

Abstract
Objective(s):
In the present study, a new series of 6-methoxy-2-arylquinoline analogues was designed and synthesized as P-glycoprotein (P-gp) inhibitors using quinine and flavones as the lead compounds.
Materials and Methods:
The cytotoxic activity of the synthesized compounds was evaluated against two human cancer cell lines including EPG85-257RDB, multidrug-resistant gastric carcinoma cells (P-gp-positive gastric carcinoma cell line), and EPG85-257P, drug-sensitive gastric carcinoma cells. Compounds showing low to moderate toxicity in the MTT test were selected to investigate their P-gp inhibition activity. Moreover, trying to explain the results of biological experiments, docking studies of the selected compounds into the homology-modeled human P-gp, were carried out. The physicochemical and ADME properties of the compounds as drug candidate were also predicted.
Results:
Most of our compounds exhibited negligible or much lower cytotoxic effect in both cancer cells. Among the series, 5a and 5b, alcoholic quinoline derivatives were found to inhibit the efflux of rhodamine 123 at the concentration of 10 μM significantly.
Conclusion:
Among the tested quinolines, 5a and 5b showed the most potent P-gp inhibitory activity in the series and were 1.3-fold and 2.1-fold stronger than verapamil, respectively. SAR data revealed that hydroxyl methyl in position 4 of quinolines has a key role in P-gp efflux inhibition of our compounds. ADME studies suggested that all of the compounds included in this study may have a good human intestinal absorption.
Keywords: Molecular docking, P-glycoprotein, P-gp inhibition, Quinoline, Synthesis
Introduction
Cancer is the cause of one-quarter of all deaths in developed countries. It is now the second leading cause of death in the United States and is anticipated to surpass heart diseases as the leading cause of death in the future (1). Although chemotherapy is the common method for treatment of different cancers, it fails to cure most cancer patients with advanced disease due to the incidence of drug resistance (2, 3). Among the stated mechanisms leading to drug resistance, the most extensively considered one is the overexpression of P-glycoprotein (P-gp), belonging to the ATP-binding cassette (ABC) family of transport proteins, which efflux the chemotherapeutic drug out of cells by the hydrolysis of ATP as the energy source (4), resulting in dropped intracellular anticancer drug concentrations and leading to drug resistance (5). Thus, the design of inhibitors of P-gp is a favorable strategy in cancer therapy. Recently many compounds that can modulate the P-gp transporter by interfering with P-gp by inhibition of drug efflux have been reported (6-9). The aim is to achieve better drug bioavailability, uptake of the drug in the targeted tissue, and more successful cancer chemotherapy through the ability to block the action of P-gp selectively.
P-gp inhibitors have huge structural diversity. They are classified into three generations according to their affinity, specificity, and toxicity. First-generation agents such as verapamil (10), quinidine (11), and tamoxifen attempt to block P-gp and have been successful in doing just that. But these compounds often produce unfavorable results in vivo because their low binding affinities request the use of high doses and consequential intolerable toxicity (6). Some antimalarial quinoline derivatives, such as chloroquine (12), mefloquine (13), and quinine (14) are also inhibitors of P-gp function. Second-generation P-gp inhibitors such as cyclosporine A, and verapamil analogue dexverapamil (15) are less toxic than the first generation agents and have also higher specificity. However, they showed unpredicted pharmacokinetic interactions and interactions with other ABC transporter proteins (6). Finally, third-generation inhibitors such as tariquidar (16) and zosuquidar have been developed to overcome the limitations of the second generation P-gp inhibitors. Flavonoids are widely distributed in plants (17) included flavones represent the third generation of P-gp inhibitors and they created a comparable effect to those of the well-known P-gp inhibitors verapamil and cyclosporine A (18). Extensive efforts have been made by different research groups to overcome multidrug resistance of tumor cells and have developed three generations of P-gp inhibitors. Nevertheless, no P-gp inhibitors have been approved for clinical application because of observed toxicity, low selectivity or pharmacokinetic interactions (19). Therefore; it is still demanding to develop new and potent P-gp inhibitors with low toxicity and high selectivity for cancer treatment.
As it can be seen in Figure 1, there are some known P-gp inhibitors possessing quinoline moiety, so quinoline ring can be an appropriate scaffold for designing P-gp inhibitors.
Figure 1.
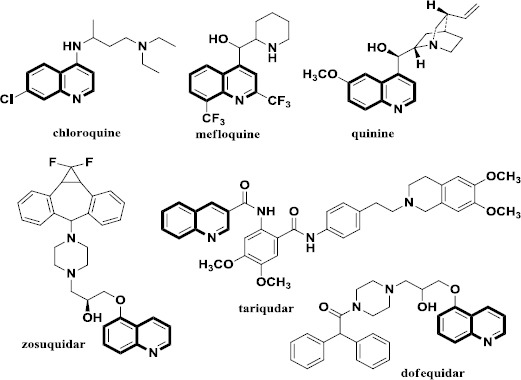
Chemical structures of reported P-gp inhibitors possessing quinoline fragment
We designed novel 2-arylquinolines as P-gp inhibitors, using quinine and flavone as the lead compounds. The rationale for the design of these compounds is depicted in Figure 2. The cytotoxic activity of the synthesized compounds was evaluated against two human cancer cell lines including EPG85-257RDB, multidrug-resistant gastric carcinoma cells (P-gp-overexpressing gastric carcinoma cell line); and EPG85-257P, drug-sensitive gastric carcinoma cells. Compounds showing low to moderate toxicity in MTT test were selected to investigate their P-gp inhibition activity. Moreover, trying to explain the results of biological experiments docking studies were carried out.
Figure 2.
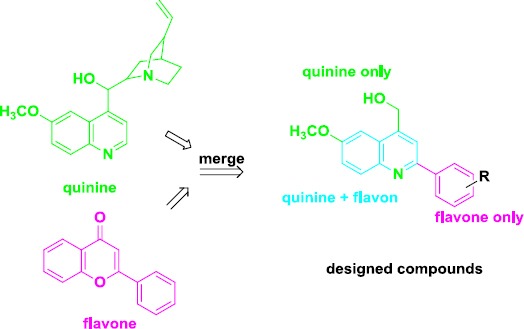
The lead compounds, quinine, and flavone and the designed molecules
Materials and Methods
General chemistry
All chemicals, reagents, and solvents used in this study were purchased from Merck AG and Aldrich Chemical. Melting points were determined with a Thomas–Hoover capillary apparatus. Infrared spectra were acquired using a Perkin Elmer Model 1420 spectrometer. Bruker FT-500 and 300 MHz instruments (Brucker Biosciences, USA) were used to acquire 1HNMR spectra and a Bruker FT-300 MHz instrument was used to acquire 13CNMR spectra with TMS as the internal standard. Chloroform-D and DMSO-D6 were used as solvents. Coupling constant (J) values are assessed in hertz (Hz) and spin multiples are given as s (singlet), d (double), t (triplet), q (quartet), and m (multiplet). Elemental analyses were performed on a Cos-Tec model EAS 4010 instrument (Cernusco, Italy) and the results are within ±0.4% of the theoretical values. The mass spectral measurements were performed on a 3200 QTRAP LCMS triple quadrupole mass spectrometer (LCMS) with an electrospray ionization (ESI) interface.
General procedure for preparation of 6-methoxy-2-arylquinoline-4-carboxylic acid (Doebner reaction)
A solution of appropriate benzaldehyde (9.45 mmol) and pyruvic acid (1.26 g, 14.3 mmol) in ethanol (5 ml) was heated for 30 min then p-anisidine (9.45 mmol) was added to the solution and refluxed overnight. After cooling, the produced precipitate was filtered and washed with ethanol and hexane and recrystallized in ethanol (20).
