

Abstract
Cytochrome P450 (P450, CYP) research provides many opportunities for the application of kinetic isotope effect (KIE) strategies. P450s collectively catalyze oxidations of more substrates than any other group of enzymes, and C-H bond cleavage is a major feature in a large fraction of these reactions. The presence of a significant primary deuterium KIE is evidence that hydrogen abstraction is at least partially rate-limiting in the reactions, and this appears to be the case in many P450 reactions. The first report of a KIE in (P450-linked) drug metabolism appeared in 1961 (for morphine N-demethylation), and in a number of cases it has been possible to modulate the in vivo metabolism or toxicity of chemicals by deuterium substitution. A number of efforts are in progress to utilize deuterium substitution to alter the metabolism of drugs in an advantageous manner.
Keywords: Cytochrome P450 (P450, CYP); Kinetic isotope effects; Hydroxylation; Intrinsic isotope effect; Deuterium; Drug metabolism; Drug development
1. Introduction and history of cytochrome P450 kinetic isotope effects
The concept and theory of the kinetic isotope effect (KIE) are long-established and treated elsewhere in this monograph. This chapter will be restricted to deuterium KIEs on catalysis by cytochrome P450 (P450, CYP) enzymes. In the general mechanism of P450 catalysis (Fig. 1), there is a single C-H bond-breaking step that could be affected by isotopic substitution, and all KIEs must be interpreted in this context. (D2O KIEs have been reported for P450 reactions but the interpretation of these effects are complicated and their meaning is less well understood (Fersht, 1999), and they will not be considered because of space limitations.)
Fig. 1.

Catalytic cycle of microsomal P450s. RH is the substrate, with a C-H bond to be broken. With the seven mitochondrial P450s and many bacterial P450s, the electrons are delivered from a ferredoxin (including adrenodoxin) instead of the diflavin NADPH-P450 reductase. In some cases, cytochrome b5 (b5) delivers an electron in step 4, but not always. Note that the only step in which C-H bond breaking occurs is 7.
P450 reactions are well-suited to the application of KIE approaches, in that a large fraction of the reactions catalyzed by these enzymes involve the breaking of C–H bonds (F. P. Guengerich, 2001; F.P. Guengerich & Macdonald, 1984; P.R. Ortiz de Montellano, 2015). Thus, the ability to determine the extent to which this step is rate-limiting in a complex cycle is very useful, and therefore many KIE studies have been done. The first report on a P450 KIE appears to be that of Elison et al. in 1961 (Elison, Rapoport, Laursen, & Elliott, 1961), who showed that substitution of deuterium in an N-methyl group of morphine slowed its metabolism and increased its potency in rats.
The interpretation of P450 KIE studies is, in one sense, simple. If a deuterium KIE is observed, the rate of C-H bond-breaking has affected the overall rate of catalysis. However, enzyme mechanisms are complex and exactly how such a change is manifested is a function of the experimental design.
2. Types of experiments
One of the most common P450 reactions is the conversion of a methyl, methylene, or methine group to an alcohol. Other common C-H bond-breaking reactions are heteroatom cleavages (e.g. N, O, halogen), where a formal hydroxylation is followed by a breakdown of a carbinolamine, hemiacetal, halohydrins, etc.
There are myriad P450 reactions (Rendic & Guengerich, 2012, 2015). Most of them can be readily analyzed by modern chromatography methods, i.e. HPLC (or UPLC) and GC. With both methods, mass spectrometry (electrospray for charged molecules, atmospheric pressure-chemical ionization (APCI) for neutral ones) generally provides the best sensitivity. With HPLC/UPLC, UV-visible and fluorescence methods also provide excellent sensitivity in many cases. Mass spectrometry, done in a full-scan mode, has the advantages of (i) verifying the identity of products and (ii) screening out overlap between molecules that are not well-resolved by chromatography. Internal standards are useful for quantitation and, in the case of GC, critical with split injection methods.
An extensive discussion of synthetic methods for generating substrate isotopologues is beyond the scope of this chapter. When N- and O-alkyl derivatives (which are often P450 substrates) are under consideration, the synthesis is generally straightforward with Gabriel methods (i.e., the reaction of amines or alcohols with alkyl halides) or (for N-alkylamines) hydride reduction of amides prepared with coupling an amine with an acyl halide or alkylchlorformate, e.g. see (Miwa, Walsh, Kedderis, & Hollenberg, 1983; Okazaki & Guengerich, 1993). The synthesis of deuterated (or tritiated) derivatives of some hydrocarbon chains can be accomplished by treatment of halides with NaBH4 (H. M. Bell, Vanderslice, & Spehar, 1969; Kim, Cha, Nagy, Yun, & Guengerich, 2014). Other molecules can be much more problematic, depending on the precursor molecules available (Gelb, Heimbrook, Mälkönen, & Sligar, 1982; Krauser & Guengerich, 2005; Yoshimoto & Guengerich, 2014).
The use of multiply-deuterated substrates is sometimes involved. For instance, the oxidation of –CD3 in a molecule is common for P450 substrates. In some cases, a perdeuterated substrate may be commercially available, e.g., medium- and long-chain fatty acids. These compounds can be problematic in the (i) there is potential for vicinal secondary KIEs and (ii) interpretations of any metabolic switching may be more complex. However, synthesis of deuterated materials is not trivial, and the placement of deuterium may often be a matter of practicality.
An issue to consider in any experimental design (and synthesis) is prochirality. This is often an issue in labeling methylene groups, in that only one of the two hydrogens may be abstracted by a P450 (White, Miller, Favreau, & Bhattacharyya,1986; Krauser & Guengerich, 2005).
Finally, atomic purity is a serious issue. The experiments discussed under sections 2.1, 2.2, and 2.3 are not very sensitive to impurities because both the “protium” and “deuterium” reactions occur with the same enzyme. The non-competitive intermolecular experiment (2.4) is, however, very sensitive to impurities. If the deuterated substrate contains an enzyme inhibitor, the apparent D(V/K) value will be too high. (Note: the conventions of Northrop, 1982 regarding DV, D(V/K), Dk, etc. are used in this chapter.) If the deuterated substrate has low atomic enrichment, then (depending on some aspects of the reaction) the reaction may preferentially occur with the protiated material and yield an apparent D(V/K) that is too low (an experiment intended to be of the 2.4 type (Fig. 2D) may partially become an experiment of type 2.2 (Fig. 1B)). For these reasons, deuterated substrates should have as high an atomic excess as possible (preferably > 98%), as analyzed by 1H-NMR (in our experience this is a more accurate method than mass spectrometry).
Fig. 2.
