

Abstract
Although it is common in untargeted metabolomics to apply reversed-phase liquid chromatography (RPLC) and hydrophilic interaction liquid chromatography (HILIC) methods that have been systematically optimized for lipids and central carbon metabolites, here we show that these established protocols provide poor coverage of semipolar metabolites due to inadequate retention. Our objective was to develop a RPLC approach that improved detection of these metabolites without sacrificing lipid coverage. We initially evaluated columns recently released by Waters under the CORTECS line by analyzing 47 small-molecule standards that evenly span the nonpolar and semipolar ranges. A RPLC method commonly used in untargeted metabolomics was considered a benchmarking reference. We found that highly nonpolar and semipolar metabolites cannot be reliably profiled with any single method due to solubility and retention limitations of the injection solvent. Instead, we optimized a multiplexed approach using the CORTECS T3 column to analyze semipolar compounds and the CORTECS C8 to analyze lipids. Strikingly, we determined that combining these methods enabled detection of 41 of the total 47 standards whereas our reference RPLC method only detected 10 of 47. We then applied credentialing to compare method performance at the comprehensive scale. The tandem method showed over a 5-fold increase in credentialing coverage relative to our RPLC benchmark. Our results demonstrate that comprehensive coverage of metabolites amenable to reversed-phase separation necessitates two reconstitution solvents and chromatographic methods. Thus, we suggest complementing HILIC methods with a dual T3 and C8 RPLC approach to increase coverage of semipolar metabolites and lipids for untargeted metabolomics.
Keywords: Metabolomics, Untargeted profiling, Mass spectrometry, Global coverage, Semipolar metabolome, Reversed-phase
Graphical Abstract
Analysis of semipolar and nonpolar metabolites necessitates two RPLC methods, which extend metabolome coverage >5 fold for untargeted profiling.
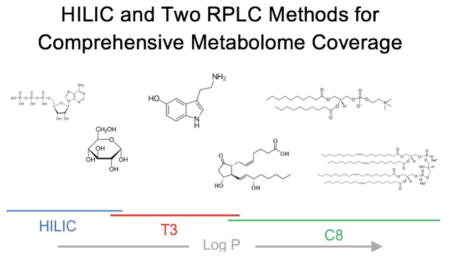
1. Introduction
The physiochemical diversity of the cellular metabolome prevents its comprehensive analysis in a single experiment [1]. Accordingly, to profile metabolites at a global scale, untargeted metabolomics requires the application of multiple extraction approaches, separation methods, and ionization technologies [2–5]. The combination of techniques that provides the best metabolome coverage has been the focus of much attention.
In theory, untargeted metabolomic protocols could be benchmarked simply by the number of metabolites detected from a complex biological sample. In practice, however, such direct comparisons have not been possible because the majority of signals detected in a typical liquid chromatography/mass spectrometry (LC/MS) untargeted metabolomic experiment cannot be chemically identified [6,7]. Without metabolite identifications, the question then becomes what metric should method comparisons be based upon.
Historically, a popular approach was to rank metabolomic protocols according to the total number of LC/MS features detected. A decade ago, counting features was state of the art and provided one of the first comprehensive benchmarks for method development in the field [8]. Now, due to improvements in software functionality and user friendliness, generating feature lists has become relatively routine and new approaches to qualitatively annotate classes of features have since emerged [9–14]. While still inferior to chemical identification, feature classification is an important advance for comprehensive benchmarking. Method comparisons based on only the total number of features are generally unreliable and, in some cases, may actually select for sub-optimal protocols. As a simple example, consider assessing a workflow that introduces contamination into the analysis. The contamination may result from carry over in the lines or columns, from a dirty piece of equipment, from plasticizers or slip agents in tubes, from solvent impurities, etc. [15]. Although workflows introducing contamination increase the total feature count, they are obviously not desirable for metabolomic experiments. Similar to contaminants, artifacts arising due to informatic error also distort the correlation between feature count and true metabolites. Notably, contaminants and artifacts have recently been shown to represent over half of the features in some untargeted metabolomic data sets [16]. Thus, experimental strategies such as credentialing that remove these types of features are essential for more accurate benchmarking of untargeted metabolomic methods at the comprehensive scale [16].
Another potential limitation of optimizing untargeted metabolomic methods by maximizing feature number is that bigger feature counts do not necessarily correlate with capturing more of the metabolome’s physiochemical diversity. A given method might extend coverage to one large class of metabolites at the cost of losing coverage of many smaller classes of metabolites, thereby sacrificing breadth for depth. Using commercial standards rationally selected a priori to evaluate detection of specific classes of metabolites (or specific metabolites) is therefore a highly complementary approach to feature counting [17]. Indeed, even though such studies are limited by the availability of standards, they provide the most robust results when assessing method performance for a targeted group of metabolites [18,19].
By using both credentialing and a set of 47 commercial standards, in this work we sought to improve our chromatographic methods for LC/MS-based untargeted metabolomics. Chromatographic separation of as many metabolites as possible is critical in untargeted metabolomics for four reasons, which we and others have outlined in more detail previously: (i) it reduces ion suppression and therefore increases sensitivity, (ii) it facilitates acquisition of non-chimeric MS/MS data for metabolite identification, (iii) better peak shapes improve the fidelity of informatic processing, and (iv) retention times can be used as a molecular identifier [20–24]. In most untargeted metabolomic approaches, hydrophilic interaction liquid chromatography (HILIC) and reversed-phase liquid chromatography (RPLC) are used in two independent experiments to achieve separation of water-soluble compounds and lipids, respectively. Previous studies systematically evaluating multiple chromatographic technologies identified the Phenomenex Luna NH2 column and the Waters XBridge C18 column as promising solutions for separating central carbon metabolites and lipids [17,25,26]. Although a variety of other HILIC and RPLC methods exist, these methods are widely used in untargeted metabolomics and will serve as our benchmarking reference here. One deficiency of the conventional approach combining HILIC and RPLC for untargeted metabolomics is that existing chromatographic methods poorly separate many metabolites in the range of intermediate polarity, which we will refer to as “semipolar”. Semipolar metabolites include classes of compounds of significant biochemical importance, such as neurotransmitters and nucleobases (Fig. 1). Yet, because previous benchmarking studies focused on central carbon metabolites or lipids and feature counting does not optimize breadth of molecular coverage, semipolar metabolites have received little attention during method development in untargeted metabolomics. Our goal was to develop a chromatographic strategy for untargeted metabolomics that retains semipolar metabolites, without sacrificing separation of the more hydrophilic and hydrophobic compounds.