6-methoxy-2-phenylquinoline-4-carboxylic acid (4a)
Yield: 23%; yellow crystalline powder; mp = 234-236 °C; IR (KBr): v (cm-1) 3501 (OH), 1691 (CO); 1H-NMR (300 MHz-DMSO-d6): δ (ppm) 3.94 (s, 3H, OCH3,), 7.49-7.61 (m, 4H, phenyl H3, H4 & H5 & quinoline H7), 8.08–8.15 (m, 2H, phenyl H2 & H6), 8.25-8.28(m, 2H, quinoline, H5 & H8), 8.47(s, 1H, quinoline H3), 13.96 (s, 1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 55.92, 104.09, 120.24, 123.00, 125.44, 127.32, 129.43, 130.01, 131.86, 135.83, 138.49, 145.23, 153.65, 158.80, 168.10. Anal. Calcd for C17H13NO3: C, 73.11; H, 4.69; N, 5.02. Found: C, 74.23; H, 4.66; N, 5.12.
2-(4-fluorophenyl)-6-methoxyquinoline-4-carboxylic acid (4b)
Yield: 13%; yellow crystalline powder; mp=226-228 °C; IR (KBr): v (cm-1) 3574 (OH), 1697 (CO); 1H-NMR (300 MHz–DMSO-d6): δ (ppm) 3.93 (s, 3H, OCH3), 7.35 (t, 2H, phenyl H2 & H6), 7.50-7.54 (dd, 1H, quinoline H7, J=9.23Hz, J=2.52Hz), 8.06-8.13 (m, 2H, H8 quinoline H5 &H8), 8.29-8.34 (m, 2H, phenyl, H3 &H5), 8.46 (s, 1H, quinoline H3),13.78 (s, 1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 55.91, 104.06, 116.42, 120.07, 123.05, 125.33, 129.65, 131.79, 135.89, 145.14, 152.64, 158.80, 161.98, 165.24, 168.04. Anal. Calcd for C17H12FNO3: C, 68.68; H, 4.07; N, 4.71. Found: C, 68.14; H, 4.21; N, 4.64.
2-(3-hydroxyphenyl)-6-methoxyquinoline-4-carboxylic acid (4c)
Yield: 17%; yellow crystalline powder; mp=288-290°C; IR (KBr): v (cm-1) 3102 (OH), 1691 (CO); 1H-NMR (300 MHz-DMSO-d6): δ (ppm) 3.93(s, 3H, OCH3) 6.89-6.93 (dd, 1H, phenyl H4, J=8.03Hz, J=1.65Hz), 7.34-7.39 (t, 1H, phenyl H5, J=7.85Hz), 7.50-7.54 (dd, 1H, quinoline H7, J=9.23Hz, J=2.85Hz), 7.64-7.67 (d, 1H, phenyl H6, J=7.82Hz), 7.69-7.71 (t, 1H, phenyl H2, J=1.65Hz) 8.05-8.08 (d, 1H, quinoline H8, J=9.2 Hz) 8.16-8.17 (d, 1H, quinoline H5, J=2.8Hz) 8.42 (s, 1H, quinoline H3) 9.64 (s, 1H, OH) 13.81 (s, 1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 55.90, 104.13, 113.81, 117.09, 118.10, 120.28, 122.94, 125.94, 130.51, 131.79, 135.66, 139.85, 145.19, 153.61, 158.32, 158.75, 168.09. Anal. Calcd for C17H13NO4: C, 69.15; H, 4.44; N, 4.74. Found: C, 69.93; H, 4.76; N, 4.65.
2-(4-hydroxyphenyl)-6-methoxyquinoline-4-carboxylic acid (4d)
Yield: 16%; yellow crystalline powder; mp=310-312°C; IR (KBr): v (cm-1) 3428 (OH), 1706 (CO);1H-NMR (300 MHz-DMSO-d6): δ (ppm) 3.89(s, 3H, OCH3), 6.92-6.96 (d, 2H, phenyl H3&H5, J=8.75Hz), 7.46-7.50(dd, 1H, quinoline H7, J=9.2Hz, J=2.85Hz), 8.00-8.03 (d, 1H, quinoline H8, J=9.2Hz), 8.10-8.13 (m, 3H, quinoline H5 & phenyl H2&H6), 8.43 (s, 1H, quinoline H3), 9.85(s, 1H, OH) 13.95 (s, 1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 55.85, 104.18, 116.16, 119.68, 122.66, 124.83, 128.81, 129.44, 131.51, 135.61, 145.18, 153.18, 158.30, 159.43, 168.23. Anal. Calcd for C17H13NO4: C, 69.15; H, 4.44; N, 4.74. Found: C, 69.43; H, 4.22; N, 4.85.
6-methoxy-2-(p-toll) quinoline-4-carboxylic acid (4e)
Yield: 18%; yellow crystalline powder; mp=145-150°C IR (KBr): v (cm-1) 2957.98 (OH), 1659 (C=O); 1H-NMR (300 MHz–DMSO-d6): δ (ppm) 2.382 (s, 3H, CH3), 3.92 (s, 3H, OCH3), 7.36-3.36 (d, 2H, phenyl H3 & H5, J =8.15 Hz), 7.47-7.51 (dd, 1H, quinoline H7, J=9.2Hz, J=2.85Hz), 8.03-8.07 (d, 1H, quinoline H8, J= 9.2 Hz), 8.12-8.15 (m, 3H, quinoline H5 & phenyl H2 & H6), 8.43 (s, 1H, quinoline H3) 13.76 (s, 1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 21.31, 55.82, 104.12, 120.03, 122.73, 125.31, 127.13, 129.94, 131.69, 135.56, 135.73, 139.53, 145.20, 153.53, 158.58, 168.16. Anal. Calcd for C18H15NO3: C, 73.71; H, 5.15; N, 4.78. Found: C, 73.27; H, 5.22; N, 4.67.
2-(4-hydroxy-3-methoxyphenyl)-6-methoxyquinoline -4-carboxylic acid (4f)
Yield: 22%; yellow crystalline powder; mp=288-290°C; IR (KBr): v (cm-1) 3574(OH), 1659 (CO); 1H- NMR (300 MHz-DMSO-d6): δ (ppm) 3.92(s, 3H, OCH3) 3.93 (s, 3H, OCH3) 6.93-6.96 (d,1H, phenyl H5, J= 8.27Hz) 7.46-7.51 (dd, 1H, quinoline H7, J=9.17Hz, J=2.82Hz) 7.67-7.72 (dd, 1H, phenyl H6) 7.841-7.847(d,1H, phenyl H2, J=1.68Hz), 7.91-7.94 (d, 1H, quinoline H8, J=9.23Hz) 8.02-8.05(s, 1H, quinoline H8, J=9.23Hz), 8.08-8.09 (d, 1H, quinoline H5, J=2.79Hz), 8.4 (s, 1H, quinoline H3), 9.47 (s, 1H, OH), 13.52(s,1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 55.86, 56.18, 104.19, 104.19, 110.90, 116.16, 119.16, 119.77, 120.58, 122.65, 124.84, 129.90, 130.21, 131.54, 135.81, 145.07, 148.48, 153.76, 158.31, 168.29. Anal. Calcd for C18H15NO5: C, 76.96; H, 5.70; N, 5.28. Found: C, 77.08; H, 5.81; N, 5.45. LC-MS (ESI): 324.0 (M-1).