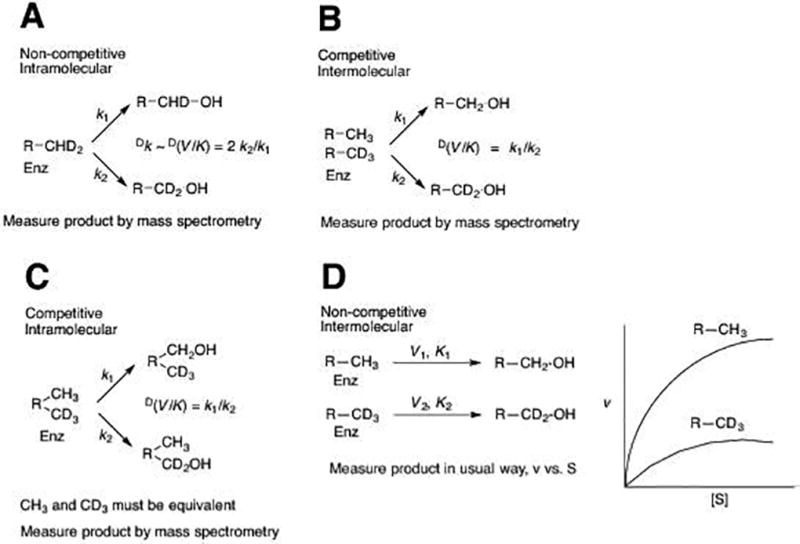
Types of deuterium KIE studies used in P450 research. (A) Non-competitive intramolecular. The factor of 2 is a statistical correction, in that there are two deuteriums and one protium available here. The factor would be ½ if the substrate were –CH2D. (B) Competitive intermolecular. (C) Competitive intramolecular. (D) Non-competitive intermolecular. In the “k1” pathways, a C–H bond is broken; in “k2” pathways a C–D bond is broken.
In that several of the experiments involve only analysis by mass spectrometry (2.1, 2.2, 2.3; Fig. 2A, 2B, 2C), some caution is in order. A critical problem is the 13C spillover (1.1% × number of carbons), which can be quite problematic. In principle these can be corrected using formulae (Biemann, 1962), although a more accurate approach is to utilize high-resolution mass spectrometry. At a resolution of 60,000–100,000, M+1 ions due to 13C and 2H are readily distinguished (Yoshimoto & Guengerich, 2014).
What a KIE means will depend on the type of experiment used to generate the data. Following are five approaches used with P450 experiments (Fig. 2).
2.1 Non-competitive intramolecular experiment
The non-competitive intramolecular experiment (Fig. 2A) works well with hydroxylations of methyl groups by P450 enzymes, which is a fairly common reaction that generates a primary alcohol. The reaction is non-competitive because a single isotopologue is being analyzed, and the deuterium atoms to be removed are equivalent with each other. The comparison of C–H bonds being broken is intramolecular. The KIE should be independent of issues of substrate concentration and only a single (mass spectrometry) measurement is required (preferably replicates).
In principle, the KIE in this approach is an estimate of the intrinsic KIE (designated Dk), i.e., the KIE for the actual C-H bond-breaking step, as in a purely chemical reaction. There are three caveats about a KIE measured using this approach:
The method works well with methyl groups but probably will not with a pro-chiral methylene, in that only one of the two hydrogen atoms may be abstracted (Krauser & Guengerich, 2005).
The KIE may not be the Dk, due to possible contribution of a secondary KIE, coming from either the geminal or vicinal deuterium substitution (which can be up to 1.4 (Fersht, 1999)), and these are multiplicative (Matsson & Westaway, 1998).
A KIE measured by this approach provides no information about how rate-limiting the C-H bond-breaking step is. The competition is between a protium and a deuterium (or tritium) atom at the same carbon, but no insight into which step in Fig. 1 limits the overall rate is provided.
2.2 Competitive intermolecular experiment
In a competitive intermolecular experiment, there is competition between two kinds of molecules, the protiated and the deuterium-labeled (Fig. 2B). In this reaction two different substrate isotopologues are used that compete for enzyme binding and the reaction is considered intermolecular relative to case 2.1. The experiment shown in Fig. 2B has two methyl hydroxylations, but in principle the comparison can be done at any position on the substrate molecule. The results, as in the case of 2.1, can easily be determined by mass spectrometry with assays done at a single substrate concentration to provide an estimate of D(V/K).
As in the case of 2.1, the resulting KIE is not a direct reflection of the rate-limiting nature of the C-H bond-breaking step. A high value, based on this argument, would indicate that the chemical step is at least partially rate limiting and also that the enzyme has fast exchange of substrates. If this value is low compared to Dk, the intrinsic KIE, this is evidence that the substrate is “locked in” the active site (low koff rate for substrate) and there is “high commitment to catalysis” (Northrop, 1982).
2.3 Competitive intramolecular experiment
This is a rather specialized experiment and can only be done with substrates that have equivalent moieties within them, e.g. N,N-dimethyl groups or methyl groups on alkanes (n-octane). The experiments cannot be done if pro-chirality is an issue. As in cases 2.1, 2.2, and 2.3, the analysis is done using mass spectrometry and yields an estimate of D(V/K).
The results are a measure of the ability of the substrate to rearrange (“tumble”) in the active site. A value much lower than the Dk for the intermolecular reaction can be interpreted in this context. However, the results can also be a reflection of the substrate koff (release and rebinding), so this behavior may not be discernable in the absence of other information such as an isotope dilution experiment using a product or non-reactive substrate trap (Gonzalez & Guengerich, 2017).
2.4 Non-competitive intermolecular experiment
This experiment requires more effort than any of the three already described (2.1, 2.2, 2.3), in that assays must be done at multiple substrate concentrations with both protiated and deuterated substrates. The experiments yield both DV and D(V/K), and non-competitive experiments are the only method to determine DV. This type of experiment is also subject to more error because of the multiplicity of measurements and the propagation of error in quotients because they are ratios, which multiply error in the numerator and divisor. Finally, those experiments are subject to two problems with impurities:
If the deuterated substrate has any inhibitory contaminant, the enzyme activity will be reduced and the observed KIE will be artificially high.
If the deuterated substrate is contaminated with protiated substrate, then (if substrate exchange is rapid) a high KIE may be attenuated because the enzyme can “pick” the protiated substrate out of the deuterated pool and use it.
Despite these inherent problems, the approach is very powerful and provides information to address the question of how rate-limiting the C-H bond-breaking step is. If the KIE (DV or D(V/K)) is > 1, then the C-H bond-breaking step is at least partially rate-limiting. If DV or D(V/K) approaches the Dk, then the C-H bond-breaking step is very rate-limiting.
2.5 Estimation of the intrinsic KIE
As already mentioned, Dk is the estimate of the KIE for the C-H bond-breaking step, as in a strictly chemical reaction. There are at least three approaches that have been used, with two used in P450 systems, to determine the extent to which the observed KIE reflects the intrinsic KIE on the chemical step.
-
Northrop’s Method. This approach, described in more detail elsewhere (Northrop, 1975, 1977, 1982), involves comparison of the deuterium and tritium V/K results.
The procedure used in 2.4 will not generate T(V/K) because complete tritium substitution is impractical and one of the other experiments is necessary. The value y can be estimated from tables (Northrop, 1977). This approach has been used for some P450 reactions (Krauser & Guengerich, 2005; Miwa, Walsh, & Lu, 1984).
The “Multiple Isotope Effect Method” utilizes a combination of 2H and 13C isotope effects (Cook & Cleland, 2007). Only a few 13C KIEs have been reported with P450 reactions, and apparently this approach to determining Dk has not been applied.