Fig. 1.

Comparison of 47 small-molecule standards between columns. The T3 demonstrated the best performance for semipolar standards. The C8 had the best performance for nonpolar standards. Combined, the two CORTECS methods had the best overall performance with 41 of the 47 standards being reliably detected. Red indicates that a standard was undetected, eluted in the void volume, or had an unreliable peak shape. Yellow indicates peak tailing or fronting, band broadening, or asymmetry (see Fig. 2 for examples).
It is convenient to quantify the lipophilicity of metabolites by using the logarithm of the partition coefficient (logP). We use the partition coefficients as calculated by Advanced Chemistry Development Labs (and reported by ChemSpider) in this work, which are defined as the ratio of the concentration of an unionized metabolite in octanol to the concentration of the unionized metabolite in water. We consider metabolites with logP values roughly from −2 to 1.5 to be challenging semipolar compounds. While some targeted methods have been optimized to retain certain classes of metabolites within this logP range, they are not well suited for untargeted metabolomics because they sacrifice separation of other metabolite classes [27]. Additionally, although a small number of compounds within this logP range can be retained by the Luna NH2 and/or XBridge C18 methods, their peak shapes are generally poor and unreliable for quantification. Here we set out to optimize a RPLC approach that enabled robust analysis of semipolar metabolites, without sacrificing lipid coverage. We hypothesized that previous RPLC methods fail to separate semipolar metabolites because they are not retained at the starting mobile phase of these methods, 95% water. Thus, we investigated the reversed-phase T3 column recently released by Waters under the CORTECS UPLC line because it is compatible with 100% aqueous mobile phases. To achieve reliable peak shapes and retention of semipolar metabolites interacting weakly with the stationary phase, we found that we needed to suspend our T3 samples in pure water [28]. This constraint of using a purely aqueous injection solvent compromises the dissolution and detection of many lipids, thereby necessitating that we split the analysis into two injections optimized for semipolar and highly nonpolar compounds. With two injections, the chromatographic gradient is not extended but rather distributed between two experiments. Next, we evaluated other reversed-phase CORTECS UPLC columns that might offer complementary coverage of highly nonpolar small molecules. We benchmarked each method with a set of 47 standards that were evenly distributed over a logP range of −2 to 32, which spans semipolar metabolites and lipids (Fig. 1 and Electronic Supplementary Material (ESM), Table S1). As predicted, the CORTECS T3 method was found to provide the best coverage of semipolar metabolites, while the CORTECS C8 method allowed for superior resolution of nonpolar species. Strikingly, the combination of the CORTECS T3 and C8 methods enabled 41 of the 47 standards to be detected with optimal quantitative reliability, whereas only 10 of the 47 standards were detected with the XBridge C18 method. Out of the methods we evaluated, we also found that the CORTECS T3 and C8 methods yielded the largest number of credentialed features from a complex biological extract. Taken together, our data suggest that combining the complementary CORTECS T3 and C8 methods with HILIC extends metabolome coverage for comprehensive metabolite profiling studies.
2. Materials and Methods
2.1. Materials
A list of the full names of the 47 chemical standards and their suppliers (as well as other materials used) are included in the ESM (Table S1).
2.2. Growing E. coli for Credentialing
Credentialed E. coli samples were generated according to our credentialing protocol as previously established and described in ESM [16].
2.3. Credentialed Metabolite Extraction
The E. coli pellets were extracted as previously described and detailed in the ESM [16]. The soluble fraction of the extracts were vacuum concentrated and reconstituted in 100 μL of water for LC/MS analysis on the CORTECS T3 or isopropanol:methanol:water (2:1:1) for all other CORTECS columns. The soluble fractions of extracts were resuspended in acetonitrile:water (1:1) for the XBridge C18 and Phenomenex Luna NH2 methods, as previously described [17,25].
2.4. Purchasing Metabolic Extracts of Credentialed E. coli
For some analyses, metabolic extracts of credentialed E. coli were obtained from Cambridge Isotope Laboratories (MSK-CRED-DD-KIT). The Credentialed E. coli Cell Extract Kit is offered as 100 μL of unlabeled and 100 μL of U-13C-labeled extract, either dried down or in solution. The cells are E. coli K12 strain MG1655 and are extracted as previously described and detailed in ESM [16]. ESM Table S2 shows the overlap between our standards and the E. coli metabolites that could be detected from the Cambridge Kit. While several of our standards could not be detected, we were able to detect metabolites spanning the semipolar and nonpolar logP range, highlighting the physiochemical diversity of the Cambridge Kit.
2.5. LC/MS Analysis
For initial column optimization and comparison, all commercial standards were diluted to 10 μM in 100% water or 100% methanol.
