

Abstract
Background
Human induced pluripotent stem cell (hiPSC)-derived neuronal cultures are a useful tool for studying the mechanisms of neurological disorders and developing novel therapeutics. While plating hiPSC-derived neuronal progenitors onto glial feeder layers prepared from rodent cortex has been reported to promote functional differentiation of neuronal networks, this has not been examined in detail.
New Method
Here we describe a method of using cryopreserved cells from primary cultures for generation of mouse astrocyte-enriched, neuron-free feeder layers that grow from 10% to 100% confluence in 1 week.
Results
Electrophysiological analysis demonstrated that compared to biochemical substrates alone, astrocyte-enriched feeder layers support more rapid differentiation of hiPSC-derived progenitors into excitable neurons that form spontaneously active networks in culture. There was a positive correlation between the degree of astroglial confluence at the time of progenitor plating and the average frequency of postsynaptic currents 3 weeks after plating. One disadvantage to plating on 100% confluent feeder layers was a high incidence of the astroglial layer with the overlying neurons detaching from the coverslips during transfer to the recording chamber.
Comparison with Existing Method(s)
Prevailing methods using primary glial feeder layers can result in possible contamination with rodent neurons and an unpredictable rate of growth. We provide a reliable method of generating mouse astroglial feeder layers from cryopreserved primary cultures to support differentiation of hiPSC-derived neurons.
Conclusions
The ability to make astrocyte-enriched feeder layers of defined confluence from cryopreserved primary cultures will facilitate the use of human stem cell derived neuronal cultures for disease modeling.
Keywords: Cell Culture, Astroglia, Human Induced Pluripotent Stem Cell, Differentiation, hiPSC Derived-Neurons
1. Introduction
Human induced pluripotent stem cells (hiPSCs) can be differentiated in vitro into a wide variety of cell types including central nervous system neurons [1]. Patient-specific iPSC-derived neuronal cultures have proven to be an important tool for exploring the molecular mechanisms of a number of neurological disorders, including Parkinson’s, amyotrophic lateral sclerosis, Huntington’s, autism, schizophrenia, and epilepsy [2–8]. A critical requirement for understanding disease associated changes in neuronal function is that the derived cells not only have a neuronal morphology but that they are also capable of firing action potentials and forming functional synaptic connections.
Recent evidence demonstrates that the plating substrate can have significant influence on the development of functional properties of iPSC-derived neurons. Common substrates on which iPSC-derived neural progenitor cells are seeded include Matrigel, poly-D-lysine (PDL) or poly-L-ornithine (PLO) with laminin, and rodent astroglia [2, 9–12]. Several studies have shown that compared to plating on cell-free extracellular matrices, co-culturing iPSC-derived neural progenitors onto rodent astroglial feeder layers promotes a greater degree of morphological development and functional maturation of neuronal excitability and synaptic transmission [13–16].
In most published protocols astroglial feeder cultures are prepared from the early postnatal rodent brain [10, 12, 15, 16]. As this tissue source contains both neurons and glia, protocols have been developed to enrich the cultures for glia and eliminate neurons. Enrichment protocols often rely on differences in neuronal and glial response to culture media supplements and adherence to the substrate [17, 18]. Harsh trituration of cortical tissue in the absence of glutamate receptor blockers can also be used to inhibit neuronal survival [19, 20]. While glial cells survive these enrichment protocols, an extended and unpredictable period of time is typically required for recovery and glial proliferation. The variability in the growth rate of primary astroglia to form feeder layers makes it difficult to coordinate their availability concurrent with the hiPSCs-derived neuronal progenitors at the appropriate stage of patterning for terminal differentiation. In addition, the possibility that some rodent neurons, even a small population, are present in the primary astroglial feeder layers complicates distinguishing between hiPSC-derived and rodent neurons in live cultures.
Cryopreservation of cells harvested from rodent primary astroglial cultures has been shown to be an effective way to eliminate neurons, while the astroglia retain the ability to proliferate when replated [21, 22]. Therefore we asked whether astroglial feeder layers generated from cryopreserved cells would support differentiation of functionally active hiPSC-derived neurons. Here we describe an efficient method using cryopreserved primary mouse astroglia to generate neuron-free, astrocyte-enriched feeder layers in 4–6 days. Immunostaining demonstrated that the feeder cultures were composed primarily of GFAP positive astrocytes with no evidence of β-III tubulin positive, GFAP negative neurons. iPSC-derived neural progenitors plated onto the astrocyte-enriched feeder layers formed spontaneously active networks of hiPSC-derived neurons within 21 days. In contrast, neural progenitors plated on biochemical substrates alone or when supplemented with glial conditioned medium were less effective in supporting functional neuronal differentiation in the same time frame. There was also a positive correlation between support layer confluence at the time of progenitor plating and the degree of synaptic connectivity. This efficient method for preparation of astrocyte-enriched cultures will be of great value for neurological disease modeling and drug screening using hiPSC-derived neuronal cultures.
2. Methods
2.1 Preparation of frozen astroglia stocks from mouse brain primary cultures
Dissection of neonatal mouse brains was performed in adherence with approved animal use protocols and was consistent with a previously published protocol [19]. Postnatal cortical rinds were digested and triturated into a single cell suspension and seeded onto PDL-coated (Sigma-Aldrich, P7886) plastic 60 mm tissue culture dishes at 1 dish per brain. Cultures were maintained in minimal essential medium (MEM; Life Technologies, 11090-081) supplemented with 10% heat-inactivated fetal bovine serum (vol/vol) (Sigma-Aldrich, G8270), 2% 1 M D-(+)-glucose (vol/vol) (Sigma-Aldrich G8270), 1% penicillin/streptomycin (vol/vol) (Life Technologies, 15070-063) until 100% confluent (approximately 8–10 days). Confluent dishes were washed once with sterile Dulbecco’s calcium- and magnesium-free phosphate-buffered saline (D-PBS; Gibco, 14190-144), then incubated for 8–10 minutes in 3 ml of 37°C TrypLE Express Enzyme (Life Technologies, 12604-021). TrypLE was then diluted with an additional 50% supplemented MEM (vol/vol) per dish before detaching the cells with gentle, repetitive pipetting. Cells were then centrifuged at 156 g for 5 min and gently resuspended at a concentration of 106 cells/ml in 4°C filtered freezing media containing 45% supplemented MEM (vol/vol), 45% conditioned stock MEM (vol/vol), and 10% dimethyl sulfoxide (vol/vol) (DMSO; Sigma-Aldrich, D2650). Aliquots of cell suspension (1.5 ml) were stored in 1.8 ml polypropylene cryovials (Sigma-Aldrich, V7634) and immediately transferred into a 2-propanol jacketed freezing container (Nalgene, 5100-0001) to slowly cool at a rate of −1°C/min to a final temperature of −80°C. The entire process of expansion and cryopreservation was defined as a single passage. Frozen cell suspension was stored for a minimum of 24 hours and a maximum of 2 months prior to use. All media were equilibrated in a 37°C incubator with 5% CO2 before use on the same day.