2-(3-hydroxy-4-methoxyphenyl)-6-methoxyquinoline -4-carboxylic acid (4g)
Yield: 15%; yellow crystalline powder; mp=283-285°C; IR (KBr): v (cm-1) 3536 (OH), 1652 (CO); 1H-NMR (300 MHz-DMSO-d6): δ (ppm) 2.55 (s, 1H, OH),3.92 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 7.50-7.60 (m, 4H, phenyl H2, H5& H5 & quinoline H7), 8.08-8.11 (d, 1H, quinoline H8, J=9.20 Hz), 8.14-8.15 (m, 2H, quinoline H5, & OH), 13.78 (s, 1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 55.87, 56.87, 104.20, 112.61, 114.03, 118.70, 119.82, 122.74, 125.02, 131.25, 131.55, 135.58, 145.17, 147.26, 149.70, 153.51, 158.41, 168.16. Anal. Calcd for C18H15NO5: C, 76.96; H, 5.70; N, 5.28. Found: C, 76.78; H, 5.96; N, 5.31. LC-MS (ESI): 324.0 (M-1).
2-(3, 4-dimethoxyphenyl)-6-methoxyquinoline-4-carboxylic acid (4h)
Yield: 19%; yellow crystalline powder; mp=251-252°C; IR (KBr): v (cm-1) 3510 (OH), 1691 (CO); 1H-NMR (300 MHz-DMSO-d6): δ (ppm) 3.85 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 7.11-7.14 (d, 1H, phenyl H5, J=9Hz), 7.48–7.52 (dd, 1H, quinoline H7, J=9.0Hz, J=2.80Hz), 7.79–7.83 (dd, 1H, phenyl H6, J=9Hz, J=2.1Hz), 7.86–7.87 (d, 1H, phenyl H2, J=2.1Hz), 8.05–8.10 (m, 2H, quinoline H5 &H8), 8.44 (s, 1H, quinoline H3), 13.94 (s, 1H, COOH); 13C-NMR (DMSO, 75 MHz): δ 55.88, 56.05, 104.15, 110.33, 112.19, 119.89, 120.25, 122.76, 124.98, 131.15, 131.65, 135.80, 145.07, 149.55, 150.77, 153.48, 158.46, 168.22. Anal. Calcd for C19H17NO5: C, 76.96; H, 5.70; N, 5.28. Found: C, 75.91; H, 5.74; N, 5.33.
General procedure for preparation of 2-aryl- 6-methoxyquinoline-4-yl) methanol
LiAlH4 (0.45 g, 12 mmol) was added to dry THF (20 ml) under a nitrogen atmosphere. Under vigorous stirring, a solution of appropriate acid (5.67 mmol) in dry THF was added dropwise to keep the reaction mixture somewhat boiling. After stirring for 5 hr at room temperature, the resultant suspension was carefully hydrolyzed with NaOH solution (10%) till no more hydrogen was formed. The solid was filtered off and washed thoroughly with chloroform. The filtrate was dried over Na2SO4 and the solvent was removed under reduced pressure to yield a brown oil which was purified by column chromatography (21).
(6-methoxy-2-phenylquinolin-4-yl) methanol (5a)
Yield: 47%; yellow crystalline powder; mp=191-193°C; 1H- NMR (300 MHz-CDCl3): δ (ppm) 3.85(s, 1H, OH), 3.89 (s, 3H, OCH3), 5.05 (s, 2H, CH2), 7.01-7.05 (dd, 1H, quinoline H7, J=9.2 Hz, J=2.7Hz), 7.19-7.36 (m, 5H, phenyl),7.99-8.05 (m, 3H, quinolineH5, H3& H8); 13C-NMR (CDCl3, 75 MHz): δ 61.27, 66.95, 100.09, 103.99, 115.58, 118.32, 126.16, 127.94, 128.05, 129.99, 13047,144.22, 153.81, 156.73. Anal. Calcd for C17H15NO2: C, 76.96; H, 5.70; N, 5.28. Found: C, 77.04; H, 5.26; N, 5.44.
(2-(4-fluorophenyl)-6-methoxyquinolin-4-yl) methanol (5b)
Yield: 36%; yellow crystalline powder; mp=185-187°C; IR (KBr): v (cm-1) 3185 (OH); 1H- NMR (300 MHz-CDCl3): δ (ppm) 3.36 (s, 1H, OH), 3.94 (s, 3H, OCH3), 5.06 (s, 2H, CH2), 7.32-7.53 (m, 4H, phenyl H2 & H6 & quinoline H5 & H7),7.99 (d, 1H, quinoline H8, J=9.17Hz), 8.10 (s, 1H, quinoline H3), 8.25-8.30(m, 2H, phenyl H3 & H5); 13C-NMR (CDCl3, 75 MHz): δ 56.02, 60.61, 102.30, 115.71, 116.31, 122.19, 126.02, 129.33, 131.60, 135.97, 143.70, 147.94, 152.81, 157.62, 161.76. Anal. Calcd for C17H14FNO2: C, 72.07; H, 4.98; N, 4.94. Found: C, 71.9; H, 5.19; N, 5.22. LC-MS (ESI): 282.0 (M-1).
3-(4-(hydroxymethyl)-6-methoxyquinolin-2-yl) phenol (5c)
Yield: 46%; yellow crystalline powder; mp=198-200°C; IR (KBr): v (cm-1) 3205(OH); 1H- NMR (300 MHz-CDCl3): δ (ppm) 3.87 (m, 3H, OCH3), 5.04(s, 2H, CH2), 6.88-6.91(m, 2H, phenyl), 7.10-7.11 (d, 1H, phenyl H2, J=2.7Hz), 7.24-7.28 (m, 2H, phenyl & quinoline H7), 7.88-7.97 (m, 4H, OH & quinolineH3, H5 & H8); Anal. Calcd for C17H15NO3: C, 72.58; H, 5.37; N, 4.98. Found: C, 71.6; H, 5.49; N, 5.12. LC-MS (ESI): 279.8 (M-1).
4-(4-(hydroxymethyl)-6-methoxyquinolin-2-yl) phenol (5d)
Yield: 21%; yellow crystalline powder; mp=203-205°C; IR (KBr): v (cm-1) 3355(OH); 1H- NMR (300 MHz-CDCl3): δ (ppm) 3.96 (m, 4H, OCH3 & OH), 5.07 (s, 2H, CH2), 6.88-6.91 (d, 2H, phenyl H3 & H5, J= 8.7 Hz), 7.33-7.34 (d, 1H, quinoline H5, J=2.7Hz), 7.41-7.45 (dd, 1H, quinoline H7, J=9.2 Hz, J=2.7Hz), 7.78(s, 1H, OH), 7.97-8.00(d, 1H, quinoline H8, J=9.2 Hz); 8.11(s,1H, quinoline H3) 8.13-8.14 (d, 2H, phenyl H2 & H6, J= 8.7Hz); 13C-NMR (CDCL3, 75 MHz): δ 55.96, 60.61, 102.36, 115.34, 116.03, 121.77, 125.60, 128.66, 130.45, 131.34, 143.76, 147.39, 153.94, 157.16, 159.06. Anal. Calcd for C17H15NO3: C, 72.58; H, 5.37; N, 4.98. Found: C, 71.8; H, 5.45; N, 4.82. LC-MS (ESI): 282.2 (M+1).