High performance LC/MS (HPLC/MS) was performed on an Agilent 6530 Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) with an electrospray ionization (ESI) source interfaced with an Agilent 1260 Capillary LC system. For RPLC separations, an XBridge C18 column (150 mm × 1.0 mm, 3.5 μm, Waters, Milford, MA) was coupled to MS detection. For HILIC separations, a Luna NH2 column (150 mm × 1.0 mm, 3 μm, Phenomenex, Torrance, CA) was coupled to MS detection. RPLC solvents were A: 100% water with 0.1% formic acid, and B: 100% acetonitrile with 0.1% formic acid. The flow rate for RPLC was 50 μL/min with the following linear gradient: 0–5 min: 2% B, 5–50 min: 2–98% B, 50–55 min: 98% B. HILIC solvents were A: 95% water and 5% acetonitrile with 10 mM ammonium acetate and 10 mM ammonium hydroxide, and B: 95% acetonitrile and 5% water. The flow rate for HILIC was 50 μL/min with the following linear gradient: 0–5 min: 100% B, 5–50 min: 100-0% B, 50–55 min: 0% B. Injection volumes were 8 μL for all HPLC/MS experiments. Mass range was set from 60–1600 m/z in both positive and negative ionization mode. MS parameters were as follows: gas, 300 °C at 9 L/min; nebulizer, 44 psi at 2000 V; sheath gas, 350 °C at 12 L/min; capillary, 3000 V; fragmentor, 100 V; scan rate, 4 scans/second. We used ultra performance LC/MS (UPLC) to analyze 4 reversed-phase columns of the Waters CORTECS UPLC line, each with identical specifications (150 mm × 2.1 mm, 1.6 μm) but differing column chemistries (C18, C18+, C8, vs T3). All columns were coupled to a Waters CORTECS UPLC VanGuard pre-column of matching column chemistry (5 mm × 2.1 mm, 1.6 μm). Metabolite analysis was performed on an Agilent 6530 Accurate-Mass Q-TOF with an ESI source interfaced with an Agilent 1290 Infinity LC system. RPLC solvents were A: 100% water with 5 mM ammonium acetate and 5 μM ammonium phosphate, and B: 90% isopropanol and 10% methanol with 5 mM ammonium acetate and 5 μM ammonium phosphate. The column compartment was maintained at 55 °C with a flow rate of 0.200 mL/min. For the C18, C18+, and C8 columns, the linear gradient was as follows during column evaluation and optimization: 0–2 min: 5% B, 2–36 min: 5–100% B, 36–40 min: 100% B, 40–45 min: 100–5% B. For the T3 column, whose column chemistry allows for 100% aqueous mobile phase, the following linear gradient was applied during column evaluation and optimization: 0–2 min: 0% B, 2–36 min: 0–100% B, 36–40 min: 100% B, 40–45 min: 100–0% B. The final, complementary linear gradient determined for the CORTECS C8 method was as follows: 0–1 min: 40% B, 16 min: 100% B, 18 min: 100% B. The final, complementary linear gradient applied for the CORTECS T3 method was as follows: 0–2 min: 0% B, 12 min: 40% B, 13.5 min: 40% B. Columns were washed with 100% B for 2 minutes and equilibrated at starting conditions for 2 minutes prior to each injection. Injection volumes for all UPLC experiments were 2 μL. All other MS settings were the same as those described above.
2.6. Data Analysis
All experiments were performed in replicates of three (n=3) per sample group. Analysis of credentialing data was performed by applying the latest version of the credentialing software, which is freely available on our laboratory website at http://pattilab.wustl.edu/software/credential/credential.php. Peak picking was accomplished by CentWave and alignment by Warpgroup [22,29].
3. Results and Discussion
Often in untargeted metabolomics, samples are analyzed by both RPLC and HILIC. While many variations of each chromatography are used, two common methods include the Phenomenex Luna NH2 column and the Waters XBridge C18 column [17,25,26]. These separation methods, which we will use as a reference benchmark here, do not retain many compounds in the logP range of −2 to 1.5 that we refer to as semipolar (see Fig. 1). The primary goal of the current study was to extend metabolome coverage in comprehensive profiling experiments by developing a chromatographic approach that enables retention of these semipolar metabolites, while still enabling analysis of nonpolar and polar molecules. Instead of attempting to extend HILIC to semipolar metabolites, here we focused on developing a RPLC method for both lipids and semipolar metabolites. As such, we chose to use 47 standards that span logP values characteristic of lipids (1.5 to 35) and semipolar metabolites (−2 to 1.5) as a metric. This gave us coverage over a range of major classes and sub-classes of lipid metabolites. Many of these compounds were derivatives of each other so that we could assess resolution of structural features such as double bond placement (e.g., diacylglycerol 18:1/18:1 versus 18:0/18:2), polar isomer effects in complex lipid head groups (e.g., glucosyl versus galactosyl cerebroside), number of fatty acid chains (e.g., fatty acid versus diacylglycerol versus triacylglycerol versus cardiolipin), and cis/trans isomers (e.g., elaidic versus oleic acid). The 47 standards were mixed such that each had a final concentration of 10 μM. A list of the standards is shown in Fig. 1 and ESM (Table S1).
3.1. Evaluation of 47 Standards with CORTECS UPLC Columns
We identified columns in the CORTECS UPLC line from Waters as promising candidates to improve RPLC in untargeted metabolomics. These columns, which to the best of our knowledge have not yet been explored for global metabolite profiling, contain among the smallest solid-core particles commercially available at this time. Compared to larger porous particles, small solid-core particles improve chromatographic efficiency, resolution, and peak capacity for methods operating at higher linear velocities [30]. The CORTECS T3, which is a C18 column with a reduced carbon load and ligand density, is particularly relevant to the current study because of its compatibility with 100% aqueous mobile phases. We examined three other CORTECS UPLC columns with varying column chemistries as possible alternatives or complements to the T3. In addition to C8 and C18, we studied the C18+. The C18+ is a general-purpose column with a positively charged surface engineered to enhance peak shape of basic analytes. A comparison of column specifications can be found in ESM (Table S3).
We first sought to assess the ability of the CORTECS columns to retain each of our 47 small-molecule standards. An organic mobile phase containing isopropanol:methanol (9:1) with 5 mM ammonium acetate and 5 μM ammonium phosphate was used in all gradients. The use of an organic mobile phase stronger than acetonitrile is necessary to elute complex lipid species such as phospholipids, triacylglycerols, and cardiolipin from the column [31]. It is common to use 10 mM ammonium acetate in RPLC methods [32], but a concentration of 5 mM proved to be less suppressing for semipolar compounds and improved retention time stability as well as peak shape for lipid species. Although ammonium phosphate has been considered incompatible with MS because of ion suppression and nonvolatility when used as a standard LC additive [33], we found that micromolar amounts of ammonium phosphate prevented peak tailing of phospholipids, triacylglycerols, and diacylglycerols (ESM, Fig. S1) while further improving the ionization of semipolar metabolites (Fig. 2A).