2.2 Preparation of astroglial feeder cultures from frozen stocks and astroglial conditioned media (GCM)
Cryovials of the frozen astroglial cell suspension were defrosted in a 37°C water bath with gentle swirling until only a small ice crystal remained. 37°C supplemented MEM (vol/vol) (sMEM) was added to the cryovial in a dropwise manner to a volume of approximately 2 ml before being transferred to a 15 ml conical with 3 ml warmed sMEM. Cells were then centrifuged at 156 g for 5 minutes and re-suspended in 37°C sMEM. Cell suspension concentration was determined using Trypan Blue exclusion staining (Trypan Blue 0.4%; Life Technologies, 15250-061). 5×104 cells were then seeded onto individual PDL-coated 12 mm round glass coverslips (Bellco Glass, 1943-10012A) contained in the wells of a 24-well plate. Cultures were maintained in sMEM in a 5% CO2 incubator at 37°C, with half the media exchanged every 2 days. All media were equilibrated in a 37°C incubator with 5% CO2 before use on the same day. A flow chart of the full procedure and the corresponding timeline are illustrated in Figure 1.
Figure 1. Culture process work flow from collection of tissue to plating of astrocyte-enriched cultures for iPSC-derived neuron differentiation.

(A) Cortical rinds are isolated from P0-P2 mouse coronal slices, triturated into a single cell suspension, and grown to confluence on PDL-coated dishes. Cells harvested from confluent cultures are resuspended in media and frozen for later use. (B) 4–6 days prior to need for astrocyte-enriched feeder layers the cells are thawed, plated onto PDL-coated coverslips, and grown to desired level of confluence. (C) iPSCs are patterned into neural progenitors, which are plated onto astrocyte-enriched feeder layers for the final stage of neuronal differentiation. (D) Timeline of astroglial cultures and neural differentiation from iPSCs.
To generate astroglial-conditioned media, frozen astroglia were seeded onto PDL-coated, tissue-treated 75 cm2 cell culture flasks (Corning, 430641U). Flasks were grown to 100% confluence over a period of 5–7 days in 30 ml total sMEM. After reaching full confluence, the MEM was exchanged for neural differentiation media (NDM), prepared in a separate protocol [9], for 24 hours, before exchanging back to sMEM for 48 hours. Flasks underwent a maximum of 2 NDM exchanges.
To study the effect of feeder layer percent confluence on iPSC-derived neurons, neural progenitors were plated in six conditions: PDL, PDL supplemented with GCM, PDL/laminin, and astroglial cultures of 20%, 50% and 100% confluence.
2.3 Immunostaining
Astroglial feeder cultures were fixed in 4% paraformaldehyde (wt/vol) (Sigma-Aldrich, P6148) for 15 minutes at room temperature and washed three times with PBS. Cells were permeabilized and blocked using PBS with 4% bovine serum albumin (wt/vol) (BSA; Sigma-Aldrich, A4919), 0.25% Triton X-100 (vol/vol) (Sigma-Aldrich, X100), and 0.02% sodium azide (wt/vol) (Sigma-Aldrich, S2002) in 1× PBS (Sigma-Aldrich, P3813) for 1 h at room temperature. Cultures were exposed to primary antibody and incubated overnight at 4°C. Coverslips were then washed three times with PBS and incubated in 5% normal goat serum (vol/vol) (ThermoFisher Scientific, 16210-064), 0.25% Triton X-100, 0.02% sodium azide in 1× PBS containing a 1:1000 dilution of secondary antibody (Alexa-Fluor; Life Technologies) for 2 h at room temperature. Coverslips were washed twice with PBS before counter-staining with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies, D9542). Fluoromount G (SouthernBiotech, 0100-01) was used to mount coverslips on glass slides. Images were taken using a Zeiss M2 Imager microscope and processed by Zen software. Primary antibodies used were β-III tubulin (Sigma-Aldrich, T8660; 1:1000 dilution) to identify neurons, glial fibrillary acidic protein (GFAP; DAKO Z0334; 1:1500 dilution) to identify astroglial cells, and DAPI (1:40000 dilution) to identify nuclei of all cells.
2.4 iPSC culture
The iPSC cell line used in this paper is Line 173 (SC173.1-SF6-2I2.M16, passages 9 through 21). Line naming followed National Stem Cell Resource Center nomenclature. Fibroblasts were isolated from a skin biopsy of a 56-year-old male donor with no known clinical diagnoses. Skin fibroblasts were reprogrammed by the National Stem Cell Resource Center (Laboratory of Phillip H. Schwartz) using a previously published protocol [20]. iPSCs were maintained using feeder-free conditions based on published protocols [20, 23].
2.5 Plating of hiPSC-derived neural progenitors onto astroglial feeder layers
The differentiation protocol was adapted from Liu et al., 2013 [9] with modifications as briefly outlined. On day 0, embryoid bodies were generated from confluent human iPSC cultures by manual scraping [24]. Detached culture fragments from 15 wells were collected in a conical tube and centrifuged at 156 g for 5 min. Pellets were gently resuspended in 15 ml human iPSC medium (hiPSCM; [9]) with DMH-1 (Tocris, 4126), SB-431542 (Stemgent, 04-0010) and rho-associated protein kinase inhibitor Y27632 (ROCK inhibitor; Stemgent, 04-0012) and then transferred to a T25 non-tissue treated flask (Thermofisher Scientific, 169900). Neural induction and patterning procedures were followed according to the original protocol [9]. On day 16, neural rosettes were isolated and collected following 1 h incubation with STEMDiff Neural Rosette Selection Reagent (STEMCELL Technologies, 05832). Cells were patterned into neural progenitor cells (NPCs) using the ventralizing small molecule purmorphamine (Stemgent, 04-0009) following the original protocol until day 25 [9].
On day 26, neurospheres of NPCs were pre-treated with ROCK inhibitor, dissociated using Accutase (Life Technologies, A1110501), centrifuged at 156 g for 5 minutes, and resuspended in fresh neural induction medium (NIM) containing ROCK inhibitor. After counting cells with Trypan Blue exclusion, 2.5×104 cells were plated on PDL, PDL/laminin, and 20%, 50%, or 100% confluent mouse astroglial feeder layers grown on PDL. 24 h after plating, 50% of the NIM was replaced by neural differentiation medium (NDM) or GCM containing 0.4 μM γ-secretase inhibitor XXI (Compound E, Calbiochem, 565790), 10 ng/ml BDNF (Peprotech, 450-02), 10 ng/ml GDNF (Peprotech, 450-10), 10 ng/ml IGF-1 (Peprotech, 100-11), and 0.5 g/ml membrane permeable cyclic AMP (Sigma-Aldrich, D0260). 48 h after plating, all media were exchanged with NDM or GCM with 0.2 μM Compound E. 96 h after plating, all media were exchanged for NDM or GCM without Compound E. From this point, 50% culture media exchange with fresh NDM or GCM without Compound E was performed every other day. All media were equilibrated in a 37°C incubator with 5% CO2 before use on the same day.
To prepare PDL- and laminin-coated (PDL/L) coverslips, 50 μl of natural mouse laminin (Invitrogen, 23017-015) was added onto each PDL-coated coverslip 2 hours prior to neural progenitor plating. PDL/L coverslips were then incubated at 37°C until use.