5-(4-(hydroxymethyl)-6-methoxyquinolin-2-yl)-2-methoxyphenol (5e)
Yield: 46%; yellow crystalline powder; mp=208-210°C; IR (KBr): v (cm-1) 3265 (OH); 1H-NMR (300 MHz-CDCl3): δ (ppm) 3.76 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 4.97 (s, 2H, CH2), 5.57 (s, 1H, OH), 6.95-6.98 (d, 1H, phenyl H5, J=8.4 Hz), 7.20–7.21 (d, 1H, phenyl H2, J=2.1Hz), 7.28–7.32 (dd, 1H, quinoline H7, J=9.0Hz, J=2.80Hz), 7.53–7.57 (dd, 1H, phenyl H6, J=9 Hz, J=2.1Hz) 7.70-7.71 (d, 1H, quinoline H5, J=2.8 Hz), 7.86-7.89 (d, 1H, quinoline H8, J=9.0Hz), 7.95 (s, 1H, quinoline H3), 8.22 (s, 1H, OH); 13C NMR (CDCl3, 75 MHz): δ 55.94, 79.64, 102.34, 112.57, 114.29, 115.46, 118.52, 121.79, 125.75, 131.39, 132.42, 143.76, 147.24, 147.40, 149.39, 153.77, 157.29. Anal. Calcd for C18H17NO4: C, 72.58; H, 5.37; N, 4.98. Found: C, 73.6; H, 5.43; N, 5.11. LC-MS (ESI): 312.2 (M+1).
General procedure for preparation of methyl 6-methoxy-2-arylquinoline-4-carboxylate (6)
6-methoxy-2-arylquinoline-4-carboxylic acid (4) (1 mmol) and potassium carbonate (5 mmol) were weighed into a round-bottom flask. Methyl iodide (5 mmol) and acetone (5 ml) were added. The reaction mixture was refluxed with magnetic stirring. The progress of the reaction was monitored by TLC. The reaction was completed after 5 hr. The solvent was evaporated in vacuo and water was added to the remaining mixture. The product was collected by suction filtration and air-dried to obtain pure product (22).
Methyl 6-methoxy-2-phenylquinoline-4-carboxylate (6a)
Yield: 98%; yellow crystalline powder; mp=165-167°C; IR (KBr): v (cm-1) 1721 (CO);1H- NMR (300 MHz-CDCl3): δ (ppm) 4.02 (s, 3H, OCH3), 4.09 (s, 3H, OCH3), 7.44-7.59 (m, 4H, phenyl H3 & H4 & H5 & quinoline H7), 8.13-8.16(d, 1H, quinoline H8, J=9.2), 8.19-8.22(m, 2H, phenyl H2 & H6), 8.26-8.27(d, 1H, quinoline H5, J=2.7Hz), 8.56 (s, 1H, quinoline H3); 13C-NMR (CDCl3, 75 MHz): δ 52.64, 55.63, 103.20, 120.77, 122.87, 125.58, 127.17, 128.58,129.32, 131.76, 133.27, 138.95, 145.74, 154.13, 159.08, 167.00. Anal. Calcd for C18H15NO3: C, 73.71; H, 5.15; N, 4.78. Found: C, 73.58; H, 5.35; N, 4.82.
Methyl 2-(4-fluorophenyl)-6-methoxyquinoline-4-carboxylate (6b)
Yield: 98%; yellow crystalline powder; mp=163-165°C; IR (KBr): v (cm-1) 1721(CO); 1H- NMR (300 MHz-CDCl3): δ (ppm) 3.99 (s, 3H, OCH3), 4.05 (s, 3H, OCH3), 7.19-7.25 (m, 2H, 4-fluorophenyl, H2& H6,),7.4-7.44 (dd, 1H, quinoline H7, J= 9 Hz, J=2.7 Hz), 8.06-8.09 (d, 1H, quinoline H8, J=9.3 Hz), 8.14-8.19 (m, 2H, phenyl H3 & H5), 8.21-8.22 (d, 1H, quinoline H5, J= 2.7 Hz), 8.45 (s, 1H, quinoline H3); 13C-NMR (CDCl3, 75 MHz): δ 52.63, 55.59, 103.18, 115.68, 115.96, 120.32, 122.95, 125.45, 128.92, 131.62, 133.27, 145.64, 152.92, 159.08, 165.39, 166.84. Anal. Calcd for C18H14FNO3: C, 69.45; H, 4.53; N, 4.50. Found: C, 69.98; H, 4.75; N, 4.62. LC-MS (ESI): 312.2 (M+1).
Methyl 6-methoxy-2-(p-tolyl) quinoline-4-carboxylate (6c)
Yield: 98%; yellow crystalline powder; mp =94-96°C; IR (KBr): v (cm-1) 1722 (CO); 1H-NMR (300 MHz-CDCl3): δ (ppm) 2.49(s, 3H, CH3), 4.01(s, 3H, OCH3), 4.09(s, 3H, OCH3), 7.29-7.34(D, 2H, phenyl H3 & H5, 8.1Hz), 7.41-7.45(dd, 1H, quinoline H7, J= 9 Hz, J=2.7 Hz), 8.09-8.14(m, 3H, phenyl H2 & H6 & quinoline H8), 8.23-8.24(d, 1H, quinoline H5), 8.43(s, 1H, quinoline H3); 13C-NMR (CDCl3, 75 MHz):: δ 21.38, 52.60, 55.59, 103.23, 120.58, 122.72, 125.41, 127.01, 129.64, 131.66, 133.15, 136.13, 139.39, 145.69, 154.09, 158.91, 167.04. Anal. Calcd for C19H17NO3: C, 74.25; H, 5.58; N, 4.56. Found: C, 74.8; H, 5.95; N, 4.28. LC-MS (ESI): 308.2 (M+1), 330.2 (M+Na).
Methyl 2-(3, 4-dimethoxyphenyl)-6-methoxyquinoline-4-carboxylate (6d)
Yield: 98%; yellow crystalline powder; mp=163-166°C; IR (KBr): v (cm-1); 1721 (CO); 1H-NMR (300 MHz-CDCl3): δ (ppm) 3.89-3.90 (s, 6H, OCH3), 3.98(s, 3H, OCH3), 4.00(s, 3H, OCH3), 4.07(s, 3H, OCH3), 4.08 (s, 3H, OCH3), 7.00-7.03 (d, 1H, phenyl H5, J=8.4 Hz), 7.28 (s, 1H, phenyl H3), 7.41-7.45 (dd, 1H, quinoline H7, J= 9.3 Hz, J=3.0Hz), 7.67-7.70 (dd, 1H, phenyl H6, J= 8.4 Hz, J=2.1 Hz), 7.87-7.88 (d, 1H, phenyl H2, J=2.1Hz), 8.10-8.13 (d, 1H, quinoline H8, J= 9.3Hz) 8.21-8.22 (d,1H, quinoline H5, J= 2.7 Hz); 13C- NMR (CDCl3, 75 MHz): δ 52.64, 55.60, 56.02, 56.08, 103.30, 109.95, 111.03, 119.86, 120.40, 122.71, 125.22, 131.51, 131.85, 133.26, 145.60, 149.47, 150.34, 153.68, 158.82, 167.07. Anal. Calcd for C20H19NO5: C, 67.98; H, 5.42; N, 3.96. Found: C, 67.7; H, 5.65; N, 4.08. . LC-MS (ESI): 354.2 (M+1), 376.2 (M+Na).
Cytotoxicity assay
General procedure
The MTT (3-[4, 5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) based assay was carried out by seeding 5000 cancer cells per 180 µl RPMI complete culture medium in each well of 96-well culture plates. The day after seeding, the culture medium was replaced with medium containing standard the anti-tumor agent daunorubicin as well as different concentrations of newly synthesized quinolines: verapamil and RPMI control (no drug). Cells were then incubated at 37°C in 5% CO2 incubator for 72 hr. Then 25 µl of MTT solution (4 mg Ml-1) were added to each well and further incubated at 37°C for 3 hr. At the end of incubation, formazan crystals were dissolved in 100 µl of DMSO and plates were read in a plate reader (Synergy H4, USA) at 540 nm. This experiment was performed in triplicate determination each time (23).