Fig. 2.

Optimizing mobile-phase buffers and RPLC. A: Adding 5 mM phosphate buffer to the mobile phase increases the signal intensities of semipolar metabolites. The ratio between signal intensity with and without phosphate buffer shows an increase in signal for all standards but histamine. B–C: EIC and TIC of serotonin shows retention on T3 but not C8. D: Mass spectra from T3 and C8 show less serotonin signal suppression on T3 relative to C8. E: EIC of phosphatidylcholine (24:0/24:0) shows optimal peak shape for C8, peak tailing for T3 and C18, and peak degradation for C18+. F: EIC for diacylglycerol (18:1/18:1) and diacylglycerol (18:0/18:2). The structural isomers start to be resolved on the T3 and C18, but not the C8.
As expected, all 15 semipolar compounds in our standard mix eluted in the void volume of the CORTECS C18 and C18+ methods, and only 2 semipolar compounds were retained by the C8 method. Elution in the void volume compromises sensitivity, as shown for serotonin in Fig. 2B–D. In contrast, all of the semipolar compounds in our standard mix were retained on the CORTECS T3 method. Although 2 of the semipolar standards did exhibit some peak tailing, all of the semipolar compounds were reliably detected with the T3 method (Fig. 1). Thus, the 100% aqueous T3 method is best suited for analysis of semipolar metabolites.
Also as expected, due to the limited solubility of nonpolar analytes in pure water, detection of standards in our mix with logP values greater than 4 was compromised when using the T3 method with a reconstitution solvent of pure water (ESM, Fig. S2). These results suggested that simultaneous analysis of semipolar metabolites and lipids could not be achieved in a single experiment. Therefore, we recognized that two complementary, truncated RPLC methods for semipolar compounds and lipids are required to maximize metabolome coverage.
Our standard mix contained 32 compounds with logP values greater than 1.5. Even when these standards were suspended in methanol (instead of pure water), the T3 column was outperformed by the C8 column. The C8 exhibited narrower peaks, minimal peak tailing, and the least run-to-run carryover relative to the other CORTECS columns tested (Fig. 2E). For the complex lipids that we evaluated, the C18+ was found to have the worst performance as shown for a representative example in Fig. 2E. We associate the peak degradation observed in C18+ experiments with the use of neutral pH solvents for mobile phases. Although using solvents with lower pH would be preferable for the C18+, acidic conditions suppress ionization of free fatty acids and were therefore avoided. It is important to note that the CORTECS T3 and C18 columns showed better resolution of positional isomers with varying double-bond locations than the C8, as shown for a diacylglycerol in Fig. 2F. However, the C8 method could resolve cis-trans isomers, as shown by the chromatogram of cis-oleic acid and trans-oleic acid (elaidic acid) in the ESM (Fig. S3). In addition, the C8 method could resolve phospholipids, fatty acids, and acylglycerols with different numbers of acyl chains, head group compositions, and acyl chain lengths (Fig. 1).
Taken together, our data identify the T3 method as having promising performance for semipolar standards and the C8 method as having superior complementary performance for nonpolar standards. While the C8 had lower selectivity for double-bond positional isomers, it expanded coverage to most major classes of nonpolar species and maintained excellent peak shape (Fig. 1). Because of its decreased hydrophobicity compared to the C18 column, the C8 column also showed significantly less run-to-run carry over and column buildup, which ultimately leads to longer column lifetime. Our analyses did not reveal any unique strength of the CORTECS C18+ or C18 methods. Thus, our next goal was to compare the CORTECS T3 and C8 methods to an established RPLC method that is widely applied in untargeted metabolomics.
3.2. Comparing XBridge C18 to CORTECS T3 and C8
Studies systematically evaluating RPLC methods in untargeted metabolomics previously identified the XBridge C18 method as a promising one-column solution to analyze nonpolar metabolites at the comprehensive level [25,26]. The XBridge C18 separation has since been applied successfully in many global profiling studies [7,34–36]. Here, we wanted to compare the performance of the established XBridge C18 method to the CORTECS T3 and C8 methods from above using our set of 47 standards. Notably, the XBridge C18 method was only able to reliably detect 10 of the 47 standards whereas the combination of the CORTECS T3 and CORTECS C8 methods enabled reliable detection of a total of 41 of the 47 standards.
All but 2 of the semipolar standards in our mix eluted in the void volume of the XBridge C18 method (Fig. 1). Standards with a logP greater than 8.5 never eluted during these experiments, thereby leaving a relatively narrow logP range for reliable analysis using the XBridge C18 method (logP 1.5 to 8.5). Many important classes of metabolites fall outside of this range including nucleobases, neurotransmitters, glycerolipids, phospholipids, glycosphingolipids, sterols, sterol esters, long-chain fatty acids, and cardiolipins. Next, we compared the peak intensity, peak width, and peak shape of standards that we could reliably detect on the XBridge C18 method to data from the CORTECS T3 and C8 methods. As shown in Fig. 3A, compounds with logP values between 1.5 and 8.5 were detected with comparable sensitivities between all three methods. We next assessed the relationship between logP and peak width to determine if peak width increased as standards become less polar (i.e., larger logP values). A comparison of this relationship across the CORTECS T3, CORTECS C8, Luna NH2, and XBridge C18 methods can be found in ESM (Fig. S4). To evaluate how the CORTECS T3 and C8 methods compared against each other, we assessed the distribution of peak width ratios for standards characterized by low logP values (1–12) and high logP values (12–32). Standards with low logP values were suspended in pure water, whereas standards with high logP values were suspended in pure methanol. The box plot in Fig. 3B shows narrower peak widths detected by the CORTECS T3 method relative to the C8 method for low logP standards, but the C8 method outperforms the T3 for more hydrophobic standards. These data motivate using the CORTECS T3 and C8 methods in sequential analyses for semipolar and nonpolar metabolome coverage, respectively.
Fig. 3.