2.6 Electrophysiological recording
Whole-cell recordings were performed on human iPSC-derived neurons between days 21–24 post plating using unpolished borosilicate glass pipettes with an open tip resistance of 4–7 MΩ (VWR International, 53432-921). External solution contained (in mM): NaCl 120, KCl 5.4, MgCl2 0.8, CaCl2 1.8, glucose 15, HEPES 20, at pH 7.2-7.4 with osmolarity between 290 and 295 mOsm. Internal solution contains (in mM): potassium gluconate 120, NaCl 20, CaCl2 0.1, MgCl2 2, EGTA 1.1, HEPES 10, Na2ATP 4.5 at pH 7.2 with osmolarity ~280 mOsm. Voltages were corrected for a −5 mV junction potential.
While holding the membrane potential at −75 mV with hyperpolarizing current, evoked action potentials (APs) were recorded from 600 ms depolarizing current injections under current-clamp, and spontaneous post-synaptic currents (sPSCs) were recorded for at least 1 minute under voltage-clamp at −75 mV. Data were acquired with an EPC7 amplifier (List Medical), a Digidata 1320A D-A converter (Axon Instruments), and a Dell Optiplex GX110 computer running pClamp8 software (Axon Instruments).
2.7 Data Analysis and Statistics
Phase-contrast micrographs were imaged with EVOS™ XL Core Cell Imaging System (ThermoFisher Scientific, AMEX1000). Visual estimations of astroglial confluence were based on n = 5 independent experiments. Fluorescent micrographs, imaged with Zeiss Axio Imager M2 and processed using Zen software, were used to quantify astroglial confluence and marker expression. Immunofluorescence analyses were based on n = 3 independent experiments.
Electrophysiological data were analyzed using Clampfit 10.6 software (Axon Instruments). Only recorded cells with a stable resting membrane potential (no greater than ± 5 mV deviation) more hyperpolarized than −30 mV, input resistance > 400 MΩ, and stable sPSC voltage-clamp recording were accepted for analysis. Evoked APs were defined as regenerative events where the first AP in the train was at least 20 mV in amplitude and any subsequent events were scored as APs if they were at least 15 mV in amplitude. Spontaneously firing cells were classified as those in which at least one spontaneous action potential (sAP) above −10 mV was observed in the first 30 seconds of recording in the absence of holding current. Mini Analysis Program 6.0.7 (Synaptosoft) was used to identify sPSC events with an amplitude threshold of 10 pA (4× RMS noise level of 2.5 pA) and an area threshold of 15 pA·ms (1.5× amplitude threshold). sPSCs identified by the software were then verified individually, with double peaks manually included. Recordings with a baseline shifted by more than 50 pA or events with a 10–90% rise time larger than 3 ms were excluded.
Statistical analyses were performed in Prism 7 (GraphPad) and figures were generated using DeltaGraph 7.1 (Red Rock Software).
3. Results
3.1 Rapid reproducible growth of feeder layers from defrosted primary astroglia
In previous studies we used astroglial feeder layers from primary cultures of neonatal mouse brains to support differentiation of hiPSC-derived neurons [20]. Significant cell loss in the primary astroglial feeder cultures was observed during the first week. Proliferation of the remaining cells typically reached confluences of 50–100% in 14–21 days when they were used plating of neuronal progenitors. The extended and variable time period made coordinating progenitor production and astroglial support layer availability challenging.
In this study we instead used cryopreserved primary astroglial cells to prepare feeder layers. Astroglial feeder layers were prepared by seeding 5×104 defrosted primary astroglial cells onto PDL-coated glass coverslips. The percentage of surface area covered by astroglial cells (% confluence) was tracked and visually estimated from phase-contrast imaging of live cultures between 2 and 10 days after plating. At day 2 post plating, a small number of cells were observed and the majority of the surface area was cell-free (Figure 2A). Astroglia proliferation was clearly apparent by day 4, with an increased number of cells and reduction in cell-free space observed. By day 8, the majority of the coverslip surface area was covered by cells. There was a steady and reproducible increase in % confluence from ~10% at 2 days after plating to ~100% by 8 days (Figure 2B). These data demonstrate the reproducible recovery and growth of defrosted primary astroglial cells, greatly facilitating coordination of hiPSC-derived neuronal progenitor plating and astroglial feeder layer availability.
Figure 2. Rapid and reproducible growth of cells in cultures prepared from stocks of frozen primary astroglial cells.

(A) Phase-contrast images of cells in live astroglial cultures at 2, 4, and 8 days after plating 5×104 cells onto PDL-coated coverslips. Scale bars represent 50 μm. (B) The percentage of surface area covered by astroglial cells was visually estimated from live cultures between 2 and 10 days after plating. Each point represents the mean ± SEM from 5 independent platings with 3 coverslips/plating.
3.2 Feeder layers are composed of primarily GFAP positive astrocytes and are neuron-free
To evaluate the contribution of astrocytes and neurons to the feeder layers prepared from the cryopreserved stocks, cultures were fixed 8 days after plating and co-stained with the nuclear marker DAPI, neuronal marker β-III tubulin, and astrocytic marker GFAP. The total number of cells in a single field was defined by the number of DAPI positive nuclei (Figure 3A). A GFAP stained image of the same field of view revealed that almost all of the nuclei were surrounded by GFAP staining indicating the majority of the cells were astrocytes (Figure 3B). While there was also one cell stained with the neuronal marker β-III tubulin (Figure 3C), the merged image revealed that the cell was also GFAP positive so it was not classified as neuronal (Figure 3D, white arrowhead). Quantitative analysis of stained cultures demonstrated that over 94% of the total cells in culture were GFAP positive astrocytes (Figure 3E). The absence of β-III tubulin positive, GFAP negative cells indicate that no mouse neurons were present in these feeder layers. The remaining 6% of the total cells fall into two groups, 2% were positive for both GFAP, and β-III tubulin possibly representing early neuroglial progenitors (Figure 3D, white arrowheads), and the remaining 4% were not stained with either GFAP or β-III tubulin (Figure 3D, pink arrowheads).
Figure 3. Feeder layers derived from cryopreserved astroglia are composed of GFAP positive astrocytes and are devoid of neurons.

An immunofluorescent image of the astroglial feeder layer from a single field of view. (A) Nuclei of cells labeled by the nuclear marker DAPI. (B) The vast majority of the cells in the feeder layer were of astrocytic identity as indicated by their staining with anti- GFAP antibodies. (C, D) A cell labeled by the neuronal marker β-III tubulin indicated by the white arrowhead is also GFAP positive indicating it was unlikely to be a neuron. A small proportion of cells were not labeled with either marker (pink arrowheads). (E) The number of total cells in each field of view was determined by counting the DAPI positive nuclei. The cells stained for GFAP alone, β-III tubulin alone, both, or neither were determined for each field were expressed as a percentage of total cells. Analysis was performed on 9 randomly selected fields from three coverslips within each plating. Bars indicate mean + SEM, n = 3 independent platings.