- The “Isotopically Sensitive Branching” method. This was developed by Jones et al. (J.P. Jones, Korzekwa, Rettie, & Trager, 1986; J. P. Jones, Rettie, & Trager, 1990) and can be applied when a reaction is irreversible and a minor product is also formed. In the work of Jones et al. (J.P. Jones et al., 1986; J. P. Jones et al., 1990) the method is based on DV results (actually only single substrate concentration results were used in the original papers). Thus, using the Northrop nomenclature, where the major product (P) comes from deuterated substrate but the minor product does not,
The approach can be used, although it becomes more problematic in cases in which there are either (a) multiple alternate products or (b) no alternate products. It is still possible for a P450 reaction to show a KIE, in that the activated complex has an alternative to productive oxygenation—the formation of reduced oxygen products, e.g, water (Gorsky, Koop, & Coon, 1984).
3. Choice of enzymatic parameters
In considering what KIE results to measure, it should be noted that several approaches measure only D(V/K) values (Fig. 2A, 2B, 2C). Many of the P450 KIE results in the literature report only KIE determinations done at a single substrate concentration, often a high one that approximates kcat conditions. However, considerably more information can be obtained about what is rate-limiting by measuring kcat and Km and thus DV and D(V/K). Exactly which steps of a model mechanism are reflected by DV, D(V/K), Dk etc. are a function of the microscopic rate constants for the multiple turnover reaction and are discussed elsewhere (Northrop, 1982; Walsh, 1979).
In general, most P450 papers, when they have considered obtaining results varying substrate concentrations, have relied on DV. However, consider the case of oxidation of ethanol to acetaldehyde by human P450 2E1 (L. C. Bell & Guengerich, 1997; Bell-Parikh & Guengerich, 1999).
The rate-limiting step occurs after product formation. Simplified, the reaction is depicted as (note that we are changing the convention of step numbers now):
where S is the substrate and P the product. Then
, and Km has units of molarity. Therefore,
and
where DV is the KIE on kcat and D(V/K) is the KIE on the ratio of kcat/Km, according to the convention of Northrop (Northrop, 1982).
The value of D(V/K) is ~3–5 (L. C. Bell & Guengerich, 1997). A plot of product vs. time indicates “burst kinetics”—rate-limiting step is after product formation. The Dk for C-H bond breaking was found to be 3.8 and the isotopic effect on product formation measured using pre-steady-state kinetic studies was estimated to be 3.2. Since DV≈ 1 due to the fact that a slow step after catalysis is limiting for V, then Dk2 + k2/k3 ≈ 1 + k2/k3. This is valid only if k2 >> k3. From the results presented here, k2 (the rate of product formation) is much faster than k3 (the rate of product release, or some other step occurring after product formation).
The above expressions for kcat and Km are reduced to
and
Thus, this paradigm includes rate-limiting product release (or something else following product formation) in the overall reaction. Km increases as k2 decreases, consistent with the observed effect of isotopic substitution and is a general case (Walsh, 1979). Further, Km is a function of kcat (and definitely not equivalent to Kd).
4. KIEs for various P450 reactions
In our own experience, almost every deuterated analog of a compound hydroxylated by P450s (at a particular site that has then been deuterated) shows at least some KIE. As discussed elsewhere (F. P. Guengerich, 2013), there are some general predictions that can be made about the tendency for P450 reactions to show kinetic deuterium isotope effects. For instance, amine N-dealkylation and aromatic hydroxylation reactions have low KIEs for mechanistic reasons and are generally not good candidates for for resolving mechanistic detail (Figs. 3, 4). However, dealkylations of ethers and amides (Hall & Hanzlik, 1990) are different, due to the much higher oxidation potentials, and high KIEs are often observed. Alkyl groups, i.e., methylenes, methines, and methyls, are possibilities, depending on what is rate limiting in the particular P450 reaction. However, care is necessary about the issue of pro-chirality of methylenes, in that the abstraction of hydrogen and oxygen rebound may, or may not be stereoselective. For example, P450 3A4 abstracts only the β-hydrogen at the C-6 carbon of testosterone (Krauser & Guengerich, 2005), so that this information must be considered in evaluating any results.
Fig. 3.

Comparisons of mechanisms of carbon hydroxylation (A) and N-dealkylation (B) (F.P. Guengerich & Macdonald, 1984).
Figure 4.

General mechanisms of aryl hydroxylation (F. P. Guengerich, 2001; P.R. Ortiz de Montellano, 2015). *H indicates a labeled hydrogen atom, i.e. deuterium or tritium. Alternate pathways (a, solid line; b, broken line) for the collapse of the tetrahedral iron-oxygen intermediate lead to the carbonyl and to the epoxide, respectively, and both can form the phenol.
Miwa et al. (Miwa et al., 1983) reported high KIEs (8.6–10.1) for amine N-dealkylation reactions catalyzed by peroxidases. However, the KIEs for such reactions are low for P450s, usually ≤ 2 (F. P. Guengerich, 1990). An explanation was provided in that amine dealkylations by both P450s and peroxidases proceed via initial 1-electron abstraction (Fig. 3), but P450s use base catalysis with FeO2+ for α-proton transfer while peroxidases do not (instead utilizing heme edge electron transfer), leaving α C-H bond breaking uncatalyzed and allowing the aminium radicals to accumulate (F. P. Guengerich, Yun, & Macdonald, 1996; Okazaki & Guengerich, 1993).
For an aryl C–H bond, the enzyme does an addition but does not break the C–H/C–D bond (Fig. 4). Therefore, one expects to see KIEs due to deuterium substitution in alkyl groups but not aryl groups. In this regard, Tomaszewski et al. (Tomaszewski, Jerina, & Daly, 1975) concluded that isomerization to phenols proceeds without detectable isotope effects (Kasperek, Bruice, Yagi, & Jerina, 1972). Arene oxide formation from deuterated aromatic substrates also proceeds without a KIE because C-H bonds are not broken in the oxidation. The general lack of KIEs in formation of phenols in aromatic substrates (Daly & Jerina, 1969; Guroff et al., 1967; Perel, Dayton, Tauriello, Brand, & Mark, 1967; M. Tanabe, Yasuda, Tagg, & Mitoma, 1967) is consistent with (but does not necessarily prove) the formation of arene oxides as intermediates.
Tanabe et al. (Masato Tanabe, Tagg, Yasuda, LeValley, & Mitoma, 1970) reported an in vitro KIE of 1.47 for the aryl oxidation of the drug zoxazolamine. However, this result could not be repeated under the same conditions by Tomaszewski et al. (Tomaszewski et al., 1975), i.e. the KIE was not > unity. Other studies have consistently shown low KIE values (little effect of deuterium substitution) for aryl oxidations, interpreted as evidence for the proposed mechanism shown in Fig. 4 (Bush & Trager, 1982; Hanzlik, Hogberg, & Judson, 1984; Korzekwa, Swinney, & Trager, 1989; P.R. Ortiz de Montellano, 1995; P. R. Ortiz de Montellano & DeVoss, 2005; Tomaszewski et al., 1975). Typical KIE values for some substituted aryl molecules have been reported in the range of 0.95–1.27 (Korzekwa et al., 1989; P.R. Ortiz de Montellano, 1995).