Comparison of peak intensities and widths between columns. A: Standards detected in XBridge C18, CORTECS T3, and CORTECS C8 experiments have similar intensities. See ESM (Table S1) for full chemical names of each standard. B: Ratio of standard peak widths detected on the CORTECS C8 and T3 methods with respect to low and high log P values.
In addition to achieving better coverage of our standard mix, the CORTECS methods have two additional advantages over the XBridge C18 method. First, the combined run time of our two complementary CORTECS methods is still ~35% shorter than one run using the XBridge C18 method (35 minutes versus 55 minutes), making them higher throughput. The gradient cutoffs, as detailed in the Experimental Section, between the two CORTECS methods were determined based on logP values of standards compared against retention time and solvent composition upon elution. Second, the CORTECS methods require only neutral salt buffers in the mobile phase (compared to acids in the XBridge C18 method). The use of salt buffers, such as ammonium acetate, significantly enhanced ionization of nonpolar compounds and retention time stability. The use of acids can decrease the signal intensity of such species by as much as 20-fold and prevent retention time reproducibility [32]. ESM Fig. S5 summarizes our comparison of the XBridge C18 method to the combined CORTECS T3 and C8 methods. Even using a shorter run time, the CORTECS methods display about a two-fold increase in peak capacity with room for improved efficiency at higher flow rates, detector sample rate permitting.
3.3. T3 and C8 Produce More Credentialed Features than XBridge C18
Although we have based our method evaluations on 47 small-molecule standards that evenly span 34 logP units, these compounds still do not capture the physiochemical diversity of the comprehensive cellular metabolome. Therefore, we sought to apply a more comprehensive analysis of each method’s performance with the credentialing technology [16]. In the credentialing workflow, two E. coli cultures are produced in parallel, with one of the cultures grown on natural-abundance glucose and the other grown solely on U-13C glucose. Prior to harvest, the cultures are mixed in distinct ratios (1:1 and 1:2 in this work). The mixed cultures are then extracted and analyzed via LC/MS. The resulting data is searched for pairs of coeluting peaks that satisfy three requirements: (i) peak intensities match mixing ratios, (ii) U-13C peaks correspond to appropriate numbers of carbons for the mass being evaluated, and (iii) the accurate mass of each peak corresponds to an integer number of carbons. By using isotopic labeling patterns in E. coli, credentialing filters features in an untargeted metabolomic data set that arise from contaminants and artifacts. Features that pass the filter are deemed to be of biological origin and are referred to as “credentialed”. Since contaminants and artifacts represent a variably significant number of features in LC/MS untargeted metabolomic data sets, counting credentialed features provides a better comprehensive metric to assess method performance than counting total features alone [16].
We analyzed credentialed E. coli samples with the CORTECS T3, CORTECS C8, and XBridge C18 methods. Relative to the XBridge C18 method, the CORTECS T3 method showed a 293% increase in credentialed features while the CORTECS C8 method showed a 384% increase (Table 1). Without identifying every credentialed feature, it is challenging to determine rigorously which classes of metabolites are uniquely detected with each method. Yet, from our standard data, we hypothesized that the increase in credentialed features in the T3 analysis compared to the XBridge C18 analysis was at least in part due to semipolar metabolites. We confirmed that this was correct by identifying 10 credentialed features as semipolar metabolites in the T3 data that were not present in the XBridge C18 data (Fig. 4A). These results show that the T3 separation enables detection of semipolar metabolites from complex biological samples that are undetected by the XBridge C18 method. It is also worth emphasizing that we were only able to detect 3 of these 10 semipolar metabolites reliably in the same samples by applying a commonly used HILIC method (Fig. 4A), reinforcing the need to combine HILIC separations with improved RPLC as developed here.
Table 1.
Comparison of credentialed features between methods # of signals *
| Method | # of signals* | # credentialed | # standards |
|---|---|---|---|
| XBridge C18 | 4,166 | 173 | 10 |
| CORTECS C8 | 13,180 | 837 | 27 |
| CORTECS T3 | 14,970 | 679 | 21 |
Note: only highly abundant features were counted
Fig. 4.

Metabolites detected from E. coli samples. A: Ten semipolar metabolites were identified from analysis with the T3 method, but were mostly undetected and poorly resolved with other methods. B: Ten complex lipids were identified from analysis with the C8 method with optimal peak shape. The lipids were undetected with the XBridge C18 and CORTECS T3 methods. See Fig. 1 for more detailed descriptions of colors.
Similarly, as noted above, the number of credentialed features in the CORTECS C8 data was greater than in the XBridge C18 data (Table 1). We surmised that the increased number of credentialed features was due to improved separation of lipids, since standards with logP values greater than 8.5 were undetected with the XBridge C18 method (Fig. 1). To explore this hypothesis, we set out to identify a subset of credentialed features that were unique to the C8 data set. The analysis revealed 10 lipids (e.g., long-chain diacylglycerols and phospholipids) that were detected and credentialed in our C8 experiment, but were not detected and credentialed in our XBridge C18 and CORTECS T3 experiments (Fig. 4B). Together with our standard data, these results indicate that the CORTECS C8 is most complementary to the CORTECS T3 out of the methods we tested here. We conclude that metabolites spanning the semipolar and nonpolar range cannot be profiled simultaneously in a single experiment with the columns that we investigated, and we suggest that comprehensive metabolome coverage is best achieved by combining HILIC with the CORTECS T3 and C8 methods.
4. Conclusions
The objective of untargeted metabolomics is to quantify a large and physiochemically diverse set of small molecules, the so-called comprehensive cellular metabolome [37]. Chromatographic separation of metabolites prior to mass spectrometry analysis improves sensitivity, facilitates structural identification, and increases the quantitative reliability of the data. Thus, finding optimal methods to separate as many compounds in the cellular metabolome as possible has been an active area of research.
