energies for reactant fragments deformed to transition state structures (M06-2X/6-31g(d)).
In summary, we have found that a readily accessible27 phosphetane is a suitable catalyst for the Cadogan indazole synthesis. The method provides a simple phosphacatalytic approach to a valuable N–N bond forming mode that has previously been accomplished via (super)stoichiometric reagent chemistry,13,28 transition metal catalysis,29 or alternative high energy azide substrates.30 Whereas previous studies involving PIII/PV=O redox cycling have focused primarily on ring strain arguments underpinning catalytic turnover of phosphine oxides by silane reductants, the results above suggest a dominant electronic component to the overall biphilic function of the phosphetane catalyst. Work continues in an effort to establish further the biphilic reactivity of phosphetanes as generalized platforms for catalytic reductive O-atom transfer.
Acknowledgments
Dedicated to Prof. Steven M. Weinreb on the occasion of his 75th birthday. This research was supported by NIH NIGMS under award number GM114547. ATR gratefully acknowledges additional support from the Alfred P. Sloan Foundation and Amgen. We thank the Buchwald laboratory (MIT) for access to equipment and chemicals.
Footnotes
Notes
The authors declare no competing financial interests.
Supporting Information
Synthetic procedures, 1H, 13C, 19F and 31P NMR spectra, computational details and cartesian coordinates. The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Rowley AG. In: Organophosphorus Reagents in Organic Synthesis. Cadogan JIG, editor. Academic Press; London: 1979. p. 295. [Google Scholar]
- 2.Maryanoff BE, Reitz AB. Chem Rev. 1989;89:863–927. [Google Scholar]
- 3.Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Chem Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 4.Appel R. Angew Chem Int Ed Engl. 1975;14:801–811. [Google Scholar]
- 5.(a) Marsden SP. In: Sustainable Catalysis. Dunn PJ, Hii KK (Mimi), Krische MJ, Williams MT, editors. John Wiley & Sons, Inc; 2013. pp. 339–361. [Google Scholar]; (b) van Kalkeren HA, van Delft FL, Rutjes FPJT. ChemSusChem. 2013;6:1615–1624. doi: 10.1002/cssc.201300368. [DOI] [PubMed] [Google Scholar]; (c) An J, Denton RM, Lambert TH, Nacsa ED. Org Biomol Chem. 2014;12:2993–3003. doi: 10.1039/c4ob00032c. [DOI] [PubMed] [Google Scholar]; (d) Voituriez A, Saleh N. Tetrahedron Letters. 2016;57:4443–4451. [Google Scholar]
- 6.Marsden SP, McGonagle AE, McKeever-Abbas B. Org Lett. 2008;10:2589–2591. doi: 10.1021/ol800921n. [DOI] [PubMed] [Google Scholar]
- 7.(a) Denton RM, An J, Adeniran B. Chem Commun. 2010;46:3025–3027. doi: 10.1039/c002825h. [DOI] [PubMed] [Google Scholar]; (b) Denton RM, An J, Adeniran B, Blake AJ, Lewis W, Poulton AM. J Org Chem. 2011;76:6749–6767. doi: 10.1021/jo201085r. [DOI] [PubMed] [Google Scholar]; (c) An J, Tang X, Moore J, Lewis W, Denton RM. Tetrahedron. 2013;69:8769–8776. [Google Scholar]
- 8.(a) O’Brien CJ, Tellez JL, Nixon ZS, Kang LJ, Carter AL, Kunkel SR, Przeworski KC, Chass GA. Angew Chem-Int Edit. 2009;48:6836–6839. doi: 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]; (b) O’Brien CJ. 20120029211 A1. Patent US. 2012 Feb 2;; (c) O’Brien CJ, Lavigne F, Coyle EE, Holohan AJ, Doonan BJ. Chem-Eur J. 2013;19:5854–5858. doi: 10.1002/chem.201300546. [DOI] [PubMed] [Google Scholar]; (d) O’Brien CJ, Nixon ZS, Holohan AJ, Kunkel SR, Tellez JL, Doonan BJ, Coyle EE, Lavigne F, Kang LJ, Przeworski KC. Chem-Eur J. 2013;19:15281–15289. doi: 10.1002/chem.201301444. [DOI] [PubMed] [Google Scholar]; (e) Coyle EE, Doonan BJ, Holohan AJ, Walsh KA, Lavigne F, Krenske EH, O’Brien CJ. Angew Chem Int Ed. 2014;53:12907–12911. doi: 10.1002/anie.201406103. [DOI] [PubMed] [Google Scholar]
- 9.(a) van Kalkeren HA, Leenders SHAM, Hommersom CRA, Rutjes FPJT, van Delft FL. Chemistry - A European Journal. 2011;17:11290–11295. doi: 10.1002/chem.201101563. [DOI] [PubMed] [Google Scholar]; (b) van Kalkeren HA, Bruins JJ, Rutjes FPJT, van Delft FL. Adv Synth Catal. 2012;354:1417–1421. [Google Scholar]; (c) Lenstra DC, Rutjes FPJT, Mecinović J. Chem Commun. 2014;50:5763–5766. doi: 10.1039/c4cc01861c. [DOI] [PubMed] [Google Scholar]