5. Examples of P450 KIE studies
The concept of inhibiting rates of P450 oxidations by C–D substitution has been largely the same since the field of P450 KIE research began (Elison et al., 1961) (which is actually one year before P450s were formally discovered (Omura & Sato, 1962)). KIE measurements have been made in many systems, but what new information has resulted? Many of the earliest studies were of the intermolecular competition type, and several investigators concluded that KIEs were absent or modest (Björkhem, 1972; M. Tanabe et al., 1967; Ullrich, 1969).
However, some early results with intramolecular reactions showed very high observed KIEs (A. B. Foster, Jarman, Stevens, Thomas, & Westwood, 1974; Hjelmeland, Aronow, & Trudell, 1977) and possibly another study as well (McMahon, Sullivan, Craig, & Pereira Jr, 1969), although the results were later reinterpreted (Hjelmeland et al., 1977). Groves et al. (Groves, McClusky, White, & Coon, 1978) considered the high intramolecular competitive KIE observed using rabbit P450 2B4 (11.5), plus earlier work, as the first definitive evidence for the radicaloid, Compound I, oxygen-rebound mechanism that is generally accepted today for hydroxylation (Fig. 3A).
Another concept in the field of P450 KIEs is that of “metabolic switching,” which Baillie (Baillie, 1981) first attributed to Mitoma et al. (Mitoma, Dehn, & Tanabe, 1971), followed by Horning et al. (Horning et al., 1975) and others. The concept has been reviewed (Miwa & Lu, 1987). Basically, a KIE on a reaction at one site of a substrate results in enhanced activity at another site of the substrate, i.e. yielding an alternate product. For instance, deuteration at the 6 β-position of testosterone attenuated the formation of 6 β-hydroxytestosterone but enhanced the production of 2 β-hydroxytestosterone (Krauser & Guengerich, 2005). The concept is that the enzyme is capable of moving to a reactive intermediate (i.e., Compound I) with the substrate but then “turns” the substrate for catalysis. The amount of total product formed from the deuterated substrate may be as great as with the protiated analog, or it may be less.
Metabolic switching is of interest in both basic and applied contexts. The existence of switching indicated that formation of activated iron-oxygen complex (i.e. FeO3+) is not reversible and, in a sense, contributes to a strong commitment to catalysis (Harada, Miwa, Walsh, & Lu, 1984). However, it should be pointed out that although the Compound I form of P450 (formally FeO3+) is formed irreversibly, it will break down to H2O if not used to catalyze substrate oxidation (Gorsky et al., 1984). In addition, it is possible to exploit metabolic switching in estimating the intrinsic KIE, Dk (section 2.5).
There are also practical aspects of metabolic switching, which are relevant to the issue of utilizing deuterium substitution in drug development (Section 7, vide infra). If the issue is attenuating rates of metabolism to improve drug half-life or lower drug clearance, then the existence of metabolic switching would abrogate a positive effect of deuteration (but only if the parent drug has pharmacological activity). Another issue is that deuteration could produce a metabolic switch to a toxic product. An example is the analgesic phenacetin (in hamsters), where deuterium substitution in the ethoxy methylene decreased liver necrosis but raised blood methemoglobin levels by 50% (S.D. Nelson et al., 1978). The authors rationalized the results as meaning that O-deethylation was blocked, so that deacetylation by another enzyme, an amidase, was favored (S. D. Nelson & Trager, 2003) and gave rise to the more toxic molecule p-phenetidine (4-ethoxyaniline) (Ross et al., 1985).
Another aspect is consideration of when KIEs are observed in P450 reactions. As already pointed out, deuterium KIEs should not be seen for P450 reactions in which no C-H bonds are broken during enzyme catalysis, e.g. epoxidation, heteroatom oxygenation, aryl hydroxylation (Fig. 4). As indicated earlier, in our experience we have found generally low KIEs for amine N-dealkylations, which we have rationalized in terms of initial 1-electron oxidation followed by proton transfer-linked rearrangement and oxygen rebound (Fig. 3B), at least in some cases (F. P. Guengerich, 1990; F. P. Guengerich et al., 1996; Okazaki & Guengerich, 1993).
The P450 reactions for which we have measured KIEs all involve mammalian enzymes and are summarized in Fig. 5. In a previous analysis of this type, there was an inverse correlation between the reaction rate and the intermolecular non-competitive KIE, with an r2 value of 0.62 (F. P. Guengerich, 2013). Even though the correlation coefficient suggested that only ~ 60% of the variability could be attributed to such a relationship, the idea that ease of C-H bond-breaking limits overall reaction rates among a plethora of P450s has some appeal. Since the earlier publication appeared (four years ago), we have added our work with P450 21A2, which has a relatively fast substrate turnover rate and also high KIEs (for both substrates, progesterone and 17 α-hydroxyprogesterone) (Pallan et al., 2015). Now any relationship is completely missing (r2 0.015). Thus, the absolute rate of a P450 reaction does not appear to provide any insight into how rate-limiting the C-H bond-breaking step is. In more recent studies we have used site-directed mutagenesis to make a series of naturally-occurring P450 21A2 variants identified in the clinic (Wang et al., 2017). Some of these have only 1/106 the catalytic activities of the wild-type enzyme but the high KIEs were seen with all. The mutations are unlikely to increase substrate binding commitments, which we found only limited evidence for, and such a result provides additional evidence that the chemical step is rate-limiting. Therefore, even with the wild-type enzyme, with a specificity constant (kcat/Km) of 107 M−1 s−1, C-H bond-breaking is at least partially rate-limiting. This could be viewed as a less-than-perfect fit of the FeO3+ entity and the C-21 hydroxylation site or simply a reflection of the symmetry of the transition state (Melander & Saunders, 1980; Westheimer, 1961).
Figure 5.
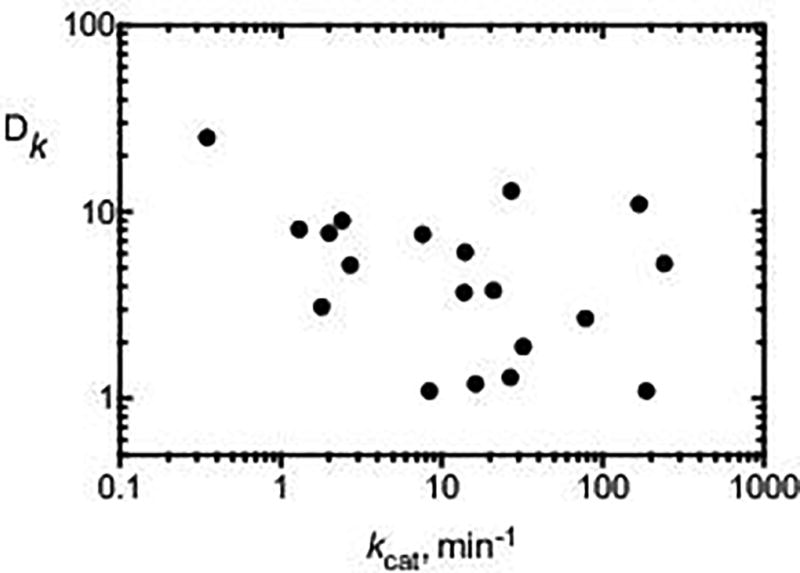
Relationship between KIE (intermolecular, non-competitive) and kcat for a number of P450 reactions, mainly with human P450s. Data points are from the indicated references (L. C. Bell & Guengerich, 1997; Bell-Parikh & Guengerich, 1999; Chowdhury, Calcutt, & Guengerich, 2010; Chowdhury, Calcutt, Nagy, & Guengerich, 2012; F. P. Guengerich, Krauser, & Johnson, 2004; F. P. Guengerich, Miller, Hanna, Sato, & Martin, 2002; Krauser & Guengerich, 2005; Pallan et al., 2015; Shinkyo & Guengerich, 2011; Yun, Miller, & Guengerich, 2000).