P-gp inhibition assay
The ability of the synthesized quinolines to inhibit the transport function of P-gp was evaluated using flowcytometry (24). EPG85-257RDB cancer cells were taken in an adjusted concentration of 1 x 106 cells. 1 mL aliquots were filled into Eppendorf centrifuge tubes. The test compounds taken from prepared stock solutions in DMSO (1.0 mg/ml) were added in a volume of 5 μl. After 10 min of inhibitor preincubation at rt, the P-gp substrate rhodamine 123 was added using 5 μl of a 0.5 mM solution in water. Incubation was continued for 45 min at 37°C. After that, the cells were centrifuged and washed twice with phosphate-buffered saline (PBS). Then they were resuspended in PBS for measurement. The non-inhibitor containing cells were treated with the same approach as the inhibitor preincubated cells. The fluorescence uptake of rhodamine 123 within the number of 104 counted cells was determined by flowcytometry using a Becton Dickinson FACScan flow cytometer.
MDR reversal studies
The MTT based assay was carried out by seeding 5 x 103 EPG85-257P and EPG85-257RDB cancer cells per 180 µl RPMI complete culture medium in each well of 96-well culture. Daunorubicin has been used in concentrations of 0.4, 2, 5, and 10 μM in both EPG85-257P and EPG85-257RDB cancer cells. Daunorubicin without compounds in the non-P-gp expressing EPG85-257P cell line and in the P-gp overexpressing EPG85-257RDB was divided in plates. Daunorubicin containing suspensions were also divided into 96-well plates and additionally supplemented with the P-gp inhibitors 5a, 5b, and verapamil at the concentration of 10 μM. Cells were then incubated at 37°C in a 5% CO2 incubator for 72 hr. Then 25 µl of MTT solution (4 mg Ml-1) were added to each well and further incubated at 37 °C for 3 hr. At the end of incubation, formazan crystals were dissolved in 100 µl of DMSO and plates were read in a plate reader (Synergy H4, USA) at 540 nm. This experiment was performed in triplicate determination each time (25).
Molecular modeling
Mode of interaction between synthesized ligands and P-gp was investigated by docking (26). 2D structure of chemicals was prepared in ChemDraw Ultra 12.0 software and 3D structures were prepared by ChemDraw ultra 12.0 software using molecular mechanic force filed pre-optimization followed by MM2 calculation. The X-ray crystal structure of P-gp (PDB ID: 4M2S) was downloaded from the Protein Data Bank (www.rcsb.org). Further modification such as polar hydrogen addition was performed by MOE software. Synthesized chemicals were docked into the binding site of tubulin by MOE software. All atoms within a (5) 0.5 Å around the co-crystallized ligand in crystal coordinates of P-gp were chosen as the binding site. The docking simulations were done employing triangle matcher placement algorithm in combination with London dG scoring function and force field as refinement method. For each compound, the top-score docking poses were chosen for final ligand-target interaction analysis employing LigX module in MOE Software.
Physicochemical and ADME Properties
The physicochemical and ADME properties of the compounds as drug candidates were predicted using Schrödinger module QikProp .8 (27) (Schrödinger, LLC, New York, 2015, USA).
Results
Synthesis
A one-step Doebner reaction was used to prepare the 6-methoxy-2-arylquinoline-4-carboxylic acid derivatives. As illustrated in Scheme 1, substituted benzaldehyde 1, pyruvic acid 2, and p-anisidine 3 were refluxed in ethanol to afford 4-carboxy quinolines and then reduction of carboxyl substituent to an alcoholic substituent was performed using LiAlH4 in dry THF.
Scheme 1.
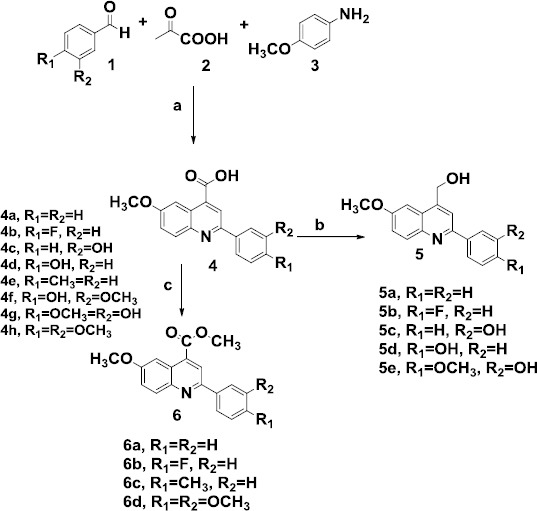
Reagents and conditions: (a) Ethanol, reflux, (b) LiAlH4, dry THF, (c) K2CO3, CH3I, Aceton, reflux
Then, by esterification of 4-quinoline carboxylic acid, novel quinoline-4-methyl esters were obtained. The compounds were characterized by nuclear magnetic resonance, infrared spectroscopy, mass spectroscopy, and elemental analysis.
Biological evaluation
In vitro cytotoxic effects
To identify ideal P-gp inhibitors reversing MDR at non-toxic concentrations, cytotoxicity of the quinoline compounds against parental sensitive EPG85-257P cells and their resistant sublines EPG85-257RDB cells which overexpress P-gp was evaluated by MTT assay. Anticancer drug daunorubicin and P-gp inhibitor verapamil were selected as controls. Most of our compounds exhibited negligible or much lower cytotoxic effect in both cancer cells. As depicted in Table 1, three of the alcoholic derivatives 5c-5e showed more cytotoxic activity with IC50 in the range of 25.34–39.64 μM. However, carboxylic and methyl carboxylate derivatives except 6c did not display significant cytotoxic activity at concentrations below 100 μM.
Table 1.
The in vitro antiproliferative activities of quinolines, verapamil and daunorubicin against EPG85-257P (drug-sensitive gastric carcinoma cells) and EPG85-257RDB (multidrug-resistant gastric carcinoma cells)
 | ||||||
|---|---|---|---|---|---|---|
| Compound | X | R1 | R2 | EPG85-257P IC50a (μM) | EPG85-257RDB IC50a (μM) | |
| 4a | COOH | H | H | >100 | >100 | |
| 4b | COOH | F | H | 96.87±21.44 | >100 | |
| 4c | COOH | H | OH | 91.43±15.87 | >100 | |
| 4d | COOH | OH | H | 85.56±7.47 | >100 | |
| 4e | COOH | CH3 | H | 72.24±8.64 | 50.23±3.23 | |
| 4f | COOH | OH | OCH3 | >100 | 82.96±9.48 | |
| 4g | COOH | OCH3 | OH | >100 | >100 | |
| 4h | COOH | OCH3 | OCH3 | >100 | >100 | |
| 5a | CH2OH | H | H | 58.36±6.21 | 59.67±2.26 | |
| 5b | CH2OH | F | H | 49.10±2.61 | 53.70±1.68 | |
| 5c | CH2OH | H | OH | 35.51±2.05 | 38.79±1.47 | |
| 5d | CH2OH | OH | H | 37.78±1.03 | 39.64 ±2.31 | |
| 5e | CH2OH | OCH3 | OH | 25.34±3.44 | 31.18±2.08 | |
| 6a | COOCH3 | H | H | 87.86±14.27 | >100 | |
| 6b | COOCH3 | F | H | >100 | >100 | |
| 6c | COOCH3 | CH3 | H | 36.21±2.11 | 75.55±9.53 | |
| 6d | COOCH3 | OCH3 | OCH3 | >100 | >100 | |
| Verapamil | 55.72±2.69 | 58.9±5.35 | ||||
| Daunorubicin | 0.037±0.003 | 0.971±0.037 | ||||
compound concentration required to inhibit tumor cell proliferation by 50%. Data are presented as the mean±SD from the dose−response curves of three independent experiments
Biological evaluation of the P-gp inhibition
Compounds did not show significant cytotoxic activity in MTT test, including all carboxylic and methyl carboxylate quinoline derivatives (4a-4h and 6a-6d) and two of alcoholic quinoline derivatives (5a and 5b) were selected to investigate their P-gp inhibition activity. P-gp inhibition was evaluated by the determination of the uptake amount of the fluorescent P-gp substrate, rhodamine 123 by EPG85-257RDB gastric carcinoma cells overexpressing P-gp in presence of the selected compounds. When tested at the concentrations of 1 and 10 μM, alcoholic quinoline derivatives (5a and 5b) were found to inhibit the efflux of rhodamine 123 in EPG85-257RDB cells (Figure 3). The P-gp efflux inhibition was also found to be concentration dependent since our compounds did not show significant P-gp inhibitory activity at the concentration of 1 μM (data not shown). P-gp inhibition folds are reported for some of the tested compounds in comparison to verapamil as the reference drug (Table 2). Among the series, 5a and 5b, alcoholic quinoline derivatives were found to inhibit the efflux of rhodamine 123 at the concentration of 10 μM significantly. Among the tested quinolines, 5a showed P-gp inhibitory activity and was 1.3-fold stronger than verapamil. 5b showed the most potent P-gp inhibitory activity and was 2.1-fold stronger than verapamil. However, the carboxylic and methyl carboxylate quinoline derivatives did not show significant P-gp inhibitory activity at a concentration below 10 μM.