3.3 Astrocyte-enriched feeder layers from cryopreserved cells facilitate the morphological and functional differentiation of iPSC-derived neurons
Previous studies have shown that primary astroglial feeder layers promote development of neuritic processes and excitability of hiPSC-derived neurons [13, 16]. One study used 50% confluent glia for their experiments to promote functional maturation [25]. However, the effect of different degrees of glia confluence was not reported. To evaluate how the presence and confluence of astrocyte-enriched feeder layers prepared from cryopreserved cells affect differentiation of hiPSC-derived neurons, progenitors were plated onto a monolayer of 20%, 50%, or 100% confluent feeder layers. These were compared to those plated on PDL, PDL supplemented with astroglial-conditioned medium (GCM), or a combination of PDL/laminin (PDL/L).
When examined at 21 days post plating, qualitative differences between the culture conditions were apparent in fluorescence images. As revealed by immunostaining with β III-tubulin antibody, typically progenitors in the PDL and GCM conditions resulted in fewer cells with neuronal morphology and relatively limited branching of the neurites (Figure 4A, B). Those plated onto PDL/laminin resulted in more cells with neurites than PDL alone (Figure 4C). The most consistent and robust differentiation of neurons with highly branched processes was observed when progenitors were plated on feeder layers enriched for GFAP-positive astroglia (Figure 4D). Cultures plated on 20% confluent astrocyte-enriched feeder layers predominantly formed a lattice pattern on the glass coverslips. These hiPSC-derived neurons were isolated into islands of various sizes and connected to each other by neurite bundles (Figure 4E). Cultures plated on 50% and 100% confluent astroglia had neurons and branching neurites more evenly distributed over the confluent astroglial layer (Figure 4F, G). Cell size was also evaluated by determining the cell capacitance through whole-cell recordings. There was no effect of feeder layer confluence on average whole cell capacitance but hiPSC-derived neurons grown without feeder layers had a significantly smaller mean capacitance than those grown on feeder layers (Table 1). Furthermore, the input resistance of the cells grown without feeder layers was higher. Both of these electrical measurements are consistent with the visual observations that feeder layers facilitate growth of hiPSC-derived neurons.
Figure 4. Astroglial feeder layers prepared from cryopreserved primary cultures facilitate the morphological differentiation of hiPSC-derived neurons.
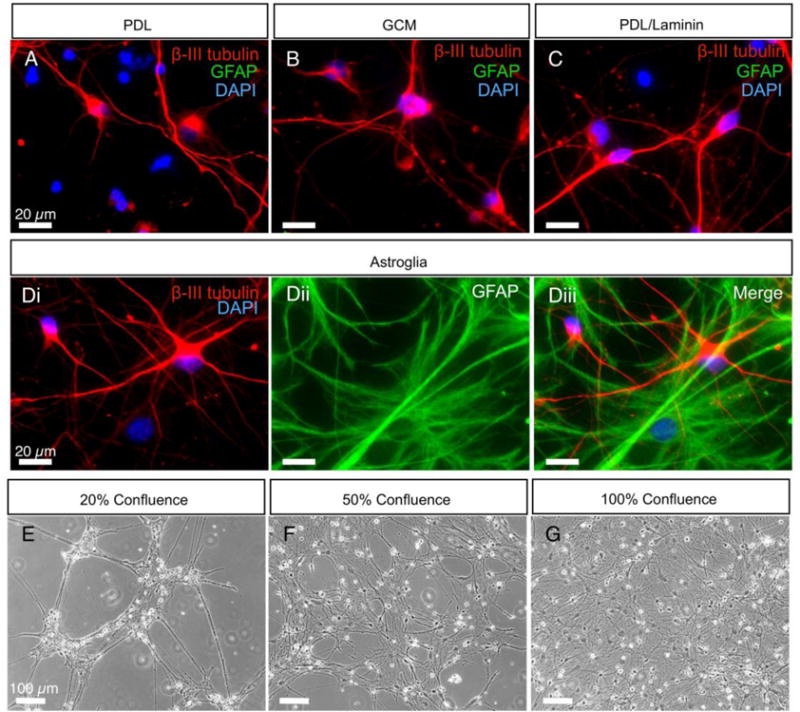
(A-C) Immunostaining of βIII-tubulin-positive and GFAP-negative derived neurons at 21–24 days after plating on PDL, PDL supplemented with GCM and PDL/laminin. (D) Immunostaining of βIII-tubulin-positive derived neurons grown on top of GFAP-positive astroglial feeder layers. Scale bars represent 20 μm. (E-G) Phase-contrast images of iPSC-derived neurons at 21–24 days after plating on astroglial feeder layers of 20%, 50% and 100% confluence, respectively. Scale bars represent 100 μm.
Table 1.
Passive membrane properties of neurons plated on PDL, PDL supplemented with GCM, PDL/laminin, and astroglial cultures of 20%, 50%, and 100% confluence.
| Cell capacitance (pF) | Input resistance (GΩ) | RMP (mV) | |
|---|---|---|---|
| PDL | 24.1 ± 2.8 (14) | 3.3 ± 0.5 (16) | −47.5 ± 2.6 (16) |
| GCM | 23.1 ± 3.4 (14) | 3.6 ± 0.5 (14) | −52.3 ± 2.6 (14) |
| PDL/laminin | 17.6 ± 1.6 (9) | 2.2 ± 0.3 (12) | −43.7 ± 2.8 (12) |
| 20% confluence | 33.6 ± 2.9 (16) c1 | 1.5 ± 0.1 (16) a3, b4 | −46.9 ± 1.8 (16) |
| 50% confluence | 31.6 ± 2.0 (16) | 1.3 ± 0.2 (17) a3, b4 | −47.3 ± 2.3 (17) |
| 100% confluence | 37.5 ± 3.5 (18) a1, b1, c3 | 1.1 ± 0.1 (18) a4, b4 | −45.0 ± 1.7 (17) |
Data reported as mean ± SEM. Number of included cells indicated in parentheses. a, b and c superscripts represent significance to PDL, GCM and PDL/laminin groups, respectively. 1, 3, and 4 superscripts denote p < 0.05, 0.001, and 0.0001, respectively (one-way ANOVA with post hoc Bonferroni test).
Active membrane properties of neurons grown in the different conditions were also evaluated by whole-cell recordings. In all six growth conditions over 75% of the hiPSC-derived neurons fired at least one action potential in response to a depolarizing stimulus (Figure 5A, B). However, the peak firing frequency was significantly higher in neurons grown on astroglial feeder layers compared to those grown in PDL, GCM, and PDL/laminin conditions, despite having similar resting membrane potentials (Figure 5C, Table 1). This is consistent with the astroglial feeder layers supporting more rapid maturation of excitability. While there was considerable variability in maximal evoked firing frequency, this was significantly higher in the neurons grown on the 20% confluent feeder layers compared to those on 50 and 100% confluent feeder layers.
Figure 5. Astroglial feeder layers facilitate maturation of intrinsic firing properties of hiPSC-derived neurons.

(A) Traces of AP firing (APs) evoked by depolarizing current injections in neurons plated on the indicated substrate at 21–24 days post plating. (B) Percentage of recorded neurons that fire ≥ 1 AP. No significant difference in % of firing APs between groups (p=0.12, Chi-square test). (C) Peak firing frequency is significantly higher in neurons grown on astroglial feeder layers compared to the other three groups (one-way ANOVA with post hoc Bonferroni test. *,**, **** denote p < 0.05, 0.01, and 0.0001 respectively. Data are represented as mean + SEM. Numbers in the parentheses represent the number of recorded neurons.