Previous studies have systematically assessed chromatographic methods to identify the best separation strategies for global analysis of lipids and central carbon metabolites by LC/MS. One limitation of this work has been the minimal attention given to metabolite classes of intermediate polarity (i.e., semipolar compounds), which we show here cannot be reliably detected with HILIC and RPLC methods commonly used in untargeted metabolomics. Using 47 standards that evenly span both the semipolar and nonpolar range in addition to our credentialing technology, we set out to optimize a RPLC approach that enables detection of semipolar metabolites and lipid species. Our results indicate that robust, comprehensive analysis of semipolar metabolites and lipids cannot be achieved with a single chromatographic method. Rather, we found that instead of profiling nonpolar metabolites with a single RPLC method, using both the CORTECS T3 and C8 RPLC columns substantially extends metabolome coverage. Our results have important implications for how the metabolome is partitioned for analysis in global profiling studies. Currently, it is common in untargeted metabolomics to perform two separate chromatographic separations for the analysis of polar and nonpolar compounds, typically with a HILIC and RPLC method, respectively. The data presented here indicate that the single-method RPLC approach is inadequate for profiling many metabolites of intermediate and low polarity. Therefore, in concert with HILIC, we suggest a combined RPLC approach using complementary CORTECS T3 and C8 methods that extend metabolome coverage without increasing analysis time.
Supplementary Material
Table S1 Chemical names and vendors for each standard used
Table S2 Diversity of Cambridge Credentialing Kit. Compounds designated as “N/A” are exogenous drugs not expected to be in E. coli. Biological compounds that were not detected are designated “ND”. These compounds may not be present in E. coli or may not be compatible with the extraction methods used. Detected metabolites spanned from logP −2.12 to 32.62, indicating the breadth of physiochemical diversity in the kit
Table S3 CORTECS column descriptions and specifications
Fig. S1 Extracted ion chromatogram of phosphatidylcholine (24:0/24:0). The addition of ammonium phosphate improves peak shape and intensity
Fig. S2 Effects of suspending standards in pure water or pure methanol. Each sample was analyzed with the T3 column. The ratios of signal intensities show enhanced sensitivity when nonpolar species are suspended in methanol compared to water
Fig. S3 Extracted ion chromatogram of elaidic acid and oleic acid. The C8 method is able to resolve these cis/trans isomers
Fig. S4 Peak widths of all standards detected on each method. Solid lines represent averages. Average peak widths on the CORTECS T3 method started narrow but increased approximately 2-fold by the time the final hydrophobic standard eluted, underperforming the CORTECS C8 method among nonpolar metabolite classes. The average peak widths of standards detected on the XBridge C18 method were approximately 3-fold wider compared to the same compounds detected on the T3 method. The CORTECS T3 method also outperformed the Luna NH2 method with regards to semipolar metabolites, producing mostly symmetrical peaks that were at least half as narrow Peak Width Comparison of Standards Across Columns
Fig. S5 General comparison of CORTECS methods to XBridge C18 method
Acknowledgments
This work was supported by the National Institutes of Health (grants: R01 ES022181, R21 CA191097, R01 GM05698); the Alfred P. Sloan Foundation; the Camille & Henry Dreyfus Foundation; the Edward Mallinckrodt, Jr. Foundation; and the Pew Scholars Program in the Biomedical Sciences.
Biographies
 Fuad Naser is a graduate student at Washington University in St. Louis. Fuad is interested in improving untargeted metabolomic analyses, both in terms of biochemical coverage and throughput.
Fuad Naser is a graduate student at Washington University in St. Louis. Fuad is interested in improving untargeted metabolomic analyses, both in terms of biochemical coverage and throughput.
 Nathaniel Mahieu is a postdoctoral researcher at Washington University in St. Louis. Nathaniel is interested in developing new computational approaches to interrogate large data sets.
Nathaniel Mahieu is a postdoctoral researcher at Washington University in St. Louis. Nathaniel is interested in developing new computational approaches to interrogate large data sets.
 Lingjue Wang is a graduate student at Washington University. His research focuses on understanding how reduction-oxidation reactions regulate cellular metabolism.
Lingjue Wang is a graduate student at Washington University. His research focuses on understanding how reduction-oxidation reactions regulate cellular metabolism.
 Jonathan Spalding is a graduate student at Washington University in St. Louis. Jonathan is developing and applying metabolomics to investigate questions in developmental biology.
Jonathan Spalding is a graduate student at Washington University in St. Louis. Jonathan is developing and applying metabolomics to investigate questions in developmental biology.
 Stephen L. Johnson is a Professor of Genetics at Washington University in St. Louis. His lab studies growth control using a variety of techniques including transposon-based clonal analysis, vital (GFP-expressing) markers, and metabolomics.
Stephen L. Johnson is a Professor of Genetics at Washington University in St. Louis. His lab studies growth control using a variety of techniques including transposon-based clonal analysis, vital (GFP-expressing) markers, and metabolomics.
 Gary J. Patti is the Michael and Tana Powell Associate Professor in the Department of Chemistry at Washington University in St. Louis. The Patti lab develops NMR- and mass spectrometry-based metabolomic technologies that can be applied to problems in mammalian physiology.
Gary J. Patti is the Michael and Tana Powell Associate Professor in the Department of Chemistry at Washington University in St. Louis. The Patti lab develops NMR- and mass spectrometry-based metabolomic technologies that can be applied to problems in mammalian physiology.
Footnotes
Conflict of Interest
G.J.P. is a scientific advisory board member for Cambridge Isotope Laboratories and a recipient of the Agilent Early Career Professor Award. No other authors have competing financial interests. The authors declare no nonfinancial conflicts of interest.