Figure 3.
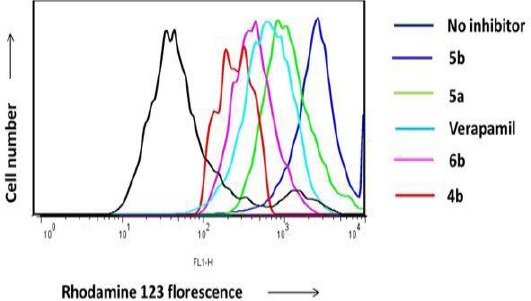
Inhibition of P-gp-mediated rhodamine 123 efflux, EPG85-257RDB cells were incubated with 0.5 mg/ml rhodamine 123 alone (black), 0.5 mg/mL rhodamine 123 and 10 μM verapamil (blue), or 0.5 mg/ml rhodamine 123 and 10 μM of the quinoline derivatives at 37°C for 30 min. Retention of rhodamine 123 fluorescence in cells after 1 hr of rhodamine 123-free efflux was measured by flowcytometry.
Table 2.
Inhibition of P-gp-mediated Rhodamine 123 efflux of selected quinolines in comparison to verapamil at the concentration of 10 μM
| Compound | P-gp inhibition fold |
|---|---|
| 4b | 0.31 |
| 6b | 0.75 |
| Verapamil | 1 |
| 5a | 1.375 |
| 5b | 2.175 |
Reversal of P-gp -mediated MDR by quinoline derivatives
The reversal of multidrug resistance by the new quinoline derivatives was evaluated in drug-resistant cancer cell line with overexpression of P-gp (EPG85-257RDB). The multidrug-resistant cancer cell lines are remarkably resistant to the corresponding substrate anticancer drugs. We determined the cytotoxicity of daunorubicin, in EPG85-257RDB, multidrug-resistant gastric carcinoma cells (P-gp-overexpressing gastric carcinoma cell line) and EPG85-257P, drug-sensitive gastric carcinoma cells. The resulting IC50 values are shown in Table 1. We found different IC50 values with 0.037 μM in sensitive cell line and 0.97 μM in the P-gp overexpressing cell line. We investigated the potential of two of our P-gp inhibitors 5a and 5b to reverse this determined MDR phenomenon of daunorubicin. As shown in Figure 4, 5a, and 5b, the new quinoline derivatives, when used at 10 μM, increased the anticancer activity of the daunorubicin in the corresponding transporter-overexpressing cell line. Both 5a and 5b showed a significant decrease in the IC50 values of the determined daunorubicin toxicity and led to a complete reversal of the MDR phenomenon with near equal IC50 values than in the parental non-P-gp expressing cell line.
Figure 4.
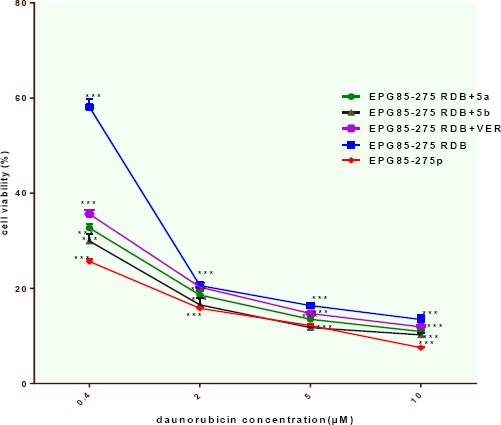
Concentration-dependent curves of daunorubicin toxicity without compounds in the non-P-gp expressing EPG85-257P cell line (red) and in the P-gp overexpressing EPG85-257RDB (blue), after preincubation with P-gp inhibitors 5a (green), 5b (black), and verapamil (purple) in the P-gp overexpressing EPG85-257RDB at concentrations of 10 μM, number (n) of experiences n=3. Different groups were compared with negative control. Each point represents mean±standard error ***P< 0.001
Figure 5.
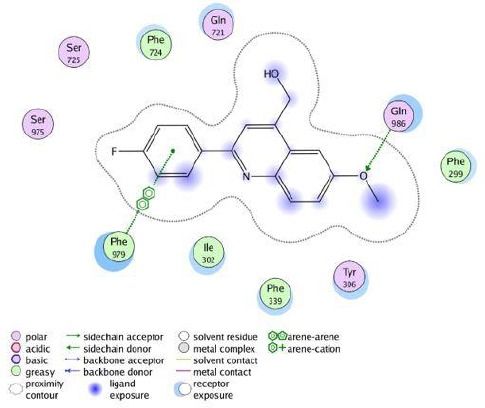
The 2D representation of the interaction between compound 5b in the crystal structure of site-1 of homology modeled human P-gp (4M2S.pdb) using LigX in MOE
Docking studies of compound 5b with homology-modeled human P-gp
As mentioned above, compound 5b demonstrated the most potent P-gp inhibitory effect. Studying ligand interaction mode of 5b into the site-1 of homology modeled human P-gp (Figure 5) by LigX module of MOE software revealed that 5b was docked well and stabilized through interactions with amino acid residues in the drug-binding pocket of P-gp. The hydrogen bond acceptor oxygen atom of methoxy moiety showed hydrogen bonding interaction with the side chain of Gln 986, phenyl ring of the 5b interacted with Phe 979 through π−π stacking. Additionally, Compound 5b was able to establish some hydrophobic interactions with the surrounding hydrophobic residues. The methyl group of the methoxy group and quinoline ring were mainly stabilized through hydrophobic contacts within the large hydrophobic pocket formed by the side chains of Phe299, Tyr306, Phe339, Ile302.