3.4 Astroglial feeder layers from cryopreserved cells support the development of spontaneous firing and synaptic activity in iPSC-derived neurons
Spontaneous firing was evaluated by gap-free current-clamp recordings for at least 1 minute in the absence of applied holding current (Figure 6A). None of the neurons grown in PDL, GCM, and PDL/laminin conditions fired spontaneous action potentials (sAPs), whereas many of the neurons on feeder layers fired sAPs (Figure 6A). The sAP firing frequency was quite variable and there was no significant association with the percent confluence of the feeder layers for the sample evaluated (Figure 6B).
Figure 6. Astroglial feeder layers support spontaneous firing and spontaneous synaptic activity in hiPSC-derived neurons.

(A) Representative traces of spontaneous action potentials (sAPs) at rest membrane potentials recorded from iPSC-derived neurons grown on indicated substrate at 21–24 days post plating. (B) sAP frequency at RMP, one-way ANOVA with post hoc Bonferroni test. (C) Overlay of two traces of spontaneous postsynaptic currents (sPSCs) recorded from neurons on indicated substrate held at −75 mV under voltage-clamp to illustrate the relative differences in sPSC frequencies. (D) sPSC frequency of neurons, one-way ANOVA test with post hoc Bonferroni test. Data are represented as mean + SEM. Numbers in the parentheses represent the number of recorded neurons. *** and **** denote p value < 0.001, and 0.0001 respectively.
Assessment of spontaneous postsynaptic activity was based on gap-free voltage-clamp recordings from a holding potential of −75 mV for at least 1 minute. There was very little synaptic activity in the neurons without astroglial feeder layers, with the average frequency of spontaneous postsynaptic currents (sPSCs) being less than 0.1 Hz, while cells grown on feeder layers exhibited significantly higher sPSC frequencies with the average frequency increasing from 0.8 Hz in the 20% confluence condition to ~7 Hz in the 100% confluence condition (Figure 6C, D). This suggests that astroglial feeder layers prepared from cryopreserved astroglia support functional development of synaptic activity in iPSC-derived neurons via physical contact, in addition to astrocyte-secreted factors [13]. One drawback to plating on 100% confluent feeder layers in our study was an increased incidence of detachment of the underlying astroglial layer from the glass coverslips during transfer to the recording chamber, which resulted loss of the overlying neurons as well. However, there was no obvious feeder layer detachment or difficulty obtaining recordings in cultures of 20% and 50% confluence.
4. Discussion and Conclusions
Our results demonstrate more rapid morphological and functional differentiation of hiPSC-derived neurons on astroglial feeder layers prepared from cryopreserved cells compared with those plated in the absence of feeder layers. This protocol offers several advantages over using primary astroglia cultures: elimination of mouse neurons without a significant increase in time or effort, the ability to store cell suspension stocks for generation of feeder cultures at a later date, and significant reduction in the number of animals required.
While previous studies have shown that primary astroglial feeder layers prepared from mouse brain tissue can support rapid differentiation of functionally active neurons from human stem cells [20, 25], one of the drawbacks is the potential for the presence of mouse neurons. These are not easily distinguished from human neurons in live cultures without introducing an exogenous marking system or requiring post-recording staining to confirm identity. Passaging glia cultures multiple times prior for use in neuronal derivation is a common practice to eliminate neurons [2, 12, 15]. While effective, this requires additional time and resources to grow glia and does not address the difficulty in synchronizing the timing of glia culture and neuronal progenitor availability. Protocols resulting in isolation of astrocytes based on differential adherence are also an option [17, 18]; however, they involve vigorous shaking and careful timing of passaging. In contrast, cryopreservation of astroglial cells efficiently removes neurons as shown by the absence of β-III tubulin positive and GFAP negative cells in the cultures, meanwhile allowing for rapid and reproducible recovery. In addition, cryopreserved astroglial cells can be banked and used at a future date to generate astroglial support layers within a week, which enables thawing of the stocks at a relatively late stage of hiPSC patterning.
The protocol described in this paper can also significantly reduce the number of mice required to generate the astroglial feeder layers and the time required to prepare them. By expanding and passaging primary cultures on dishes for cryopreservation, the same number of newborn mice can be used to prepare 2–5 times as many astrocyte-enriched cultures as those from primary tissue. The efficacy of later passages has not been tested systematically but initial studies suggested that later passages were not as reliable in supporting functional maturation. Whether the astroglial passage number definitively affects the rate of functional development in derived neurons, however, will require additional investigation. Additionally, although the current protocol uses an initial glial seeding density of 5×104 cells, lower seeding densities can also be used, for example 2.5×104 cells. This typically requires an extra two days of culturing but effectively doubles the number of coverslips generated from a single vial of cryopreserved cells. When taken together, this protocol contributes to resource management and the ability to reproducibly generate neuron-free astroglial feeder layers.
It has been shown that a variety of biochemical substrates including PDL, PLO, fibronectin, integrins, and Matrigel can influence survival, proliferation and differentiation iPSC-derived neurons [16, 26–30]. A combination of PDL/laminin has also been used to support functional differentiation of iPSC-derived neurons [31–35]. A study using high-resolution multielectrode arrays (MEAs) have shown increased spontaneous firing rate in iPSC-derived neurons without the use of astrocytes [36]. While this may indicate that astrocytes are not required for network maturation, future MEA studies may benefit by further evaluating synaptic maturation using functional analyses in neurons cultured with and without astrocytes [37, 38]. In addition, astroglial feeder layers with confluences between 20–100% are more effective at promoting maturation of functional properties than PDL supplemented with GCM. This indicates that astrocytes in our cultures may possibly regulate the functional development of derived neurons through a contact-dependent mechanism [13, 16].
Plating neural progenitors onto astroglial cultures resulted in cultures where ~50% of the neurons fired spontaneous action potentials and 100% received postsynaptic input. This indicates the astroglial cells support formation of spontaneously active neural networks in culture. Future studies using MEAs and multisite calcium imaging will be useful in evaluating the spatiotemporal changes in neuronal network dynamics that occur in these cultures. Developmental changes in spontaneous burst firing rate have been detected using MEA to detect spikes in hiPSC-derived neurons co-cultured with rodent primary astrocytes [38, 39].
The consequence of plating on fully confluent astroglia in our system is the loss of neurons when they are transferred to the recording chamber. In these cultures, the astroglia occupy the vast majority (over 95%) of the coverslip’s surface area while the derived neurons on top are not themselves confluent. The underlying astrocytes behave as a sheet and when coverslips are removed from their culture well the astroglial sheet can lift off the coverslip removing the network of neurons with it. Additional studies will be necessary to determine the underlying cause of this detachment.
For studies focused on functional analysis of excitability and synaptic transmission in iPSC-derived neurons carrying different disease causing mutations, our data indicate that 50% confluence provides an appropriate balance for functional studies, supporting spontaneously active neuronal networks while preserving the integrity of cell layer during transfer that is required for electrophysiological recording. Finally, the ability to reliably generate astroglial cultures from the same stock allows enhanced control of the terminal differentiation environment, important for assaying differences between neurons derived from different iPSC clones or lines.