6. In vivo KIEs with P450s
Although the first application of KIEs to P450 drug metabolism in 1961 involved in vivo work (Elison et al., 1961), far fewer studies have been done in vivo than in vitro. Another example involves (methylene) dideuteration of the barbiturate butethal, which doubled the sleeping time when the compound was administered to mice (M. Tanabe, Yasuda, LeValley, & Mitoma, 1969). Early work in the in vivo area was reviewed by Baillie (Baillie, 1981).
One example of a large KIE was seen in our own work on the further metabolism of a product of the calcium channel blocker nifedipine (Funaki, Soons, Guengerich, & Breimer, 1989). P450s catalyze the oxidative demethylation of methyl esters, with high KIE values (F. P. Guengerich, 1987; F. P. Guengerich, Peterson, & Bocker, 1988). The strong effect of deuteration on pharmacokinetic parameters provides evidence that the in vitro ester cleavage is due to oxidation and not hydrolysis.
In several cases large kinetic isotope effects had been observed in cell culture or in vivo (in animals), including procarbazine, tamoxifen, N-nitrosodimethylamine, pulegone, methylene chloride, chloroform, bromoform, ethylene dibromide, furosemide, N-methylformamide, 3,3′-iminodiproprionitrile, N-(3,5-didhlorophenyl)succinimide, 3-methylcholanthrene, butylated hydroxytoluene, phenacetin, and 3-methylindole, and these are all related to altered biological effects such as toxicity, as reviewed by Baillie (Baillie, 1981) and by Nelson and Trager (S. D. Nelson & Trager, 2003). As an example, deuterated N,N-dimethylnitrosamine is 3-fold less carcinogenic (than the protiated version) in rats (Keefer, Lijinsky, & Garcia, 1973). The role of P450 oxidation in the phenomenon was established by in vitro (microsomal) N-demethylation kinetics (Wade et al., 1987) and in vivo measurement of methylated DNA adducts (Swann, Mace, Angeles, & Keefer, 1983). Another cancer-related example is ethylene dibromide (1,2-dibromethane), which is oxidized by P450s. However, its genotoxicity is related to another pathway, glutathione conjugation (F. P. Guengerich, 2005). Deuteration of ethylene dibromide blocks oxidation and increases the genotoxicity in rats (White, Gandolfi, Bowden, & Sipes, 1983), as does administration of the P450 2E1/2A6 inhibitor disulfiram (Wong, Winston, Hong, & Plotnick, 1982).
7. Consideration of P450 KIEs in drug development
The first application of KIEs to P450 drug metabolism was that of Elison et al. in 1961 (Elison et al., 1961) on the N-demethylation of morphine, which is recognized today as a P450 reaction. Subsequent work on the P450-catalyzed O-demethylation of anisoles was published by Mitoma et al. (Mitoma, Yasuda, Tagg, & Tanabe, 1967) and Foster et al. 1974 (A. B. Foster et al., 1974). In a review article in 1984 (Allan B. Foster, 1984), Foster concluded that “Two promising areas involve the attenuation of the metabolism pathways which generate the active metabolites for prodrugs and, where overall metabolism cannot be retarded, the deflection of metabolism away from pathways leading to metabolites with toxic properties or other undesirable biological activity towards innocuous pathways.” As pointed out in 2009 (Yarnell, 2009), “The strategy is far from new.” That 2009 article also cites a 2006 paper by Sepracor on tramadol (Shao, Abolin, Hewitt, Koch, & Varney, 2006). In the 56 years since the reports of Elison et al. (Elison et al., 1961), there have been numerous theoretical and fundamental studies of KIEs as well as efforts to modify in vivo pharmacokinetics.
In 2009–2010 there was a renewed interest in deuterated analogs of known pharmacologically active compounds. Deuteration strategy was highlighted in 2009 as the “latest retro rage in the pharmaceutical world” (Yarnell, 2009) and several companies were established (e.g., Concert, Dueteria, Auspex (now part of Teva), Protia). Concert advertised that this approach allows it to “rapidly create novel, differentiated compounds with substantially reduced R&D risk, time and expense.” The concept is that of routine optimization by deuterating at known metabolic hotspots. For a 2016 review of progress in the commercialization see (Halford, 2016). In April 2017 the US Food and Drug Administration (FDA) approved deutetrabenazine (Austedo, from Auspex-Teva) for the treatment of chorea associated with Huntington's disease.
8. Summary and conclusions
P450s are very appropriate for KIE studies because of their proclivity for C–H bond-breaking reactions in the oxidations that they catalyze. KIE studies have provided important insights into how P450s catalyze reactions, and today there is considerable interest in utilizing deuterium substitution (and KIEs) in drug development. What does the future hold?
Many of the reported KIEs for P450 reactions are larger than the classical limit of ~ 7 (Fig. 5) but to the author’s knowledge no reports of serious efforts at studying tunneling have appeared. Some of the approaches to studying tunneling are technically difficult, but there are probably some good candidates. Archebacterial P450s would seem to be good candidates because of their broader temperature range for comparing activation energies for protiated and deuterated substrates (Klinman, 2013), although only limited information is available regarding their substrates and redox partners.
In principle, one could examine thousands of more P450 reactions with KIE studies to determine how rate-limiting the C-H bond breaking step is. This would not be very practical nor would the results be that enlightening. Nevertheless, many individual reactions catalyzed by P450s are of inherent interest, and KIE analysis is a relatively rapid method (assuming ease of deuterated synthesis) of establishing if C–H bond-breaking is a rate-limiting step. In some cases, this information is important in the development of P450 systems for industrial catalysis. The potential for development of drugs with altered metabolism properties has already been discussed.
Acknowledgments
Research in this area in the author’s laboratory is supported by National Institutes of Health grants R01 GM 118122 and R01 GM103937. Thanks are extended to K. Trisler for help in preparation of the manuscript and to M. J. Reddish and to M. E. Harris for comments on the draft and initial submission.