Calculated Physicochemical and ADME Properties
The calculated physicochemical and ADME properties of all the newly synthesized quinolines are listed in Table 3. In particular, predicted brain/blood partition coefficient (CNS), aqueous solubility (QPlogS), lipophilicity (QPlogPo/w), Percent human-oral absorption, human ether-a-go-go-related gene (QPlogHERG), cell permeability (QPPCaco), blood−brain barrier (BBB) penetration (QPlogBB), and cell permeability to the blood-brain barrier (QPPMDCK) have been predicted using the Schrödinger Software Release 2015, which sorted the molecules on the basis of GI absorption, octanol/water partition coefficient, BBB permeability, and blockage of HERG K+ channels. All the newly synthesized quinolines were passed through Lipinski’s rule of five filters. All of the compounds present suitable values of both lipophilicities (which is expressed as the logarithm of the partition coefficient between n-octanol and water, clogP<3.6) and aqueous solubility (QPlogS>-5). In silico predictions suggested that all compounds included in this study may have a good human intestinal absorption at least higher than 76%, the risk of hERG channel inhibition might be low (QPlogHERG≥-5.3), but among the most active compounds there is a risk of BBB penetration, and carboxylic acid derivatives 4a-4h may not penetrate the cells well, because of their low predicted Caco-2 cell permeability (QPPCaco<500).
Table 3.
The calculated physicochemical and ADME properties of quinolines
| molecule | mol_MW | QPlogSa | QPlogPo/wb | PercentHuman OralAbsorptionc | QPlogHERGd | QPPCacoe | QPlogBBf |
|---|---|---|---|---|---|---|---|
| 4a | 279.295 | -3.689 | 3.252 | 89.836 | -3.443 | 281.83 | -0.515 |
| 4b | 297.285 | -3.979 | 3.513 | 91.648 | -3.225 | 292.199 | -0.383 |
| 4c | 295.294 | -3.393 | 2.562 | 76.868 | -3.228 | 89.377 | -1.046 |
| 4d | 295.294 | -3.393 | 2.562 | 76.868 | -3.228 | 89.377 | -1.046 |
| 4e | 293.321 | -4.191 | 3.586 | 92.074 | -3.297 | 292.192 | -0.517 |
| 4f | 325.32 | -3.683 | 2.7 | 78.382 | -3.108 | 97.856 | -1.094 |
| 4g | 325.32 | -3.455 | 2.713 | 81.164 | -2.991 | 138.582 | -0.915 |
| 4h | 339.347 | -4.671 | 3.454 | 91.523 | -3.254 | 300.636 | -0.644 |
| 5a | 265.311 | -3.636 | 3.303 | 100 | -5.241 | 3929.986 | -0.056 |
| 5b | 283.301 | -3.982 | 3.526 | 100 | -5.125 | 3929.909 | 0.053 |
| 5c | 281.31 | -3.369 | 2.562 | 100 | -5.127 | 1202.102 | -0.612 |
| 5d | 281.31 | -3.369 | 2.562 | 100 | -5.127 | 1202.158 | -0.612 |
| 5e | 311.321 | -3.848 | 3.374 | 90.834 | -3.267 | 1292.182 | -0.574 |
| 6a | 295.337 | -3.868 | 3.395 | 100 | -5.176 | 3929.953 | -0.128 |
| 6b | 311.31 | -3.976 | 3.452 | 100 | -5.164 | 3929.957 | -0.112 |
| 6c | 307.337 | -3.619 | 2.692 | 100 | -5.026 | 1279.208 | -0.666 |
| 6d | 353.363 | -4.084 | 3.507 | 100 | -5.167 | 3929.958 | -0.198 |
Predicted aqueous solubility(acceptable rang: –6.5 – 0.5);
Predicted octanol/water partition coefficient (acceptable rang: -2.0–6.5);
Predicted human oral absorption on 0-100% scale (acceptable rang: <25% is poor, >80% is high);
Predicted IC50 value for blockage of HERG K+ channels (concern below -5);
Predicted Caco-2 cell permeability in nm/s (acceptable rang: <25 is poor, >500 is great);
Predicted brain/blood partition coefficient (concern value is -3.0 to -1.2
Discussion
A new series of 6-methoxy-2-arylquinolines analogues was designed and synthesized as P-glycoprotein (P-gp) inhibitors. The cytotoxic activity of the synthesized compounds was evaluated against two human cancer cell lines including EPG85-257RDB, (P-gp-positive gastric carcinoma cell line) and EPG85-257P. Compounds showing low to moderate cytotoxicity in the MTT test were selected to investigate their P-gp inhibition activity. Comparison of the P-gp efflux inhibition of 5b with those of its corresponding carboxylic and methyl carboxylate derivatives 4b and 6b indicates that hydroxyl methyl in position 4 of quinolines has a key role in P-gp efflux inhibition of our compounds. It is not surprising since the same substitution pattern of our alcoholic quinoline derivatives and quinine as the lead compound. We calculated binding energy (E-score, KJ/mol) of quinolines (4b, 6b, 5a, and 5b) by MOE software and found a relationship between P-gp inhibition activity with binding energy and predicted Caco-2 cell permeability (QPPCaco) of compounds (Table 4). Compound 5b with the least binding energy score showed the most P-gp inhibition activity compared to other compounds (5a, 4a, and 6b), it shows that 5b has more affinity to bind to P-gp. Surprisingly compound 4b with less binding energy score compared to 6b, showed less P-gp inhibition activity. As predicted Caco-2 cell permeability (QPPCaco) of 4b is less than that of 6b, this result can be due to the poor ability of carboxylic derivative 4b to penetrate the cell membrane because of its high polarity.
Table 4.
P-gp inhibition fold, binding energy (E score), and Predicted cell permeability (QPPCaco) of compounds 4b, 6b, 5a, and 5b
 | ||||||
|---|---|---|---|---|---|---|
| Compound | X | R1 | R2 | P-gp inhibition fold (10 μM) | E score (KJ/mol) | QPPCaco |
| 4b | COOH | F | H | 0.31 | -10.73 | 292.199 |
| 6b | COOCH3 | F | H | 0.75 | -9.88 | 3929.957 |
| 5a | CH2OH | H | H | 1.375 | -11.08 | 3929.986 |
| 5b | CH2OH | F | H | 2.175 | -11.55 | 3929.909 |
Conclusion
Among the tested quinolines, 5a and 5b showed the most potent P-gp inhibitory activity in the series and were 1.3-fold and 2.1-fold stronger than verapamil, respectively. Molecular docking studies of 5b into the homology-modeled human P-gp, displayed possible mode of interaction between this compound and P-gp. Both 5a and 5b showed a significant decrease in the IC50 values of the determined daunorubicin toxicity and led to a complete reversal of the MDR phenomenon with near equal IC50 values than in the parental non-P-gp expressing cell line. SAR data revealed that hydroxyl methyl in position 4 of quinolines has a key role in P-gp efflux inhibition of our compounds. Compounds with less binding energy exhibited more P-gp inhibition activity except for carboxylic acid derivatives which might not penetrate the cell membrane because of their high polarity. ADME studies suggested that all the compounds included in this study may have a good human intestinal absorption.