References
- 1.Haggarty J, Burgess KEV. Recent advances in liquid and gas chromatography methodology for extending coverage of the metabolome. Curr Opin Biotechnol. 2017;43:77–85. doi: 10.1016/j.copbio.2016.09.006. doi: http://dx.doi.org/10.1016/j.copbio.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Vinayavekhin N, Homan EA, Saghatelian A. Exploring Disease through Metabolomics. ACS Chem Biol. 2010;5:91–103. doi: 10.1021/cb900271r. [DOI] [PubMed] [Google Scholar]
- 3.Weckwerth W. Metabolomics in Systems Biology. Annu Rev Plant Biol. 2003;54:669–689. doi: 10.1146/annurev.arplant.54.031902.135014. [DOI] [PubMed] [Google Scholar]
- 4.Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, Young N, Xia J, Knox C, Dong E, Huang P, Hollander Z, Pedersen TL, Smith SR, Bamforth F, Greiner R, McManus B, Newman JW, Goodfriend T, Wishart DS. The Human Serum Metabolome. PLoS One. 2011;6:e16957. doi: 10.1371/journal.pone.0016957. https://doi.org/10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masson P, Alves AC, Ebbels TMD, Nicholson JK, Want EJ. Optimization and Evaluation of Metabolite Extraction Protocols for Untargeted Metabolic Profiling of Liver Samples by UPLC-MS. Anal Chem. 2010;82:7779–7786. doi: 10.1021/ac101722e. [DOI] [PubMed] [Google Scholar]
- 6.Stanstrup J, Gerlich M, Dragsted LO, Neumann S. Metabolite profiling and beyond: approaches for the rapid processing and annotation of human blood serum mass spectrometry data. Anal Bioanal Chem. 2013;405:5037–5048. doi: 10.1007/s00216-013-6954-6. [DOI] [PubMed] [Google Scholar]
- 7.Benton HP, Ivanisevic J, Mahieu NG, Kurczy ME, Johnson CH, Franco L, Rinehart D, Valentine E, Gowda H, Ubhi BK, Tautenhahn R, Gieschen A, Fields MW, Patti GJ, Siuzdak G. Autonomous Metabolomics for Rapid Metabolite Identification in Global Profiling. Anal Chem. 2015;87:884–891. doi: 10.1021/ac5025649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nordström A, Want E, Northen T, Lehtiö J, Siuzdak G. Multiple Ionization Mass Spectrometry Strategy Used To Reveal the Complexity of Metabolomics. Anal Chem. 2008;80:421–429. doi: 10.1021/ac701982e. [DOI] [PubMed] [Google Scholar]
- 9.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng Z, Liu X, Dai W, Yin P, Zhou L, Huang Q, Lin X, Xu G. Ion Fusion of High-Resolution LC–MS-Based Metabolomics Data to Discover More Reliable Biomarkers. Anal Chem. 2014;86:3793–3800. doi: 10.1021/ac500878x. [DOI] [PubMed] [Google Scholar]
- 11.Kuhl C, Tautenhahn R, Böttcher C, Larson TR, Neumann S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal Chem. 2012;84:283–289. doi: 10.1021/ac202450g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daly R, Rogers S, Wandy J, Jankevics A, Burgess KEV, Breitling R. MetAssign: probabilistic annotation of metabolites from LC–MS data using a Bayesian clustering approach. Bioinformatics. 2014;30:2764–2771. doi: 10.1093/bioinformatics/btu370. http://dx.doi.org/10.1093/bioinformatics/btu370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W, Chang J, Lei Z, Huhman D, Sumner LW, Zhao PX. MET-COFEA: A Liquid Chromatography/Mass Spectrometry Data Processing Platform for Metabolite Compound Feature Extraction and Annotation. Anal Chem. 2014;86:6245–6253. doi: 10.1021/ac501162k. [DOI] [PubMed] [Google Scholar]
- 14.Broeckling CD, Afsar FA, Neumann S, Ben-Hur A, Prenni JE. RAMClust: A Novel Feature Clustering Method Enables Spectral-Matching-Based Annotation for Metabolomics Data. Anal Chem. 2014;86:6812–6817. doi: 10.1021/ac501530d. [DOI] [PubMed] [Google Scholar]
- 15.Yao C-H, Liu G-Y, Yang K, Gross RW, Patti GJ. Inaccurate quantitation of palmitate in metabolomics and isotope tracer studies due to plastics. Metabolomics. 2016;12:143. doi: 10.1007/s11306-016-1081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahieu NG, Huang X, Chen Y-J, Patti GJ. Credentialing Features: A Platform to Benchmark and Optimize Untargeted Metabolomic Methods. Anal Chem. 2014;86:9583–9589. doi: 10.1021/ac503092d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajad SU, Lu W, Kimball EH, Yuan J, Peterson C, Rabinowitz JD. Separation and quantitation of water soluble cellular metabolites by hydrophilic interaction chromatography-tandem mass spectrometry. J Chromatogr A. 2006;1125:76–88. doi: 10.1016/j.chroma.2006.05.019. doi: http://dx.doi.org/10.1016/j.chroma.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Contrepois K, Jiang L, Snyder M. Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)–Mass Spectrometry. Mol Cell Proteomics. 2015;14:1684–1695. doi: 10.1074/mcp.M114.046508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R, Watson DG, Wang L, Westrop GD, Coombs GH, Zhang T. Evaluation of mobile phase characteristics on three zwitterionic columns in hydrophilic interaction liquid chromatography mode for liquid chromatography-high resolution mass spectrometry based untargeted metabolite profiling of Leishmania parasites. J Chromatogr A. 2014;1362:168–179. doi: 10.1016/j.chroma.2014.08.039. doi: http://dx.doi.org/10.1016/j.chroma.2014.08.039. [DOI] [PubMed] [Google Scholar]
- 20.Patti GJ. Separation strategies for untargeted metabolomics. J Sep Sci. 2011;34:3460–3469. doi: 10.1002/jssc.201100532. [DOI] [PubMed] [Google Scholar]
- 21.Nikolskiy I, Mahieu NG, Chen Y-J, Tautenhahn R, Patti GJ. An Untargeted Metabolomic Workflow to Improve Structural Characterization of Metabolites. Anal Chem. 2013;85:7713–7719. doi: 10.1021/ac400751j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahieu NG, Spalding JL, Patti GJ. Warpgroup: increased precision of metabolomic data processing by consensus integration bound analysis. Bioinformatics. 2016;32:268–275. doi: 10.1093/bioinformatics/btv564. http://dx.doi.org/10.1093/bioinformatics/btv564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu T, Park Y, Johnson JM, Jones DP. apLCMS-adaptive processing of high-resolution LC/MS data. Bioinformatics. 2009;25 doi: 10.1093/bioinformatics/btp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conley CJ, Smith R, Torgrip RJO, Taylor RM, Tautenhahn R, Prince JT. Massifquant: open-source Kalman filter-based XC-MS isotope trace feature detection. Bioinformatics. 2014;30:2636–2643. doi: 10.1093/bioinformatics/btu359. http://dx.doi.org/10.1093/bioinformatics/btu359. [DOI] [PubMed] [Google Scholar]