References
- Baillie TA. The use of stable isotopes in pharmacological research. Pharmacological Reviews. 1981;33(2):81–132. [PubMed] [Google Scholar]
- Bell HM, Vanderslice CW, Spehar A. The reduction of organic halogen compounds by sodium borohydride. Journal of Organic Chemistry. 1969;34:3923–3926. [Google Scholar]
- Bell LC, Guengerich FP. Oxidation kinetics of ethanol by human cytochrome P450 2E1. Rate-limiting product release accounts for effects of isotopic hydrogen substitution and cytochrome b5 on steady-state kinetics. The Journal of Biological Chemistry. 1997;272(47):29643–29651. doi: 10.1074/jbc.272.47.29643. [DOI] [PubMed] [Google Scholar]
- Bell-Parikh LC, Guengerich FP. Kinetics of cytochrome P450 2E1-catalyzed oxidation of ethanol to acetic acid via acetaldehyde. The Journal of Biological Chemistry. 1999;274(34):23833–23840. doi: 10.1074/jbc.274.34.23833. [DOI] [PubMed] [Google Scholar]
- Biemann K. Mass Spectrometry, Organic Chemical Applications. New York: McGraw-Hill; 1962. pp. 223–225. [Google Scholar]
- Björkhem I. On the rate-limiting step in microsomal hydroxylation of steroids. European Journal of Biochemistry. 1972;27(2):354–363. doi: 10.1111/j.1432-1033.1972.tb01845.x. [DOI] [PubMed] [Google Scholar]
- Bush ED, Trager WF. Evidence against an abstraction or direct insertion mechanism for cytochrome P-450 catalysed meta hydroxylations. Biochemical and Biophysical Research Communications. 1982;104:626–632. doi: 10.1016/0006-291x(82)90683-0. [DOI] [PubMed] [Google Scholar]
- Chowdhury G, Calcutt MW, Guengerich FP. Oxidation of N-nitrosoalkylamines by human cytochrome P450 2A6: Sequential oxidation to aldehydes and carboxylic acids and analysis of reaction steps. The Journal of Biological Chemistry. 2010;285(11):8031–8044. doi: 10.1074/jbc.M109.088039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury G, Calcutt MW, Nagy LD, Guengerich FP. Oxidation of methyl and ethyl nitrosamines by cytochrome P450 2E1 and 2B1. Biochemistry. 2012;51(50):9995–10007. doi: 10.1021/bi301092c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. New York: Garland Science Publishing; 2007. pp. 277–280. [Google Scholar]
- Daly J, Jerina D. Migration of deuterium during aryl hydroxylation. 3. Effect of ortho- and meta- substituents. Archives of Biochemistry and Biophysics. 1969;134(1):266–268. doi: 10.1016/0003-9861(69)90280-x. [DOI] [PubMed] [Google Scholar]
- Elison C, Rapoport H, Laursen R, Elliott HW. Effect of deuteration of N-CH3 group on potency and enzymatic N-demethylation of morphine. Science. 1961;134(3485):1078–1079. doi: 10.1126/science.134.3485.1078. [DOI] [PubMed] [Google Scholar]
- Fersht A. Structure and Mechanism in Protein Science. New York: Freeman; 1999. pp. 98–99. [Google Scholar]
- Foster AB. Deuterium isotope effects in studies of drug metabolism. Trends in Pharmacological Sciences. 1984;5:524–527. [Google Scholar]
- Foster AB, Jarman M, Stevens JD, Thomas P, Westwood JH. Isotope effects in O- and N-demethylations mediated by rat liver microsomes: an application of direct insertion electron impact mass spectrometry. Chemico-Biological Interactions. 1974;9(5):327–340. doi: 10.1016/0009-2797(74)90128-8. [DOI] [PubMed] [Google Scholar]
- Funaki T, Soons PA, Guengerich FP, Breimer DD. In vivo oxidative cleavage of a pyridine-carboxylic acid ester metabolite of nifedipine. Biochemical Pharmacology. 1989;38(23):4213–4216. doi: 10.1016/0006-2952(89)90517-0. [DOI] [PubMed] [Google Scholar]
- Gelb MH, Heimbrook DC, Mälkönen P, Sligar SG. Stereochemistry and deuterium isotope effects in camphor hydroxylation by the cytochrome P450cam monoxygenase system. Biochemistry. 1982;21:370–377. doi: 10.1021/bi00531a026. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Guengerich FP. Kinetic processivity of the two-step oxidations of pregnenolone and progesterone to androgens by human cytochrome P450 17A1. The Journal of Biological Chemistry. 2017;292 doi: 10.1074/jbc.M117.794917. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsky LD, Koop DR, Coon MJ. On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450: Products of oxygen reduction. The Journal of Biological Chemistry. 1984;259:6812–6817. [PubMed] [Google Scholar]
- Groves JT, McClusky GA, White RE, Coon MJ. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450: Evidence for a carbon radical intermediate. Biochemical and Biophysical Research Communications. 1978;81:154–160. doi: 10.1016/0006-291x(78)91643-1. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Oxidative cleavage of carboxylic esters by cytochrome P-450. The Journal of Biological Chemistry. 1987;262(18):8459–8462. [PubMed] [Google Scholar]
- Guengerich FP. Low kinetic hydrogen isotope effects in the dehydrogenation of 1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-3,5-pyridinedicarboxylic acid dimethyl ester (nifedipine) by cytochrome P-450 enzymes are consistent with an electron/proton/electron transfer mechanism. Chemical Research in Toxicology. 1990;3(1):21–26. doi: 10.1021/tx00013a004. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chemical Research in Toxicology. 2001;14(6):611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Activation of alkyl halides by glutathione transferases. Methods in Enzymology. 2005;401:342–353. doi: 10.1016/S0076-6879(05)01021-9. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Kinetic deuterium isotope effects in cytochrome P450 oxidation reactions. Journal of Labelled Compounds and Radiopharmaceuticals. 2013;56(9–10):428–431. doi: 10.1002/jlcr.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP, Krauser JA, Johnson WW. Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry. 2004;43(33):10775–10788. doi: 10.1021/bi0491393. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Macdonald TL. Chemical mechanisms of catalysis by cytochromes P-450: A unified view. Accounts of Chemical Research. 1984;17:9–16. [Google Scholar]
- Guengerich FP, Miller GP, Hanna IH, Sato H, Martin MV. Oxidation of methoxyphenethylamines by cytochrome P450 2D6. Analysis of rate-limiting steps. The Journal of Biological Chemistry. 2002;277(37):33711–33719. doi: 10.1074/jbc.M205146200. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Peterson LA, Bocker RH. Cytochrome P-450-catalyzed hydroxylation and carboxylic acid ester cleavage of Hantzsch pyridine esters. The Journal of Biological Chemistry. 1988;263(17):8176–8183. [PubMed] [Google Scholar]
- Guengerich FP, Yun CH, Macdonald TL. Evidence for a 1-electron oxidation mechanism in N-dealkylation of N,N-dialkylanilines by cytochrome P450 2B1. Kinetic hydrogen isotope effects, linear free energy relationships, comparisons with horseradish peroxidase, and studies with oxygen surrogates. The Journal of Biological Chemistry. 1996;271(44):27321–27329. doi: 10.1074/jbc.271.44.27321. [DOI] [PubMed] [Google Scholar]