- 10.(a) Harris JR, Haynes MT, Thomas AM, Woerpel KA. J Org Chem. 2010;75:5083–5091. doi: 10.1021/jo1008367. [DOI] [PubMed] [Google Scholar]; (b) Kosal AD, Wilson EE, Ashfeld BL. Angew Chem-Int Edit. 2012;51:12036–12040. doi: 10.1002/anie.201206533. [DOI] [PubMed] [Google Scholar]; (c) Fourmy K, Voituriez A. Org Lett. 2015;17:1537–1540. doi: 10.1021/acs.orglett.5b00426. [DOI] [PubMed] [Google Scholar]; (d) Werner T, Hoffmann M, Deshmukh S. Eur J Org Chem. 2015;2015:3286–3295. [Google Scholar]
- 11.Kirby AJ, Warren SG. The Organic Chemistry of Phosphorus. Elsevier; Amsterdam: 1967. p. 20. [Google Scholar]
- 12.(a) Dunn NL, Ha M, Radosevich AT. J Am Chem Soc. 2012;134:11330–11333. doi: 10.1021/ja302963p. [DOI] [PubMed] [Google Scholar]; (b) Reichl KD, Dunn NL, Fastuca NJ, Radosevich AT. J Am Chem Soc. 2015;137:5292–5295. doi: 10.1021/jacs.5b01899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao W, Yan PK, Radosevich AT. J Am Chem Soc. 2015;137:616–619. doi: 10.1021/ja511889y. [DOI] [PubMed] [Google Scholar]
- 13.(a) Cadogan JIG, Cameron-Wood M, Mackie RK, Searle RJG. J Chem Soc. 1965:4831–4837. [Google Scholar]; (b) Cadogan JIG. Synthesis. 1969;1969:11–17. [Google Scholar]; (c) Cadogan JIG, Todd MJ. J Chem Soc C. 1969:2808–2813. [Google Scholar]
- 14.Sundberg RJ. J Org Chem. 1965;30:3604–3610. [Google Scholar]
- 15.Genung NE, Wei L, Aspnes GE. Org Lett. 2014;16:3114–3117. doi: 10.1021/ol5012423. [DOI] [PubMed] [Google Scholar]
- 16.a) Marsi KL. J Am Chem Soc. 1969;91:4724. [Google Scholar]; b) Marsi KL. J Org Chem. 1974;39:265. [Google Scholar]
- 17.A catalytic mechanism proceeding via initial imine reduction is excluded by the observation that reaction of N-(2-nitrobenzyl)aniline under standard catalytic conditions gives an incomparably low yield of indazole 2. See SI for details.
- 18.For theoretical studies and discussion of related downstream events, see: Davies IW, Guner VA, Houk KN. Org Lett. 2004;6:743–746. doi: 10.1021/ol0364273.
- 19.(3+1) Additions are rare, but precedented: Xiong Y, Yao S, Driess M. Organometallics. 2010;29:987–990.May A, Roesky HW, Herbst-Irmer R, Freitag S, Sheldrick GM. Organometallics. 1992;11:15–16.
- 20.(a) Thompson QE. J Am Chem Soc. 1961;83:845–851. [Google Scholar]; (b) Stephenson LM, McClure DE. J Am Chem Soc. 1973;95:3074–3076. [Google Scholar]
- 21.(a) Ess DH, Houk KN. J Am Chem Soc. 2007;129:10646–10647. doi: 10.1021/ja0734086. [DOI] [PubMed] [Google Scholar]; (b) Fernández I, Bickelhaupt FM. Chem Soc Rev. 2014;43:4953–4967. doi: 10.1039/c4cs00055b. [DOI] [PubMed] [Google Scholar]
- 22.We have previously reported on the phosphaperi-cyclic reactivity of phosphetanes as ω2s-components, see ref. 12b.
- 23.Chesnut DB, Quin LD, Wild SB. Heteroatom Chem. 1997;8:451–457. [Google Scholar]
- 24.Hudson RF, Brown C. Acc Chem Res. 1972;5:204–211. [Google Scholar]
- 25.Westheimer FH. Acc Chem Res. 1968;1:70–78. [Google Scholar]
- 26.Sella A, Basch H, Hoz S. J Am Chem Soc. 1996;118:416–420. [Google Scholar]
- 27.(a) McBride JJ, Jungermann E, Killheffer JV, Clutter RJ. J Org Chem. 1962;27:1833–1836. [Google Scholar]; (b) Marinetti A, Carmichael D. Chem Rev. 2002;102:201–230. doi: 10.1021/cr990135r. [DOI] [PubMed] [Google Scholar]
- 28.Sun F, Feng X, Zhao X, Huang Z-B, Shi D-Q. Tetrahedron. 2012;68:3851–3855. [Google Scholar]
- 29.(a) Akazome M, Kondo T, Watanabe Y. J Chem Soc, Chem Commun. 1991:1466–1467. [Google Scholar]; (b) Akazome M, Kondo T, Watanabe YJ. Org Chem. 1994;59:3375–3380. [Google Scholar]; (c) Kumar MR, Park A, Park N, Lee S. Org Lett. 2011;13:3542–3545. doi: 10.1021/ol201409j. [DOI] [PubMed] [Google Scholar]; (d) Okuro K, Gurnham J, Alper H. Tetrahedron Letters. 2012;53:620–622. [Google Scholar]; (e) Moustafa AH, Malakar CC, Aljaar N, Merisor E, Conrad J, Beifuss U. Synlett. 2013;24:1573–1577. [Google Scholar]
- 30.(a) Stokes BJ, Vogel CV, Urnezis LK, Pan M, Driver TG. Org Lett. 2010;12:2884–2887. doi: 10.1021/ol101040p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu J, Cheng Y, Yang Y, Rao Y. Chem Commun. 2011;47:10133–10135. doi: 10.1039/c1cc13908h. [DOI] [PubMed] [Google Scholar]