- 25.Ivanisevic J, Zhu Z-J, Plate L, Tautenhahn R, Chen S, O’Brien PJ, Johnson CH, Marletta MA, Patti GJ, Siuzdak G. Toward ‘Omic Scale Metabolite Profiling: A Dual Separation–Mass Spectrometry Approach for Coverage of Lipid and Central Carbon Metabolism. Anal Chem. 2013;85:6876–6884. doi: 10.1021/ac401140h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yanes O, Tautenhahn R, Patti GJ, Siuzdak G. Expanding Coverage of the Metabolome for Global Metabolite Profiling. Anal Chem. 2011;83:2152–2161. doi: 10.1021/ac102981k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tufi S, Lamoree M, de Boer J, Leonards P. Simultaneous analysis of multiple neurotransmitters by hydrophilic interaction liquid chromatography coupled to tandem mass spectrometry. J Chromatogr A. 2015;1395:79–87. doi: 10.1016/j.chroma.2015.03.056. doi: http://dx.doi.org/10.1016/j.chroma.2015.03.056. [DOI] [PubMed] [Google Scholar]
- 28.Keunchkarian S, Reta M, Romero L, Castells C. Effect of sample solvent on the chromatographic peak shape of analytes eluted under reversed-phase liquid chromatogaphic conditions. J Chromatogr A. 2006;1119:20–28. doi: 10.1016/j.chroma.2006.02.006. doi: http://dx.doi.org/10.1016/j.chroma.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Tautenhahn R, Böttcher C, Neumann S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics. 2008;9:504. doi: 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fekete S, Oláh E, Fekete J. Fast liquid chromatography: The domination of core–shell and very fine particles. J Chromatogr A. 2012;1228:57–71. doi: 10.1016/j.chroma.2011.09.050. doi: http://dx.doi.org/10.1016/j.chroma.2011.09.050. [DOI] [PubMed] [Google Scholar]
- 31.Cajka T, Fiehn O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal Chem. 2016;88:524–545. doi: 10.1021/acs.analchem.5b04491. [DOI] [PubMed] [Google Scholar]
- 32.Cajka T, Fiehn O. Increasing lipidomic coverage by selecting optimal mobile-phase modifiers in LC–MS of blood plasma. Metabolomics. 2016;12:34. doi: 10.1007/s11306-015-0929-x. [DOI] [Google Scholar]
- 33.Cappiello A, Famiglini G, Rossi L, Magnani M. Use of nonvolatile buffers in liquid chromatography/mass spectrometry: advantages of capillary-scale particle beam interfacing. Anal Chem. 1997;69:5136–5141. doi: 10.1021/ac970765y. [DOI] [PubMed] [Google Scholar]
- 34.Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, Ivanisevic J, Cho K, Wick EC, Hechenbleikner EM, Uritboonthai W, Goetz L, Casero RA, Pardoll DM, White JR, Patti GJ, Sears CL, Siuzdak G. Metabolism Links Bacterial Biofilms and Colon Carcinogenesis. Cell Metab. 2015;21:891–897. doi: 10.1016/j.cmet.2015.04.011. doi: http://dx.doi.org/10.1016/j.cmet.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cotter DG, Ercal B, Huang X, Leid JM, d’Avignon DA, Graham MJ, Dietzen DJ, Brunt EM, Patti GJ, Crawford PA. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J Clin Invest. 2014;124:5175–5190. doi: 10.1172/JCI76388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ivanisevic J, Epstein AA, Kurczy ME, Benton PH, Uritboonthai W, Fox HS, Boska MD, Gendelman HE, Siuzdak G. Brain Region Mapping Using Global Metabolomics. Chem Biol. 2014;21:1575–1584. doi: 10.1016/j.chembiol.2014.09.016. doi: http://dx.doi.org/10.1016/j.chembiol.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zamboni N, Saghatelian A, Patti GJ. Defining the Metabolome: Size, Flux, and Regulation. Mol Cell. 2015;58:699–706. doi: 10.1016/j.molcel.2015.04.021. doi: http://dx.doi.org/10.1016/j.molcel.2015.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Chemical names and vendors for each standard used
Table S2 Diversity of Cambridge Credentialing Kit. Compounds designated as “N/A” are exogenous drugs not expected to be in E. coli. Biological compounds that were not detected are designated “ND”. These compounds may not be present in E. coli or may not be compatible with the extraction methods used. Detected metabolites spanned from logP −2.12 to 32.62, indicating the breadth of physiochemical diversity in the kit
Table S3 CORTECS column descriptions and specifications
Fig. S1 Extracted ion chromatogram of phosphatidylcholine (24:0/24:0). The addition of ammonium phosphate improves peak shape and intensity
Fig. S2 Effects of suspending standards in pure water or pure methanol. Each sample was analyzed with the T3 column. The ratios of signal intensities show enhanced sensitivity when nonpolar species are suspended in methanol compared to water
Fig. S3 Extracted ion chromatogram of elaidic acid and oleic acid. The C8 method is able to resolve these cis/trans isomers
Fig. S4 Peak widths of all standards detected on each method. Solid lines represent averages. Average peak widths on the CORTECS T3 method started narrow but increased approximately 2-fold by the time the final hydrophobic standard eluted, underperforming the CORTECS C8 method among nonpolar metabolite classes. The average peak widths of standards detected on the XBridge C18 method were approximately 3-fold wider compared to the same compounds detected on the T3 method. The CORTECS T3 method also outperformed the Luna NH2 method with regards to semipolar metabolites, producing mostly symmetrical peaks that were at least half as narrow Peak Width Comparison of Standards Across Columns
Fig. S5 General comparison of CORTECS methods to XBridge C18 method