Acknowledgment
We are grateful to Research Deputy of Mashhad University of Medical Sciences, Mashhad, Iran, for financial support of this research as part of the thesis of Sayyed Mohammad Aboutorabzadeh.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Cozzi P. The discovery of a new potential anticancer drug: a case history. Farmaco. 2003;58:213–220. doi: 10.1016/S0014-827X(03)00014-4. [DOI] [PubMed] [Google Scholar]
- 3.Liang XJ, Chen C, Zhao Y, Wang PC. Circumventing tumor resistance to chemotherapy by nanotechnology. Methods Mol Biol. 2010;596:467–488. doi: 10.1007/978-1-60761-416-6_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capparelli E, Zinzi L, Cantore M, Contino M, Perrone MG, Luurtsema G, et al. SAR studies on tetrahydroisoquinoline derivatives: the role of flexibility and bioisosterism to raise potency and selectivity toward P-glycoprotein. J Med Chem. 2014;57:9983–9994. doi: 10.1021/jm501640e. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Wong IL, Li FX, Yang C, Liu Z, Jiang T, et al. Optimization of permethyl ningalin B analogs as P-glycoprotein inhibitors. Bioorg Med Chem. 2015;23:5566–5573. doi: 10.1016/j.bmc.2015.07.027. [DOI] [PubMed] [Google Scholar]
- 6.Chen CY, Liu NY, Lin HC, Lee CY, Hung CC, Chang CS. Synthesis and bioevaluation of novel benzodipyranone derivatives as P-glycoprotein inhibitors for multidrug resistance reversal agents. Eur J Med Chem. 2016;118:219–229. doi: 10.1016/j.ejmech.2016.03.070. [DOI] [PubMed] [Google Scholar]
- 7.Kuriakose J, Hrycyna CA, Chmielewski J. Click chemistry-derived bivalent quinine inhibitors of P-glycoprotein-mediated cellular efflux. Bioorg Med Chem lett. 2012;22:4410–4412. doi: 10.1016/j.bmcl.2012.04.125. [DOI] [PubMed] [Google Scholar]
- 8.Lopes-Rodrigues V, Oliveira A, Correia-da-Silva M, Pinto M, Lima RT, Sousa E, et al. A novel curcumin derivative which inhibits P-glycoprotein, arrests cell cycle and induces apoptosis in multidrug resistance cells. Bioorg Med Chem. 2017;25:581–596. doi: 10.1016/j.bmc.2016.11.023. [DOI] [PubMed] [Google Scholar]
- 9.Zeslawska E, Kincses A, Spengler G, Nitek W, Wyrzuc K, Kiec-Kononowicz K, et al. The 5-aromatic hydantoin-3-acetate derivatives as inhibitors of the tumour multidrug resistance efflux pump P-glycoprotein (ABCB1): Synthesis, crystallographic and biological studies. Bioorg Med Chem. 2016;24:2815–2822. doi: 10.1016/j.bmc.2016.04.055. [DOI] [PubMed] [Google Scholar]
- 10.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Research. 1981;41:1967–1972. [PubMed] [Google Scholar]
- 11.Tsuruo T, Iida H, Kitatani Y, Yokota K, Tsukagoshi S, Sakurai Y. Effects of quinidine and related compounds on cytotoxicity and cellular accumulation of vincristine and adriamycin in drug-resistant tumor cells. Cancer Research. 1984;44:4303–4307. [PubMed] [Google Scholar]
- 12.Shiraishi N, Akiyama S, Kobayashi M, Kuwano M. Lysosomotropic agents reverse multiple drug resistance in human cancer cells. Cancer Lett. 1986;30:251–259. doi: 10.1016/0304-3835(86)90049-2. [DOI] [PubMed] [Google Scholar]
- 13.Riffkin CD, Chung R, Wall DM, Zalcberg JR, Cowman AF, Foley M, et al. Modulation of the function of human MDR1 P-glycoprotein by the antimalarial drug mefloquine. Biochem Pharmacol. 1996;52:1545–1552. doi: 10.1016/s0006-2952(96)00556-4. [DOI] [PubMed] [Google Scholar]
- 14.Avendano C, Menendez JC. Inhibitors of multidrug resistance to antitumor agents (MDR) Curr Med Chem. 2002;9:159–193. doi: 10.2174/0929867023371175. [DOI] [PubMed] [Google Scholar]
- 15.Warner E, Hedley D, Andrulis I, Myers R, Trudeau M, Warr D, et al. Phase II study of dexverapamil plus anthracycline in patients with metastatic breast cancer who have progressed on the same anthracycline regimen. Clin Cancer Res. 1998;4:1451–1457. [PubMed] [Google Scholar]
- 16.Pusztai L, Wagner P, Ibrahim N, Rivera E, Theriault R, Booser D, et al. Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer. 2005;104:682–691. doi: 10.1002/cncr.21227. [DOI] [PubMed] [Google Scholar]
- 17.Salar Bashi D, Fazly Bazzaz BS, Sahebkar A, Karimkhani MM, Ahmadi A. Investigation of optimal extraction, antioxidant, and antimicrobial activities of Achillea biebersteinii and A wilhelmsii. Pharm Biol. 2012;50:1168–1176. doi: 10.3109/13880209.2012.662235. [DOI] [PubMed] [Google Scholar]
- 18.Bansal T, Jaggi M, Khar RK, Talegaonkar S. Emerging significance of flavonoids as P-glycoprotein inhibitors in cancer chemotherapy. J Pharm Pharm Sci. 2009;12:46–78. doi: 10.18433/j3rc77. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Pan M, Dai Y, Liu B, Cui J, Shi W, et al. Design, synthesis and biological evaluation of LBM-A5 derivatives as potent P-glycoprotein-mediated multidrug resistance inhibitors. Bioorg Med Chem. 2016;24:2287–2297. doi: 10.1016/j.bmc.2016.03.065. [DOI] [PubMed] [Google Scholar]
- 20.Malayeri SO, Abnous K, Arab A, Akaberi M, Mehri S, Zarghi A, et al. Design, synthesis and biological evaluation of 7-(aryl)-2,3-dihydro-[1,4]dioxino[2,3-g]quinoline derivatives as potential Hsp90 inhibitors and anticancer agents. Bioorg Med Chem. 2017;25:1294–1302. doi: 10.1016/j.bmc.2016.12.050. [DOI] [PubMed] [Google Scholar]
- 21.Shobeiri N, Rashedi M, Mosaffa F, Zarghi A, Ghandadi M, Ghasemi A, et al. Synthesis and biological evaluation of quinoline analogues of flavones as potential anticancer agents and tubulin polymerization inhibitors. Eur J Med Chem. 2016;114:14–23. doi: 10.1016/j.ejmech.2016.02.069. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y, Chen Z, Liu Y, Yu L, Zhou L, Yang S, et al. Quinoline-4-methyl esters as human nonpancreatic secretory phospholipase A(2) inhibitors. Bioorg Med Chem. 2011;19:3361–3366. doi: 10.1016/j.bmc.2011.04.039. [DOI] [PubMed] [Google Scholar]
- 23.Ghodsi R, Azizi E, Grazia Ferlin M, Pezzi V, Zarghi A. Design, synthesis and biological evaluation of 4-(imidazolylmethyl)-2-aryl-quinoline derivatives as aromatase inhibitors and anti-breast cancer agents. Lett Drug Des Discov. 2016;13:89–97. [Google Scholar]
- 24.Baumert C, Günthel M, Krawczyk S, Hemmer M, Wersig T, Langner A, et al. Development of small-molecule P-gp inhibitors of the N-benzyl 1,4-dihydropyridine type: Novel aspects in SAR and bioanalytical evaluation of multidrug resistance (MDR) reversal properties. Bioorg Med Chem. 2013;21:166–177. doi: 10.1016/j.bmc.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 25.Li XQ, Wang L, Lei Y, Hu T, Zhang FL, Cho CH, et al. Reversal of P-gp and BCRP-mediated MDR by tariquidar derivatives. Eur J Med Chem. 2015;101:560–572. doi: 10.1016/j.ejmech.2015.06.049. [DOI] [PubMed] [Google Scholar]
- 26.Hosseinzadeh H, Mazaheri F, Ghodsi R. Pharmacological effects of a synthetic quinoline, a hybrid of tomoxiprole and naproxen, against acute pain and inflammation in mice: a behavioral and docking study. Iran J Basic Med Sci. 2017;20:446–450. doi: 10.22038/IJBMS.2017.8588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abdizadeh T, Kalani MR, Abnous K, Tayarani-Najaran Z, Khashyarmanesh BZ, Abdizadeh R, et al. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur J Med Chem. 2017;132:42–62. doi: 10.1016/j.ejmech.2017.03.024. [DOI] [PubMed] [Google Scholar]