Highlights.
Feeder layers from frozen mouse glia are astrocyte-enriched and neuron free.
Feeder layers reproducibly reach 50% confluence in 5–7 days.
Confluence of 20–100% supports functional differentiation of hiPSC-derived neurons
50% confluence promotes hiPSC-derived neural networks and retains integrity
Acknowledgments
We would like to thank Martin A. Smith, PhD, for his helpful input and Olga Safrina, PhD, for making primary mouse astroglial cultures. Thanks to Philip Schwartz, PhD, for providing iPSCs. This work was supported by University of California, Irvine California Institute for Regenerative Medicine Postdoctoral Fellowship (TG2-01152) (RJS); California State University, Long Beach California Institute for Regenerative Medicine Grant (PF, ATP) (TB1-01182); National Institutes of Health R01 Grant (NS083009).
Appendix A: Detailed Methods
The initial steps and preparation of the protocol have been adapted from Hilgenberg and Smith, 2007.
Preparation of Glass Coverslips
In a laminar flow hood use forceps to place autoclaved 12 mm glass coverslips (Bellco Glass) into the center 8 wells on a 24 well culture plate.
Repeat for the number of required plates.
- Leave the culture plate lids open and expose to the coverslips/plates to UV for a minimum of 30 minutes.
- Perimeter wells are not used to mitigate contamination.
Coating Coverslips with PDL or PDL/laminin (PDL/L)
Dilute a 10× aliquot to 1× by adding 9 ml sterile H2O.
- Coat each 12 mm round coverslip with a 150 μl 1× PDL droplet or flood with 2 ml 1× PDL per 60 mm dish. Keep overnight at room temperature.
- Desired plating area can be adjusted by varying volume of PDL used.
- The entire coverslip can also be coated via submersion in 400 μl PDL.
Rinse each coverslip 3× with sterile H2O and air dry in the laminar flow hood.
To coat with PDL/L, add 50 μl of natural mouse laminin solution (per coverslip) onto dried PDL-coated coverslips 2 hours prior to plating. Incubate PDL/L coverslips at 37°C.
Mouse Brain Dissection and Primary Culture (adapted from Hilgenberg and Smith, 2007)
Cut out block of 4% agar and glue it onto the microtome Vibraslicer support block using super glue (cyanoacrylate).
Decapitate P0-P2 mouse pups according to a protocol approved by an Institutional Animal Care and Use Committee.
Remove skin and skull and place brain in a 35 mm petri dish filled with 4°C dissecting solution (DS).
Remove olfactory bulbs and cerebellum using scalpel, razor, or flat-ended spatula.
Place thin layer of super glue on support block perpendicular to the 4% agar.
Pick up brain and transfer to support block. Glue brain in place with rostral side up and ventral surface resting on agar.
Insert support block into plastic chamber and submerge tissue with 4°C DS.
Cut 400 μm sections, beginning from olfactory bulb. Once the cortex is reached, adjust and begin cutting 600 μm coronal sections.
Transfer brain slices to 35 mm petri dish using the reverse end of unplugged 10 ml borosilicate glass pipet and air bladder.
Remove meninges from slices. Dissect out cortical rinds using 27 gauge (0.5 inches in length) syringe needles.
Section cortical rinds into small pieces, approximately 0.5–1.0 mm in length.
Filter cleared ES through a 0.2 μm PES filter into a 35 mm petri dish.
Transfer the cortical tissue pieces to petri dish containing filtered ES.
Incubate at 37°C for 30 min.
Transfer cortical pieces from ES into 10 ml cold DS.
Centrifuge tissue for 1 min at 349 g.
Remove supernatant and gently re-suspend pellet in an additional 10 ml cold DS. Allow tissue chunks to settle to bottom. Repeat steps 17 and 18 twice.
Transfer tissue to 15 ml conical tube containing filtered 37°C and CO2-equilibrated supplemented MEM (sMEM). Use 1 ml sMEM per brain.
- Triturate tissue with a flame polished glass Pasteur pipette to generate a single cell suspension.
- Flame polish Pasteur pipettes with a Bunsen burner for 5–10 sec or until diameter has been reduced.
- Prepare several Pasteur pipettes of varying diameter.
Add 3 ml equilibrated sMEM to PDL-coated 60 mm dishes.
Add 1 ml cell suspension to each pre-flooded 60 mm dish. Gently swirl dishes to disperse cells evenly. Maintain cultures at 37°C and 5% CO2.
Allow to adhere for 24–48 hours before starting scheduled feeding.
Feed cultures every 2 days with 50% exchange of fresh equilibrated sMEM.
Cryopreservation of Primary Glial Cultures
Transfer an empty 2-propanol jacketed freezing container to a −20°C freezer.
Aspirate all sMEM from 100% confluent primary cultures (60 mm dishes) and retain media. Gently wash dishes once with 2 ml Ca2+ and Mg2+ free D-PBS with attention not to disturb cells.
- Use retained conditioned sMEM to make freezing media (45% vol/vol conditioned sMEM, 45% vol/vol fresh sMEM, 10% vol/vol DMSO).
- Use 1.5 ml freezing media for each 60 mm dish.
Filter freezing media through a 0.2 μm PES filter and maintain media on ice.
- Add 3 ml of 37°C TrypLE to coat bottom of dish. Place in incubator for 8–10 minutes. Observe cells under a microscope.
- Cells should begin to round up and detach from the bottom of the dish. If not detached, return dish back to incubator.
- Periodically check cultures until cells are detaching.
Dilute TrypLE by adding 2 ml of fresh sMEM to each dish and gently pipet the solution to dislodge cells. Collect all media and cells into a 15 ml conical tube.
Centrifuge cells at 156 g for 5 minutes.
Re-suspend pellet in ice cold, filtered freezing media.
Make 1.5 ml aliquots into labeled cryovials (1 dish per cryovial) and transfer into the chilled 2-propanol jacketed freezing container. Immediately transfer freezing container to an −80°C freezer for overnight storage.
Remove aliquots from the freezing container and store at −80°C for a maximum of 2 months.
Defrosting Cryopreserved Glia
24 hours prior to plating, prepare PDL-coated 12 mm glass coverslips as described.
After drying, flood each well to be plated with 500 μl of sMEM and place in 37°C, 5% CO2 incubator.
Take a cryovial of frozen cells from −80°C storage and quickly defrost in a 37°C water bath until only a small ice crystal is left in the vial.
Slowly add 500 μl of filtered equilibrated sMEM in drop-wise fashion to center of cryovial.
Transfer cells from cryovial to a 15 ml conical tube filled with 3 ml fresh 37°C sMEM. Gently mix cells by pipetting without introducing air bubbles.
Centrifuge cells for 5 minutes at 156 g.
Aspirate supernatant and re-suspend cell pellet in 1 ml fresh 37°C sMEM without introducing air bubbles.
- Perform Trypan Blue exclusion cell counting to assess density of viable cells
- Each cryovial should yield approximately 1.2×106 viable cells.