- Guroff G, Daly JW, Jerina DM, Renson J, Witkop B, Udenfriend S. Hydroxylation-induced migration: The NIH shift. Science. 1967;157:1524–1530. doi: 10.1126/science.157.3796.1524. [DOI] [PubMed] [Google Scholar]
- Halford B. Deuterium switcheroo breathes life into old drugs. Chemical & Engineering News. 2016;94(27):32–36. [Google Scholar]
- Hall LR, Hanzlik RP. Kinetic deuterium isotope effects on the N-demethylation of tertiary amides by cytochrome P-450. The Journal of Biological Chemistry. 1990;265:12349–12355. [PubMed] [Google Scholar]
- Hanzlik RP, Hogberg K, Judson CM. Microsomal hydroxylation of specifically deuterated monosubstituted benzenes. Evidence for direct aromatic hydroxylation. Biochemistry. 1984;23:3048–3055. doi: 10.1021/bi00308a031. [DOI] [PubMed] [Google Scholar]
- Harada N, Miwa GT, Walsh JS, Lu AYH. Kinetic isotope effects on cytochrome P-450-catalyzed oxidation reactions: Evidence for the irreversible formation of an activated oxygen intermediate of cytochrome P-448. The Journal of Biological Chemistry. 1984;259:3005–3010. [PubMed] [Google Scholar]
- Hjelmeland LM, Aronow L, Trudell JR. Intramolecular determination of primary kinetic isotope effects in hydroxylations catalyzed by cytochrome P-450. Biochemical and Biophysical Research Communications. 1977;76:541–549. doi: 10.1016/0006-291x(77)90758-6. [DOI] [PubMed] [Google Scholar]
- Horning MG, Haegele KD, Sommer KR, Nowlin J, Stafford M, Thenot J-P. In: Oak Brook IL, Klein ER, Klein PD, editors. Metabolic switching of drug pathways as a consequence of deuterium substitution, Proceedings of the 2nd International Conference on Stable Isotopes; October 20–23, 1975; Washington: Springfield, VA: Energy Research and Development Administration; National Technical Information Service; 1975. [Google Scholar]
- Jones JP, Korzekwa KR, Rettie AE, Trager WF. Isotopically sensitive branching and its effect on the observed intramolecular isotope effects in cytochrome P-450 catalyzed reactions: A new method for the estimation of intrinsic isotope effects. Journal of the American Chemical Society. 1986;108:7074–7078. [Google Scholar]
- Jones JP, Rettie AE, Trager WF. Intrinsic isotope effects suggest that the reaction coordinate symmetry for the cytochrome P-450 catalyzed hydroxylation of octane is isozyme independent. Journal of Medicnal Chemistry. 1990;33(4):1242–1246. doi: 10.1021/jm00166a024. [DOI] [PubMed] [Google Scholar]
- Kasperek GJ, Bruice TC, Yagi H, Jerina DM. Differentiation between the concerted and stepwise mechanisms for aromatization (NIH-shift) of arene epoxides. Journal of the Chemical Society, Chemical Communications. 1972;(13):784–785. [Google Scholar]
- Keefer LK, Lijinsky W, Garcia H. Deuterium isotope effect on the carcinogenicity of dimethylnitrosamine in rat liver. Journal of the National Cancer Institute. 1973;51:299–302. doi: 10.1093/jnci/51.1.299. [DOI] [PubMed] [Google Scholar]
- Kim D, Cha GS, Nagy LD, Yun CH, Guengerich FP. Kinetic analysis of lauric acid hydroxylation by human cytochrome P450 4A11. Biochemistry. 2014;53(39):6161–6172. doi: 10.1021/bi500710e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman JP. Importance of protein dynamics during enzymatic C-H bond cleavage catalysis. Biochemistry. 2013;52(12):2068–2077. doi: 10.1021/bi301504m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzekwa KR, Swinney DC, Trager WF. Isotopically labeled chlorobenzenes as probes for the mechanism of cytochrome P-450 catalyzed aromatic hydroxylation. Biochemistry. 1989;28:9019–9027. doi: 10.1021/bi00449a010. [DOI] [PubMed] [Google Scholar]
- Krauser JA, Guengerich FP. Cytochrome P450 3A4-catalyzed testosterone 6β-hydroxylation stereochemistry, kinetic deuterium isotope effects, and rate-limiting steps. The Journal of Biological Chemistry. 2005;280(20):19496–19506. doi: 10.1074/jbc.M501854200. [DOI] [PubMed] [Google Scholar]
- Matsson O, Westaway KC. Secondary deuterium kinetic isotope effects and transition state structure. Advances in Physical and Organic Chemistry. 1998;31:143–248. [Google Scholar]
- McMahon RE, Sullivan HR, Craig JC, Pereira WE., Jr The microsomal oxygenation of ethyl benzene: Isotopic, stereochemical, and induction studies. Archives of Biochemistry and Biophysics. 1969;132(2):575–577. doi: 10.1016/0003-9861(69)90401-9. [DOI] [PubMed] [Google Scholar]
- Melander L, Saunders WH. Reaction Rates of Isotopic Molecules. New York: John Wiley & Sons; 1980. [Google Scholar]
- Mitoma C, Dehn RL, Tanabe M. In vitro metabolic studies on propyl p-nitrophenyl ether. Biochim Biophys Acta. 1971;237(1):21–27. doi: 10.1016/0304-4165(71)90026-2. [DOI] [PubMed] [Google Scholar]
- Mitoma C, Yasuda DM, Tagg J, Tanabe M. Effect of deuteration of the O-CH3 group on the enzymic demethylation of o-nitroanisole. Biochim Biophys Acta. 1967;136(3):566–567. doi: 10.1016/0304-4165(67)90016-5. [DOI] [PubMed] [Google Scholar]
- Miwa GT, Lu AYH. Kinetic isotope effects and 'metabolic switching' in cytochrome P450-catalyzed reactions. BioEssays. 1987;7:215–219. doi: 10.1002/bies.950070506. [DOI] [PubMed] [Google Scholar]
- Miwa GT, Walsh JS, Kedderis GL, Hollenberg PF. The use of intramolecular isotope effects to distinguish between deprotonation and hydrogen atom abstraction mechanisms in cytochrome P-450- and peroxidase-catalyzed N-demethylation reactions. The Journal of Biological Chemistry. 1983;258:14445–14449. [PubMed] [Google Scholar]
- Miwa GT, Walsh JS, Lu AYH. Kinetic isotope effects on cytochrome P-450-catalyzed oxidation reactions: The oxidative O-dealkylation of 7-ethoxycoumarin. The Journal of Biological Chemistry. 1984;259:3000–3004. [PubMed] [Google Scholar]
- Nelson SD, Garland WA, Mitchell JR, Vaishnav Y, Statham CN, Buckpitt AR. Deuterium isotope effects on the metabolism and toxicity of phenacetin in hamsters. Drug Metabolism and Disposition. 1978;6:363–367. [PubMed] [Google Scholar]
- Nelson SD, Trager WF. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metabolism and Disposition. 2003;31(12):1481–1498. doi: 10.1124/dmd.31.12.1481. [DOI] [PubMed] [Google Scholar]
- Northrop DB. Steady-state analysis of kinetic isotope effects in enzymic reactions. Biochemistry. 1975;14:2644–2651. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- Northrop DB. Determining the absolute magnitude of hydrogen isotope effects. In: Cleland WW, O'Leary MH, Northrop DB, editors. Isotope Effects on Enzyme-Catalyzed Reactions (Proceedings of the Sixth Annual Harry Steenbock Symposium) Baltimore, London, and Tokyo: University Park Press; 1977. pp. 122–152. [Google Scholar]