Remove pre-flooded plates from incubator and inject cell suspension containing 5.0×104 Trypan Blue negative cells over center of coverslip. Gently swirl plates to disperse.
Allow cells to settle for 24 hours before starting normal feeding schedule.
Feed cells with 50% exchange of fresh 37°C sMEM every 2 days.
Preparing Astroglia-Conditioned MEM (GCM) (adapted from Hilgenberg and Smith, 2007)
Coat a tissue-treated 75 cm2 culture flasks with 30 ml 1× PDL and let sit for at least 1 min.
Transfer 1× PDL to second flasks and repeat for a total of 4 flasks.
Rinse flasks 3 times with 30 ml sterile H2O.
Leave flasks to air dry for 2 min.
Defrost astroglial cryovials and plate cells in PDL-coated flasks (1–3 cryovials per flask). Fill flask to volume with 30 ml total sMEM.
Allow astroglia to grow to full confluence over a period of 5–7 days.
After reaching full confluence, exchange MEM for neural differentiation medium (NDM) (Liu et al., 2013).
Allow 24 incubation with NDM before exchanging back with sMEM for 48 hours.
Exchange NDM for a maximum of two times for each flask.
Appendix B: Preparation of Stock Solutions
Poly-D-Lysine
Dissolve 10 mg of stock poly-D-lysine in 10 ml sterile H2O to make 10× stock solution.
Filter the 10× stock solution through a 0.2 μm PES syringe filter and make 1.0 ml aliquots in 15 ml conical tubes. Store at −20°C for up to 3 months.
Before using, defrost 1.0 ml 10× stock PDL in a 37°C water bath. Dilute the 10× aliquot to 1× by adding 9 mL sterile H2O.
Dissection Solution
- Solution A (Buffered Saline)
- Weigh out all ingredients and dissolve in 400 ml distilled and autoclaved H2O. Bring final volume to 500 ml.
- Place in a clean bottle and autoclave.
- Store at 4°C and label as “DS Solution A”.
- Solution B (HEPES)
- Dissolve in 200 ml distilled and autoclaved H2O and bring final volume to 250 ml.
- Place in a clean bottle and autoclave.
- Store at 4°C and label “DS Solution B”.
- Working Solution (Final Dissecting Solution)
- Add components in order, as described below and adjust pH to 7.4 with 1N NaOH.
- Vacuum filter and autoclave before use.
Table 1.
Preparation of Solution A.
| Name | Source (cat. no.) | FW | For 500 ml | Concentration |
|---|---|---|---|---|
| NaCl | Sigma (S-9625) | 58.45 | 80.0 g | 137 mM |
| KCl | Sigma (P-4504) | 74.56 | 4.0 g | 5.4 mM |
| Na2HPO4 | Sigma (S-0876) | 141.96 | 0.24 g | 0.17 mM |
| KH2PO4 | Sigma (P-5379) | 136.09 | 0.3 g | 0.22 mM |
Table 2.
Preparation of Solution B.
| Name | Source (cat. no.) | FW | For 250 ml | Concentration |
|---|---|---|---|---|
| HEPES | Sigma (H-3375) | 283.3 | 20.97 g | 9.9 mM |
Table 3.
Preparation of dissecting solution.
| Name | Source (cat. no.) | Amount |
|---|---|---|
| Distilled autoclaved H2O | – | 400 ml |
| Stock Solution A | – | 25 ml |
| Stock Solution B | – | 14 ml |
| D-(+)-glucose | Sigma (G-8270) | 3.0 g |
| Sucrose | Sigma (S-0389) | 7.5 g |
Supplemented Glia Minimum Essential Media (sMEM)
- In a laminar flow hood, add to 435 ml of MEM in order
- 10 ml of filtered 1M Glucose solution (1.8 g Glucose in 10 ml MEM)
- 5 ml Penicillin/Streptomycin
- 50 ml heat-inactivated Fetal Bovine Serum (HI-FBS)
Mix and prepare 40 ml aliquots. Store at −20°C. Filter prior to use.
Table 4.
Preparation of sMEM.
| Name | Source (cat. no.) | Amount |
|---|---|---|
| MEM | Gibco (11090-500ml) | 435 ml |
| 1M D-(+)-glucose | Sigma (G-8270) | 10 ml |
| Penicillin/streptomycin | Gibco (15070) | 5 ml |
| HI-FBS | Atlanta Biologicals (S11150) | 50 ml |
Enzyme Solution (ES)
- In a laminar flow hood, add to 5 ml of Dissecting Solution in order:
- 150 μl L-cysteine (0.8 mg in 150 μl of DS)
- 50 units of papain
- 7 μl 0.1N NaOH
Mix well and place in 37°C water or bead bath. ES will be ready for use when solution becomes clear. Filter immediately before use using a 0.2 μm PES filter.
Table 5.
Preparation of ES.
| Name | Source (cat. no.) | Amount |
|---|---|---|
| Dissecting Solution | – | 5 ml |
| L-cysteine | Sigma (C-8152) | 150 μl |
| Papain | Worthington (LS003126) | 50 U |
| 0.1N NaOH | – | 7 μl |
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Srikanth P, Young-Pearse TL. Stem cells on the brain: modeling neurodevelopmental and neurodegenerative diseases using human induced pluripotent stem cells. J Neurogenet. 2014;28:5–29. doi: 10.3109/01677063.2014.881358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473:221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byers B, Lee HL, Pera R Reijo. Modeling Parkinson’s disease using induced pluripotent stem cells. Curr Neurol Neurosci Rep. 2012;12:237–242. doi: 10.1007/s11910-012-0270-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaye JA, Finkbeiner S. Modeling Huntington’s disease with induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:50–64. doi: 10.1016/j.mcn.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim C-S, Yang J-e, Lee Y-K, Lee K, Lee J-A, Kaang B-K. Understanding the molecular basis of autism in a dish using hiPSCs-derived neurons from ASD patients. Molecular Brain. 2015;8:1–17. doi: 10.1186/s13041-015-0146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marchetto MC, Brennand KJ, Boyer LF, Gage FH. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet. 2011;20:R109–115. doi: 10.1093/hmg/ddr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parent JM, Anderson SA. Reprogramming patient-derived cells to study the epilepsies. Nat Neurosci. 2015;18:360–366. doi: 10.1038/nn.3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sances S, Bruijn LI, Chandran S, Eggan K, Ho R, Klim JR, Livesey MR, Lowry E, Macklis JD, Rushton D, Sadegh C, Sareen D, Wichterle H, Zhang SC, Svendsen CN. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat Neurosci. 2016;16:542–553. doi: 10.1038/nn.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Liu H, Sauvey C, Yao L, Zarnowska ED, Zhang SC. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat Protocols. 2013;8:1670–1679. doi: 10.1038/nprot.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Y, Pasca SP, Portmann T, Goold C, Worringer KA, Guan W, Chan KC, Gai H, Vogt D, Chen YJ, Mao R, Chan K, Rubenstein JL, Madison DV, Hallmayer J, Froehlich-Santino WM, Bernstein JA, Dolmetsch RE. A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. eLIFE. 2016;5:e13073. doi: 10.7554/eLife.13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Topol A, Tran NN, Brennand KJ. A Guide to Generating and Using hiPSC Derived NPCs for the Study of Neurological Diseases. J Vis Exp. 2015:e52495. doi: 10.3791/52495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, Xu W, Yang N, Danko T, Chen L, Wernig M, Sudhof TC. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson MA, Weick JP, Pearce RA, Zhang SC. Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci. 2007;27:3069–3077. doi: 10.1523/JNEUROSCI.4562-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muratore CR, Srikanth P, Callahan DG, Young-Pearse TL. Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS One. 2014;9:e105807. doi: 10.1371/journal.pone.0105807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pré D, Nestor MW, Sproul AA, Jacob S, Koppensteiner P, Chinchalongporn V, Zimmer M, Yamamoto A, Noggle SA, Arancio O. A time course analysis of the electrophysiological properties of neurons differentiated from human induced pluripotent stem cells (iPSCs) PLoS One. 2014;9:e103418. doi: 10.1371/journal.pone.0103418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang X, Zhou L, Wagner AM, Marchetto MC, Muotri AR, Gage FH, Chen G. Astroglial cells regulate the developmental timeline of human neurons differentiated from induced pluripotent stem cells. Stem Cell Res. 2013;11:743–757. doi: 10.1016/j.scr.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. J Vis Exp. 2013:e50079. doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilgenberg LG, Smith MA. Preparation of dissociated mouse cortical neuron cultures. J Vis Exp. 2007:562. doi: 10.3791/562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stover AE, Brick DJ, Nethercott HE, Banuelos MG, Sun L, O’Dowd DK, Schwartz PH. Process-based expansion and neural differentiation of human pluripotent stem cells for transplantation and disease modeling. J Neurosci Res. 2013;91:1247–1262. doi: 10.1002/jnr.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gómez-Lechón MJ, Iborra FJ, Azorín I, Guerri C, Renau-Piqueras J. Cryopreservation of rat astrocytes from primary cultures. J Tissue Cult Methods. 1992;14:73–77. [Google Scholar]
- 22.Yoshida T, Takeuchi M. Primary culture and cryopreservation of mouse astrocytes under serum-free conditions. Cytotechnology. 1991;5:99–106. doi: 10.1007/BF00365426. [DOI] [PubMed] [Google Scholar]
- 23.Stover AE, Schwartz PH. Adaptation of human pluripotent stem cells to feeder-free conditions in chemically defined medium with enzymatic single-cell passaging. Methods Mol Biol. 2011;767:137–146. doi: 10.1007/978-1-61779-201-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Son MY, Kim HJ, Kim MJ, Cho YS. Physical passaging of embryoid bodies generated from human pluripotent stem cells. PLoS One. 2011;6:e19134. doi: 10.1371/journal.pone.0019134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JE, O’Sullivan ML, Sanchez CA, Hwang M, Israel MA, Brennand K, Deerinck TJ, Goldstein LS, Gage FH, Ellisman MH, Ghosh A. Investigating synapse formation and function using human pluripotent stem cell-derived neurons. Proc Natl Acad Sci U S A. 2011;108:3005–3010. doi: 10.1073/pnas.1007753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Liu M, Yan Y, Yang ST. Neural differentiation from pluripotent stem cells: The role of natural and synthetic extracellular matrix. World journal of stem cells. 2014;6:11–23. doi: 10.4252/wjsc.v6.i1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Luo X, Leighton J. Extracellular Matrix and Integrins in Embryonic Stem Cell Differentiation. Biochemistry Insights. 2015;8:15–21. doi: 10.4137/BCI.S30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flanagan LA, Rebaza LM, Derzic S, Schwartz PH, Monuki ES. Regulation of Human Neural Precursor cells by Laminin and Integrins. J Neurosci Res. 2006;83 doi: 10.1002/jnr.20778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma W, Tavakoli T, Derby E, Serebryakova Y, Rao MS, Mattson MP. Cell-extracellular matrix interactions regulate neural differentiation of human embryonic stem cells. BMC Dev Biol. 2008;8:90. doi: 10.1186/1471-213X-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harper MM, Ye EA, Blong CC, Jacobson ML, Sakaguchi DS. Integrins contribute to initial morphological development and process outgrowth in rat adult hippocampal progenitor cells. J Mol Neurosci. 2010;40:269–283. doi: 10.1007/s12031-009-9211-x. [DOI] [PubMed] [Google Scholar]
- 31.Berry BJ, Akanda N, Smith AS, Long CJ, Schnepper MT, Guo X, Hickman JJ. Morphological and functional characterization of human induced pluripotent stem cell-derived neurons (iCell Neurons) in defined culture systems. Biotechnol Prog. 2015;31:1613–1622. doi: 10.1002/btpr.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell reports. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bardy C, van den Hurk M, Kakaradov B, Erwin JA, Jaeger BN, Hernandez RV, Eames T, Paucar AA, Gorris M, Marchand C, Jappelli R, Barron J, Bryant AK, Kellogg M, Lasken RS, Rutten BPF, Steinbusch HWM, Yeo GW, Gage FH. Predicting the functional states of human iPSC-derived neurons with single-cell RNA-seq and electrophysiology. Mol Psychiatry. 2016;21:1573–1588. doi: 10.1038/mp.2016.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartfield EM, Yamasaki-Mann M, Ribeiro Fernandes HJ, Vowles J, James WS, Cowley SA, Wade-Martins R. Physiological Characterisation of Human iPS-Derived Dopaminergic Neurons. PLoS One. 2014;9:e87388. doi: 10.1371/journal.pone.0087388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahairaki V, Ryu J, Peters A, Chang Q, Li T, Park TS, Burridge PW, Talbot CC, Jr, Asnaghi L, Martin LJ, Zambidis ET, Koliatsos VE. Induced pluripotent stem cells from familial Alzheimer’s disease patients differentiate into mature neurons with amyloidogenic properties. Stem cells and development. 2014;23:2996–3010. doi: 10.1089/scd.2013.0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amin H, Maccione A, Marinaro F, Zordan S, Nieus T, Berdondini L. Electrical Responses and Spontaneous Activity of Human iPS-Derived Neuronal Networks Characterized for 3-month Culture with 4096-Electrode Arrays. Front Neurosci. 2016;10 doi: 10.3389/fnins.2016.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boehler MD, Wheeler BC, Brewer GJ. Added astroglia promote greater synapse density and higher activity in neuronal networks. Neuron Glia Biol. 2007;3:127–140. doi: 10.1017/S1740925X07000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Odawara A, Saitoh Y, Alhebshi AH, Gotoh M, Suzuki I. Long-term electrophysiological activity and pharmacological response of a human induced pluripotent stem cell-derived neuron and astrocyte co-culture. Biochem Biophys Res Commun. 2014;443:1176–1181. doi: 10.1016/j.bbrc.2013.12.142. [DOI] [PubMed] [Google Scholar]
- 39.Odawara A, Katoh H, Matsuda N, Suzuki I. Physiological maturation and drug responses of human induced pluripotent stem cell-derived cortical neuronal networks in long-term culture. Sci Rep. 2016;6:26181. doi: 10.1038/srep26181. [DOI] [PMC free article] [PubMed] [Google Scholar]