- Northrop DB. Deuterium and tritium kinetic isotope effects on initial rates. Methods in Enzymology. 1982;87:607–625. doi: 10.1016/s0076-6879(82)87032-8. [DOI] [PubMed] [Google Scholar]
- Okazaki O, Guengerich FP. Evidence for specific base catalysis in N-dealkylation reactions catalyzed by cytochrome P450 and chloroperoxidase. Differences in rates of deprotonation of aminium radicals as an explanation for high kinetic hydrogen isotope effects observed with peroxidases. The Journal of Biological Chemistry. 1993;268(3):1546–1552. [PubMed] [Google Scholar]
- Omura T, Sato R. A new cytochrome in liver microsomes. The Journal of Biological Chemistry. 1962;237:1375–1376. [PubMed] [Google Scholar]
- Ortiz de Montellano PR. Oxygen activation and reactivity. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 2. New York: Plenum Press; 1995. pp. 245–303. [Google Scholar]
- Ortiz de Montellano PR. Substrate oxidation. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 4. New York: Springer; 2015. pp. 111–176. [Google Scholar]
- Ortiz de Montellano PR, DeVoss JJ. Substrate oxidation by cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. New York: Plenum Publishers; 2005. pp. 183–245. [Google Scholar]
- Pallan PS, Wang C, Lei L, Yoshimoto FK, Auchus RJ, Waterman MR, Guengerich FP, Egli M. Human cytochrome P450 21A2, the major steroid 21-hydroxylase: Structure of the enzyme-progesterone substrate complex and rate-limiting C-H bond cleavage. The Journal of Biological Chemistry. 2015;290(21):13128–13143. doi: 10.1074/jbc.M115.646307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perel JM, Dayton PG, Tauriello CL, Brand L, Mark LC. Metabolic studies with deuterated phenobarbital. Journal of Medicinal Chemistry. 1967;10(3):371–374. doi: 10.1021/jm00315a019. [DOI] [PubMed] [Google Scholar]
- Rendic S, Guengerich FP. Contributions of human enzymes in carcinogen metabolism. Chemical Research in Toxicology. 2012;25(7):1316–1383. doi: 10.1021/tx300132k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rendic S, Guengerich FP. Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chemical Research in Toxicology. 2015;28(1):38–42. doi: 10.1021/tx500444e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross D, Larsson R, Andersson B, Nilsson U, Lindquist T, Lindeke B, Moldéus P. The oxidation of p-phenetidine by horseradish peroxidase and prostaglandin synthase and the fate of glutathione during such oxidations. Biochemical Pharmacology. 1985;34:343–351. doi: 10.1016/0006-2952(85)90042-5. [DOI] [PubMed] [Google Scholar]
- Shao L, Abolin C, Hewitt MC, Koch P, Varney M. Derivatives of tramadol for increased duration of effect. Bioorganic & Medicinal Chemistry Letters. 2006;16(3):691–694. doi: 10.1016/j.bmcl.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Shinkyo R, Guengerich FP. Cytochrome P450 7A1 cholesterol 7 α-hydroxylation: Individual reaction steps in the catalytic cycle and rate-limiting ferric iron reduction. The Journal of Biological Chemistry. 2011;286(6):4632–4643. doi: 10.1074/jbc.M110.193409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann PF, Mace R, Angeles RM, Keefer LK. Deuterium isotope effect on metabolism of N-nitrosodimethylamine in vivo in rat. Carcinogenesis. 1983;4:821–825. doi: 10.1093/carcin/4.7.821. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Tagg J, Yasuda D, LeValley SE, Mitoma C. Pharmacologic and metabolic studies with deuterated zoxazolamine. Journal of Medicinal Chemistry. 1970;13(1):30–32. doi: 10.1021/jm00295a008. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Yasuda D, LeValley S, Mitoma C. The pharmacologic effect of deuterium substitution on 5-n-butyl-5-ethylbarbituric acid. Life Sciences. 1969;8(19):1123–1128. doi: 10.1016/0024-3205(69)90166-0. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Yasuda D, Tagg J, Mitoma C. Absence of isotope effects in the microsomal hydroxylation of acetanilide. Biochemical Pharmacology. 1967;16(11):2230–2233. doi: 10.1016/0006-2952(67)90024-x. [DOI] [PubMed] [Google Scholar]
- Tomaszewski JE, Jerina DM, Daly JW. Deuterium isotope effects during formation of phenols by hepatic monoxygenases. Evidence for an alternative to arene oxide pathway. Biochemistry. 1975;14(9):2024–2031. doi: 10.1021/bi00680a033. [DOI] [PubMed] [Google Scholar]
- Ullrich V. On the hydroxylation of cyclohexane in rat liver microsomes. Hoppe-Seyler's Zeitschrift fur Physiologische Chemie. 1969;350:357–365. doi: 10.1515/bchm2.1969.350.1.357. [DOI] [PubMed] [Google Scholar]
- Wade D, Yang CS, Metral CJ, Roman JM, Hrabie JA, Riggs CW, Anjo T, Keefer LK, Mico BA. Deuterium isotope effect on denitrosation and demethylation of N-nitrosodimethylamine by rat liver microsomes. Cancer Research. 1987;47:3373–3377. [PubMed] [Google Scholar]
- Walsh C. Enzymatic Reaction Mechanisms. San Francisco: W. H. Freeman Co; 1979. pp. 118–121. [Google Scholar]
- Wang C, Pallan PS, Zhang W, Lei L, Yoshimoto FK, Waterman MR, Egli M, Guengerich FP. Functional analysis of human cytochrome P450 variants involved in congenital adrenal hyperplasia. The Journal of Biological Chemistry. 2017:292. doi: 10.1074/jbc.M117.792465. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westheimer FH. The magnitude of the primary kinetic isotope effect for compounds of hydrogen and deuterium. Chemical Reviews. 1961;61(3):265–273. [Google Scholar]
- White RD, Gandolfi AJ, Bowden GT, Sipes IG. Deuterium isotope effect on the metabolism and toxicity of 1,2-dibromoethane. Toxicology and Applied Pharmacology. 1983;69:170–178. doi: 10.1016/0041-008x(83)90297-1. [DOI] [PubMed] [Google Scholar]
- White RE, Miller JP, Favreau LV, Bhattacharyya A. Stereochemical dynamics of aliphatic hydroxylation by cytochrome P-450. Journal of the American Chemical Society. 1986;108:6024–6031. doi: 10.1021/ja00279a059. [DOI] [PubMed] [Google Scholar]
- Wong LCK, Winston JM, Hong CB, Plotnick H. Carcinogenicity and toxicity of 1,2-dibromoethane in the rat. Toxicology and Applied Pharmacology. 1982;63:155–165. doi: 10.1016/0041-008x(82)90036-9. [DOI] [PubMed] [Google Scholar]
- Yarnell AT. Heavy-hydrogen drugs turn heads, again. Chemical & Engineering News. 2009;87(25):36–39. [Google Scholar]
- Yoshimoto FK, Guengerich FP. Mechanism of the third oxidative step in the conversion of androgens to estrogens by cytochrome P450 19A1 steroid aromatase. Journal of the American Chemical Society. 2014;136(42):15016–15025. doi: 10.1021/ja508185d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CH, Miller GP, Guengerich FP. Rate-determining steps in phenacetin oxidations by human cytochrome P450 1A2 and selected mutants. Biochemistry. 2000;39(37):11319–11329. doi: 10.1021/bi000869u. [DOI] [PubMed] [Google Scholar]